

Yersinia pestis causes a rapid, lethal disease referred to as plague. Y. pestis actively inhibits the innate immune system to generate a noninflammatory environment during early stages of infection to promote colonization. The ability of Y. pestis to create this early noninflammatory environment is in part due to the action of seven Yop effector proteins that are directly injected into host cells via a type 3 secretion system (T3SS).
KEYWORDS: Yersinia pestis, granules and degranulation, leukotriene, neutrophils, plague, type 3 secretion
ABSTRACT
Yersinia pestis causes a rapid, lethal disease referred to as plague. Y. pestis actively inhibits the innate immune system to generate a noninflammatory environment during early stages of infection to promote colonization. The ability of Y. pestis to create this early noninflammatory environment is in part due to the action of seven Yop effector proteins that are directly injected into host cells via a type 3 secretion system (T3SS). While each Yop effector interacts with specific host proteins to inhibit their function, several Yop effectors either target the same host protein or inhibit converging signaling pathways, leading to functional redundancy. Previous work established that Y. pestis uses the T3SS to inhibit neutrophil respiratory burst, phagocytosis, and release of inflammatory cytokines. Here, we show that Y. pestis also inhibits release of granules in a T3SS-dependent manner. Moreover, using a gain-of-function approach, we discovered previously hidden contributions of YpkA and YopJ to inhibition and that cooperative actions by multiple Yop effectors are required to effectively inhibit degranulation. Independent from degranulation, we also show that multiple Yop effectors can inhibit synthesis of leukotriene B4 (LTB4), a potent lipid mediator released by neutrophils early during infection to promote inflammation. Together, inhibition of these two arms of the neutrophil response likely contributes to the noninflammatory environment needed for Y. pestis colonization and proliferation.
INTRODUCTION
Plague is the human disease caused by infection with the bacterial pathogen Yersinia pestis (1). Depending upon the route of inoculation, plague can manifest in three forms (1). Primary bubonic, pneumonic, or septicemic plague arises when bacteria are inoculated into the skin, lungs, or bloodstream, respectively. Upon infection with Y. pestis, mean time to death without medical intervention can range from 3 days for primary pneumonic or septicemic plague to 7 days for bubonic plague. A hallmark of Y. pestis infection is the lack of inflammation during early stages of colonization. During pneumonic plague in mice, a minimal inflammatory response is observed for the first 24 to 36 h of infection (2–5). Beginning at ∼48 h postinfection, the inflammatory response to Y. pestis changes, resulting in a significant increase in inflammatory mediators, including monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor alpha (TNF-α), interleukin 12p70 (IL-12p70), gamma interferon (IFN-γ), and IL-6 (2, 5). This coincides with an influx of immune cells, especially neutrophils, into the lungs, resulting in a rapid pneumonia (2, 5). Similarly, inflammation is delayed in bubonic plague and does not occur until after Y. pestis has begun to proliferate in the draining lymph node and disseminate (6–8). The ability of Y. pestis to actively inhibit innate immune responses is a key virulence mechanism for Y. pestis (2, 3, 5, 6, 8, 9).
Normally, neutrophils are recruited in response to a variety of stimuli derived from damaged or activated host cells via damage-associated molecular patterns (DAMPs), cytokines, chemokines, or complement products (10–12). Microbial components, such as lipopolysaccharide, peptidoglycan, or N-formylmethionine-leucyl-phenylalanine peptides (fMLF), known as pathogen-associated molecular patterns (PAMPs), can also stimulate the recruitment of neutrophils (13). Upon stimulation, neutrophils traverse the vasculature to reach the site of infection. Upon arrival at the site of infection, neutrophil antimicrobial responses are multifactorial and are comprised of phagocytosis, induction of the respiratory burst, degranulation, and release of neutrophil extracellular traps (NETs) (13). Combined efforts from each of these responses make neutrophils very adept at killing microorganisms. Phagocytosis is important for clearing many bacterial infections, although some pathogens have acquired virulence factors that inhibit uptake by neutrophils (14, 15). In such situations, neutrophils rely upon extracellular release of antimicrobial mechanisms to effectively clear the infection.
One mechanism utilized by neutrophils to combat extracellular pathogens is the release of antimicrobial cargo contained in preformed granules (a process referred to as degranulation or graded exocytosis) (16). Degranulation occurs in a regulated manner to coordinate release or modification of cytokines, chemokines, and signaling ligands/receptors to facilitate neutrophil transmigration and chemotaxis with release of antimicrobial components that can directly restrict pathogen growth. Neutrophils contain four different granule subtypes, and mobilization of each granule is tightly controlled and dependent on the intensity of stimulation to coordinate functional responses (17). Neutrophil degranulation is hierarchical, with secretory vesicles being the first subtype to undergo exocytosis, followed by gelatinase granules. Degranulation of specific and azurophilic granules, both loaded with toxic antimicrobial cargo, is more limited and requires stronger stimulation to promote granule mobilization (13). Tightly graded control of granule release ensures that contents are released at the correct location to diminish collateral damage to the host.
The ability of neutrophils to mediate inflammatory responses has become more appreciated (18). Neutrophils release a variety of cytokines and chemokines, as well as other immune modulatory factors that contribute to the cellular communication network during inflammation (19, 20). One of the most potent modulators released by neutrophils is leukotriene B4 (LTB4). Not only is it important for recruitment of additional neutrophils to the site of infection (21–23), but LTB4 also enhances the antimicrobial responses of both neutrophils and macrophages, including phagocytosis, respiratory burst, degranulation, and the release of inflammatory cytokines (24–28). Importantly, LTB4 production is not dependent on transcriptional regulation (29). Therefore, LTB4 is produced more rapidly than other chemoattractants, such as IL-8, and it is pivotal in mounting a swift inflammatory response (26, 28, 30–32). Moreover, release of LTB4 is independent of degranulation (33), suggesting that regulation of LTB4 release also differs from degranulation.
Although neutrophils are extremely capable of restricting microbial colonization, Y. pestis encodes a variety of virulence factors to evade recognition and killing by neutrophils (34–38). The Ysc type 3 secretion system (T3SS) secretes seven Yersinia outer protein (Yop) effectors directly into host cells and is paramount for inhibition and evasion of neutrophil responses (4, 9, 34, 39–43). Moreover, several in vivo studies have demonstrated that neutrophils are the primary cell type that Y. pestis interacts with during early stages of infection (4, 36, 44). Once injected into neutrophils, Y. pestis Yop effectors interact with specific host factors to disrupt multiple host signaling pathways. YpkA, YopE, YopH, and YopT disrupt the actin cytoskeleton via interactions with host Rac, Rho, and focal adhesion complex proteins (41–43, 45–59). YopH has also been shown to inhibit host cell calcium flux (49, 60), while YopJ inhibits mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) cascades (40, 60–63). Together, these Yop effectors have been shown to effectively inhibit neutrophil phagocytosis, respiratory burst, and cytokine/chemokine release (34, 40, 61, 63). Importantly, the Yop translocon pore and effects of Yop effectors on host proteins can trigger inflammasome activation, which should lead to inflammatory responses (64–67). However, YopM and YopK function to inhibit inflammasome activation and subsequent inflammatory responses (64, 65, 67–72). Together, the Yop effectors allow Y. pestis to actively inhibit the inflammatory response.
Recently it was shown that Yersinia pseudotuberculosis inhibits neutrophil degranulation in a T3SS-dependent manner, which was dependent on the actions of YopE and YopH (73). Here, we show T3SS-dependent inhibition of neutrophil degranulation by Y. pestis, as well as roles of both YopE and YopH in inhibition. However, using a gain-of-function approach with a library of Y. pestis strains only expressing one Yop effector, we were able to identify additional Yop effectors contributing to inhibition of degranulation that have not been previously observed. Moreover, we show for the first time that Y. pestis actively inhibits production of LTB4 by human neutrophils, and we identify the Yop effectors contributing to this inhibition.
RESULTS
Y. pestis inhibits neutrophil degranulation in a T3SS-dependent manner.
Degranulation is a highly regulated but quick response that generally occurs within minutes after encountering a stimulus. Multiple studies have provided an understanding of the contents of the different granules that are released during degranulation (e.g., albumin is released during degranulation of secretory vesicles; gelatinase is released during degranulation of gelatinase granules), and the increased expression of receptors displayed on the neutrophil cell surface upon granule fusion with the plasma membrane (e.g., CD66b is displayed after degranulation of specific granules; CD63 is displayed after degranulation of azurophilic granules) (reviewed by Cowland and Borregaard [74]). Importantly, using these markers, degranulation of each granule subtype in response to different stimuli can be reliably monitored. Recently, it has been shown that Y. pseudotuberculosis inhibits degranulation by human neutrophils (73). To determine whether Y. pestis similarly inhibits degranulation, human neutrophils were infected with Y. pestis CO92 or with a strain lacking the pCD1 plasmid encoding the Ysc T3SS [Y. pestis CO92 T3(−)]. At a multiplicity of infection (MOI) of 10 or 100, minimal, if any, release of the four granule subtypes was observed in response to Y. pestis CO92 (Fig. 1). Similarly, at an MOI of 10, infection with Y. pestis CO92 T3(−) did not result in degranulation. However, at an MOI of 100, Y. pestis CO92 T3(−) caused significant release of all four granule subtypes compared to infection with Y. pestis CO92 (Fig. 1 and Fig. S1A and B). Infection with a Y. pestis KIM derivative with and without the pCD1 plasmid recapitulated the phenotypes observed for Y. pestis CO92 and CO92 T3(−), respectively. Together, these data indicate that degranulation is inhibited by Y. pestis in a T3SS-dependent manner.
