

Revealing the mechanisms by which bacteria establish long-lasting colonization in the gastrointestinal tract is an area of intensive investigation. The obligate intracellular bacterium Chlamydia is known to colonize mouse colon for long periods. A colonization-deficient mutant strain of this intracellular bacterium is able to regain long-lasting colonization in gamma interferon (IFN-γ) knockout mice following intracolon inoculation.
KEYWORDS: Chlamydia, IFN-γ, innate lymphoid cells, RORγt, obligate intracellular pathogen
ABSTRACT
Revealing the mechanisms by which bacteria establish long-lasting colonization in the gastrointestinal tract is an area of intensive investigation. The obligate intracellular bacterium Chlamydia is known to colonize mouse colon for long periods. A colonization-deficient mutant strain of this intracellular bacterium is able to regain long-lasting colonization in gamma interferon (IFN-γ) knockout mice following intracolon inoculation. We now report that mice deficient in conventional T lymphocytes or recombination-activating gene (Rag) failed to show rescue of mutant colonization. Nevertheless, antibody depletion of IFN-γ or genetic deletion of interleukin 2 (IL-2) receptor common gamma chain in Rag-deficient mice did rescue mutant colonization. These observations suggest that colonic IFN-γ, responsible for inhibiting the intracellular bacterial mutant, is produced by innate lymphoid cells (ILCs). Consistently, depletion of NK1.1+ cells in Rag-deficient mice both prevented IFN-γ production and rescued mutant colonization. Furthermore, mice deficient in transcriptional factor RORγt, but not chemokine receptor CCR6, showed full rescue of the long-lasting colonization of the mutant, indicating a role for group 3-like ILCs. However, the inhibitory function of the responsible group 3-like ILCs was not dependent on the natural killer cell receptor (NCR1), since NCR1-deficient mice still inhibited mutant colonization. Consistently, mice deficient in the transcriptional factor T-bet only delayed the clearance of the bacterial mutant without fully rescuing the long-lasting colonization of the mutant. Thus, we have demonstrated that the obligate intracellular bacterium Chlamydia maintains its long-lasting colonization in the colon by evading IFN-γ from group 3-like ILCs.
INTRODUCTION
Bacteria can establish long-lasting colonization in the gastrointestinal (GI) tracts of mammalian hosts when the bacterium-host interactions have achieved homeostasis (1, 2). A critical host component responding to bacterial colonization in the GI tract is the innate lymphoid cells (ILCs) (3–6]). On one hand, ILCs promote the anatomical containment of bacteria in the GI tract (7, 8); on the other, bacteria have to evade the effector mechanisms of the same ILCs in order to establish and maintain long-lasting colonization, although the exact evasion mechanisms are not clear. Illuminating the mechanisms by which ILCs restrict bacteria or bacteria evade ILC-mediated immunity is an area of extensive investigations (9, 10), since information thus obtained may be used to improve human health (11). Both pathogenic (12, 13) and commensal (14–16) bacterial species have been used to investigate bacterium-ILC interactions (6). Gene-deficient mice have been routinely used to probe bacterium-gut ILC interactions (17, 18). Besides extracellular and facultative intracellular bacteria (6), the obligate intracellular parasite Toxoplasma gondii has also been shown to interact with ILCs (19, 20), although the mechanisms of the T. gondii-ILC interactions remain largely unknown. Knowledge on the interactions of the obligate intracellular bacterium Chlamydia with ILCs is limited, although natural killer (NK) cells have been shown to play important roles in chlamydial infection (21–29). These previous studies did not differentiate NK cells from non-NK ILCs. Thus, there is a lack of information on how non-NK ILCs interact with Chlamydia or obligate intracellular bacteria in general. The goal of the current study was to use Chlamydia as a model to investigate how an obligate intracellular bacterium interacts with host ILCs by taking advantage of a colon colonization-deficient clone in combination with gene knockout mice.
Chlamydia muridarum is an obligate intracellular bacterium that is able to establish long-lasting colonization in the mouse colon but without causing any significant pathology to the GI tract (30–32). However, when C. muridarum is inoculated into extra-GI tissues, it may cause pathologies (33–36). Interestingly, depending on the tissue order of first exposure to Chlamydia, chlamydial colonization in the GI tract may either exacerbate or prevent chlamydial pathogenicity in extra-GI tissues. Thus, when naive mice are first exposed to Chlamydia in the GI tract, the GI tract Chlamydia may function as an oral vaccine to prevent subsequent chlamydial infections elsewhere (37, 38). However, when Chlamydia is first inoculated into the mouse lower genital tract, the chlamydial organisms may both ascend to the upper genital tract (39) and spread to the GI tract (40). The genital to GI tract spreading is likely via blood circulation (41). It has been proposed that chlamydial organisms that spread to the GI tract may induce responses that exacerbate pathogenicity of the genital Chlamydia in the upper genital tract (42). Thus, investigating how Chlamydia achieves long-lasting colonization in the GI tract may also provide important information for both developing chlamydial oral vaccines and understanding chlamydial pathogenic mechanisms in the upper genital tract in addition to gaining insights into bacterial interactions with gut ILCs.
ILCs consist of multiple subsets, including the “killer” ILCs that are also called natural killer cells and the “helper-like” ILCs that are further grouped into subsets of ILC1s, ILC2s, and ILC3s based on their cell surface markers, cytokine profiles, and the transcriptional requirements during differentiation (43, 44). Although ILCs lack receptors for directly recognizing either classical pathogen molecular patterns or antigenic epitopes, bacteria have been shown to activate ILCs indirectly via the help of accessory cells (10, 45), and the ILC-derived effectors then regulate bacterial colonization in the GI tract (7, 13, 46, 47). ILC1s secrete the cytokine gamma interferon (IFN-γ) and express their signature transcriptional factor T-bet (48), while ILC2s may secrete interleukin 5 (IL-5) and IL-13 and express GATA3 (49). However, ILC3s appear to be more diverse despite their common signature cytokines IL-22 and IL-17 and transcriptional factor retinoic acid-receptor-related orphan receptor γt (RORγt) (50–52). In general, ILC3s are composed of at least three subpopulations, including lymphoid tissue inducer (LTi) cells that express chemokine receptor CCR6 (arising early during ontogeny and the earliest discovered ILC3) and CCR6-negative ILC3s, which are either negative for natural cytotoxicity receptor 1 (NCR1) or NKp46 (NCR-ILC3s) or positive for NCR1 (NCR1+ ILC3) (53). ILC3s are highly adaptable to local tissue environments (54). For example, local IL-23 may induce NCR1-ILC3s to secrete IL-17 and IFN-γ (55) and IL-23R+ ILC3s may participate in colitis induction (55–57), while IL-12 and/or IL-15 may induce ILC3s to become NCR1+ILC3s by upregulating T-bet, leading to secretion of both IL-22 and IFN-γ (58, 59). In the absence of T-bet, NCR1+ILC3s are greatly reduced (60). The T-bet- and RORγt-coexpressing ILCs are ILC3-like and sometimes designated ex-RORγt+ ILC3 (58, 59). These cells have been shown to both protect the epithelial barrier and promote enterocolitis during Salmonella enterica infection (59), and they may also contribute to the development of ileitis induced by Toxoplasma gondii (20). Various ILC3 to ILC1 transition subpopulations have also been detected in mucosal tissues (61). It remains unknown whether and how different ILC subsets or transition subpopulations regulate the long-lasting colonization of the obligate intracellular bacterium Chlamydia in the mouse colon.
In the current study, we have taken advantage of a colon colonization-deficient mutant strain of the obligate intracellular bacterium Chlamydia to assess whether mice with a given immune component deleted genetically or depleted with a blocking antibody can show rescue of the colonization of the mutant. We found that mice deficient in conventional T lymphocytes or a recombination-activating gene (Rag) lacking both T and B cells failed to show rescue of the mutant colonization. However, additional antibody depletion of IFN-γ or genetic deletion of IL-2 receptor common gamma chain in Rag knockout mice did rescue, indicating that IFN-γ, responsible for inhibiting the intracellular bacterial mutant, is produced by ILCs. Indeed, depletion of NK1.1+ cells in Rag knockout mice both prevented IFN-γ production and rescued mutant colonization. The inhibitory function of the responsible NK1.1+ ILCs not only requires both IL-7 and IL-15 signaling but also is also dependent on RORγt, a signature transcriptional factor of Th17 and ILC3s. The observations together have demonstrated for the first time that the obligate intracellular bacterium Chlamydia is able to evade group 3 ILC-derived IFN-γ for maintaining its long-lasting colonization in the colon.
