

Abstract
Background
Accumulating evidence suggests a regulatory role of Wnt proteins in innate immune responses. However, the effects of Wnt3a signaling on TLR4-mediated inflammatory responses are controversial and the signaling crosstalk between TLR4 and Wnt3a remains uncertain.
Methods
Gain- and Loss- of function approaches were utilized to determine the function of Wnt3a signaling in TLR4-mediated inflammatory responses. Cytokine production at protein and mRNA levels and phosphorylation of signaling molecules were measured by ELISA, qRT-PCR, and Western Blot, respectively. Endotoxemia mouse model was employed to assess the effect of Wnt3a on systemic inflammatory cytokine levels and neutrophil infiltration.
Results
LPS stimulation leads to an increase of Wnt3a expression and its downstream molecule, Dvl3, in primary monocytes. Inhibition or silence of Wnt3a or Dvl3 significantly increases the production of pro-inflammatory cytokines (IL-12, IL-6, TNFα), robustly reduces β-catenin accumulation, and enhances the phosphorylation of NF-κB P65 and its DNA binding activity. These results were confirmed by multiple gain- and loss- of function approaches including specific siRNA and ectopic expression of Dvl3, GSK3β, and β-catenin in monocytes. Moreover, in vivo relevance was established in a murine endotoxin model, in which Wnt3a inhibition enhances the inflammatory responses by augmenting the systemic pro-inflammatory cytokine levels and neutrophil infiltration.
Conclusions
TLR4 activation promotes Wnt3a-Dvl3 signaling, which act as rheostats to restrain the intensity of inflammation through regulating GSK3β-β-catenin signaling and NF-κB activity.
General significance
Wnt3a-Dvl3-β-catenin signaling axis could be a potential interventional target for manipulating the direction and intensity of inflammatory responses.
Introduction
Evolution builds and reshapes our immune system to adapt to the continuous irritation from the external stimulus. While microbial pathogens try to seek a conductive environment to benefit their survival, our immune system also develops a complicated mechanism against invading microbes, mainly through inflammatory immune response. Inflammation responses are divided into pro-inflammatory and anti-inflammatory responses: the former initiates a series of immune responses to eradicate invaded microbes of substances, while the latter sentinels and restrains the intensity of inflammation to protect the host from the potential collateral tissue damage inflicted by overwhelming inflammation [1, 2]. Although there are a plethora of studies characterizing the pro-inflammatory responses, the anti-inflammatory mechanisms are underappreciated, which is frequently regarded as only a passive and negative feedback such as the production of anti-inflammatory cytokines (IL-10, IL-1Ra, and TGF-beta) or the synthesis of some inflammation suppressors, i.e., IκB, suppressor of cytokine signaling (SOCS) to aid the resolution of inflammation [2, 3]. In this regard, studies have shown that anti-inflammatory responses could also be an active process and initiated simultaneously to restrain the duration and magnitude of pro-inflammatory responses [1, 4]. Apart from the seminal work on the function of PI3K-GSK3β in TLR4-mediated inflammatory responses [5], multiple other anti-inflammatory mechanisms such as mitogen-activated protein kinase phosphatase-1 (MKP-1) [6], Insulin receptor substrate 1 (IRS-1), A20 [7, 8], Phosphoinositide 3-kinase (PI3K), β-arrestin [9], JAK3, and SGK1 [10, 11] were reported to suppress the progression of inflammatory responses. Considering the importance of the homeostasis between pro- and anti- inflammatory responses, identification of additional anti-inflammatory mechanisms and delineation of related signaling will yield novel insights on inflammation regulation and provide more potential interventional targets for the control of inflammatory diseases.
Wnt proteins are a group of secreted, cysteine-rich, palmitoylated glycoproteins that were originally defined as regulators of embryogenesis, cell proliferation and migration [12, 13]. There are at least three Wnt pathways, including canonical, alternative, and Wnt/planar cell (PCP) pathways. Activation of the canonical Wnt signaling pathway has previously been shown to recruit Dishevelled (Dvl/Dsh) protein and enhance phosphorylation of GSK3β, thus influencing β-catenin degradation and NF-κB-dependent gene transcription [14–16]. In an alternative Wnt cascade, activation of phospholipase C (PLC) leads to Ca++ influx, PKC activation and NFAT- and NF-κB-mediated transcription events [14, 16–18]. In the PCP pathway, small G-proteins, such as Rac1 and RhoA, are activated and, subsequently, MAPK/JNK1/2, which results in AP-1-driven transcriptional activity [14, 19, 20]. Recent studies have reported the functional roles for Wnt signaling, especially canonical signaling, in the regulation of inflammatory responses [14, 21]. However, the regulatory effects of Wnt3a-β-catenin signaling are controversial depending on different contexts. For example, Wnt3a-Fzd1 interaction has been shown to induce β-catenin and suppress pro-inflammatory cytokine production in M. tuberculosis or LPS-stimulated epithelial cells [14, 22]. The other specific example is that S. typhimurium induced β-catenin accumulation suppresses inflammation through physical interactions with NF-κB, in a similar manner to IκB [23]. In contrast, a recent study using LPS-stimulated human umbilical vein endothelial cells reported an opposite effect of Wnt3a-β-catenin signaling, showing Wnt3a and β-catenin are required for LPS induced inflammatory responses in endothelial cells as well as epithelial cells. Wnt3a inhibitor, LGK974, or its suppressor, Dkk1, abolishes LPS-induced inflammatory cytokines [24]. In addition, Schaale et al pointed out that TLR signaling could modify the expression and activity of Wnt signaling pathways, and through which to maintain the homeostasis of TLR-mediated inflammatory responses [14]. However, more evidence of the interaction between TLR4 and Wnt3a signaling is required to support this hypothesis.
As a key component of Wnt signaling, Dvls function as a multivalent scaffold to form dynamic protein aggregates, so-called “signalsomes”, which propagate signals from the Wnt-frizzled receptor to multiple downstream elements. Dvl has been reported to be involved in canonical-, non-canonical- Wnt signaling, Wnt-calcium signaling, Wnt-RYK signaling, and Wnt-atypical PKC signaling through different mechanisms such as degradation of β-catenin, Diaam 1-Rho A axis, or Rac-1 axis [25]. Despite all three isoforms of Dvl (Dvl1, Dvl2, and Dvl3) proteins are engaged in Wnt3a-mediated canonical signaling, Dvl3 is more sensitive to Wnt3a stimulation and overexpression of Dvl3 is Wnt3a-mimetic [25, 26]. Moreover, Dvl3 allelic variations could interact with pro-inflammatory cytokines and through which involved in the progression of some diseases like major depressive disorder [27]. Dvl proteins also can stimulate the formation of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) by sequential actions of PtdIns 4-kinase type II (PI4KIIa) and PtdIns-4-phosphate 5-kinase type I (PIP5KI) [28, 29]. Although PtdIns(4,5)P2-mediated activation of Akt signaling has been demonstrated with a gatekeeping function in inflammation and innate immunity [30, 31], the potential immunomodulatory function of Dvl3 has never been reported in previous studies. Thus, the interaction between TLR4 and Wnt3a activation, as well as the effects of Wnt3a/Dvl3 signaling on TLR4-mediated inflammatory responses, remain uncertain.
In this study, we demonstrated the anti-inflammatory function of Wnt3a-Dvl3 signaling in innate immune cells and revealed for the first time that TLR4 activation enhances the expression of Wnt3a and Dvl3. TLR4-mediated signaling model, Wnt3a-Dvl3-GSK3β-β-catenin, act as a rheostat to restrain the activity of NF-κB. Moreover, we confirmed these results in a mouse endotoxemia model. These findings suggest that Wnt3a-Dvl3 signaling could be an interventional target for manipulating the direction and intensity of inflammatory responses, depending on clinical necessity.