FIG 1.
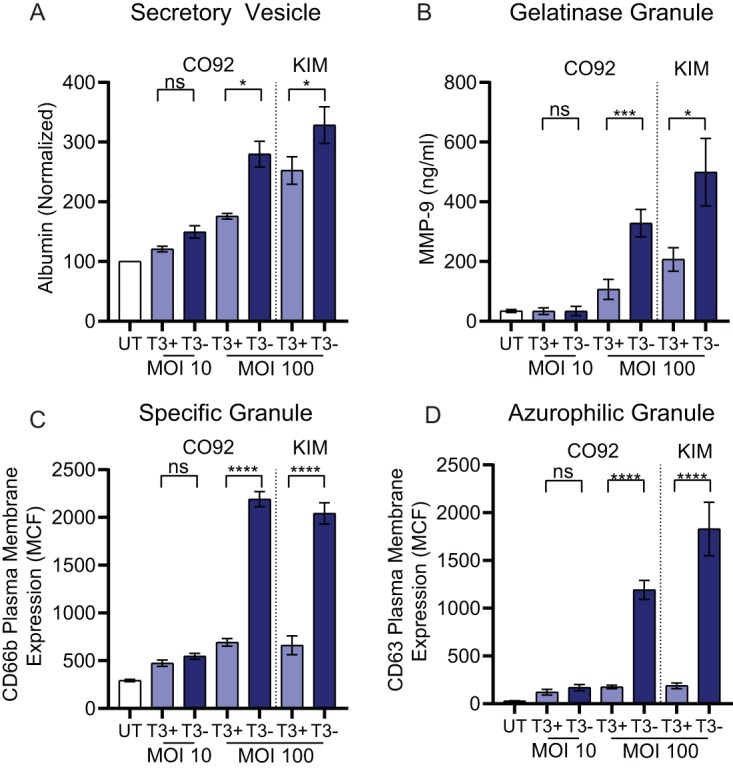
Y. pestis inhibits degranulation in a T3SS-dependent manner. Human neutrophils (4 × 106) were infected with Y. pestis CO92 or Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively) at indicated multiplicities of infection (MOIs) (10 or 100). Degranulation was measured after 30 min of infection for (A) secretory vesicles, (B) gelatinase, (C) specific, and (D) azurophilic granules. UT, untreated cells. Mean ± standard error of the mean (SEM) from 5 biologically independent experiments. One-way analysis of variance (ANOVA) with Sidak’s post hoc test; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. Representative flow cytometry histograms for specific and azurophilic granules are shown in Fig. S1.
Cooperative inhibition of neutrophil degranulation by Yop effectors revealed through gain-of-function approach.
The Ysc T3SS delivers seven effector proteins into targeted host cells (75). To determine if a single Yop effector is responsible for inhibiting neutrophil degranulation, human neutrophils were infected with a library of Y. pestis KIM1001 strains containing in-frame deletions of one yop gene (Table S1). While Y. pestis is able to inhibit release of all four granule subtypes (Fig. 1), specific and azurophilic granules contain most of the antimicrobial components produced by neutrophils, and are typically released at the site of infection, where neutrophils would come into direct contact with Y. pestis. We therefore focused on the ability of Y. pestis Yop effectors to inhibit release of these two granule subtypes. Moreover, comparing the expression of degranulation markers after incubation with Y. pestis T3(−) for 30 and 60 min indicated that degranulation peaked by 30 min postinfection (Fig. S1C and D). Therefore, degranulation was monitored at 30 min postinfection for subsequent experiments. As shown in Fig. 1, infection with the Y. pestis KIM1001 T3(−) strain resulted in significant release of both specific and azurophilic granules compared to infection with Y. pestis KIM1001 (Fig. 2). Mutants lacking any single yop gene retained the ability to inhibit release of either granule, with surface expression of degranulation markers similar to that observed for Y. pestis KIM1001. Similar results were observed with individual yop deletion mutants in the Y. pestis CO92 background (data not shown). These data suggest that more than one Yop effector protein is able to inhibit neutrophil granule release (i.e., functional redundancy in the system).
FIG 2.

Deletion of individual Yop effector proteins does not alter neutrophil degranulation response to Y. pestis infection. Human neutrophils (4 × 106) were infected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively) or with strains lacking ypkA (ΔA), yopE (ΔE), yopH (ΔH), yopJ (ΔJ), yopK (ΔK), yopM (ΔM), or yopT (ΔT); MOI = 100. Degranulation was measured after 30 min of infection for (A) specific or (B) azurophilic granules. Mean ± SEM from 5 biologically independent experiments. One-way ANOVA with Dunnett’s post hoc test to T3+; ****, P < 0.0001.
Recently, Palace et al. developed a library of Y. pestis strains that only express one Yop effector (62). This library allows for the study of individual Yop effectors without the presence of the other six, which could confound data interpretation due to phenotypical masking by functionally redundant proteins. To determine whether individual Yop effectors inhibit degranulation, neutrophils were infected with strains from this library and monitored for exocytosis of specific and azurophilic granules (Fig. 3). While strains expressing YopE, YopH, or YopT trended toward decreased specific granule exocytosis, none of the mutants demonstrated statistically significant decreases in exocytosis compared to the T3(−) strain (Fig. 3A). Similar trends were observed for exocytosis of azurophilic granules for strains expressing YopE and YopH, but surprisingly, the strain expressing only YopT caused increased release of azurophilic granules (Fig. 3B). These data indicate that while there is functional redundancy for inhibiting degranulation by neutrophils, the effector proteins also work in a cooperative manner during Y. pestis infection to effectively inhibit exocytosis of specific and azurophilic granules.
FIG 3.

Individual Yop effector proteins are unable to completely inhibit degranulation. Human neutrophils (4 × 106) were infected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively) or with strains expressing only ypkA (+A), yopE (+E), yopH (+H), yopJ (+J), yopK (+K), yopM (+M), or yopT (+T); MOI = 100. Degranulation was measured after 30 min of infection for (A) specific or (B) azurophilic granules. Mean ± SEM from 5 biologically independent experiments. One-way ANOVA with Dunnett’s post hoc test to T3−: **, P < 0.01; ****, P < 0.0001.
YopE, YopH, YopJ, and YpkA act cooperatively to inhibit degranulation of specific and azurophilic granules.
To determine which Yop effectors act cooperatively to inhibit degranulation, a coinfection approach with two strains of Y. pestis expressing different individual Yop effectors was employed. Neutrophils were infected with a 1:1 mixture of two Y. pestis strains, each expressing different Yop proteins (final MOI = 100; MOI of 50 for each strain). Exocytosis of specific and azurophilic granules was compared to that of cells infected with Y. pestis KIM1001 T3(−) or a 1:1 mixture of Y. pestis KIM1001 and Y. pestis KIM1001 T3(−). As expected, coinfections with Y. pestis KIM1001 expressing all of the Yop proteins significantly decreased exocytosis of both specific and azurophilic granules compared to infection with only Y. pestis KIM1001 T3(−) (Fig. 4 and 5). Coinfection with two strains expressing only one Yop protein revealed that cooperative actions by four effectors were sufficient to inhibit degranulation of both specific and azurophilic granules. Coinfection with strains expressing YopH and YopE, YopH and YpkA, YopH and YopJ, or YopE and YopJ was sufficient to inhibit degranulation of both granules to levels similar to coinfection with Y. pestis KIM1001 and Y. pestis KIM1001 T3(−) (Fig. 4 and 5). Coinfection with YopH and YopK appeared to also sufficiently inhibit release of azurophilic granules. For specific granules, coinfection with YopH and YopK, YopH and YopT, or YopE and YopT showed intermediate phenotypes. Coinfection with YopT could reverse the ability of YopH and YopE to partially inhibit degranulation of azurophilic granules (Fig. 5A and B), reflecting the enhanced degranulation previously observed in single YopT infection (Fig. 3B). However, coinfection with YopJ or YpkA appeared to inhibit the YopT enhanced degranulation phenotype (Fig. 5C and D). Together, these data confirm previously reported roles for YopH and YopE in inhibition of degranulation (73), and also revealed previously hidden contributions of YpkA, YopJ, and YopK.
FIG 4.

At least two Yop effector proteins are required to fully inhibit specific granule release. Human neutrophils (4 × 106) were coinfected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively) or with strains expressing only ypkA (+A), yopE (+E), yopH (+H), yopJ (+J), yopK (+K), yopM (+M), or yopT (+T) mixed at a 1:1 ratio with strains expressing only (A) yopH (+H), (B) yopE (+E), (C) yopJ (+J), or (D) ypkA (+A); MOI of each strain = 50 for a combined MOI of 100. Specific granule release was measured after 30 min of infection. Mean ± SEM from 5 biologically independent experiments. One-way ANOVA with Dunnett’s post hoc test. Gray bars are significantly different than T3− (P ≤ 0.05); purple bars are significantly different from T3−/T3+ (P ≤ 0.05); hatched bars are not significantly different than T3− or T3−/T3+.