RESULTS
IFN-γ but not adaptive immunity is necessary for preventing a mutant strain of the intracellular bacterium Chlamydia from achieving long-lasting colonization in the colon.
As we showed previously (62), the wild-type intracellular bacterium C. muridarum can colonize mouse colon for long periods, while a mutant clone of this bacterium designated G28.51.1 failed to do so (Fig. 1). Following intracolon inoculation with wild-type C. muridarum, mice continuously shed live organisms in rectal swabs for 28 days and live organisms were mainly recovered from colon on day 28. However, the mutant-infected mice only shed live organisms in rectal swabs up to day 7, and no live mutant organisms were recovered from any GI tract tissues on day 28. Thus, we defined this mutant as a colon colonization-deficient mutant. Interestingly, when the mutant was inoculated into mice deficient in IFN-γ, live organisms were continuously shed in rectal swabs for 28 days and recovered from the large intestine tissues on day 28 (Fig. 2), indicating that this mutant is susceptible to IFN-γ inhibition. However, the mutant failed to establish long-lasting colonization in the colons of mice deficient in CD8+ T cells with or without depletion of CD4+ T cells or mice deficient in both T cell receptor beta and delta chains (TCRβ/δ) (Fig. 2), demonstrating that conventional T lymphocytes are not required for inhibition of the mutant. These observations together suggest that IFN-γ, responsible for inhibiting mutant colonization in the colon, may be produced by nonadaptive immune cells, although the inhibition reached the maximal level on and after day 7.
FIG 1.
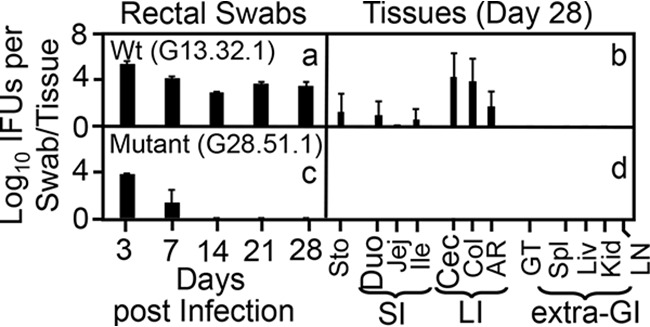
Comparison of intracellular bacteria with and without mutations for colonizing mouse gastrointestinal tract following intracolonic inoculation. Wild-type (Wt) intracellular bacterium Chlamydia muridarum without (clone G13.32.1, inoculation dose at 2 × 105 inclusion-forming units [IFU] [a and b]) and with (clone G28.51.1, inoculation dose at 1 × 107 IFU [c and d]) mutations were used to infect groups of C57BL/6J mice (n = 3 to 5) via intracolonic inoculation. On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (a and c), or on day 28, mouse tissues were harvested (b and d), as indicated along the x axis for monitoring live chlamydial organisms. The results ae expressed as log10 IFU per swab or tissue as shown along the y axis. Mouse tissues include different segments of the gastrointestinal (GI) tract, such as stomach (Sto), duodenum (Duo), jejunum (Jej), ileum (Ile, grouped as SI for small intestine), cecum (Cec), colon (Col), and anorectum (AR, grouped as LI for large intestine), and genital tract (GT), spleen (Spl), liver (Liv), kidney (Kid), and mesenteric lymph nodes (LN, grouped as extra-GI tissues), as listed along the x axis. Note the lack of long-lasting colonization by the mutant intracellular bacterial strain in the mouse colon. Data are from two independent experiments. P < 0.01, Wilcoxon rank sum (area-under curves between Wt and mutant-infected groups).
FIG 2.
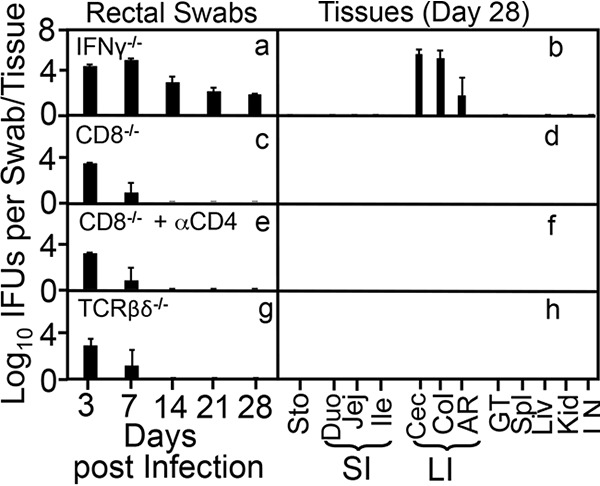
Deficiency in IFN-γ but not conventional T lymphocytes rescues the ability of the intracellular bacterial mutant to establish long-lasting colonization in the large intestine. Mice deficient in IFN-γ (IFNγ−/− [a and b]) but not CD8+ T cells (CD8−/− [c and d]), CD8+ T cells plus depletion of CD4+ T cells (IFNγ−/− + αCD4 [e and f]), or all T cells expressing T cell receptor β (TCR β) or TCRδ chain (TCRβδ−/− [g and h]) were intracolonically inoculated with the intracellular bacterium mutant clone G28.51.1 at an inoculation dose of 1 × 107 IFU. On days 3 and 7 and weekly thereafter, rectal swabs were taken (a, c, e, and g), or on day 28, mouse tissues were harvested (b, d, f, and h), as indicated along the x axis for monitoring live chlamydial organisms as shown along the y axis. Mouse tissues include different segments of gastrointestinal tract and extra-GI tract tissues as described in the Fig. 1 legend. Note that while mice deficient in IFN-γ showed rescue of long-term colonization of the mutant, the deficiency in CD4+ and CD8+ T lymphocytes, or T cells expressing TCRβ or TCRδ is insufficient to rescue the mutant. P < 0.01, Wilcoxon rank sum (area-under curves between IFN-γ knockout group and any other groups [n = 4 to 6 from 2 or 3 independent experiments]).
IFN-γ, required for inhibiting colonization of the intracellular bacterial mutant in the colon, is produced by ILCs.
To determine whether IFN-γ, responsible for inhibiting the intracellular bacterial mutant, is produced by innate immune cells, we compared the mutant colonizations among mice deficient in recombination-activating gene 1 (Rag1−/−), deficient in Rag2 plus interleukin 2 receptor (IL-2R) common gamma chain (Rag2−/− IL-2Rγc−/−), deficient in Rag1 and treated with a control (Ctrl) immunoglobulin (IgG), or deficient in Rag1 and treated with an anti-IFN-γ neutralization antibody (Rag1−/− + αIFN-γ). As shown in Fig. 3, mutant colonization was restricted within 7 days in Rag1−/− mice, validating the above-stated conclusion that innate immune cells are required for inhibiting mutant colonization, as Rag1−/− mice lack T and B cells. More importantly, additional deficiency of IL-2Rcγ in Rag2−/− mice rescued the mutant, indicating that innate lymphoid cells (ILCs) are responsible for inhibiting the mutant. This is because IL-2Rcγ is expressed only by lymphoid cells and all IL-2Rcγ-expressing cells in Rag2−/− mice are ILCs. The depletion of IFN-γ with an anti-IFN-γ neutralization antibody, but not a control immunoglobulin, rescued mutant colonization in Rag1−/− mice, further confirming that IFN-γ produced by ILCs is responsible for inhibiting the mutant.
FIG 3.
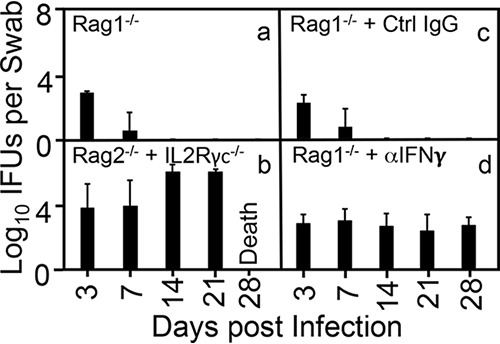
Deficiency in innate but not adaptive immunity is required for rescuing the colonization of the intracellular bacterial mutant. Mice deficient in recombination-activating gene 1 (Rag1−/− [a]), Rag2 plus interleukin 2 (IL-2) receptor common gamma chain (Rag2−/− + IL-2γc−/− [b]), mice deficient in Rag1 and treated with a control (Ctrl) immunoglobulin (Rag1−/− + Ctrl IgG [c]), and mice deficient in Rag1 and treated with an anti-IFN-γ neutralization antibody (Rag1−/− + αIFN-γ [d]) were infected intracolonically with the mutant clone (G28.51.1). On days 3 and 7 and weekly thereafter after the intracolonic inoculation as shown along the x axis, rectal swabs were taken for monitoring live chlamydial organism shedding. The results are expressed as log10 IFU per swab as shown along the y axis. Note that either additional genetic deficiency in IL-2γc or neutralization of IFN-γ rescued the ability of the intracellular bacterial mutant to colonize the colons of Rag1−/− mice. P < 0.01, Wilcoxon rank sum (area-under curves between Rag1−/− or Rag1−/− with IgG treatment group and Rag1−/− + Ctrl IgG or Rag1−/− + αIFN-γ groups, respectively [n = 3 to 6 per group from 3 independent experiments]).