Material and Method
Mice and reagents
8 weeks C57BL/ mice were from The Jackson Laboratory (Bar Harbor, ME, USA) and housed in a specific pathogen-free facility at the University of Louisville with approved all animal protocols from University of Louisville Institutional Animal Care and Use Committee. Ultrapure LPS from Escherichia coli 0111:B4 were from InvivoGen (San Diego, CA, USA). Wnt3a, Wnt7 and Dvl3 antibodies were from Abcam (Cambridge, UK). Dvl2 antibody was from ThermoFisher Scientific (Rockford, IL). All other antibodies were from Cell Signaling Technology (Danvers, MA, USA). Monocyte isolation kits was from Miltenyi Biotec (Auburn, CA). RPMI Complete (RPMI 1640 medium supplemented with 10 % FBS, 50 μM 2-ME, 1 mM sodium pyruvate, 2 mM l-glutamine, 20 mM HEPES, 50 U/ml penicillin, and 50 μg/ml streptomycin) was from Invitrogen Life Technologies (Carlsbad, CA). Enhanced chemiluminescence kits were from Thermo Scientific (Rockford, IL). Cytokine ELISA kits (TNFα, IL-6 and IL-12/23 p40) were from eBioscience (San Diego, CA). The small molecular inhibitors, N-(6-methyl-2-benzothiazolyl)-2-[(3,4,6,7-tetrahydro-4-oxo-3-phenylthieno[3,2-d] pyrimidin-2-yl) thio]-acetamide (IWP-2) and benzoic acid, 2-phenoxy-,2-[(5-methyl-2-furanyl) methylene] hydrazide (PNU-74654), were from Tocris Biosciences (Minneapolis, MN). The GSK3 inhibitor, SB216763, was from Sigma-Aldrich (St. Louis, MO). Human Monocytes Nucleofector kit, including pCMV-GFP, was from Amaxa AG (Koln, Germany), while SMARTpool siRNA was from Dharmacon (Lafayette, CO). The NF-κB p65 (Ser529/Ser536) inhibitory peptide set (p65 inhibitory peptide sequence, 5′-DRQIKIWFQNRRMKWKKNGLLSGDEDFSS-3′; control sequence, 5′-DRQIKIWFQNRRMKWKK-3′) was from Novus Biologicals (Littleton, CO, USA). All plasmids including pcDNA3-S33Y β-catenin (Cat. No #19286) with Flag tag, pcDNA3 Flag HA (Cat. No #10792), which was constructed with HA and Flag tags, HA GSK3β S9A pcDNA3 (Cat. No #14754), M77 pIRESpuro-Glue-hDsh3 (Cat. No #15102), and pIRESpuro-GLUE (Cat. No #15100) were from Addgene (Watertwon, MA, USA).
Preparation of human monocytes and measurement of the cytokine release by human monocytes
PBMCs were obtained from healthy donors as per protocols approved by the University of Louisville, Institutional Review Board, Human Subjects Protection Program. Primary human monocytes were purified from anonymized, citrated whole blood by indirect magnetic monocyte isolation, as we have previously reported [10]. This procedure routinely results in >95 % pure CD14+cells, as shown by flow cytometry. Human monocytes were cultured at 37 °C and 5 % CO2 atmosphere in complete RPMI plus or minus stimulating agents, as described below. Primary human monocytes were stimulated with or without inhibitor, siRNA, exogenous plasmids and/or LPS (1ug/ml), as described in the figure legends. Organic solvent controls (0.01 % DMSO) were included, as appropriate. Cell-free supernatants were harvested after 24 h stimulation with LPS and assayed for cytokine levels by ELISA, according to the manufacturer’s instructions.
Transfection of siRNA, exogenous plasmids and measurement of Wnt3a signaling components by Western blot
Transfection of human monocytes was carried out by electroporation using an Amaxa 4D-Nucleofector device (Lonza, Cologne, Germany) according to the manufacturer’s protocols. Briefly, purified monocytes (4 × 106) were resuspended in 100 μl Nucleofector Solution (Amaxa Human Monocyte Nucleofector Kit; Lonza) with 2 μg siRNA duplexes or ectopic plasmids for each target. Immediately after electroporation, 400 μl prewarmed M-199 containing 10% fetal calf serum (FCS) was added to cells that were then transferred into culture plates containing prewarmed M-199 with 10% FCS. At 48 h post-transfection, cells were exposed to LPS (1 μg/ml) with or without other treatments. Cell lysates were prepared as previously described [32], and the levels of total Wnt3a, Dvl3, GSK3β and β-catenin were assessed by Western blot. Images were acquired using the ImageQuant LAS 100 (GE Healthcare BioScience, Pittsburgh, PA, USA). For experiments using inhibitors for Wnt3a (100nM IWP-2 or 1μM PNU-74654), control cells were pretreated for 2 h with 0.01% DMSO (organic solvent control) prior to LPS (1 μg/ml) stimulation.
RT-PCR and NF-κB p65 nuclear-binding assay
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA), and real-time RT-PCR was performed using an Applied Biosystems 7500 system (Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control, and fold increase was calculated according to ΔΔCT method. Nuclear lysates were obtained from human monocytes using a nuclear/cytosolic isolation kit (Active Motif, Carlsbad, CA, USA). Nuclear lysates were analyzed for DNA-binding levels of NF-κB p65 using TransAM NF-κB p65 (Active Motif, Carlsbad, CA, USA) and performed according to the manufacturer’s protocol.
Endotoxin model and immunohistochemistry analysis
C57BL/6 mice were injected intraperitoneally with a sublethal dose of E. coli 0111:B4 LPS (1 mg/kg) with or without the pretreatment of IWP-2 (10 mg/kg) for 2 h. Control mice were injected with vehicle only. After 4 h and 24 h intraperitoneal injection of vehicle or E. coli LPS, blood was collected, and plasma was isolated to measure the levels of TNFα, IL-6, and IL-12P40. Lung was harvested 4 h and 24 h post-injection of LPS but from separate groups. Also, the lung tissue samples were transferred to ice-cold PBS, and processed for myeloperoxidase determination and immunofluorescence analysis. The analyses of tissue myeloperoxidase were performed using a murine MPO-specific enzyme-linked immunosorbent assay (ELISA; HyCult Biotechnology) according to the manufacturer’s instructions. Plasma levels of TNFα, IL-6, and IL-12 were determined by ELISA. Lung tissue samples from septic mice with or without treatment with IWP-2 were fixed in 4% formaldehyde, embedded in optimal cutting temperature compound (Fisher Scientific; Pittsburgh, PA, USA), and stored at −80°C. Serial sections (8 μm thick) were cut and stained with hematoxylin and eosin (H&E) to evaluate the inflammatory cell infiltration and tissue damage. Immunofluorescence staining was performed to assess infiltrated neutrophils. Slides were rehydrated, blocked, and incubated for 1 h at room temperature with primary FITC-conjugated antibodies to mouse Ly6G, a specific neutrophil marker (FITC conjugate; LifeSpan BioSciences, Incorporated, Seattle, WA, USA). The specificity of staining was confirmed by using appropriate FITC-conjugated isotype controls or normal rabbit IgG followed by Alexa Fluor 594 goat anti-rabbit IgG. Images were captured using a fluorescence microscope (Eclipse E800; Nikon, Tokyo, Japan) and processed by Neurolucida (MBF Bioscience, Williston, VT, USA).
Statistical analyses
Statistical significance between groups was evaluated by the analysis of variance and the Tukey multiple comparison test using the InStat program (GraphPad, San Diego, CA). Differences between groups were considered significant at the level of P ≤ 0.05.