FIG 5.

At least two Yop effector proteins are required to fully inhibit azurophilic granule release. Human neutrophils (4 × 106) were coinfected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively) or with strains expressing only ypkA (+A), yopE (+E), yopH (+H), yopJ (+J), yopK (+K), yopM (+M), or yopT (+T) mixed at a 1:1 ratio with strains expressing only (A) yopH (+H), (B) yopE (+E), (C) yopJ (+J), or (D) ypkA (+A); MOI of each strain = 50 for a combined MOI of 100. Azurophilic granule release was measured after 30 min of infection. Mean ± SEM from 5 biologically independent experiments. One-way ANOVA with Dunnett’s post hoc test. Gray bars are significantly different than T3− (P ≤ 0.05); purple bars are significantly different than T3−/T3+ (P ≤ 0.05); hatched bars are not significantly different than T3− or T3−/T3+.
Y. pestis inhibits LTB4 response of human neutrophils.
LTB4 is a potent chemoattractant released by neutrophils independent of degranulation, and it contributes to early inflammation in response to infection (22, 33). As inhibition of inflammation is a hallmark of Y. pestis infection, we next asked whether Y. pestis inhibits release of LTB4 by human neutrophils. Neutrophils were infected with Y. pestis KIM1001 or Y. pestis KIM1001 T3(−), and the level of LTB4 released into the supernatant was compared to that released by untreated neutrophils (Fig. 6A). Infection with Y. pestis KIM1001 did not result in significant release of LTB4 compared to untreated neutrophils. However, when neutrophils were infected with Y. pestis KIM1001 T3(−), a significant increase in LTB4 secretion was observed (Fig. 6A; P < 0.01). To determine if these differences in LTB4 levels were sufficient to alter chemotaxis of naive neutrophils, conditioned supernatants from infected neutrophils were used in a chemotaxis assay and compared to supernatant from untreated neutrophils (Fig. 6B). Naive neutrophils exposed to buffer or fMLF, a known chemoattractant, were used as controls. The numbers of naive neutrophils migrating toward the conditioned supernatant from untreated and Y. pestis KIM1001-infected neutrophils were not significantly different. However, in direct correlation with the elevated levels of LTB4 in the conditioned supernatant, significantly more neutrophils migrated toward the supernatant collected from cells infected with Y. pestis KIM1001 T3(−) (Fig. 6B; P < 0.01). Pretreatment of naive neutrophils with an inhibitor that blocks signaling through the LTB4 high-affinity receptor BLT1 eliminated chemotaxis toward the conditioned supernatant but not toward fMLF (Fig. 6C). These results indicate that the presence of LTB4 in the conditioned supernatant was promoting chemotaxis.
FIG 6.

Y. pestis inhibits human neutrophil LTB4 response. Human neutrophils (4 × 106) were infected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively); MOI = 100. (A) Release of LTB4 was measured after 30 min of infection in supernatant. (B and C) Chemotaxis of naive neutrophils in response to conditioned supernatant (B) without or (C) with pretreatment of the BLT1 inhibitor LY293111. (D) LTB4 concentrations in the supernatant or (E) cell lysates of neutrophils infected with strains expressing only ypkA (+A), yopE (+E), yopH (+H), yopJ (+J), yopK (+K), yopM (+M), or yopT (+T). Mean ± SEM from 5 biologically independent experiments. One-way ANOVA with Dunnett’s post hoc test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. (D and E) Gray bars are significantly different than T3− (P ≤ 0.05); purple bars are significantly different from T3−/T3+ (P ≤ 0.05); hatched bars are not significantly different from T3− or T3−/T3+.
Next, we used the library of Y. pestis mutants expressing only one Yop effector to ask whether individual effector proteins are sufficient to inhibit LTB4 release. In contrast to the data observed for inhibition of specific and azurophilic granule release, four of the seven Yop effectors (YpkA, YopE, YopH, and YopJ) were able to inhibit LTB4 release to levels similar to those of Y. pestis KIM1001 (Fig. 6D). Moreover, infection with the strain only expressing YopT also substantially decreased the amount of LTB4 released from the neutrophils, although to a lesser degree than the other four effectors. Finally, to determine if Y. pestis infection inhibits synthesis or release of LTB4, intracellular levels of LTB4 from infected neutrophils were measured. Similar to the results observed for conditioned supernatants, significantly lower amounts of intracellular LTB4 were detected in cells infected with Y. pestis KIM1001 and mutants expressing YpkA, YopE, YopH, YopJ, and YopT (Fig. 6E). Together, these data indicate that Y. pestis actively inhibits synthesis of LTB4 from human neutrophils in a T3SS-dependent manner, multiple Yop effectors are sufficient to inhibit LTB4 synthesis, and the inhibition of LTB4 release by infected neutrophils negatively impacts the chemotactic activity of naive neutrophils to respond to the infection.
Disruption of the host cytoskeleton inhibits LTB4 release in response to Y. pestis infection.
Although different mechanisms are used by YpkA, YopE, YopH, and YopT, all four proteins have been shown to affect actin cytoskeletal rearrangement in host cells (41, 46, 76–78). Because of this common effect, we hypothesized that Y. pestis disruption of the actin cytoskeleton could inhibit LTB4 release. If true, the release of LTB4 observed during infection with Y. pestis KIM1001 T3(−) could be blocked by artificially disrupting the actin cytoskeleton. To test this hypothesis, human neutrophils were incubated with latrunculin A, a chemical inhibitor of actin polymerization, prior to infection with Y. pestis, and LTB4 released into the supernatant was measured. As previously observed, significantly higher levels of LTB4 were secreted by neutrophils treated with the vehicle and infected with Y. pestis KIM1001 T3(−) than by vehicle-treated neutrophils infected with Y. pestis KIM1001 (Fig. 7A; P < 0.01). However, treatment with latrunculin A resulted in loss of LTB4 release in response to the strain lacking the T3SS, supporting that actin cytoskeleton disruption by Yop effectors can inhibit the LTB4 response in neutrophils.
FIG 7.

Inhibition of cytoskeletal rearrangement or MAPK signaling inhibits LTB4 release. Human neutrophils (4 × 106 for LatA treatment or 8 × 106 for Western blots) were infected with Y. pestis KIM1001 with or without the pCD1 plasmid encoding the T3SS (T3+ or T3−, respectively); MOI = 100. (A) Concentration of LTB4 in supernatant from infected neutrophils with indicated Y. pestis strains after pretreatment with vehicle control (LatA−) or latrunculin A (LatA+) prior to infection. (B) Concentration of LTB4 in culture supernatants after infection with indicated Y. pestis strains after pretreatment with vehicle control (untreated), the TAK1 inhibitor (5Z)-7-oxozeaenol [(5Z)-7-Oxo], or the ERK inhibitor U0126. (C) Phosphorylation of p38 and (D) ERK during infection with indicated strains after pretreatment with vehicle control (untreated), the TAK1 inhibitor (5Z)-7-oxozeaenol [(5Z)-7-Oxo], or the ERK inhibitor U0126. (E) Phosphorylation of ERK during infection with indicated Y. pestis strains. T3+, Y. pestis KIM1001; T3−, Y. pestis KIM1001 T3(−); +J, KIM1001 expressing only yopJ; UT, uninfected. (A and B) Mean ± SEM from 5 biologically independent experiments. (C, D, and E) Mean relative expression calculated from 3 biologically independent Western blots. One-way ANOVA with Sidak’s post hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Disruption of MAPK signaling inhibits LTB4 synthesis in response to Y. pestis infection.