NK1.1+ ILCs are responsible for producing IFN-γ to inhibit the intracellular bacterial mutant from colonizing the mouse colon.
NK1.1, initially identified as a natural killer (NK) cell receptor (63), is also expressed by subsets of other nonkiller innate lymphoid cells, including type 1 (ILC1) (48) and type 3 (ILC3) (50). To determine whether NK1.1+ ILCs are responsible for producing IFN-γ and inhibiting mutant colonization, Rag1−/− mice with and without depletion with an anti-NK1.1 antibody were compared for their susceptibilities to mutant colonization (Fig. 4). Following an intracolon inoculation, Rag1−/− mice showed significant reductio of mutant colonization on day 7 and completely cleared the mutant by day 14. However, depletion of NK1.1+ cells in Rag1−/− mice fully rescued mutant colonization in the colon, demonstrating that NK1.1+ cells are responsible for inhibiting the mutant. Furthermore, when these mice were sacrificed for monitoring both live bacteria and IFN-γ in cecum and colon tissues (Fig. 5), as expected, significant levels of mutant organisms were recovered from NK1.1+ cell-depleted mice but not mice treated with a control antibody. This rescue correlated with decreased IFN-γ in the same tissue samples. Thus, the above-desribed observations have together demonstrated that either ILC1s or ILC3s are responsible for the IFN-γ-mediated inhibition of mutant colonization in the colon.
FIG 4.
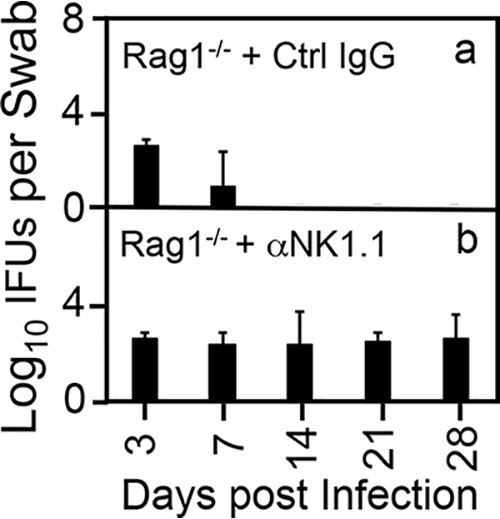
Depletion of NK1.1+ cells rescues the ability of the mutant intracellular bacterium to achieve long-lasting colonization in the colon of Rag1-deficient mice. Rag1−/− mice treated with a control mouse IgG (Ctrl IgG [a]) or a mouse anti-NK1.1 monoclonal antibody (αNK1.1 [b]) were infected via intracolonic inoculation with 1 × 107 IFU of the mutant intracellular bacterial clone G28.51.1. On days 3 and 7 and weekly thereafter after intracolon inoculation as shown along the x axis, rectal swabs were taken for monitoring live chlamydial organism shedding. The results are expressed as log10 IFU per swab as shown along the y axis. Note that αNK1.1 antibody treatment successfully rescued intracellular bacterial shedding until day 28 postinfection. P < 0.05, Wilcoxon rank sum (area-under curves for IFU comparison [n = 3 to 5 from 3 independent experiments]).
FIG 5.
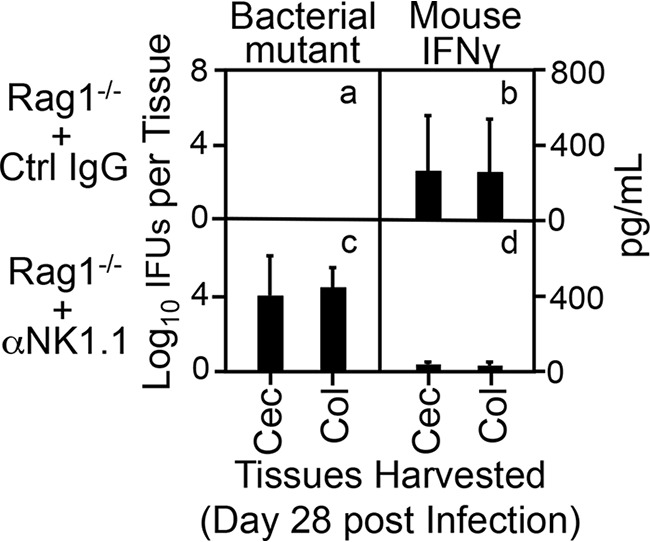
Depletion of NK1.1+ cells blocks IFN-γ production in the colons of Rag1-deficient mice. Rag1−/− mice treated and infected as described in the Fig. 4 legend were sacrificed on day 28 after infection for titrating both live organisms (bacterial mutant [a and c], left y axis) and IFN-γ (b and d, right y axis) from cecum (Cec) and colon (Col). Note that mice treated with a control mouse IgG completely cleared the bacterial mutant from both cecum and colon tissues (a), which correlated with the rise of IFN-γ in the same tissues (b). However, mice treated with an anti-NK1.1 antibody showed rescue of the mutant colonization in the colon (c), which correlated with the blockade of IFN-γ in the same tissues (d). P < 0.01, Wilcoxon rank sum (area-under curves for IFU comparison between panels a and c and IFN-γ between panels b and d [n = 3 to 6 from two independent experiments]).
The ILCs required for inhibiting mutant colonization in the colon are dependent on RORγt, a signature transcriptional factor of ILC3.
We further tested whether the NK1.1+ ILCs responsible for inhibiting the intracellular bacterial mutant are dependent on IL-7 and/or IL-15 signaling, since these cytokines have been shown to promote the development and maturation of ILCs (64, 65) in addition to their roles in promoting T cell homeostasis (66) and memory T cell development (67). As shown in Fig. 6, deficiency in either the IL-7 or IL-15 signaling pathway fully rescued mutant colonization in mouse colon. Following intracolon inoculation, live organisms of the intracellular bacterial mutant were continuously recovered from the rectal swabs of mice deficient in receptors for either cytokine during the entire observation period. On day 28, live organisms were detected in the large intestine tissues only, including cecum, colon, and anorectal tissues, indicating that the replication of the intracolonically inoculated mutant organisms was restricted to the large intestine despite the gene deficiency. These observations demonstrated that both cytokine signaling pathways are required for the NK1.1+ ILCs to exhibit an antibacterial effect.
FIG 6.
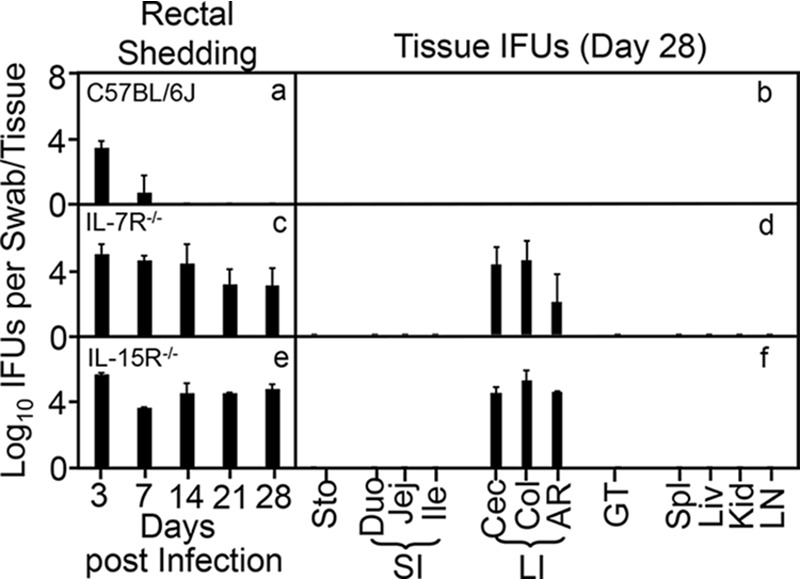
Both IL-7 signaling and IL-15 signaling are required to clear the intracellular bacterial mutant from the colon. Wild-type C57BL/6J mice (a), IL-7 receptor knockout mice (IL-7R−/− [b]), and IL-15 receptor knockout mice (IL-15R−/− [c]) were infected with 1 × 107 IFU of the mutant intracellular bacterial clone G28.51.1 via intracolonic inoculation. On days 3 and 7 and weekly thereafter, rectal swabs were taken (a, c, and e), or on day 28, mouse tissues were harvested (b, d, and f), as indicated along the x axis, for monitoring live chlamydial organisms as shown along the y axis. Mouse tissues include different segments of gastrointestinal tract and extra-GI tract tissues as described in the Fig. 1 legend. Note that both IL-7R−/− mice (b) and IL-15R−/− knockout mice (c) rescued the ability of the mutant to colonize the GI tract. P < 0.01, Wilcoxon rank sum (area-under curves for IFU comparison [n = 3 to 5 from 2 independent experiments]).