Results
Inhibition of Wnt3a-Dvl3 enhances the production of LPS-induced inflammatory cytokines in innate immune cells
Since the effects of Wnt3a on TLR-mediated inflammation are controversial [14, 22, 24] and its downstream signaling remains less clear, here, we first inhibit Wnt3a activity using two different chemical inhibitors, IWP-2 and PNU-74654, to test the effects of Wnt3a on TLR4-mediated inflammatory cytokine production. We found that Wnt3a inhibition significantly enhances the production of IL-12P40, IL-6 and TNFα in E. coil LPS stimulated human monocytes, which is also confirmed in THP1 cell lines (Fig. 1A, B). As we know, IWP-2 targets Wnt-dependent phosphorylation of LRP6 and Dvl and, thus, inhibits β-catenin accumulation while PNU-74654 binds to β-catenin and, subsequently, prevents the interactions between β-catenin and transcription factors. To avoid the potential non-specific influences of these chemical inhibitors, siRNA was utilized to knock down Wnt3a and we observed that Wnt3a deficiency significantly increases LPS-induced inflammatory cytokines in human monocytes (Fig. 1C, D). Due to the pivotal role of Dvl3 in canonical and non-canonical Wnt signaling pathways [25, 27], and its unknown influences on TLR4-mediated inflammation, we next silenced Dvl3 using siRNA (Fig. 1C) and examined the potential influences of Dvl3 on LPS-induced inflammatory cytokine levels in monocytes. As showed in figure 1E and F, silence of Dvl3 leads to a significant increase of pro-inflammatory cytokine production at both message and protein levels in LPS stimulated human monocytes. These results demonstrated the anti-inflammatory property of Dvl3 for the first time, and consolidated that the Wnt3a-Dvl3 signaling inhibits rather than promotes LPS-induced inflammatory cytokine in innate immune cells.
Figure 1. Inhibition of Wnt3a-Dvl3 enhances the production of LPS-induced inflammatory cytokines in innate immune cells.
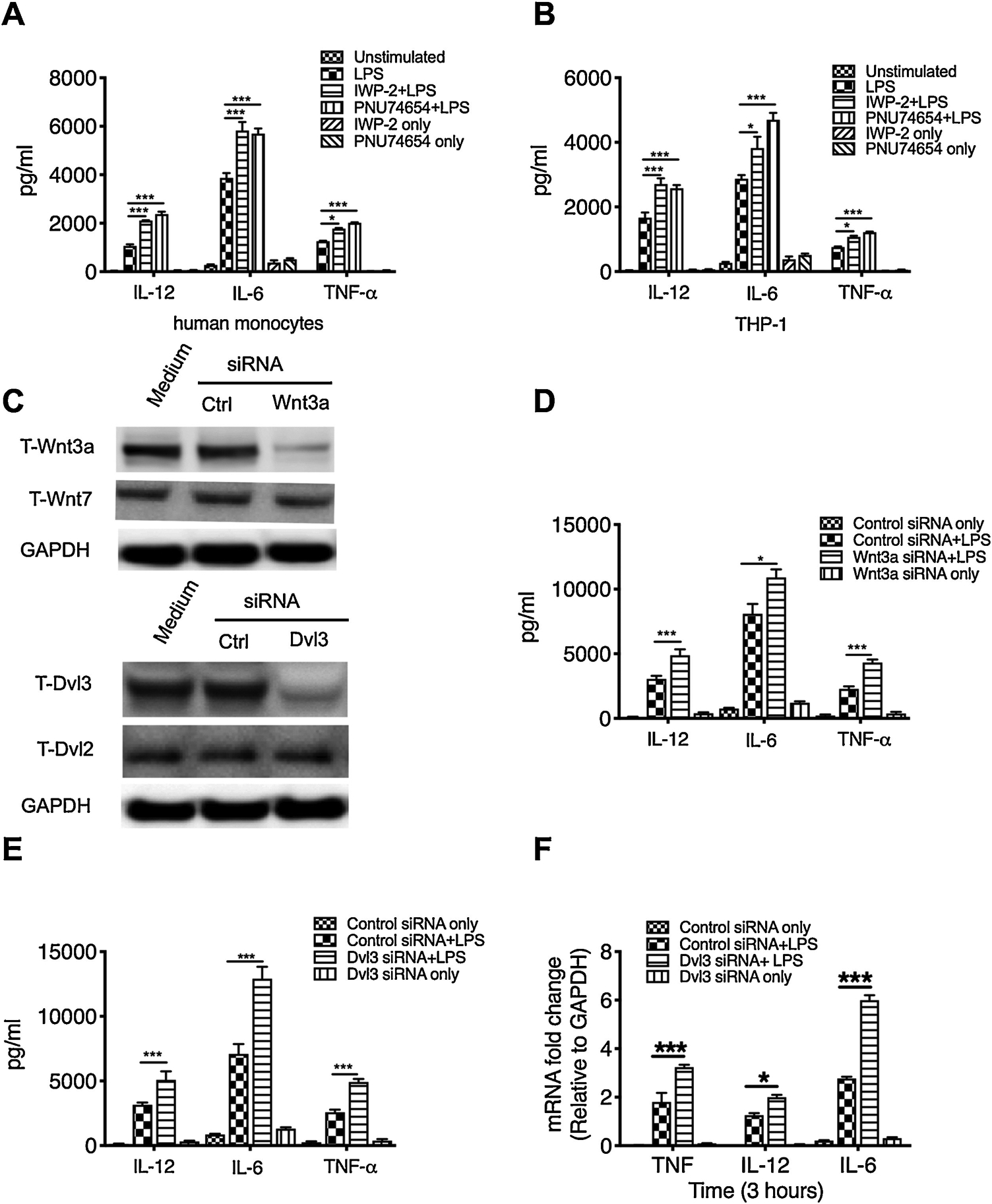
Purified human monocytes and THP1 cells were pretreated with different Wnt3a inhibitors (100nM IWP-2 or 1μM PNU-74654) for 2 hours or transfected with pre-validated siRNA targeting Wnt3a or Dvl3 for 48 hours, then stimulated with E. coil LPS (1μg/ml). After 24 hours stimulation, the cell-free supernatants were collected and the production of IL-12P40, IL-6, and TNFα by monocytes (A) and THP1 (B) was measured by ELISA. (C) Silencing of wnt3a or dvl3 gene in human monocytes robustly reduces the expression of Wnt3a and Dvl3, but didn’t affect other proteins (Wnt7 or Dvl2). D and E, the production of IL-12P40, IL-6, and TNFα in LPS stimulated human monocytes with or without the pre-treatment of Wnt3a siRNA (C) or Dvl3 siRNA (E). The mRNA levels of inflammatory cytokines were measured by qRT-PCR in Dvl3 siRNA-treated monocytes after 3 hours stimulation with LPS (F). *, and *** indicates statistically significant at P<0.05, and P<0.001, respectively. Data represent the arithmetic mean±S.D. of three independent experiments.
LPS challenge enhances the expression of Dvl3 and Wnt3a in monocytes
Along with pro-inflammatory signaling pathways, a plethora of anti-inflammatory signaling pathways are simultaneously activated upon TLR4 activation [4, 10]. To determine if TLR4 activation affects the activity of Wnt3a-Dvl3 signaling, we next examine the expression of Wnt3a-Dvl3 and its downstream molecules in LPS-stimulated human monocytes. We find that LPS indeed induce a transient increase of Wnt3a, Dvl3, and β-catenin, a downstream molecule involved in the Wnt3a-mediated canonical signaling pathway (Fig. 2A to C), which are all waned away after 4 hours of LPS challenge. To further examine the influences of TLR4 activation on the expression of Wnt3a and Dvl3, we next examined their mRNA levels in LPS stimulated human monocytes. We didn’t observe the significant change of mRNA for both Wnt3a and Dvl3 after 2 h challenge with LPS (Figure 2G), suggesting that LPS-enhanced Wnt3a and Dvl3 expression is through other mechanisms. GSK3β has been shown to be critical for β-catenin degradation and involved in both the PI3K-Akt and the Wnt3a canonical signaling pathways [33, 34]. Considering the influences of different GSK3β compartmentalization on the consequences of signaling pathways[35, 36], we next test the phosphorylation of Akt and NF-κB activation, representing by phosphorylation of P65 at serine 536, using the same blot. As showed in figure 2D, we found that LPS stimulation leads to the phosphorylation of AKT, GSK3β, and NF-κB P65, and also alters the expression of total IκB. Moreover, the increased phosphorylation of Akt and GSK3β are a little delay than the increase of total IκB, suggesting other downstream molecules rather than IκB employed by GSK3β to suppress the activity of NF-κB. Since the decrease of NF-κB phosphorylation occurs simultaneously with the enhancement of GSK3β and β-catenin phosphorylation, our results indicates β-catenin could be a suppressor of NF-κB activity in LPS stimulated monocytes. Thus, we next utilize Wnt3a and Dvl3 siRNA to examine Wnt3a-mediated GSK3β-β-catenin in LPS stimulated monocytes. We found that silence of either Wnt3a or Dvl3 robustly attenuates the increased expression of β-catenin, phosphorylation of GSK3β and enhances phosphorylation of NF-κB in LPS-stimulated monocytes (Fig. 2E, F), which suggests Wnt3a-Dvl3 indeed regulate, at least in part, the activity and GSK3β-β-catenin, and subsequently restrain NF-κB transcription activity. These results demonstrated Wnt3-Dvl3 and its downstream signaling components are inducible, and act as an anti-inflammatory mechanism in LPS-stimulated innate immune cells.