YopJ does not directly impact the host cell cytoskeleton, but it is a potent inhibitor of MAPK signaling (79–82). Since MAPK signaling has been shown to control LTB4 synthesis in other models (79–86), we hypothesized that YopJ inhibition of LTB4 synthesis is mediated by disruption of MAPK signaling. In vitro data indicate that YopJ can interact with multiple kinases in this pathway, including MAP3K (e.g., the TGF-β activating kinase [TAK1]) and MAP2K (e.g., mitogen-activated kinase kinase 6 [MEK6]) (72, 79, 86–88). Because TAK1 represents the earliest point in MAPK signaling targeted by YopJ, we tested whether treatment of neutrophils with a TAK1 chemical inhibitor was sufficient to inhibit LTB4 synthesis in response to Y. pestis KIM1001 T3(−). As expected, when cells were exposed to the drug vehicle, we observed a significant increase in LTB4 release by neutrophils infected with Y. pestis KIM1001 T3(−) compared to that by neutrophils infected with Y. pestis KIM1001 (Fig. 7B; untreated, P ≤ 0.001). However, addition of the TAK1-specific inhibitor (5Z)-7-oxozeaenol [(5Z)-7-oxo] inhibited this response by neutrophils, and no difference in LTB4 concentration was observed in the supernatants of neutrophils infected with Y. pestis KIM1001 or Y. pestis KIM1001 T3(−) [Fig. 7B; (5Z)-7-oxo]. TAK1 signaling is upstream of the MAPKs ERK and p38, but has not been shown to activate JNK in neutrophils (87). To determine which MAPK was impacted by inhibition of TAK1 signaling, cell lysates from infected neutrophils were harvested, and the levels of phosphorylated p38 and ERK were measured by Western blotting. Compared to untreated neutrophils, we observed no difference in the phosphorylation of p38 during Y. pestis infection in the presence of the TAK1 inhibitor [Fig. 7C; untreated versus (5Z)-7-oxo]. However, while phosphorylation of ERK was significantly increased in untreated cells during infection with Y. pestis KIM1001 T3(−), chemical inhibition of TAK1 signaling resulted in decreased ERK phosphorylation [Fig. 7D; (5Z)-7-oxo], indicating that TAK1-mediated activation of LTB4 synthesis during Y. pestis KIM1001 T3(−) infection is through the ERK signaling pathway. To confirm that ERK signaling mediates LTB4 production in response to Y. pestis KIM1001 T3(−), neutrophils were treated with the ERK-specific inhibitor U0126 prior to Y. pestis infection. Similarly to treatment with the TAK1 inhibitor, blocking ERK signaling with U0126 inhibited the release of LTB4 in response to the Y. pestis KIM1001 T3(−) strain (Fig. 7B; U0126). Western blot analysis confirmed that U0126 specifically inhibited ERK phosphorylation and not p38 phosphorylation during Y. pestis KIM1001 T3(−) infection (Fig. 7C and D; U0126). Importantly, infection with Y. pestis KIM1001 expressing only YopJ recapitulated the inhibition of ERK phosphorylation observed during infection with Y. pestis expressing all of the Yop effectors (Fig. 7E), demonstrating that YopJ is sufficient to inhibit ERK signaling during Y. pestis infection. Together, these data indicate that inhibition of ERK signaling in neutrophils by YopJ is sufficient to inhibit LTB4 synthesis during Y. pestis infection.
DISCUSSION
Through the T3SS and other virulence factors, Y. pestis is able to actively evade and inhibit the mammalian innate immune response, which allows the bacterium to colonize the host (75, 89, 90). Previous work has demonstrated targeting of resident and arriving neutrophils by Y. pestis for T3SS injection, which inhibits neutrophil antibacterial mechanisms that would otherwise result in bacterial killing (4, 34, 36, 39, 44, 63, 91–93). Specifically, Y. pestis has been shown to inhibit phagocytosis (9, 34), reactive oxygen species production (9, 34, 40), and production of cytokines (63) by neutrophils. Our study further expands the understanding of how Y. pestis impairs the inflammatory response of host neutrophils by inhibition of neutrophil degranulation and LTB4 synthesis.
Work in the closely related species Y. pseudotuberculosis demonstrated that the T3SS actively inhibits neutrophil degranulation via the contributions of YopE and YopH (73). Our data, and a recent report from Eichelberger et al. (117), demonstrate that Y. pestis also utilizes these two effector proteins to inhibit neutrophil degranulation. However, by using a gain-of-function technique, we were also able to identify the contributions of YopJ and YpkA to the inhibition of specific and azurophilic granule exocytosis. Moreover, and importantly, we were able to show that multiple Yop effectors must act cooperatively to inhibit degranulation. The likely reasons YopJ and YpkA contributions were missed previously are because (i) four different protein combinations can inhibit degranulation of both specific and azurophilic granules, and (ii) while four proteins are involved, the bacterium requires either YopH or YopE (i.e., YopJ and YpkA cannot inhibit without YopE or YopH). Therefore, using a conventional loss-of-function deletion approach, a yopE yopH double mutant will have a phenotype, while any other double mutation combination will not, leading to the erroneous conclusion that YopE and YopH are redundant and sufficient to inhibit degranulation. These data also suggest the potential for hidden contributions of Yop effectors to other previously described phenotypes identified via loss-of-function mutational approaches. For example, while YopJ has been linked to inhibition of IL-8 by neutrophils, a yopJ mutant does not release as much IL-8 as a T3SS-deficient mutant, suggesting cooperative actions by other Yop effectors (63). Identification of other Yop effectors involved in inhibition could be performed using a similar gain-of-function approach to that described here.
Previous work has shown that YopE and YpkA target Rac signaling (54, 94, 95), YopH targets the focal adhesion complex (41), and YopJ targets the MAPK signaling pathway (59, 61, 79, 80, 82–84). All three of these host factors are key nodes in signaling pathways shown to be integral to regulating neutrophil granule release (96, 97). However, based on data from infections with single gain-of-function mutants, inhibition of one of these pathways by an individual Yop effector is not sufficient to inhibit degranulation. This suggests that individual signaling pathways may not be completely inhibited by the effector, or alternatively, that loss of signaling through one pathway can be compensated for in the neutrophil by signaling through the other pathways. While the latter hypothesis may be supported by our observation that YopE and YpkA, which both target the same node/pathway, are not able to inhibit degranulation, our data do not rule out the former, as some degree of signaling through this node may still occur during coinfection with the YopE and YpkA strains. To overcome this hurdle, Y. pestis evolved to inhibit all three signaling pathways, with inhibition of at least two being sufficient to inhibit degranulation (an example of cellular process redundancy [98]). Importantly, the signaling pathways affected by these nodes are also important for other neutrophil antimicrobial mechanisms (16, 99–101). Therefore, by targeting these host factors, Y. pestis is able to simultaneously inhibit multiple arms of the neutrophil response to subvert the functions of host neutrophils.
While the contributions of YopH, YopE, YpkA, and YopJ to inhibition of degranulation were conserved for specific and azurophilic granules, coinfections with YopH and YopK only appeared to inhibit the release of azurophilic granules. Based on the described function of YopK, which is thought to primarily regulate the translocation of other Yop effectors into the host cell to evade inflammasome recognition (69), we were surprised that YopK enhanced inhibition during coinfection with YopH. While YopK is thought to act as a gatekeeper, regulating the translocation of the other effectors from inside the cell (102), it has not been shown to regulate the transport of effectors through the T3SS of other bacteria during coinfection of a cell (i.e., transcomplementation). While it is possible that during coinfection YopK is transregulating the levels of YopH translocated by other bacteria, it is not clear how this would enhance inhibition of degranulation of azurophilic granules or why this would not also impact specific granules. Alternatively, it is possible that YopK has other yet-to-be defined functions in the host cells, beyond its role as a gatekeeper, that contribute to this phenotype, and future studies with YopK should be open to this possibility.
While YopE, YopH, YpkA, and YopT disrupt the host actin cytoskeleton, translocation of YopT by itself resulted in a phenotype that differed from the other three—enhanced azurophilic granule exocytosis (Fig. 5B). Johnson et al. described Gem-interacting protein (GMIP), through RhoA GAP activity, controling actin remodeling around the secretory Rab27a-JCF1 positive subpopulation of azurophilic granules to facilitate exocytosis (103). Inhibition of actin polymerization by regulation of RhoA and ROCK activity releases the barrier that limits granule exocytosis (97). Therefore, inactivation of RhoA by YopT is likely responsible for this phenotype. However, since this phenotype is specific for YopT, this suggests that YopT targeting of RhoA is spatially or temporally distinct from that of the other Yop effectors, that YopE and YpkA do not target RhoA during neutrophil infection, or that different mechanisms of RhoA inactivation by individual Yop effectors (e.g., protease cleavage versus GAP activity) may result in different degrees/rates of inactivation. Importantly, the action of the other Yop effectors inhibits this enhanced degranulation response in the context of wild-type (WT) Y. pestis infection to protect the bacterium from release of azurophilic granules.
Individually, YpkA, YopE, YopH, YopJ, and YopT all appear to be sufficient to inhibit LTB4 synthesis. Synthesis of LTB4 requires activation and relocalization of the enzyme 5-lipooxygenase (5-LO) to a membrane such as the nucleus or endoplasmic reticulum or to recently described cytosolic structures called lipidosomes (23, 104, 105). In this active state, 5-LO rapidly converts arachidonic acid to LTA4, which is followed by conversion to LTB4 by LTA4 hydrolase (23, 106). The mechanisms leading to 5-LO translocation are not well defined. Moreover, whether the rate-limiting step for initiation of LTB4 synthesis is relocalization to membranes or bringing 5-LO in proximity to 5-LO activating protein (FLAP) is still uncertain. However, 5-LO is known to associate with two actin-interacting proteins, growth factor receptor-bound protein 2 (Grb2) and coactosin-like protein (CLP) (107). These interactions suggest that 5-LO translocation or interactions with FLAP require the actin cytoskeleton. This is further supported by our data, as four out of the five effectors that inhibit LTB4 synthesis also disrupt the actin cytoskeleton. Moreover, treatment with the actin inhibitor latrunculin A also inhibited LTB4 synthesis in response to Y. pestis T3(−). However, it is possible that individual effectors may inhibit the synthesis process at different steps, and identifying which steps are inhibited during Y. pestis infection is a future direction of our studies.