Since T-bet is a signature transcriptional factor for ILC1 (68), while RORγt is one for ILC3 (51), we compared the bacterial mutant colonizations in mice with and without deficiency in T-bet or RORγt (Fig. 7). We found that mice deficient in T-bet only showed mutant colonization extended by one more week, up to day 14. By day 21, the mutant was completely cleared, suggesting that T-bet is only partially required for inhibiting the mutant during the first 2 weeks after inoculation. Thus, we conclude that ILC1s are not essential for inhibiting the mutant. To our surprise, mice deficient in RORγt showed full rescue of colonization of the mutant, indicating that the NK1.1+ ILCs responsible for inhibiting the mutant represent ILC3-like cells.
FIG 7.
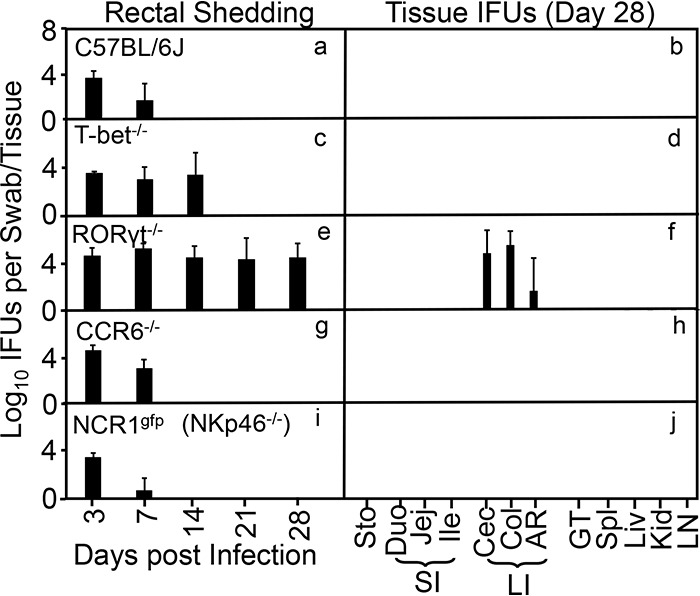
RORγt is essential for rescuing the intracellular bacterium mutant to achieve long-lasting colonization in mouse colon. Wild-type C57BL/6J mice (a and b), T-bet knockout mice (T-bet−/− [c and d]), RORγt knockout mice (RORγt−/− [e and f]), CCR6 knockout mice (CCR6−/− [g and h]), and NCR1 knockout mice (NCR1gfp/gfp or NKp46−/− [i and j]) were intracolonically infected with the intracellular bacterium mutant at 1 × 107 IFU. On days 3 and 7 and weekly thereafter, rectal swabs were taken (panels a, c, e, g, and i), or on day 28, mouse tissues were harvested (b, d, f, h, and j), as indicated along the x axis for monitoring live chlamydial organisms as shown along the y axis. Mouse tissues include different segments of gastrointestinal tract and extra-GI tract tissues as described in the Fig. 1 legend. Note that T-bet−/− only extended the colonization of the mutant for an additional 1 week while RORγt−/− fully rescued the mutant colonization, although neither CCR6 nor NKp46 was required for clearing the mutant. P < 0.01, Wilcoxon rank sum (area-under curves for IFU comparison between C57BL6/J and RORγt−/− mice [n = 3 to 5 from 2 or 3 independent experiments).
Furthermore, the inhibitory function of ILC3 cells does not depend on the chemokine receptor CCR6, since mice deficient in CCR6 failed to show rescue of mutant colonization (Fig. 7). Although the failure of CCR6−/− mice to rescue the mutant bacteria may not necessarily exclude the contribution of LTi-like ILC3s, it may suggest that non-LTi ILC3s may be responsible for the inhibition of the mutant. The non-LTi ILC3s can be divided into two subsets based on their expression of natural cytotoxicity receptor 1 (NCR1) or natural killer cell p46 (NKp46) (69). We then monitored the colonization of the intracellular bacterial mutant in NCRgfp/gfp mice, since these mice are genetically deficient in NCR1 and lack ILC1s and NCR1-dependent TRAIL expression (70, 71). We found that in NCRgfp/gfp mice the mutant colonization was inhibited as efficiently as in wild-type mice, suggesting that ILC1s are not critical for inhibiting mutant colonization. Thus, we can conclude that IFN-γ-producing ILC3-like cells are responsible for preventing the intracellular bacterial mutant from establishing long-lasting colonization in the colon.
DISCUSSION
In the present study, we have used Chlamydia as a model to investigate how an obligate intracellular bacterium interacts with host ILCs, which has allowed us to reveal a mechanism by which such a bacterium achieves long-lasting colonization in the colon and obtain new information for understanding chlamydial pathogenesis (42) and vaccine development (37). By taking advantage of a colon colonization-deficient mutant bacterium and using mouse gene knockouts and antibody depletion for rescuing mutant colonization, we have demonstrated that Chlamydia is able to cooperate with IFN-γ-producing ILCs to maintain long-lasting colonization in the mouse colon. First, the colonization-deficient chlamydial mutant regained long-lasting colonization in IFN-γ knockout mice following intracolon inoculation, indicating that IFN-γ is a critical effector molecule that regulates chlamydial colonization in the colon. It is necessary for wild-type Chlamydia to evolve functional genes for maintaining a homeostatic relationship with mouse gut IFN-γ-mediated immunity in order to establish long-lasting colonization in the colon. Second, mice deficient in conventional T lymphocytes failed to show rescue of the mutant, suggesting that the responsible colonic IFN-γ is produced by innate cells. The failure of mice deficient in the Rag1 gene, which is required for developing adaptive immune cells, to show rescue of the mutant colonization further validated the conclusion that innate immune cells are sufficient for inhibiting the mutant. Third, antibody depletion of IFN-γ or genetic deletion of IL-2Rcγ in Rag-deficient mice fully rescued mutant colonization, suggesting that the innate immune cells responsible for producing colonic IFN-γ are ILCs. This is because IL-2Rcγ is only expressed by lymphoid cells and all IL-2Rcγ-expressing cells in Rag2−/− mice are ILCs. Furthermore, depletion of NK1.1+ cells in Rag1-deficient mice both prevented IFN-γ production and rescued mutant colonization, suggesting that the responsible ILCs are either NK cells or the nonkiller ILC1s or ILC3s. This is because these cells are responsible for expressing NK1.1 in Rag1−/− mice. Finally, the inhibitory function of the responsible ILCs is dependent on both IL-7 and IL-15 signaling, since deficiency in either receptor fully rescued the mutant colonization. These cytokine receptor-mediated signaling pathways are required for the development and differentiation of different ILCs (43, 65, 72).