Figure 2. LPS challenge enhances the expression of Wnt3a-Dvl3 signaling in monocytes.
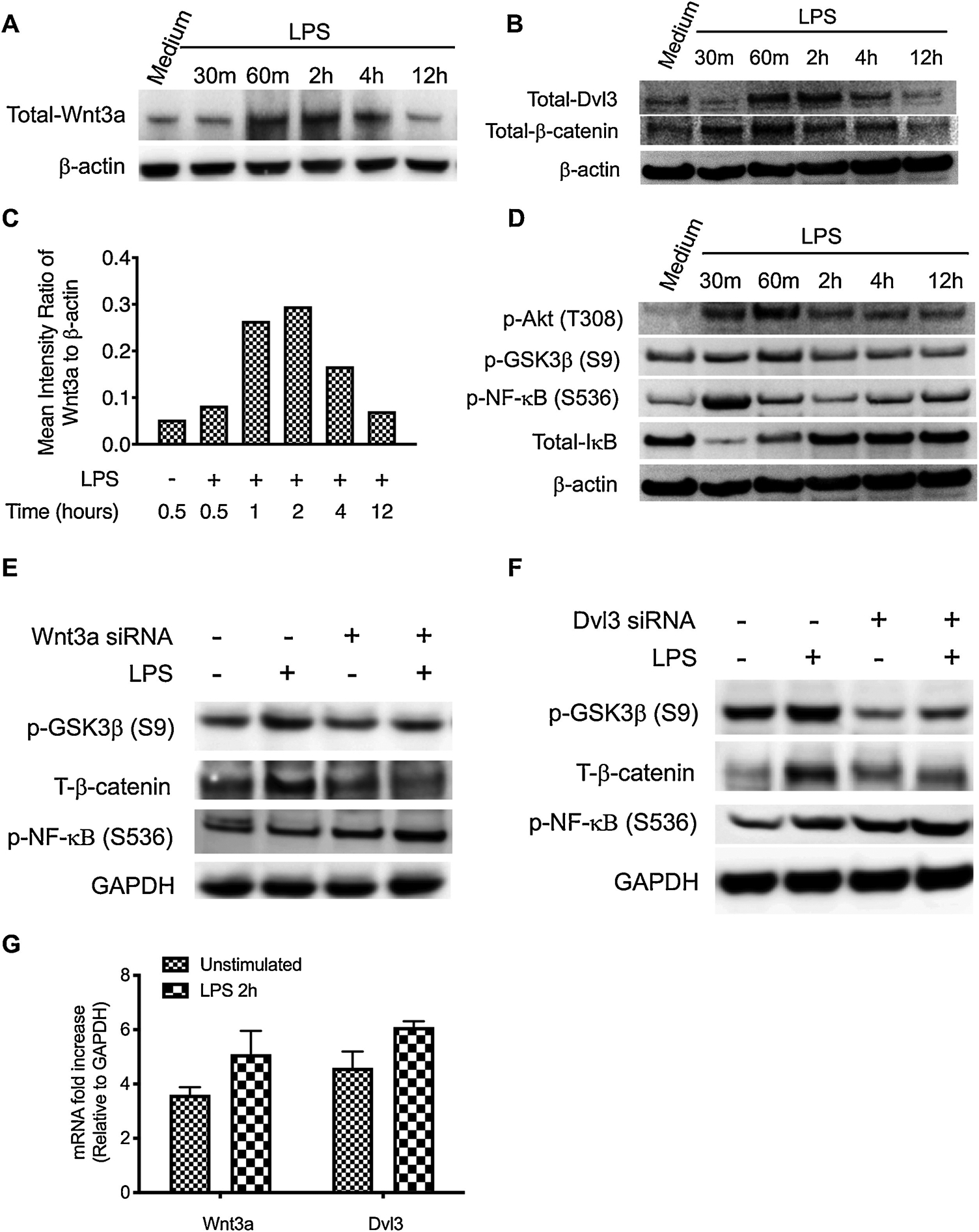
Purified human monocytes were stimulated with E. coil LPS (1 μg/ml) over 12 h time course with or without the pretreatment of Wnt3a or Dvl3 siRNA. (A, B) Total cell lysates were probed for the levels of Wnt3a (A), Dvl3 (B), β-catenin (Ser 9) (B), and β-actin by Western Blot. (C) Densitometry scans were performed to calculate the ratio of Wnt3a to β-actin. (D) The same blot was restriped and probed for the levels of phosphorylation of Akt (T308), GSK3β (S9), NF-κB (S536), and total IκB. For E and F, cells were pretreated with Wnt3a or Dvl3a siRNA for 48 hours and then stimulated with LPS. After 60min stimulation, the total cell lysates were collected and probed for the levels of phopho-GSK3β and total-β-catenin by Western Blot, GAPDH was also probed as loading control. (G) The mRNA levels of Wn3a and Dvl3 were detected by qRT-PCR after 2 h stimulation with LPS. Data are representative of three (for E to G) and five (for A, B, and D) independent experiments.
Wnt3a mediated anti-inflammatory is dependent on the expression of Dvl3
Our data have shown that LPS stimulation enhances the expression of Wnt3a and Dvl3, which suppresses the production of subsequent inflammatory cytokines in monocytes. To delineate Wnt3a-mediated anti-inflammatory signaling model, we next determine if Dvl3 relay the anti-inflammatory signaling from Wnt3a upon the challenge of LPS. To this end, a plasmid encoding Dvl3 was transfected into human monocytes (Fig. 3A) and examine the outcomes of Wnt3a inhibition in LPS-stimulated monocytes. We found ectopic overexpression of Dvl3 significantly reduces the expression of IL-12p40, IL-6, and TNFα in LPS stimulated human monocytes, as compared with the control group transfected with empty vector (Fig. 3B to D; p<0.05). Moreover, overexpression of Dvl3 leads to a substantial increase in GSK3β phosphorylation and elevates the expression of β-catenin upon the challenge of LPS in human monocytes (Fig. 3E and F). All these results suggested the anti-inflammatory signals of Wnt3a are relayed through Dvl3, which will affect the activity of downstream GSK3β/β-catenin in LPS stimulated human monocytes.
Figure 3. Anti-inflammatory function of Wnt3a is dependent on the expression of Dvl3.