In addition to disruption of the host cytoskeleton, we have shown that Y. pestis is able to inhibit LTB4 via YopJ disruption of ERK signaling. While YopJ inhibition of MAPK signaling has been extensively studied in the context of macrophages (79, 80), to our knowledge, this is the first time YopJ inhibition of MAPK phosphorylation has been confirmed in neutrophils. Specifically, our data demonstrates that both ERK and p38 phosphorylation are inhibited during Y. pestis infection of neutrophils in a T3SS-dependent manner and that inhibition of TAK1-ERK signaling axis by YopJ is sufficient to inhibit LTB4 synthesis (Fig. 7). In primary human neutrophils, TAK1 can differentially signal through ERK and p38, and phosphorylation of these MAPKs is dependent on the stimulus encountered by the neutrophils (87). For example, stimulation of neutrophils with lipopolysaccharide (LPS) results in TAK1-mediated phosphorylation of both ERK and p38, while stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) results in TAK1-mediated regulation of the MEK/ERK axis (90). Importantly, signaling via the TAK1-ERK pathway has also been shown to mediate LTB4 synthesis by neutrophils in response to other chemoattractant factors (90), supporting our findings that targeting ERK signaling by YopJ contributes to inhibition of LTB4 synthesis during Y. pestis infection. Importantly, MAPK signaling not only regulates LTB4 synthesis in neutrophils but also induction of the respiratory burst, production of cytokines, and degranulation (87, 108–112). Therefore, targeting of MAPK signaling by YopJ and inhibition of TAK1-ERK-mediated signaling allows Y. pestis to disrupt many arms of the neutrophil response simultaneously.
In conclusion, Y. pestis is well adapted to surviving within the hostile host environment. Through this work, we found that neutrophils can only undergo granule exocytosis in response to Y. pestis infection when the T3SS is absent. In addition, the data presented here support previously described roles for YopE and YopH in inhibition of degranulation (73), while uncovering previously unidentified roles for YopJ and YpkA, which cooperatively work with YopE and YopH. Given these new data, we can also update the current model for inhibition of degranulation by Y. pestis to include the information that the bacterium needs to inhibit two of three signaling pathways to completely inhibit neutrophil degranulation. Moreover, Y. pestis also inhibits the synthesis of the potent chemoattractant LTB4. Without LTB4, neutrophil recruitment to the site of infection would be impaired. Moreover, as LTB4 also stimulates macrophages toward enhanced phagosomal degradation of microorganisms (28) and promotes dendritic cell activation of T-cell responses (113–115), both of these important mechanisms to coordinate early antimicrobial responses by host innate immune cells are likely impaired during Y. pestis infection. Inhibition of neutrophil degranulation and LTB4 production likely contributes to Y. pestis subverting the innate immune response and maintaining a noninflammatory host environment early during infection.
MATERIALS AND METHODS
Bacterial growth conditions.
Bacterial strains used in these studies are listed in Table S1. Prior to infection, Y. pestis was cultured for 15 to 18 h at 26°C in Difco brain heart infusion (BHI) broth (BD Biosciences) with aeration. Cultures were diluted 1:10 in fresh BHI broth containing 20 mM MgCl2 and 20 mM Na-oxalate and cultured at 37°C for 2 h with aeration to induce expression of the T3SS. Bacteria were centrifuged and resuspended in LPS-free Krebs-Ringer phosphate buffer (pH 7.2) containing 0.2% dextrose (Krebs) buffer for infection.
Human neutrophil isolation.
Use of human neutrophils was approved by the University of Louisville’s Institutional Review Board (IRB) guidelines (approval no. 96.0191). Neutrophils were isolated from peripheral blood of healthy, medication-free donors as described previously (10). Neutrophil isolations yielded ≥95% purity with ≥97% viability by Trypan blue exclusion staining and were used within 1 h of isolation.
Human neutrophil infection.
Neutrophils (4 × 106; for Western blotting, 8 × 106 cells were used) were resuspended in Krebs buffer and, where indicated, incubated at room temperature (RT) for 30 min with 1 μM latrunculin A (catalog [cat.] no. 428021; Sigma), 20 μM U0126 (cat. no. 70970; Cayman), 50 nM LY293111 (cat. no. 10009768; Cayman), or 3 μM (5Z)-7-oxozeaenol (cat. no. 17459; Cayman). Neutrophils were infected at a multiplicity of infection (MOI) of 10 or 100 and incubated for 30 min in a 37°C water bath with gentle agitation. Coinfections were performed at a final MOI of 100 (50 for each strain), and bacteria were mixed together prior to adding to the cells. For specific and azurophilic granule exocytosis, the increases in plasma membrane expression of CD66b and CD63, respectively, were measured by flow cytometry as previously described (16). For CD63 plasma membrane expression, human albumin (cat. no. 521302; Alpha Therapeutic Corporation) was added to the cells (final concentration, 250 μg/ml) and incubated for an additional 5 min prior to staining. To measure release of gelatinase granules, secretory vesicles, or LTB4, separate samples were centrifuged, and cell-free supernatants were transferred to new tubes containing Halt phosphatase and protease inhibitor cocktail (cat. no. 78442; ThermoFisher Scientific) and stored at −80°C. Intracellular LTB4 was measured by lysing neutrophils in ultrapure H2O with phosphatase and protease inhibitors.
Measurement of exocytosis by flow cytometry and enzyme-linked immunosorbent assay.
Neutrophils were stained with fluorescein isothiocyanate (FITC)-labeled anti-CD63 (cat. no. 215-040; Ancell) or FITC-labeled anti-CD66b (cat. no. 305104; BioLegend) as markers for azurophilic and specific granules, respectively. As antibody isotype controls, neutrophils were separately labeled with FITC-labeled anti-IgM (cat. no. 401108; BioLegend) or FITC-labeled anti-IgG1 (cat. no. 400108; BioLegend) on ice for 45 min before washing with FTA buffer (BD Biosciences) plus 0.05% sodium azide and fixing with 1% paraformaldehyde (PFA). Mean cellular fluorescence intensity (MCF) was measured using a fluorescence-activated cell sorting (FACS) Aria flow cytometer (BD Biosciences) with isotype control values subtracted as previously described (73). Albumin (cat. no. ab108788; abcam), MMP-9/gelatinase (cat. no. EK0465; Boster), and LTB4 (cat. no. 520111; Cayman) levels were measured by enzyme-linked immunosorbent assay (ELISA) following the manufacturer’s protocols.
Chemotaxis assay.
Supernatants from infected neutrophils were filtered using a 0.2 μm syringe filter to generate conditioned supernatants. Naive neutrophils (1 × 106 cells/ml) were loaded into the upper chamber of a 24-well Transwell plate (Corning). The lower chambers were filled with Krebs buffer, 100 nM fMLF (Sigma), or the conditioned supernatants. After incubation for 30 min at 37°C, neutrophils that migrated from the upper chamber to the lower side of the Transwell membranes were fixed and stained with Hema 3 (ThermoFisher) and counted by microscopy as described previously (116).
Western blotting.
After 30 min of infection, cell pellets were obtained by centrifugation (6,000 × g for 30 s). Pellets were lysed using ice-cold lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Triton X-100, 0.5% [vol/vol] Nonidet P-40, 20 mM NaF, 20 mM NaVO3, 1 mM EDTA, 1 mM EGTA, 5 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM diisopropylfluorophosphate [DFP], 21 μg/ml aprotinin, and 5 μg/ml leupeptin). Lysates were mixed with Laemmli loading buffer and boiled for 10 min prior to snap cooling. Lysates were run on a 10% SDS-PAGE gel and immunoblotted with antibodies to phospho-ERK1/2, total ERK1/2, phospho-p38 MAPK, or total p38 MAPK (Cell Signaling) diluted 1:2,000 in 10 ml of Tris-buffered saline plus 0.1% Tween 20 (TBST) plus 5% bovine serum albumin (BSA). The appropriate secondary antibodies were used at 1:50,000 (cat. no. A9169; Sigma-Aldrich; cat. no. 31430; ThermoFisher Scientific). SuperSignal West Femto maximum-sensitivity substrate (cat. no. 34095; ThermoFisher Scientific) was used to detect antigen-antibody binding. Densitometry was performed using ImageJ software to quantify bands, normalized using the total protein form.
Statistics.
Degranulation and LTB4 data are the mean of five biologically independent experiments. Phosphorylation data are the mean of three biologically independent experiments. For all, neutrophils were harvested from both male and female donors and infections were performed on different days. Where appropriate, one-way analysis of variance (ANOVA) with Dunnett’s or Sidak’s post-test, as indicated in individual figure legends, was used for statistical analysis and performed using Prism 8 (GraphPad).
Supplementary Material
ACKNOWLEDGMENTS
We thank Terri Manning, Irina Miralda, and Keith C. Klaes for isolation of human neutrophils and for helpful discussions.