We have further demonstrated that the IFN-γ-producing ILCs responsible for inhibiting the bacterium mutant colonization in colon are RORγt+ ILC3-like cells but not ILC1s. Both the killer ILC1s (conventional NK cells) and the helper ILC1s are considered the major sources of innate IFN-γ (73, 74). Innate IFN-γ produced during chlamydial infection in the airway or genital tract has been attributed to NK cells (21–29). However, these previous studies did not attempt to differentiate NK cells from other ILCs. Thus, the relative contributions of distinct ILC subsets to the production of IFN-γ upon chlamydial infection remained unclear. In the current study, we found that mice deficient in RORγt, a signature transcriptional factor of ILC3s, were able to show full rescue of chlamydial mutant colonization. In contrast, deficiency in T-bet, a transcriptional factor of ILC1s, only extended mutant colonization for 1 week, after which the T-bet-deficient mice were no longer able to show rescue of the mutant. Together, these observations demonstrate for the first time that the colonic IFN-γ responsible for regulating the long-lasting colonization of the obligate intracellular bacterium Chlamydia is produced by ILC3-like cells. Furthermore, the ILC1s were not critical for controlling mutant colonization, as NCR1-deficient mice showed inhibition of mutant colonization. NCR1, recognizing complement factor P as a ligand, has been shown to promote mouse survival during lethal Neisseria meningitidis infections (75). This finding is consistent with the above-mentioned observation that mice deficient in T-bet, a transcriptional factor required for expression of NCR1 (69, 70), only showed delayed clearance of the mutant for a week without full rescue of long-lasting colonization. The fact that inhibition of mutant colonization is dependent on RORγt but independent of NKp46 has led us to both exclude the contribution ILC1s and narrow down the responsible IFN-γ-producing effector cells to a unique subset of ILC3s. The failure of CCR6−/− mice to show rescue of colonization suggests that CCR6 is not required for ILC3s to control intracellular bacterium colonization, as LTi-like ILC3s express CCR6 (76). Thus, despite the roles of CCR6+ cells (including many non-ILC cells) in intestinal mucosal immunity (77), CCR6 expression is not required for inhibiting the intracellular bacterium mutant colonization.
The IFN-γ-producing CCR6− NCR1− RORγt+ ILC3 subset cells have been implicated in microbial interactions with the host. Many intestinal RORγt+ ILC3s can be negative for both NCR1 and CCR6 (52, 78). ILC3s are extremely flexible and can produce different cytokine profiles in response to signals from local tissues (54, 58, 60, 61). For example, IL-23 has been shown to induce NCR1− RORγt+ ILC3s to secrete both IL-17 and IFN-γ (55), which may participate in microbial induction of colitis (55–57). Although the colitis-associated NCR1− RORγt+ ILC3s may coexpress T-bet, it remains unclear whether T-bet coexpression is necessary for these cells to produce IFN-γ (55). In contrast, T-bet seems to be necessary for the function of NCR1+ RORγt+ ILC3s (58, 60, 61). These NCR1+ ILC3s have been shown to protect the epithelial barrier and promote enterocolitis during Salmonella enterica infection (59) as well as contribute to the development of ileitis induced by Toxoplasma gondii (20). In the current study, we have shown that IFN-γ-producing RORγt+ ILC3s are responsible for regulating the long-lasting colonization of the obligate intracellular bacterium Chlamydia in the colon. Since the chlamydial colonization in the colon is nonpathological (37), the current study has provided the first example for the IFN-γ-producing RORγt+ ILC3 subset to interact with gut bacteria under nonpathological conditions, which may provide a suitable platform for uncovering new mechanisms by which gut microbiota interact with ILCs. Microbiota are known to confer colonization resistance to pathogenic microbial infections (79–81). One of the mechanisms is by maintaining the production of IFN-γ in the mucosal tissue (80). However, it is not clear whether ILCs are required for microbiota to maintain IFN-γ-mediated colonization resistance, although microbiota have been shown to induce regulatory ILCs for attenuating gut inflammation (9, 16). Here, we have presented evidence that the nonpathological long-lasting colonization of the obligate intracellular bacterium Chlamydia is regulated by IFN-γ-producing RORγt+ ILC3-like cells, which suggests that this subset of ILC3s may contribute to microbiota-induced IFN-γ-mediated colonization resistance.
The IFN-γ deficiency-dependent rescue of the ability of the chlamydial mutant clone G28.51.1 to colonize the mouse colon has motivated us to test whether the mutant is more susceptible to IFN-γ-induced cell-autonomous inhibition of chlamydial growth in a cell culture system. This is because parental wild-type Chlamydia muridarum is known to evade interferon-induced cell-autonomous mechanisms (82). To our surprise, the mutant did not show any significant increase in susceptibility to IFN-γ inhibition (data not shown). These results suggest that Chlamydia may be able to evade immunity mediated by IFN-γ produced by CCR6− NCR1− RORγt+ ILC3s via indirect mechanisms in addition to the cell-autonomous mechanisms. Microbiota have been shown to use their metabolites, such as short-chain fatty acids, to induce T regulatory cells (Tregs) (83–85) and suppress Th17/ILC3 (15), which may enable microbiota evasion of antimicrobial immunity. Thus, efforts are under way to both optimize the in vitro inhibition experimental conditions and explore non-cell-autonomous mechanisms in mice.
It is worth noting that besides clone G28.51.1 (86, 87), there are other C. muridarum mutants that have been shown to be defective in colonizing the GI tract and susceptible to IFN-γ inhibition (88, 89). A common property of these mutants is that they all carry mutations in chromosomal genes. It will be interesting to test whether all these chromosomal gene mutants are defective in colonizing the large intestine and are susceptible to inhibition by IFN-γ produced by ILC3-like cells. These experiments will likely reveal how the obligate intracellular bacterium C. muridarum has learned to use multiple chromosomal genes to deal with innate IFN-γ in order to achieve long-lasting colonization in the colon. We have recently shown that a C. muridarum mutant deficient in the plasmid-encoded pGP3 remains resistant to IFN-γ and can maintain long-lasting colonization in the colon (62) but is highly susceptible to gastric acid killing (90). Thus, it appears that the obligate intracellular bacterium C. muridarum may have acquired the plasmid for aiding its journey to the large intestine, where it may then switch on its chromosomal genes for living a long life.
MATERIALS AND METHODS
Intracellular bacterial strains.
The obligate intracellular bacterial organisms used in the current study were wild-type Chlamydia muridarum clone G13.32.1 and mutant clone G28.51.1 (86, 87). G13.32.1 and G28.51.1 are isogenic to each other, with two mutations in G28.51.1 genome: a substitution mutation in gene tc0237 (a guanine at chromosomal position 277313 is replaced with a cytosine [G277313C]), resulting in an amino acid substitution at codon 117 from glutamine (Q) to glutamic acid (E), designated mutation TC0237Q117E, and another mutation in gene tc0668 (a guanine at chromosomal position 797979 is replaced with an adenine [G797979A]), resulting in a stop codon at codon 216 originally coding for glycine (G), designated mutation TC0668G216*. The wild-type G13.32.1 and mutant G28.51.1 clones were found to achieve similar colonizations in the mouse genital tract (87), but G28.51.1 was significantly defective in colonizing the mouse GI tract following intragastric inoculation (62, 91). Both of these intracellular bacterium clones were grown in HeLa cells (human cervical carcinoma epithelial cells; ATCC CCL-2) for density gradient purification into elementary bodies (EBs) and stored in aliquots at –80°C until use as described previously (40).
Mouse infection.
The mouse experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals, endorsed by the National Institutes of Health (92). The protocol was approved by the Committee on the Ethics of Laboratory Animal Experiments of the University of Texas Health Science Center at San Antonio.
The following 5- to 7-week-old female mice used in the current study were all from Jackson Laboratories, Inc., Bar Harbor, ME: wild-type mice (C57BL/6J, stock number 000664) or mice deficient in IFN-γ (B6.129S7-Ifngtm1Ts/J or IFN-γ−/−, 002287), CD8 (B6.129S2-Cd8atm1Mak/J or CD8−/−, 002665), both T cell receptor β and δ chains (B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J or TCRβδ−/−, 002122), recombination-activating gene 1 (B6.129S7-Rag1tm1Mom/J or Rag1−/−, 002216), recombination-activating gene 2 plus interleukin 2 (IL-2) receptor common gamma chain (C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J or Rag2−/− γc−/−, 014593), IL-7 receptor (B6.129S7-Il7rtm1Imx/J or IL-7R−/−, 002295), IL-15 receptor (B6;129 × 1-Il15ratm1Ama/J or IL-15R−/−, 003723) T-bet (B6.129S6-Tbx21tm1Glm/J or T-bet−/−, 004648), RORγt (B6.129P2-Rorctm1Litt/J or RORγt−/−, 007571), CCR6 (B6.129P2-Ccr6tm1Dgen/J or CCR6−/−, 005793), or NCR1 (B6;129-Ncr1tm1Oman/J or NCR1gfp/gfp or NKp46−/−, 022739).
All mice were inoculated with C. muridarum EBs at 2 × 105 to 1 × 107 inclusion-forming units (IFU) per mouse via intracolon inoculation as described previously (62). Briefly, EBs were diluted in 50 μl of SPG (220 mM sucrose, 12.5 mM phosphate, 4 mM l-glutamic acid [pH 7.5]) buffer that contained the desired number of IFU and organisms as indicated for individual experiments and delivered to the colon using a straight ball-tipped needle designed for mouse oral gavage (N-PK 020; Braintree Scientific, Inc., Braintree, MA). After inoculation, mice were monitored for live organism shedding in rectal swabs or sacrificed for titrating live organisms in designated organs/tissues.