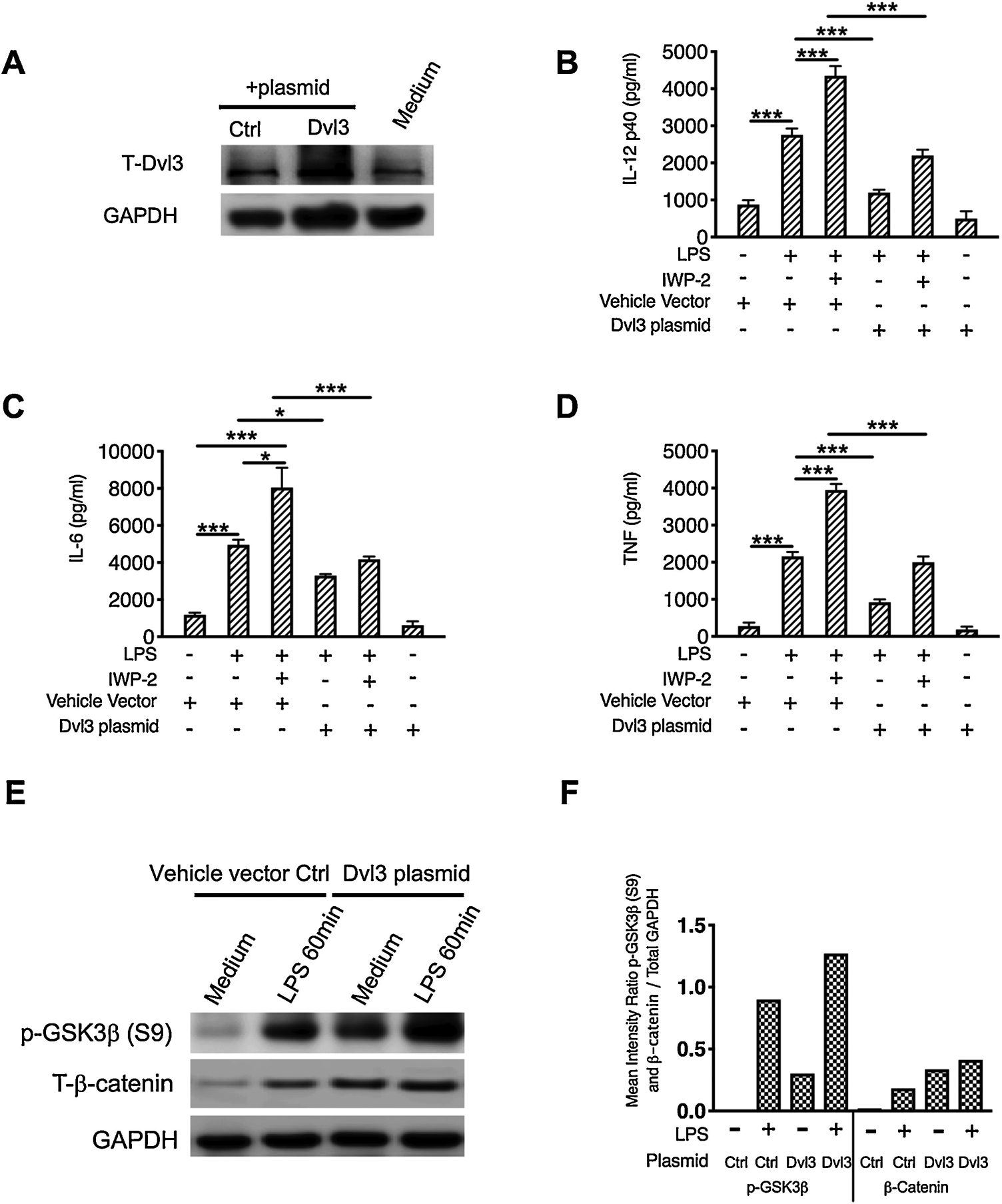
Purified human monocytes were transfected with a plasmid encoding Dvl3 or empty vector control plasmid for 48 hours, then pre-treated Wnt3a inhibitor, IWP-2, for 2 hours, followed by the stimulation of LPS for 24 hours. (A) The expression of total Dvl3 were measured by Western Blot to examine the transfection efficacy. The cell-free supernatant was collected and production of IL-12P40 (B), IL-6 (C), and TNF (D) were measured by ELISA. E and F, Cell lysates were prepared after stimulation with LPS for 60 min, and 15μg of total protein was analyzed by Western Blot using antibodies to GSK3β or β-catenin, then stripped and re-probed with an antibody to total GAPDH to ensure equal protein loading. (F) Densitometry scans were performed by calculating the ratio of phosphor-GSK3β or β-catenin to GAPDH. *, and *** indicates statistically significant at P<0.05, and P<0.001, respectively. Data represent the arithmetic mean±S.D. of three independent experiments.
Wnt3a-Dvl3 suppresses inflammatory responses through downstream GSK3β/β-catenin signaling
In canonical Wnt signaling, Wnt3a-Dvl3 activation has been demonstrated to enhance GSK3β phosphorylation and consequently lead to the accumulation of β-catenin. Since previous studies reported the opposite effects of Wnt3a-β-catenin on the inflammation responses [14, 22, 24, 37], we next examine the role of β-catenin in LPS induced inflammation in human monocytes and define if GSK3β/β-catenin relays the anti-inflammatory signaling of Wnt3a-Dvl3. Using pre-validated β-catenin siRNA, we find β-catenin deficiency results in a significant increase of IL-12P40, IL-6, and TNFα in LPS stimulated cells, which mimics the effect of Wnt3a inhibitor (Fig. 4A, B). In contrast, overexpression of the β-catenin mutant (S33Y), which is unable to be degraded upon the phosphorylation of GSK3β, could abrogate the ability of Wnt3a inhibitor to enhance LPS-induced inflammatory cytokines (Fig 4C, D). Moreover, overexpression of GSK3β constitutively active mutant (S9A) eliminates LPS induced β-catenin and thus mimics the effects of Wnt3a inhibitor or β-catenin siRNA on the production of inflammatory cytokines in LPS stimulated human monocytes (Fig. 4E, F). Of note, the influences of GSK3β in this study on inflammatory cytokines are consistent with the results of our previous studies, demonstrating the anti-inflammatory function of Akt-GSK3β signaling [11, 32, 38]. In addition, siRNA mediated silencing of Dvl3 reduces GSK3β phosphorylation, and leads to a substantial decrease of β-catenin in LPS stimulated monocytes (Fig. 4G). Combined with our previous results showing overexpression of Dvl3 enhances phosphorylation of GSK3β and robustly elevates the level of β-catenin (Fig. 3E), these findings demonstrated that β-catenin is a suppressor of inflammatory responses in LPS stimulated monocytes.
Figure 4. Wnt3a-Dvl3 suppresses inflammatory responses through downstream GSK3β-β-catenin signaling.