This work was supported by NIH grants GM125504 and AI119557 (M.B.L.), by a Collaborative Research Grant from the University of Louisville’s Center for Predictive Medicine for Biodefense and Emerging Infectious Disease (M.B.L. and S.M.U.), and in part by a grant from the Jewish Heritage Fund for Excellence Research Enhancement Grant Program at the University of Louisville School of Medicine (M.B.L.).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. doi: 10.1128/CMR.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price PA, Jin J, Goldman WE. 2012. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci U S A 109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. 2013. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog 9:e1003679. doi: 10.1371/journal.ppat.1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bubeck SS, Cantwell AM, Dube PH. 2007. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun 75:697–705. doi: 10.1128/IAI.00403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comer JE, Sturdevant DE, Carmody AB, Virtaneva K, Gardner D, Long D, Rosenke R, Porcella SF, Hinnebusch BJ. 2010. Transcriptomic and innate immune responses to Yersinia pestis in the lymph node during bubonic plague. Infect Immun 78:5086–5098. doi: 10.1128/IAI.00256-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebbane F, Gardner D, Long D, Gowen BB, Hinnebusch BJ. 2005. Kinetics of disease progression and host response in a rat model of bubonic plague. Am J Pathol 166:1427–1439. doi: 10.1016/S0002-9440(10)62360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosio CF, Jarrett CO, Gardner D, Hinnebusch BJ. 2012. Kinetics of innate immune response to Yersinia pestis after intradermal infection in a mouse model. Infect Immun 80:4034–4045. doi: 10.1128/IAI.00606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudte SC, Hinnebusch BJ, Shannon JG. 2017. Characterization of Yersinia pestis interactions with human neutrophils in vitro. Front Cell Infect Microbiol 7:358. doi: 10.3389/fcimb.2017.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB Jr, Henson PM. 1985. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol 119:101–110. [PMC free article] [PubMed] [Google Scholar]
- 11.Mocsai A, Walzog B, Lowell CA. 2015. Intracellular signalling during neutrophil recruitment. Cardiovasc Res 107:373–385. doi: 10.1093/cvr/cvv159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kienle K, Lämmermann T. 2016. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev 273:76–93. doi: 10.1111/imr.12458. [DOI] [PubMed] [Google Scholar]
- 13.Mayadas TN, Cullere X, Lowell CA. 2014. The multifaceted functions of neutrophils. Annu Rev Pathol 9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nauseef WM. 2007. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi SD, Malachowa N, DeLeo FR. 2018. Neutrophils and bacterial immune evasion. J Innate Immun 10:432–441. doi: 10.1159/000487756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uriarte SM, Rane MJ, Luerman GC, Barati MT, Ward RA, Nauseef WM, McLeish KR. 2011. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol 187:391–400. doi: 10.4049/jimmunol.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin C, Heit B. 2018. Armed for destruction: formation, function and trafficking of neutrophil granules. Cell Tissue Res 371:455–471. doi: 10.1007/s00441-017-2731-8. [DOI] [PubMed] [Google Scholar]
- 18.Scapini P, Cassatella MA. 2014. Social networking of human neutrophils within the immune system. Blood 124:710–719. doi: 10.1182/blood-2014-03-453217. [DOI] [PubMed] [Google Scholar]
- 19.Tecchio C, Cassatella MA. 2016. Neutrophil-derived chemokines on the road to immunity. Semin Immunol 28:119–128. doi: 10.1016/j.smim.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamassia N, Bianchetto-Aguilera F, Arruda-Silva F, Gardiman E, Gasperini S, Calzetti F, Cassatella MA. 2018. Cytokine production by human neutrophils: revisiting the dark side of the moon. Eur J Clin Invest 48(Suppl 2):e12952. doi: 10.1111/eci.12952. [DOI] [PubMed] [Google Scholar]
- 21.el-Ahmady O, Mansour M, Zoeir H, Mansour O. 1997. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Ann Clin Biochem 34:160–164. doi: 10.1177/000456329703400205. [DOI] [PubMed] [Google Scholar]
- 22.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. 2005. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol 174:589–594. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 23.Radmark O, Samuelsson B. 2010. Regulation of the activity of 5-lipoxygenase, a key enzyme in leukotriene biosynthesis. Biochem Biophys Res Commun 396:105–110. doi: 10.1016/j.bbrc.2010.02.173. [DOI] [PubMed] [Google Scholar]
- 24.Mancuso P, Nana-Sinkam P, Peters-Golden M. 2001. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect Immun 69:2011–2016. doi: 10.1128/IAI.69.4.2011-2016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nam YH, Min A, Kim SH, Lee YA, Kim KA, Song KJ, Shin MH. 2012. Leukotriene B(4) receptors BLT1 and BLT2 are involved in interleukin-8 production in human neutrophils induced by Trichomonas vaginalis-derived secretory products. Inflamm Res 61:97–102. doi: 10.1007/s00011-011-0425-3. [DOI] [PubMed] [Google Scholar]
- 26.Tavares NM, Araujo-Santos T, Afonso L, Nogueira PM, Lopes UG, Soares RP, Bozza PT, Bandeira-Melo C, Borges VM, Brodskyn C. 2014. Understanding the mechanisms controlling Leishmania amazonensis infection in vitro: the role of LTB4 derived from human neutrophils. J Infect Dis 210:656–666. doi: 10.1093/infdis/jiu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. 1996. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol 157:5221–5224. [PubMed] [Google Scholar]
- 28.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. 2005. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood 106:1067–1075. doi: 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam B, Gagnon L, Austen K, Soberman R. 1990. The mechanism of leukotriene B4 export from human polymorphonuclear leukocytes. J Biol Chem 265:13438–13441. [PubMed] [Google Scholar]
- 30.Prado MKB, Locachevic GA, Zoccal KF, Paula-Silva FWG, Fontanari C, Ferreira JC, Pereira PAT, Gardinassi LG, Ramos SG, Sorgi CA, Darini ALC, Faccioli LH. 2017. Leukotriene B4 is essential for lung host defence and alpha-defensin-1 production during Achromobacter xylosoxidans infection. Sci Rep 7:17658. doi: 10.1038/s41598-017-17993-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandt SL, Klopfenstein N, Wang S, Winfree S, McCarthy BP, Territo PR, Miller L, Serezani CH. 2018. Macrophage-derived LTB4 promotes abscess formation and clearance of Staphylococcus aureus skin infection in mice. PLoS Pathog 14:e1007244. doi: 10.1371/journal.ppat.1007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Min A, Lee YA, Kim KA, Shin MH. 2018. BLT1-mediated O-GlcNAcylation is required for NOX2-dependent migration, exocytotic degranulation and IL-8 release of human mast cell induced by Trichomonas vaginalis-secreted LTB4. Microbes Infect 20:376–384. doi: 10.1016/j.micinf.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Majumdar R, Tavakoli Tameh A, Parent CA. 2016. Exosomes mediate LTB4 release during neutrophil chemotaxis. PLoS Biol 14:e1002336. doi: 10.1371/journal.pbio.1002336. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Spinner JL, Cundiff JA, Kobayashi SD. 2008. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect Immun 76:3754–3760. doi: 10.1128/IAI.00385-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinnebusch BJ, Jarrett CO, Callison JA, Gardner D, Buchanan SK, Plano GV. 2011. Role of the Yersinia pestis Ail protein in preventing a protective polymorphonuclear leukocyte response during bubonic plague. Infect Immun 79:4984–4989. doi: 10.1128/IAI.05307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merritt PM, Nero T, Bohman L, Felek S, Krukonis ES, Marketon MM. 2015. Yersinia pestis targets neutrophils via complement receptor 3. Cell Microbiol 17:666–687. doi: 10.1111/cmi.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spinner JL, Carmody AB, Jarrett CO, Hinnebusch BJ. 2013. Role of Yersinia pestis toxin complex family proteins in resistance to phagocytosis by polymorphonuclear leukocytes. Infect Immun 81:4041–4052. doi: 10.1128/IAI.00648-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol 7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 39.Shannon JG, Hasenkrug AM, Dorward DW, Nair V, Carmody AB, Hinnebusch BJ. 2013. Yersinia pestis subverts the dermal neutrophil response in a mouse model of bubonic plague. mBio 4:e00170-13. doi: 10.1128/mBio.00170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spinner JL, Seo KS, O’Loughlin JL, Cundiff JA, Minnich SA, Bohach GA, Kobayashi SD. 2010. Neutrophils are resistant to Yersinia YopJ/P-induced apoptosis and are protected from ROS-mediated cell death by the type III secretion system. PLoS One 5:e9279. doi: 10.1371/journal.pone.0009279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rolan HG, Durand EA, Mecsas J. 2013. Identifying Yersinia YopH-targeted signal transduction pathways that impair neutrophil responses during in vivo murine infection. Cell Host Microbe 14:306–317. doi: 10.1016/j.chom.