Mouse treatments.
In some experiments, mice were treated with a rat anti-IFN-γ (clone XMG1.2, IgG1/κ) or a mouse anti-NK1.1 (clone PK136, IgG2a/κ) antibody (both were purchased from Bio X Cell, West Lebanon, NH). The control groups were similarly treated with rat IgG (code 012-000-002) or normal mouse IgG (code 015-000-002; both from Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). The antibody depletion treatment was carried out in Rag1−/− mice in order to assess the roles of innate IFN-γ and NK1.1+ cells in controlling G28.51.1 organism colonization. The treatment began on 5 days (−5) prior to intracolon infection, followed by −2 days, 2 days after infection, and twice a week thereafter throughout the experiments. Each injection was administered intraperitoneally with 400 μg of IgG in 400 μl of phosphate-buffered saline (PBS) as described previously (93).
Titration of live chlamydial organisms from mouse swabs and tissues.
For monitoring live organism shedding from GI tracts, rectal swabs were taken on day 3 postinoculation and weekly thereafter. Each swab was soaked in 0.5 ml of SPG buffer and vortexed with glass beads to release infectious EBs for quantitation. For titration of live chlamydial organisms recovered from mouse tissues, various organs/tissues were harvested on designated days after inoculation as specified for individual experiments. Each organ or tissue segment was transferred to a tube containing 0.5 ml (for each segment of the genital tract tissue) or 2 ml (for each remaining tissue/organ) of SPG buffer. The organs and tissue segments were homogenized in cold SPG buffer.
The live C. muridarum organisms released into swab suspensions or tissue supernatants were titrated on HeLa cells in duplicate as described previously (94). The total numbers of IFU/swab or tissue were converted into log10 values for calculating the mean and standard deviation across mice of the same group at each time point. The detection limits of the above titration method are 10 IFU per swab and 40 IFU per tissue sample. This is because we always used 50 μl of the total volume (500 μl for swab and 2,000 μl for tissue), starting with neat tissue homogenates without dilution and then following with serial dilutions to titrate the number of IFU of a given sample. We counted the entire titration culture well for chlamydial inclusions when the inclusion density was low, allowing the detection of a single IFU per 50-μl sample.
Immunofluorescence assay.
An immunofluorescence assay was used for titrating live organisms as described previously (37). Briefly, HeLa cells grown on coverslips were fixed with paraformaldehyde (Sigma) and permeabilized with saponin (Sigma). After being washed and blocked, the cell samples were subjected to a combination of antibody and chemical staining. Hoechst (blue; Sigma) was used to visualize nuclear DNA. A rabbit antichlamydial antibody (raised by immunization with C. muridarum EBs [data not shown]) and a goat anti-rabbit IgG conjugated with Cy2 (green) (Jackson ImmunoResearch Laboratories, Inc.) were used to visualize chlamydial inclusions. The cell samples after immunolabeling were used for counting inclusions under an Olympus AX-70 fluorescence microscope equipped with multiple filter sets (Olympus, Melville, NY).
Measurement of cytokines in mouse tissues using ELISA.
An enzyme-linked immunosorbent assay (ELISA) kit for measuring mouse IFN-γ from cecum and colon tissues was purchased from R&D Systems, Inc. (catalog no. MIF00). The cecum and colon tissues homogenized in SPG buffer were added with a protease inhibitor cocktail (catalog no. 78430, 100× stock; Thermo Fisher Scientific, Waltham, MA) at a final concentration of 2×. The neat homogenates were 2-fold serially diluted with PBS containing 2× inhibitor cocktail, and the diluted samples were applied to 96-well plates precoated with a capture antibody. IFN-γ binding was detected with a detection antibody plus detection reagent. The absorbance at 450 nm was detected with a Synergy H4 microplate reader (BioTek, Winooski, VT), and the results were expressed as picograms per milliliter based on the standard curve obtained from the plate.
Statistics.
The numbers of live organisms expressed as IFU were compared between groups using a Wilcoxon rank sum test.
ACKNOWLEDGMENT
This work was supported in part by grants from the U.S. National Institutes of Health (R01AI121989 and R01AI047997 to G.Z. and R21AI135574 to A.T.).
REFERENCES
- 1.Atarashi K, Umesaki Y, Honda K. 2011. Microbiotal influence on T cell subset development. Semin Immunol 23:146–153. doi: 10.1016/j.smim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonnenberg GF, Artis D. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Britanova L, Diefenbach A. 2017. Interplay of innate lymphoid cells and the microbiota. Immunol Rev 279:36–51. doi: 10.1111/imr.12580. [DOI] [PubMed] [Google Scholar]
- 5.Castellanos JG, Longman RS. 2019. Innate lymphoid cells link gut microbes with mucosal T cell immunity. Gut Microbes 2019:1–6. doi: 10.1080/19490976.2019.1638725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim M, Kim CH. 2016. Colonization and effector functions of innate lymphoid cells in mucosal tissues. Microbes Infect 18:604–614. doi: 10.1016/j.micinf.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D. 2012. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, Ling L, Leiner I, Pamer EG. 2015. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe 18:27–37. doi: 10.1016/j.chom.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, Cortez VS, Caparon MG, Donia MS, Gilfillan S, Cella M, Gordon JI, Hsieh CS, Colonna M. 2017. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science 357:806–810. doi: 10.1126/science.aah5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. 2014. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brussow H. 2019. Probiotics and prebiotics in clinical tests: an update. F1000Res 8(F1000 Faculty Rev):1157. doi: 10.12688/f1000research.19043.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koroleva EP, Halperin S, Gubernatorova EO, Macho-Fernandez E, Spencer CM, Tumanov AV. 2015. Citrobacter rodentium-induced colitis: a robust model to study mucosal immune responses in the gut. J Immunol Methods 421:61–72. doi: 10.1016/j.jim.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, Eberl G, Littman DR, Heikenwalder M, Tumanov AV, Nedospasov SA. 2013. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 342:1243–1246. doi: 10.1126/science.1243364. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Zheng N, Luo Q, Jiang L, He B, Yuan X, Shen L. 2019. Probiotics Lactobacillus reuteri abrogates immune checkpoint blockade-associated colitis by inhibiting group 3 innate lymphoid cells. Front Immunol 10:1235. doi: 10.3389/fimmu.2019.01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SH, Cho BH, Kiyono H, Jang YS. 2017. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep 7:3980. doi: 10.1038/s41598-017-02729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamoto N, Amiya T, Aoki R, Taniki N, Koda Y, Miyamoto K, Teratani T, Suzuki T, Chiba S, Chu PS, Hayashi A, Yamaguchi A, Shiba S, Miyake R, Katayama T, Suda W, Mikami Y, Kamada N, Ebinuma H, Saito H, Hattori M, Kanai T. 2017. Commensal Lactobacillus controls immune tolerance during acute liver injury in mice. Cell Rep 21:1215–1226. doi: 10.1016/j.celrep.2017.10.022. [DOI] [PubMed] [Google Scholar]
- 17.Martínez-López M, Iborra S, Conde-Garrosa R, Mastrangelo A, Danne C, Mann ER, Reid DM, Gaboriau-Routhiau V, Chaparro M, Lorenzo MP, Minnerup L, Saz-Leal P, Slack E, Kemp B, Gisbert JP, Dzionek A, Robinson MJ, Rupérez FJ, Cerf-Bensussan N, Brown GD, Bernardo D, LeibundGut-Landmann S, Sancho D. 2019. Microbiota sensing by Mincle-Syk axis in dendritic cells regulates interleukin-17 and -22 production and promotes intestinal barrier integrity. Immunity 50:446–461.e9. doi: 10.1016/j.immuni.2018.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashiguchi M, Kashiwakura Y, Kojima H, Kobayashi A, Kanno Y, Kobata T. 2015. Peyer’s patch innate lymphoid cells regulate commensal bacteria expansion. Immunol Lett 165:1–9. doi: 10.1016/j.imlet.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Ivanova DL, Denton SL, Fettel KD, Sondgeroth KS, Munoz Gutierrez J, Bangoura B, Dunay IR, Gigley JP. 2019. Innate lymphoid cells in protection, pathology, and adaptive immunity during apicomplexan infection. Front Immunol 10:196. doi: 10.3389/fimmu.2019.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Begue B, Ruemmele F, Combadiere C, Di Santo JP, Buzoni-Gatel D, Cerf-Bensussan N. 2012. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 37:108–121. doi: 10.1016/j.immuni.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Onsrud M, Qvigstad E. 1984. Natural killer cell activity after gynecologic infections with chlamydia. Acta Obstet Gynecol Scand 63:613–615. doi: 10.3109/00016348409155547. [DOI] [PubMed] [Google Scholar]