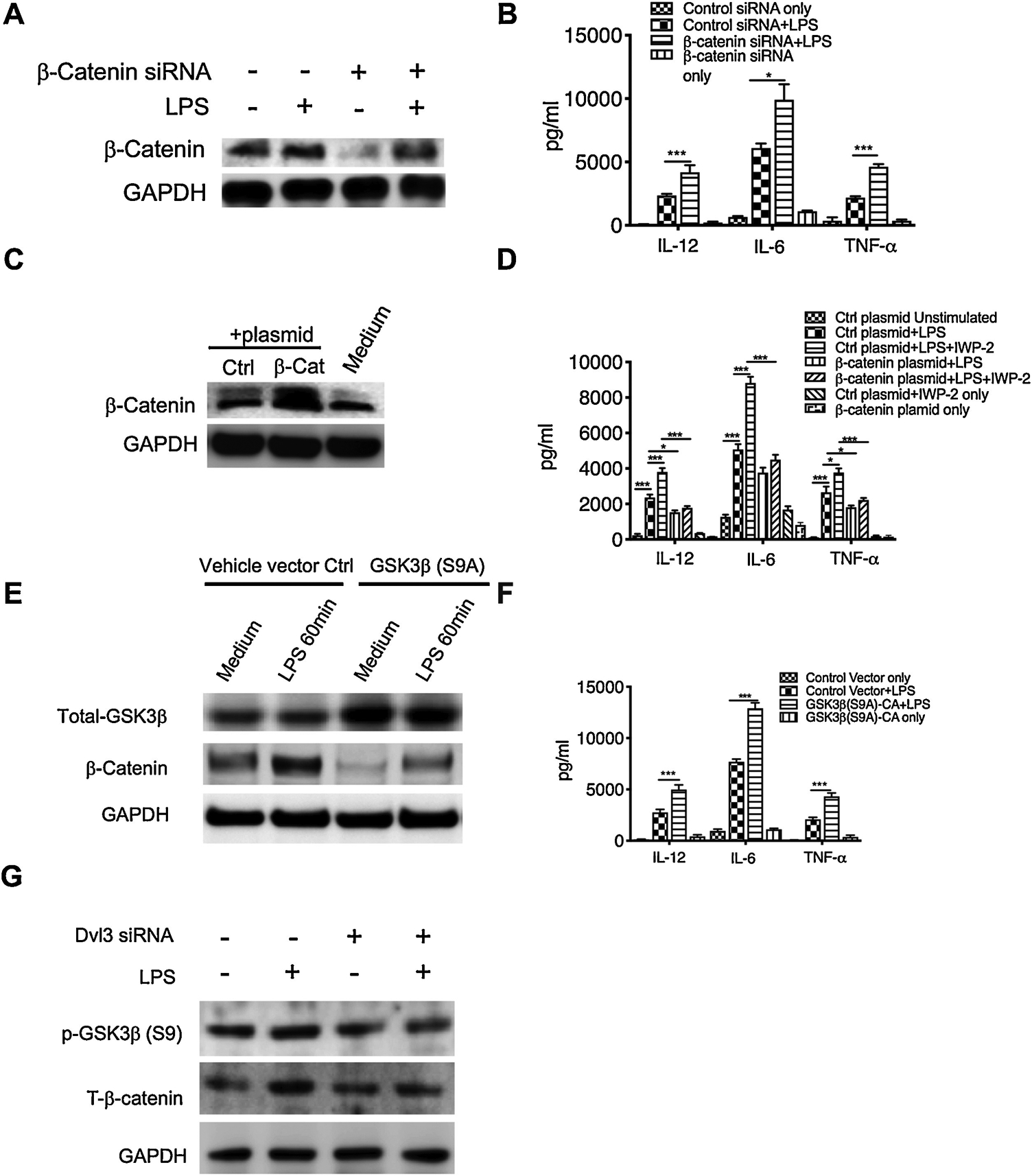
A to D, purified human monocytes were transfected with β-catenin specific siRNA (A) or a plasmid encoding β-catenin mutant (S33Y) (C), no-specific scramble siRNA and empty vector plasmid were also transfected as controls. After 48 hours, cell lysates were collected to test the efficiency of transfection by the expression of total β-catenin (A, C) and the separate groups were stimulated with LPS for another 24 hours. (B, D) The production of IL-12P40, IL-6 and TNFα in β-catenin siRNA-treated (B) or plasmid encoding mutant β-catenin-treated (D) human monocytes. E to G, the same procedure as in A to D was used to treat purified human monocytes with the plasmid encoding GSK3β mutant (S9A) (E), or the siRNA of Dvl3 (G). (E) Expression of total GSK3β was measured by Western Blot to test the transfection efficiency. (F) The production of IL-12P40, IL-6 and TNFα in human monocytes upon the overexpression of constitutive active GSK3β mutant. (G) The expression of phospho-GSK3 (S9) and total β-catenin in LPS stimulated monocytes with or without the pretreatment of Dvl3 siRNA. All results are the average of at least three independent experiments. Error bars represent standard deviation. *, and *** indicates statistically significant at P<0.05, and P<0.001, respectively. Data represent the arithmetic mean±S.D. of three independent experiments.
Inhibition of Dvl3 mediated-decrease of β-catenin enhances activity of NF- κB and the transcription of inflammatory cytokines
Multiple inflammatory signaling pathways such as NF-κB and MAPK and a plethora of transcription factors are involved in regulating TLR4-mediated inflammatory cytokines [30]. Since Wnt/β-catenin signaling has been reported to interact with NF-κB and MAPK signaling [39, 40], we next to examine which kind of transcriptional factor(s) is the target of Wnt3a-Dvl3 signaling. We find inhibition of Wnt3a or silencing of dvl3 indeed leads to a significant increase of NF-κB activity, representing by the phosphorylation of NF-κB at Ser 536 in human monocytes (Fig. 5A, B), as compared to that of controls. In contrast, overexpression of Dvl3 (Fig. 3E) or β-catenin mutant (S33Y) (Fig. 4C) abrogates the influences of Wnt3a inhibition on the DNA binding activity of NF-κB (Fig. 5C). To further confirm NF-κB is the target of Wnt3a/Dvl3-β-catenin signaling, we next utilize NF-κB inhibitory peptide to block the activity of NF-κB and we found that NF-κB inhibition significantly diminishes the ability of IWP-2 to increase the production of IL-12P40, IL-6, and TNFα in LPS-stimulated human monocytes, as compared to the cells treated with control peptide (Fig. 5D to F). These results demonstrated NF-κB is a major, at least one of, downstream effector of Wnt3a-Dvl3, indicated that Wnt3a-Dvl3/β-catenin restrains the levels of inflammatory cytokines through modifying the activity of NF-κB in LPS-stimulated monocytes.
Figure 5. Inhibition of Dvl3 mediated decreases β-catenin enhances activity of NF-κB and the transcription of inflammatory cytokines.
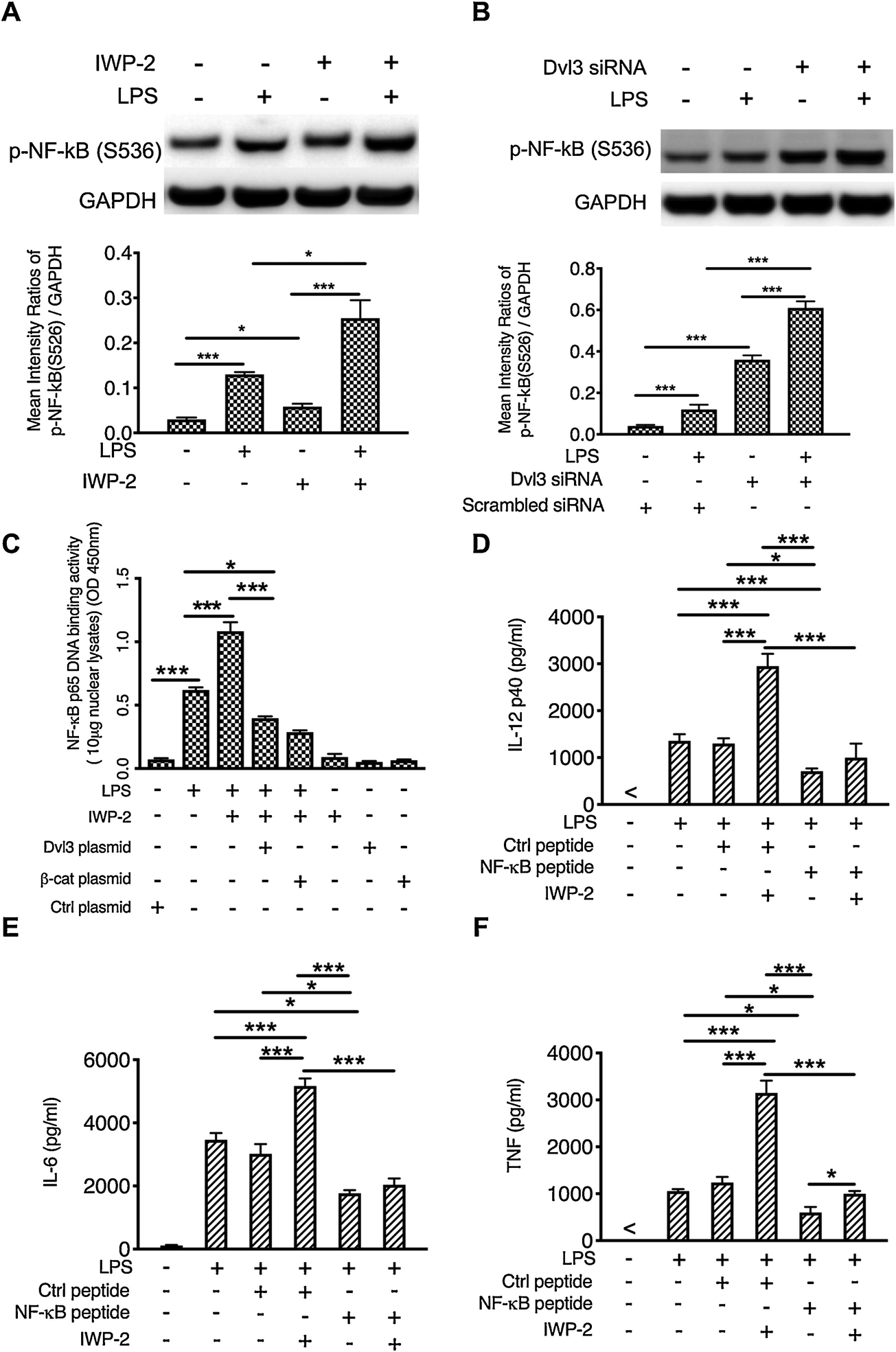
(A, B) Phosphorylation of NF-κB (S536) in LPS-stimulated human monocytes with or without the pretreatment of Wnt3a inhibitor or Dvl3 siRNA, and densitometry scans were performed and analyzed by calculating the ratio of phosphor-NF-kB to GAPDH. (C) Overexpression of Dvl3 (Fig. 3E) or β-catenin mutant (S33Y) (Fig. 4C) abrogates the influences of Wnt3a inhibition on the DNA binding activity of NF-κB (Fig. 5C), as compared to the controls. (D to F) The production of IL-12P40 (D), IL-6 (E) and TNFα (F), in LPS stimulated human monocytes in the presence and absence of NF-κB inhibitory peptide. *, and *** indicates statistically significant at P<0.05, and P<0.001, respectively. Data represent the arithmetic mean±S.D. of three biological replicates.