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Songsungthong W, Higgins MC, Rolan HG, Murphy JL, Mecsas J. 2010. ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol 12:988–1001. doi: 10.1111/j.1462-5822.2010.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiley DJ, Nordfeldth R, Rosenzweig J, DaFonseca CJ, Gustin R, Wolf-Watz H, Schesser K. 2006. The Ser/Thr kinase activity of the Yersinia protein kinase A (YpkA) is necessary for full virulence in the mouse, mollifying phagocytes, and disrupting the eukaryotic cytoskeleton. Microb Pathog 40:234–243. doi: 10.1016/j.micpath.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. 2005. Plague bacteria target immune cells during infection. Science 309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groves E, Rittinger K, Amstutz M, Berry S, Holden DW, Cornelis GR, Caron E. 2010. Sequestering of Rac by the Yersinia effector YopO blocks Fcγ receptor-mediated phagocytosis. J Biol Chem 285:4087–4098. doi: 10.1074/jbc.M109.071035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee WL, Grimes JM, Robinson RC. 2015. Yersinia effector YopO uses actin as bait to phosphorylate proteins that regulate actin polymerization. Nat Struct Mol Biol 22:248–255. doi: 10.1038/nsmb.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singaravelu P, Lee WL, Wee S, Ghoshdastider U, Ding K, Gunaratne J, Grimes JM, Swaminathan K, Robinson RC. 2017. Yersinia effector protein (YopO)-mediated phosphorylation of host gelsolin causes calcium-independent activation leading to disruption of actin dynamics. J Biol Chem 292:8092–8100. doi: 10.1074/jbc.M116.757971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viboud GI, Mejia E, Bliska JB. 2006. Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell Microbiol 8:1504–1515. doi: 10.1111/j.1462-5822.2006.00729.x. [DOI] [PubMed] [Google Scholar]
- 49.Andersson K, Magnusson KE, Majeed M, Stendahl O, Fallman M. 1999. Yersinia pseudotuberculosis-induced calcium signaling in neutrophils is blocked by the virulence effector YopH. Infect Immun 67:2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Black DS, Montagna LG, Zitsmann S, Bliska JB. 1998. Identification of an amino-terminal substrate-binding domain in the Yersinia tyrosine phosphatase that is required for efficient recognition of focal adhesion targets. Mol Microbiol 29:1263–1274. doi: 10.1046/j.1365-2958.1998.01014.x. [DOI] [PubMed] [Google Scholar]
- 51.Cantwell AM, Bubeck SS, Dube PH. 2010. YopH inhibits early pro-inflammatory cytokine responses during plague pneumonia. BMC Immunol 11:29. doi: 10.1186/1471-2172-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dave MN, Silva JE, Elicabe RJ, Jerez MB, Filippa VP, Gorlino CV, Autenrieth S, Autenrieth IB, Di Genaro MS. 2016. Yersinia enterocolitica YopH-deficient strain activates neutrophil recruitment to Peyer’s patches promoting clearance of the virulent strain. Infect Immun 84:3172–3181. doi: 10.1128/IAI.00568-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montagna LG, Ivanov MI, Bliska JB. 2001. Identification of residues in the N-terminal domain of the Yersinia tyrosine phosphatase that are critical for substrate recognition. J Biol Chem 276:5005–5011. doi: 10.1074/jbc.M009045200. [DOI] [PubMed] [Google Scholar]
- 54.Barz C, Abahji TN, Trulzsch K, Heesemann J. 2000. The Yersinia Ser/Thr protein kinase YpkA/YopO directly interacts with the small GTPases RhoA and Rac-1. FEBS Lett 482:139–143. doi: 10.1016/s0014-5793(00)02045-7. [DOI] [PubMed] [Google Scholar]
- 55.Dukuzumuremyi JM, Rosqvist R, Hallberg B, Akerstrom B, Wolf-Watz H, Schesser K. 2000. The Yersinia protein kinase A is a host factor inducible RhoA/Rac-binding virulence factor. J Biol Chem 275:35281–35290. doi: 10.1074/jbc.M003009200. [DOI] [PubMed] [Google Scholar]
- 56.Iriarte M, Cornelis GR. 1998. YopT, a new Yersinia Yop effector protein, affects the cytoskeleton of host cells. Mol Microbiol 29:915–929. doi: 10.1046/j.1365-2958.1998.00992.x. [DOI] [PubMed] [Google Scholar]
- 57.Ke Y, Tan Y, Wei N, Yang F, Yang H, Cao S, Wang X, Wang J, Han Y, Bi Y, Cui Y, Yan Y, Song Y, Yang X, Du Z, Yang R. 2015. Yersinia protein kinase A phosphorylates vasodilator-stimulated phosphoprotein to modify the host cytoskeleton. Cell Microbiol 17:473–485. doi: 10.1111/cmi.12378. [DOI] [PubMed] [Google Scholar]
- 58.Navarro L, Koller A, Nordfelth R, Wolf-Watz H, Taylor S, Dixon JE. 2007. Identification of a molecular target for the Yersinia protein kinase A. Mol Cell 26:465–477. doi: 10.1016/j.molcel.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 59.Wiley DJ, Shrestha N, Yang J, Atis N, Dayton K, Schesser K. 2009. The activities of the Yersinia protein kinase A (YpkA) and outer protein J (YopJ) virulence factors converge on an eIF2α kinase. J Biol Chem 284:24744–24753. doi: 10.1074/jbc.M109.010140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mecsas J. 2019. Unraveling neutrophil-Yersinia interactions during tissue infection. F1000Res 8:1046. doi: 10.12688/f1000research.18940.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lemaitre N, Sebbane F, Long D, Hinnebusch BJ. 2006. Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not required for virulence in a rat model of bubonic plague. Infect Immun 74:5126–5131. doi: 10.1128/IAI.00219-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palace SG, Proulx MK, Szabady RL, Goguen JD. 2018. Gain of function analysis reveals important virulence roles for the Yersinia pestis type III secretion system effectors YopJ, YopT, and YpkA. Infect Immun 86:e00318-18. doi: 10.1128/IAI.00318-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spinner JL, Hasenkrug AM, Shannon JG, Kobayashi SD, Hinnebusch BJ. 2016. Role of the Yersinia YopJ protein in suppressing interleukin-8 secretion by human polymorphonuclear leukocytes. Microbes Infect 18:21–29. doi: 10.1016/j.micinf.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng Y, Lilo S, Brodsky IE, Zhang Y, Medzhitov R, Marcu KB, Bliska JB. 2011. A Yersinia effector with enhanced inhibitory activity on the NF-κB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog 7:e1002026. doi: 10.1371/journal.ppat.1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, Chae JJ, Bliska JB. 2016. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20:296–306. doi: 10.1016/j.chom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S. 2013. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9:e1003400. doi: 10.1371/journal.ppat.1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, Shin S, Wei L, Parker M, Zhang J, Oberst A, Green DR, Brodsky IE. 2014. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc Natl Acad Sci U S A 111:7385–7390. doi: 10.1073/pnas.1403252111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. 2016. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog 12:e1006035. doi: 10.1371/journal.ppat.1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peters KN, Anderson DM. 2012. Modulation of host cell death pathways by Yersinia species and the type III effector YopK. Adv Exp Med Biol 954:229–236. doi: 10.1007/978-1-4614-3561-7_29. [DOI] [PubMed] [Google Scholar]
- 70.Philip NH, Zwack EE, Brodsky IE. 2016. Activation and evasion of inflammasomes by Yersinia. Curr Top Microbiol Immunol 397:69–90. doi: 10.1007/978-3-319-41171-2_4. [DOI] [PubMed] [Google Scholar]
- 71.Rosadini CV, Zanoni I, Odendall C, Green ER, Paczosa MK, Philip NH, Brodsky IE, Mecsas J, Kagan JC. 2015. A single bacterial immune evasion strategy dismantles both MyD88 and TRIF signaling pathways downstream of TLR4. Cell Host Microbe 18:682–693. doi: 10.1016/j.chom.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E. 2018. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taheri N, Fahlgren A, Fallman M. 2016. Yersinia pseudotuberculosis blocks neutrophil degranulation. Infect Immun 84:3369–3378. doi: 10.1128/IAI.00760-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cowland JB, Borregaard N. 2016. Granulopoiesis and granules of human neutrophils. Immunol Rev 273:11–28. doi: 10.1111/imr.12440. [DOI] [PubMed] [Google Scholar]
- 75.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 76.Wei C, Wang Y, Du Z, Guan K, Cao Y, Yang H, Zhou P, Wu F, Chen J, Wang P, Zheng Z, Zhang P, Zhang Y, Ma S, Yang R, Zhong H, He X. 2016. The Yersinia type III secretion effector YopM is an E3 ubiquitin ligase that induced necrotic cell death by targeting NLRP3. Cell Death Dis 7:e2519. doi: 10.1038/cddis.2016.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng LW, Anderson DM, Schneewind O. 1997. Two independent type III secretion mechanisms for YopE in Yersinia enterocolitica. Mol Microbiol 24:757–765. doi: 10.1046/j.1365-2958.1997.3831750.x. [DOI] [PubMed] [Google Scholar]
- 78.Fahlgren A, Westermark L, Akopyan K, Fallman M. 2009. Cell type-specific effects of Yersinia pseudotuberculosis virulence effectors. Cell Microbiol 11:1750–1767. doi: 10.1111/j.1462-5822.2009.01365.x. [DOI] [PubMed] [Google Scholar]