- 22.Williams DM, Schachter J, Grubbs B. 1987. Role of natural killer cells in infection with the mouse pneumonitis agent (murine Chlamydia trachomatis). Infect Immun 55:223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng CT, Rank RG. 1998. Role of NK cells in early host response to chlamydial genital infection. Infect Immun 66:5867–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rottenberg ME, Gigliotti Rothfuchs A, Gigliotti D, Ceausu M, Une C, Levitsky V, Wigzell H. 2000. Regulation and role of IFN-gamma in the innate resistance to infection with Chlamydia pneumoniae. J Immunol 164:4812–4818. doi: 10.4049/jimmunol.164.9.4812. [DOI] [PubMed] [Google Scholar]
- 25.Shekhar S, Peng Y, Gao X, Joyee AG, Wang S, Bai H, Zhao L, Yang J, Yang X. 2015. NK cells modulate the lung dendritic cell-mediated Th1/Th17 immunity during intracellular bacterial infection. Eur J Immunol 45:2810–2820. doi: 10.1002/eji.201445390. [DOI] [PubMed] [Google Scholar]
- 26.Hu VH, Luthert PJ, Derrick T, Pullin J, Weiss HA, Massae P, Mtuy T, Makupa W, Essex D, Mabey DC, Bailey RL, Holland MJ, Burton MJ. 2016. Immunohistochemical analysis of scarring trachoma indicates infiltration by natural killer and undefined CD45 negative cells. PLoS Negl Trop Dis 10:e0004734. doi: 10.1371/journal.pntd.0004734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radomski N, Franzke K, Matthiesen S, Karger A, Knittler MR. 2019. NK cell-mediated processing of Chlamydia psittaci drives potent anti-bacterial Th1 immunity. Sci Rep 9:4799. doi: 10.1038/s41598-019-41264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sim MJW, Rajagopalan S, Altmann DM, Boyton RJ, Sun PD, Long EO. 2019. Human NK cell receptor KIR2DS4 detects a conserved bacterial epitope presented by HLA-C. Proc Natl Acad Sci U S A 116:12964–12973. doi: 10.1073/pnas.1903781116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao L, Gao X, Bai H, Joyee AG, Wang S, Yang J, Zhao W, Yang X. 2019. The important role of dendritic cell (DC) in iNKT-mediated modulation of NK cell function in Chlamydia pneumoniae lung infection. Mediators Inflamm 2019:4742634. doi: 10.1155/2019/4742634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igietseme JU, Portis JL, Perry LL. 2001. Inflammation and clearance of Chlamydia trachomatis in enteric and nonenteric mucosae. Infect Immun 69:1832–1840. doi: 10.1128/IAI.69.3.1832-1840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeruva L, Spencer N, Bowlin AK, Wang Y, Rank RG. 2013. Chlamydial infection of the gastrointestinal tract: a reservoir for persistent infection. Pathog Dis 68:88–95. doi: 10.1111/2049-632X.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Zhang Q, Zhang T, Zhang Y, Zhu C, Sun X, Zhang N, Xue M, Zhong G. 2016. The Chlamydia muridarum organisms fail to auto-inoculate the mouse genital tract after colonization in the gastrointestinal tract for 70 days. PLoS One 11:e0155880. doi: 10.1371/journal.pone.0155880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect Immun 62:2094–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect Immun 70:2741–2751. doi: 10.1128/iai.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhou Z, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. doi: 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shekhar S, Peng Y, Wang S, Yang X. 2018. CD103+ lung dendritic cells (LDCs) induce stronger Th1/Th17 immunity to a bacterial lung infection than CD11b(hi) LDCs. Cell Mol Immunol 15:377–387. doi: 10.1038/cmi.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Zhu C, Zhang T, Tian Q, Zhang N, Morrison S, Morrison R, Xue M, Zhong G. 2018. Nonpathogenic colonization with chlamydia in the gastrointestinal tract as oral vaccination for inducing transmucosal protection. Infect Immun 86:e00630-17. doi: 10.1128/IAI.00630-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu C, Lin H, Tang L, Chen J, Wu Y, Zhong G. 2018. Oral Chlamydia vaccination induces transmucosal protection in the airway. Vaccine 36:2061–2068. doi: 10.1016/j.vaccine.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 39.Campbell J, Huang Y, Liu Y, Schenken R, Arulanandam B, Zhong G. 2014. Bioluminescence imaging of Chlamydia muridarum ascending infection in mice. PLoS One 9:e101634. doi: 10.1371/journal.pone.0101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q, Huang Y, Gong S, Yang Z, Sun X, Schenken R, Zhong G. 2015. In vivo and ex vivo imaging reveals a long-lasting chlamydial infection in the mouse gastrointestinal tract following genital tract inoculation. Infect Immun 83:3568–3577. doi: 10.1128/IAI.00673-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai J, Zhang T, Wang L, Shao L, Zhu C, Zhang Y, Failor C, Schenken R, Baseman J, He C, Zhong G. 2016. Intravenous inoculation with Chlamydia muridarum leads to a long-lasting infection restricted to the gastrointestinal tract. Infect Immun 84:2382–2388. doi: 10.1128/IAI.00432-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong G. 2018. Chlamydia spreading from the genital tract to the gastrointestinal tract—a two-hit hypothesis. Trends Microbiol 26:611–623. doi: 10.1016/j.tim.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eberl G, Colonna M, Di Santo JP, McKenzie AN. 2015. Innate lymphoid cells: a new paradigm in immunology. Science 348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. 2018. Innate lymphoid cells: 10 years on. Cell 174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 45.Castleman MJ, Dillon SM, Purba CM, Cogswell AC, Kibbie JJ, McCarter MD, Santiago ML, Barker E, Wilson CC. 2019. Commensal and pathogenic bacteria indirectly induce IL-22 but not IFNgamma production from human colonic ILC3s via multiple mechanisms. Front Immunol 10:649. doi: 10.3389/fimmu.2019.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fung TC, Artis D, Sonnenberg GF. 2014. Anatomical localization of commensal bacteria in immune cell homeostasis and disease. Immunol Rev 260:35–49. doi: 10.1111/imr.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Kim TJ, Wroblewska JA, Tesic V, Upadhyay V, Weichselbaum RR, Tumanov AV, Tang H, Guo X, Tang H, Fu YX. 2018. Type 3 innate lymphoid cell-derived lymphotoxin prevents microbiota-dependent inflammation. Cell Mol Immunol 15:697–709. doi: 10.1038/cmi.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiao Y, Huntington ND, Belz GT, Seillet C. 2016. Type 1 innate lymphoid cell biology: lessons learnt from natural killer cells. Front Immunol 7:426. doi: 10.3389/fimmu.2016.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duerr CU, Fritz JH. 2016. Regulation of group 2 innate lymphoid cells. Cytokine 87:1–8. doi: 10.1016/j.cyto.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 50.Killig M, Glatzer T, Romagnani C. 2014. Recognition strategies of group 3 innate lymphoid cells. Front Immunol 5:142. doi: 10.3389/fimmu.2014.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montaldo E, Juelke K, Romagnani C. 2015. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol 45:2171–2182. doi: 10.1002/eji.201545598. [DOI] [PubMed] [Google Scholar]
- 52.Withers DR, Hepworth MR. 2017. Group 3 innate lymphoid cells: communications hubs of the intestinal immune system. Front Immunol 8:1298. doi: 10.3389/fimmu.2017.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. 2019. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis 10:315. doi: 10.1038/s41419-019-1540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nussbaum K, Burkhard SH, Ohs I, Mair F, Klose CSN, Arnold SJ, Diefenbach A, Tugues S, Becher B. 2017. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J Exp Med 214:2331–2347. doi: 10.1084/jem.20162031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. 2010. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neurath MF. 2019. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev 45:1–8. doi: 10.1016/j.cytogfr.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 57.Eken A, Singh AK, Treuting PM, Oukka M. 2014. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol 7:143–154. doi: 10.1038/mi.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, Diefenbach A. 2010. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A. 2013. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494:261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- 60.Sciumé G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, O’Shea JJ. 2012. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209:2331–2338. doi: 10.1084/jem.20122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cella M, Gamini R, Secca C, Collins PL, Zhao S, Peng V, Robinette ML, Schettini J, Zaitsev K, Gordon W, Bando JK, Yomogida K, Cortez V, Fronick C, Fulton R, Lin LL, Gilfillan S, Flavell RA, Shan L, Artyomov MN, Bowman M, Oltz EM, Jelinsky SA, Colonna M. 2019. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol 20:980–991. doi: 10.1038/s41590-019-0425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koprivsek JJ, Zhang T, Tian Q, He Y, Xu H, Xu Z, Zhong G. 2019. Distinct roles of chromosome- versus plasmid-encoded genital tract virulence factors in promoting Chlamydia muridarum colonization in the gastrointestinal tract. Infect Immun 87:e00265-19. doi: 10.1128/IAI.00265-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yokoyama WM, Ryan JC, Hunter JJ, Smith HR, Stark M, Seaman WE. 1991. cDNA cloning of mouse NKR-P1 and genetic linkage with LY-49. Identification of a natural killer cell gene complex on mouse chromosome 6. J Immunol 147:3229–3236. [PubMed] [Google Scholar]