Inhibition of Wnt3-Dvl3 signaling aggravates TLR4-mediated inflammatory responses and enhances neutrophil infiltration in the murine model of endotoxic shock
Pro-inflammatory cytokines, especially TNFα, play a central role in the initiation and development of LPS-induced endotoxic shock [41]. Because we found that Wnt3a inhibition enhanced production of TNFα, IL-12, and IL-6 in human monocytes, we next investigated if Wnt3a inhibition would enhance the production of these proinflammatory cytokines on a systemic level and thus exacerbate the tissue damage in LPS-induced endotoxic shock model. As shown in figure 6 A, B, mice pretreated with Wnt3a inhibitor IWP-2 exhibited significant higher levels of IL-12, IL-6, and TNFα in serum after 4 h stimulation with LPS, as compared to the mice treated with LPS only. After the mice were stimulated with LPS for 24 h, both control mice and Wnt3a inhibitors-treated mice all exhibited an enormous decrease of proinflammatory cytokine production, as compared to that of mice treated with LPS for 4 h (Fig. 6B). Moreover, the production of TNFα and IL-12 did not display a significant difference between the mice with or without the pretreatment of inhibitors at 24 h post-injection of LPS (Fig. 6B), suggesting that Wnt3a might be involved only in the early stage of LPS-induced inflammatory responses. Because overwhelming neutrophil accumulation significantly contribute to the development of endotoxin-mediated multiple organ failure[42], we next examined if Wnt3a inhibition affects organ damage and neutrophil infiltration upon LPS challenge. To this end, we collected mouse lungs after 24 h challenge with LPS in the presence and absence of IWP-2, and stain the tissue sections with H&E (Fig. 6C) and FITC-conjugated anti-Ly6G antibody (Fig. 6D). We find that mice pretreated with IWP-2 displayed severe organ injury, including a widespread disruption of tissue architecture, an extensive area of cell cytoplasmic vacuolization after 24 h LPS challenge, and broad areas of inflammatory cells infiltration in lung (Fig. 6C, D). On the other hand, mild tissue and hemorrhage were found in the lung of the mice upon LPS treatment only (Fig. 6C). Consistent with this result, Wnt3a-inhibited mice exhibited higher neutrophil infiltration in lung tissues compared to the mice treated with LPS alone (Fig. 6E). Neutrophils are prone to be sequestered at microcapillaries in several organs such as the lungs and liver during LPS-induced endotoxemia. Tissue infiltration by neutrophils helps to combat disseminated bacterial infections but also frequently results in protease-mediated tissue damage. Since neutrophils activation leads to the release of secretory granule content, especially, the pro-inflammatory enzyme myeloperoxidase (MPO), we also examined the production of MPO in the lung tissue samples. As showed in figure 6F, after 4 h challenge with LPS, the secretion of MPO was significantly increased in the lung tissue as compared with the PBS-treated control. Moreover, IWP-2 pre-treatment induces more MPO secretion in LPS challenged mice as compared to LPS treated only (P<0.05) (Fig. 6F). The similar results were also observed after 24 h challenge with LPS (Fig. 6F), indicating increased MPO could be the reason for the more severe tissue damage in IWP-2 treated mice. Together, these results have demonstrated that Wnt3 inhibition systemically enhances the production of pro-inflammatory cytokine and myeloperoxidase levels, which could exacerbate the severity of septic shock, indicating that targeting Wnt3a canonical signaling might be a novel interventional strategy to control the intensity of inflammation.
Figure 6. Inhibition of Wnt3a aggravates TLR4-mediated inflammatory responses and enhances neutrophil infiltration in the murine model of endotoxic shock.
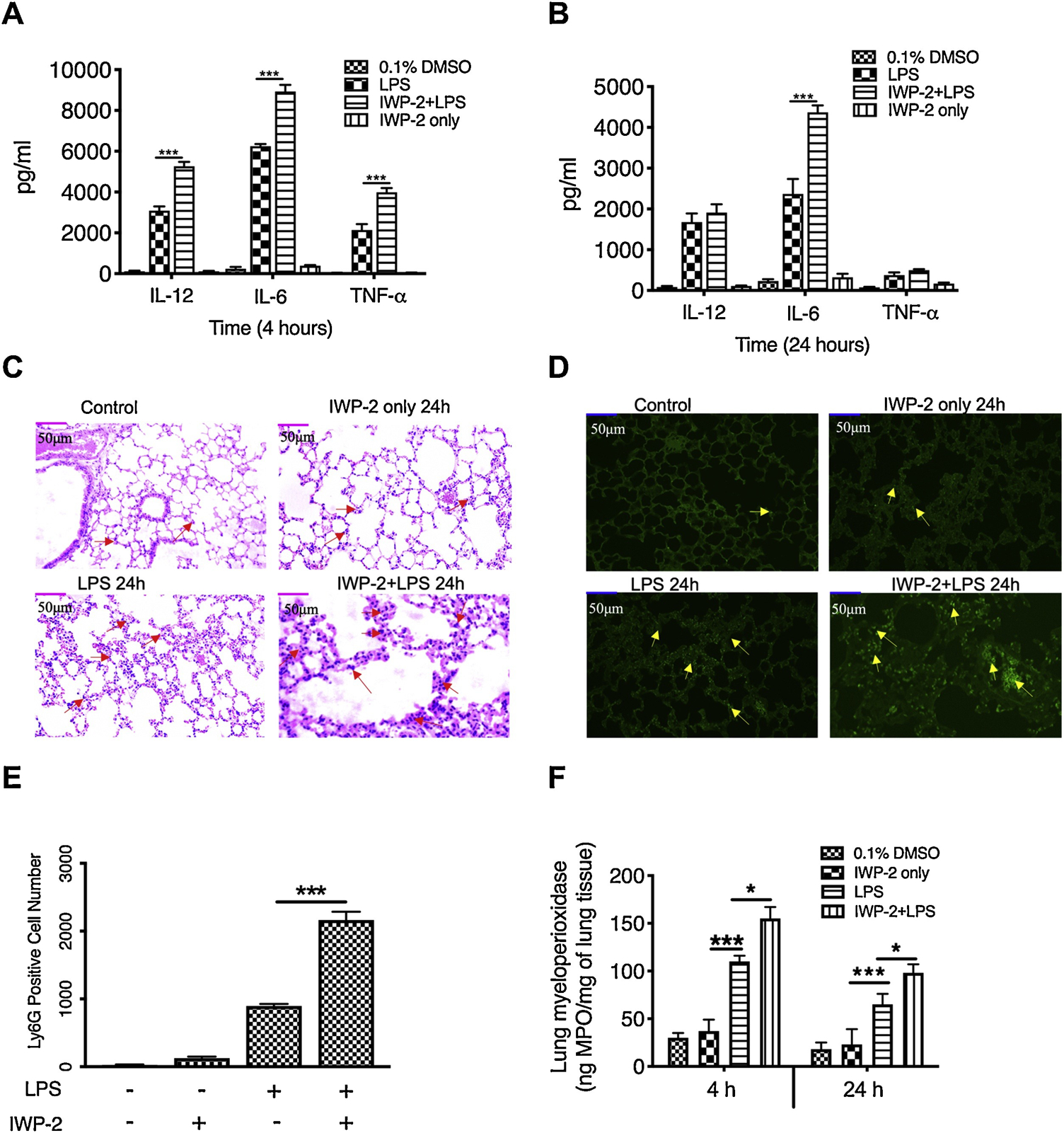
IL-12P40, IL-6 and TNFα levels at 4 hours (A) or 24 hours (B) post-injection of LPS in C57BL/6 mice with or without IWP-2 (15mg/kg) pretreatment. (C) H&E staining of serial section of lung from mice injected with PBS (Control), IWP-2, LPS, or LPS plus IWP-2 for 24 hours showing the infiltration of inflammatory cells (area with red arrows) and tissue damage (×20 magnification). (D) Polymorphonuclear neutrophil infiltration was assessed by staining tissue section of lung with FITC-conjugated Ly6G (yellow arrow; magnification, ×20). (A)-(B) and (C)-(D) are separate, and each group has a total of 6 mice for analysis. (E) Levels of infiltrated neutrophils presented by the average number of Ly6G-positive cells in 5 different views. (F) The amount of myeloperoxidase in lung tissues at 4 h and 24 h after different treatments. Data represent the arithmetic means±SD of 3 biological replicates. “*” and “***” indicate statistically significant at P<0.05 and P<0.001, respectively. Data represent the arithmetic mean±S.D. of three biological replicates.