- 79.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 80.Mukherjee S, Orth K. 2008. In vitro signaling by MAPK and NFκB pathways inhibited by Yersinia YopJ. Methods Enzymol 438:343–353. doi: 10.1016/S0076-6879(07)38024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, Dixon JE. 1999. Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science 285:1920–1923. doi: 10.1126/science.285.5435.1920. [DOI] [PubMed] [Google Scholar]
- 82.Sweet CR, Conlon J, Golenbock DT, Goguen J, Silverman N. 2007. YopJ targets TRAF proteins to inhibit TLR-mediated NF-κB, MAPK and IRF3 signal transduction. Cell Microbiol 9:2700–2715. doi: 10.1111/j.1462-5822.2007.00990.x. [DOI] [PubMed] [Google Scholar]
- 83.Meijer LK, Schesser K, Wolf-Watz H, Sassone-Corsi P, Pettersson S. 2000. The bacterial protein YopJ abrogates multiple signal transduction pathways that converge on the transcription factor CREB. Cell Microbiol 2:231–238. doi: 10.1046/j.1462-5822.2000.00049.x. [DOI] [PubMed] [Google Scholar]
- 84.Orth K, Xu Z, Mudgett MB, Bao ZQ, Palmer LE, Bliska JB, Mangel WF, Staskawicz B, Dixon JE. 2000. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290:1594–1597. doi: 10.1126/science.290.5496.1594. [DOI] [PubMed] [Google Scholar]
- 85.Palmer LE, Hobbie S, Galan JE, Bliska JB. 1998. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-α production and downregulation of the MAP kinases p38 and JNK. Mol Microbiol 27:953–965. doi: 10.1046/j.1365-2958.1998.00740.x. [DOI] [PubMed] [Google Scholar]
- 86.Paquette N, Conlon J, Sweet C, Rus F, Wilson L, Pereira A, Rosadini CV, Goutagny N, Weber AN, Lane WS, Shaffer SA, Maniatis S, Fitzgerald KA, Stuart L, Silverman N. 2012. Serine/threonine acetylation of TGFβ-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc Natl Acad Sci U S A 109:12710–12715. doi: 10.1073/pnas.1008203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sylvain-Prevost S, Ear T, Simard FA, Fortin CF, Dubois CM, Flamand N, McDonald PP. 2015. Activation of TAK1 by chemotactic and growth factors, and its impact on human neutrophil signaling and functional responses. J Immunol 195:5393–5403. doi: 10.4049/jimmunol.1402752. [DOI] [PubMed] [Google Scholar]
- 88.Mittal R, Peak-Chew SY, McMahon HT. 2006. Acetylation of MEK2 and IκB kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci U S A 103:18574–18579. doi: 10.1073/pnas.0608995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pha K, Navarro L. 2016. Yersinia type III effectors perturb host innate immune responses. World J Biol Chem 7:1–13. doi: 10.4331/wjbc.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Plano GV, Schesser K. 2013. The Yersinia pestis type III secretion system: expression, assembly and role in the evasion of host defenses. Immunol Res 57:237–245. doi: 10.1007/s12026-013-8454-3. [DOI] [PubMed] [Google Scholar]
- 91.Anderson DP. 1997. Adjuvants and immunostimulants for enhancing vaccine potency in fish. Dev Biol Stand 90:257–265. [PubMed] [Google Scholar]
- 92.Paczosa MK, Fisher ML, Maldonado-Arocho FJ, Mecsas J. 2014. Yersinia pseudotuberculosis uses Ail and YadA to circumvent neutrophils by directing Yop translocation during lung infection. Cell Microbiol 16:247–268. doi: 10.1111/cmi.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stasulli NM, Eichelberger KR, Price PA, Pechous RD, Montgomery SA, Parker JS, Goldman WE. 2015. Spatially distinct neutrophil responses within the inflammatory lesions of pneumonic plague. mBio 6:e01530-15. doi: 10.1128/mBio.01530-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Andor A, Trulzsch K, Essler M, Roggenkamp A, Wiedemann A, Heesemann J, Aepfelbacher M. 2001. YopE of Yersinia, a GAP for Rho GTPases, selectively modulates Rac-dependent actin structures in endothelial cells. Cell Microbiol 3:301–310. doi: 10.1046/j.1462-5822.2001.00114.x. [DOI] [PubMed] [Google Scholar]
- 95.Lee WL, Singaravelu P, Wee S, Xue B, Ang KC, Gunaratne J, Grimes JM, Swaminathan K, Robinson RC. 2017. Mechanisms of Yersinia YopO kinase substrate specificity. Sci Rep 7:39998. doi: 10.1038/srep39998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abdel-Latif D, Steward M, Macdonald DL, Francis GA, Dinauer MC, Lacy P. 2004. Rac2 is critical for neutrophil primary granule exocytosis. Blood 104:832–839. doi: 10.1182/blood-2003-07-2624. [DOI] [PubMed] [Google Scholar]
- 97.Ramadass M, Catz SD. 2016. Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol Rev 273:249–265. doi: 10.1111/imr.12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ghosh S, O’Connor TJ. 2017. Beyond paralogs: the multiple layers of redundancy in bacterial pathogenesis. Front Cell Infect Microbiol 7:467. doi: 10.3389/fcimb.2017.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Immler R, Simon SI, Sperandio M. 2018. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest 48(Suppl 2):e12964. doi: 10.1111/eci.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stoiber W, Obermayer A, Steinbacher P, Krautgartner WD. 2015. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules 5:702–723. doi: 10.3390/biom5020702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Futosi K, Fodor S, Mócsai A. 2013. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17:638–650. doi: 10.1016/j.intimp.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dewoody R, Merritt P, Marketon M. 2013. Regulation of the Yersinia type III secretion system: traffic control. Front Cell Infect Microbiol 3:4. doi: 10.3389/fcimb.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnson JL, Monfregola J, Napolitano G, Kiosses WB, Catz SD. 2012. Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effector JFC1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol Biol Cell 23:1902–1916. doi: 10.1091/mbc.E11-12-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hegde B, Bodduluri SR, Satpathy SR, Alghsham RS, Jala VR, Uriarte SM, Chung DH, Lawrenz MB, Haribabu B. 2018. Inflammasome-independent leukotriene B4 production drives crystalline silica-induced sterile inflammation. J Immunol 200:3556–3567. doi: 10.4049/jimmunol.1701504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, Bozza PT. 2002. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular loci involved in eicosanoid metabolism. J Immunol 169:6498–6506. doi: 10.4049/jimmunol.169.11.6498. [DOI] [PubMed] [Google Scholar]
- 106.Radmark O, Samuelsson B. 2009. 5-Lipoxygenase: mechanisms of regulation. J Lipid Res 50 Suppl:S40–5. doi: 10.1194/jlr.R800062-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lepley RA, Fitzpatrick FA. 1994. 5-Lipoxygenase contains a functional Src homology 3-binding motif that interacts with the Src homology 3 domain of Grb2 and cytoskeletal proteins. J Biol Chem 269:24163–24168. [PubMed] [Google Scholar]
- 108.Forsberg M, Löfgren R, Zheng L, Stendahl O. 2001. Tumour necrosis factor-alpha potentiates CR3-induced respiratory burst by activating p38 MAP kinase in human neutrophils. Immunology 103:465–472. doi: 10.1046/j.1365-2567.2001.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR. 2007. The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. Am J Physiol Cell Physiol 292:C1690–C1700. doi: 10.1152/ajpcell.00384.2006. [DOI] [PubMed] [Google Scholar]
- 110.Werz O, Klemm J, Samuelsson B, Rådmark O. 2000. 5-Lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc Natl Acad Sci U S A 97:5261–5266. doi: 10.1073/pnas.050588997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ward RA, Nakamura M, McLeish KR. 2000. Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J Biol Chem 275:36713–36719. doi: 10.1074/jbc.M003017200. [DOI] [PubMed] [Google Scholar]
- 112.Downey GP, Butler JR, Tapper H, Fialkow L, Saltiel AR, Rubin BB, Grinstein S. 1998. Importance of MEK in neutrophil microbicidal responsiveness. J Immunol 160:434–443. [PubMed] [Google Scholar]
- 113.Zhou J, Lai W, Yang W, Pan J, Shen H, Cai Y, Yang C, Ma N, Zhang Y, Zhang R, Xie X, Dong Z, Gao Y, Du C. 2018. BLT1 in dendritic cells promotes Th1/Th17 differentiation and its deficiency ameliorates TNBS-induced colitis. Cell Mol Immunol 15:1047–1056. doi: 10.1038/s41423-018-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toda A, Terawaki K, Yamazaki S, Saeki K, Shimizu T, Yokomizo T. 2010. Attenuated Th1 induction by dendritic cells from mice deficient in the leukotriene B4 receptor 1. Biochimie 92:682–691. doi: 10.1016/j.biochi.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 115.Pires-Lapa MA, Koga MM, da Silva IA Jr, Filgueiras LR, Jancar S. 2019. Leukotriene B4 modulation of murine dendritic cells affects adaptive immunity. Prostaglandins Other Lipid Mediat 141:34–39. doi: 10.1016/j.prostaglandins.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 116.Armstrong CL, Miralda I, Neff AC, Tian S, Vashishta A, Perez L, Le J, Lamont RJ, Uriarte SM. 2016. Filifactor alocis promotes neutrophil degranulation and chemotactic activity. Infect Immun 84:3423–3433. doi: 10.1128/IAI.00496-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eichelberger KR, Jones GS, Goldman WE. 2019. Inhibition of neutrophil primary granule release during Yersinia pestis pulmonary infection. mBio 10:e02759-19. doi: 10.1128/mBio.02759-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.