- 64.Kang J, Coles M. 2012. IL-7: the global builder of the innate lymphoid network and beyond, one niche at a time. Semin Immunol 24:190–197. doi: 10.1016/j.smim.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinette ML, Bando JK, Song W, Ulland TK, Gilfillan S, Colonna M. 2017. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat Commun 8:14601. doi: 10.1038/ncomms14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim HR, Hwang KA, Park SH, Kang I. 2008. IL-7 and IL-15: biology and roles in T-cell immunity in health and disease. Crit Rev Immunol 28:325–339. doi: 10.1615/critrevimmunol.v28.i4.40. [DOI] [PubMed] [Google Scholar]
- 67.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F, Lupo-Stanghellini MT, Mavilio F, Mondino A, Bicciato S, Recchia A, Bonini C. 2013. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Marotel M, Fauteux-Daniel S, Mathieu A-L, Viel S, Marçais A, Walzer T. 2018. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol 48:738–750. doi: 10.1002/eji.201747299. [DOI] [PubMed] [Google Scholar]
- 69.Viant C, Rankin LC, Girard-Madoux MJ, Seillet C, Shi W, Smyth MJ, Bartholin L, Walzer T, Huntington ND, Vivier E, Belz GT. 2016. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal 9:ra46. doi: 10.1126/scisignal.aaf2176. [DOI] [PubMed] [Google Scholar]
- 70.Sheppard S, Schuster IS, Andoniou CE, Cocita C, Adejumo T, Kung SKP, Sun JC, Degli-Esposti MA, Guerra N. 2018. The murine natural cytotoxic receptor NKp46/NCR1 controls TRAIL protein expression in NK cells and ILC1s. Cell Rep 22:3385–3392. doi: 10.1016/j.celrep.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Y, Dong W, Zhang Y, Caligiuri MA, Yu J. 2018. Dependence of innate lymphoid cell 1 development on NKp46. PLoS Biol 16:e2004867. doi: 10.1371/journal.pbio.2004867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colonna M. 2018. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity 48:1104–1117. doi: 10.1016/j.immuni.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piersma SJ, Pak-Wittel MA, Lin A, Plougastel-Douglas B, Yokoyama WM. 2019. Activation receptor-dependent IFN-gamma production by NK cells is controlled by transcription, translation, and the proteasome. J Immunol 203:1981–1988. doi: 10.4049/jimmunol.1900718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perchet T, Petit M, Banchi EG, Meunier S, Cumano A, Golub R. 2018. The Notch signaling pathway is balancing type 1 innate lymphoid cell immune functions. Front Immunol 9:1252. doi: 10.3389/fimmu.2018.01252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narni-Mancinelli E, Gauthier L, Baratin M, Guia S, Fenis A, Deghmane AE, Rossi B, Fourquet P, Escaliere B, Kerdiles YM, Ugolini S, Taha MK, Vivier E. 2017. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol 2:eaam9628. doi: 10.1126/sciimmunol.aam9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhong C, Zheng M, Zhu J. 2018. Lymphoid tissue inducer—a divergent member of the ILC family. Cytokine Growth Factor Rev 42:5–12. doi: 10.1016/j.cytogfr.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin YL, Ip PP, Liao F. 2017. CCR6 deficiency impairs IgA production and dysregulates antimicrobial peptide production, altering the intestinal flora. Front Immunol 8:805. doi: 10.3389/fimmu.2017.00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cording S, Medvedovic J, Lecuyer E, Aychek T, Dejardin F, Eberl G. 2018. Mouse models for the study of fate and function of innate lymphoid cells. Eur J Immunol 48:1271–1280. doi: 10.1002/eji.201747388. [DOI] [PubMed] [Google Scholar]
- 79.Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, Kutter L, Alcantara MA, Byndloss MX, Tiffany CR, Walker GT, Faber F, Zhu Y, Bronner DN, Byndloss AJ, Tsolis RM, Zhou H, Baumler AJ. 2019. Commensal Enterobacteriaceae protect against Salmonella colonization through oxygen competition. Cell Host Microbe 25:128–139.e5. doi: 10.1016/j.chom.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 80.Thiemann S, Smit N, Roy U, Lesker TR, Galvez EJC, Helmecke J, Basic M, Bleich A, Goodman AL, Kalinke U, Flavell RA, Erhardt M, Strowig T. 2017. Enhancement of IFNgamma production by distinct commensals ameliorates Salmonella-induced disease. Cell Host Microbe 21:682–694.e5. doi: 10.1016/j.chom.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 81.Caballero S, Kim S, Carter RA, Leiner IM, Susac B, Miller L, Kim GJ, Ling L, Pamer EG. 2017. Cooperating commensals restore colonization resistance to vancomycin-resistant Enterococcus faecium. Cell Host Microbe 21:592–602.e4. doi: 10.1016/j.chom.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Finethy R, Coers J. 2016. Sensing the enemy, containing the threat: cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol Rev 40:875–893. doi: 10.1093/femsre/fuw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. 2013. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 84.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. 2013. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, Begum-Haque S, Kasper DL, Kasper LH. 2015. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes 6:234–242. doi: 10.1080/19490976.2015.1056973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen C, Zhou Z, Conrad T, Yang Z, Dai J, Li Z, Wu Y, Zhong G. 2015. In vitro passage selects for Chlamydia muridarum with enhanced infectivity in cultured cells but attenuated pathogenicity in mouse upper genital tract. Infect Immun 83:1881–1892. doi: 10.1128/IAI.03158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Conrad TA, Gong S, Yang Z, Matulich P, Keck J, Beltrami N, Chen C, Zhou Z, Dai J, Zhong G. 2016. The chromosome-encoded hypothetical protein TC0668 is an upper genital tract pathogenicity factor of Chlamydia muridarum. Infect Immun 84:467–479. doi: 10.1128/IAI.01171-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morrison SG, Giebel AM, Toh EC, Spencer HJ III, Nelson DE, Morrison RP. 2018. Chlamydia muridarum genital and gastrointestinal infection tropism is mediated by distinct chromosomal factors. Infect Immun 86:e00141-18. doi: 10.1128/IAI.00141-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Giebel AM, Hu S, Rajaram K, Finethy R, Toh E, Brothwell JA, Morrison SG, Suchland RJ, Stein BD, Coers J, Morrison RP, Nelson DE, Giebel AM, Hu S, Rajaram K, Finethy R, Toh E, Brothwell JA, Morrison SG, Suchland RJ, Stein BD, Coers J, Morrison RP, Nelson DE. 2019. Genetic screen in Chlamydia muridarum reveals role for an interferon-induced host cell death program in antimicrobial inclusion rupture. mBio 10:e00385-19. doi: 10.1128/mBio.00385-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang T, Huo Z, Ma J, He C, Zhong G, Zhang T, Huo Z, Ma J, He C, Zhong G. 2019. The plasmid-encoded pGP3 promotes Chlamydia evasion of acidic barriers in both stomach and vagina. Infect Immun 87:e00844-18. doi: 10.1128/IAI.00844-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shao L, Zhang T, Liu Q, Wang J, Zhong G. 2017. Chlamydia muridarum with mutations in chromosomal genes tc0237 and/or tc0668 is deficient in colonizing the mouse gastrointestinal tract. Infect Immun 85:e00321-17. doi: 10.1128/IAI.00321-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 93.Lin H, He C, Koprivsek JJ, Chen J, Zhou Z, Arulanandam B, Xu Z, Tang L, Zhong G. 2019. Antigen-specific CD4(+) T cell-derived gamma interferon is both necessary and sufficient for clearing Chlamydia from the small intestine but not the large intestine. Infect Immun 87:e00055-19. doi: 10.1128/IAI.00055-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]