Discussion
Despite the functional role of Wnt3a signaling in immune response has been widely investigated, recent studies reported controversial effects of wnt3a and β-catenin on LPS-induced inflammatory responses [14, 22, 24, 37]. Moreover, the downstream signaling module Wnt3a-mediated to regulate inflammation and the interaction between TLRs and Wnt3a signaling pathways remain uncertain. In this study, we demonstrated the anti-inflammatory function of Wnt3a and β-catenin in vitro and in vivo, and revealed for the first time that TLR4 activation enhances the expression of Wnt3a and Dvl3, which is key to maintain the TLR4-mediated inflammation at proper level. Moreover, we found that Wnt3a-Dvl3 functions as a negative regulator of TLR4-mediated inflammation is through controlling the phosphorylation of GSK3β, the accumulation of β-catenin, and subsequent suppression of NF-κB activity. Interestingly, the increase of Wnt3a and downstream canonical signaling components is mainly at the early stage of LPS stimulation, indicating TLR4-enhanced Wnt3a-Dvl3 could be a novel innate sentinel mechanism to restrain the overwhelming inflammation and subsequent collateral tissue damages it inflicted (Fig. 7).
Figure 7. Schematic model of how LPS-mediated Wnt3a-Dvl3 signaling restrains production of pro-inflammatory cytokine in innate immune cells and protects against lethal endotoxemia.
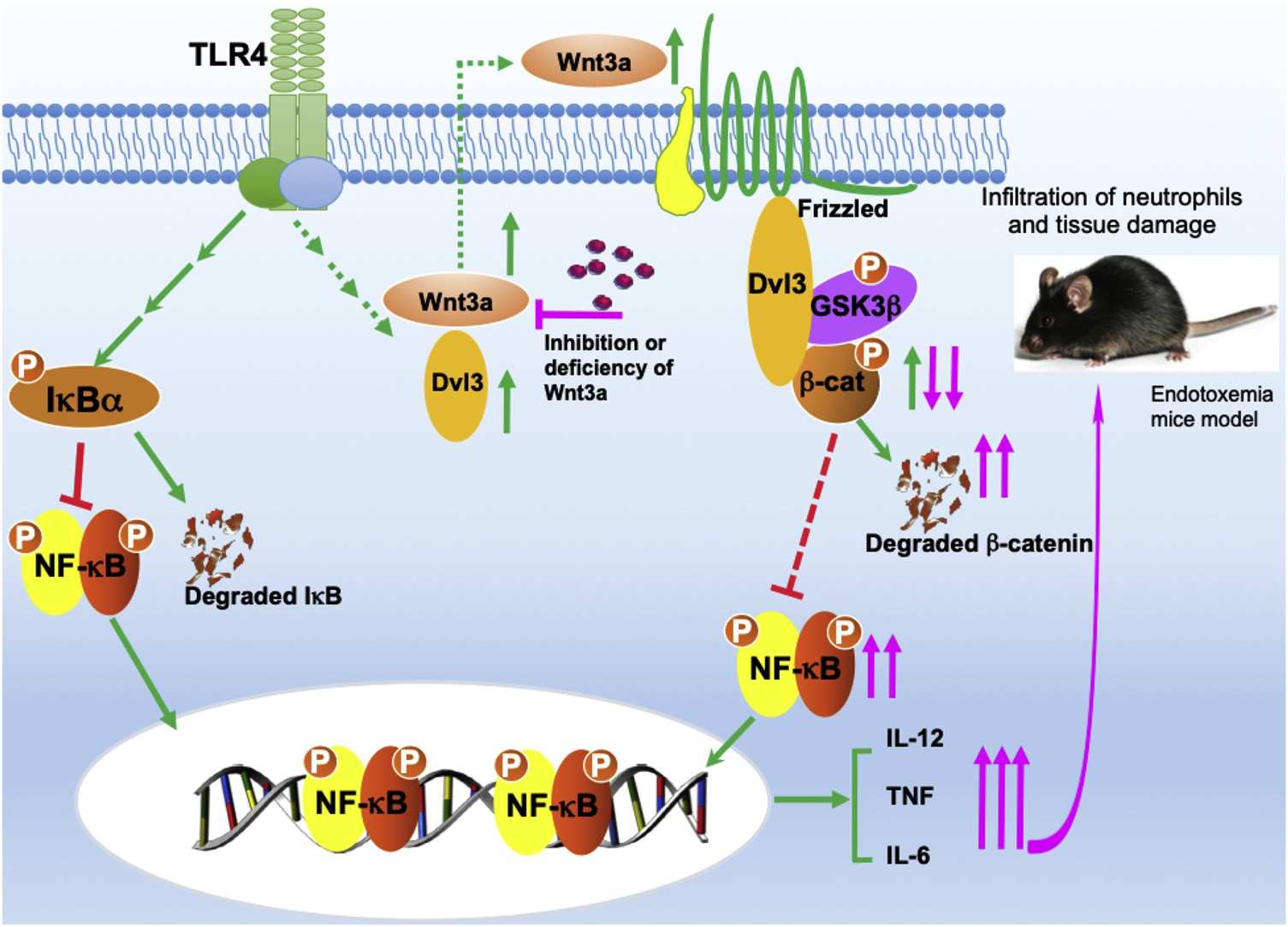
While TLR4 activation enhances the transcriptional activity of NF-κB, leading to the increase of inflammatory cytokines, it also promotes expression of Wnt3a and Dvl3 by some undetermined mechanisms. Increased Wnt3a and Dvl3 phospho-inactivates GSK3β that will reduce degradation of β-catenin, leading to its accumulation, and by which suppresses NF-κB activity, ultimately restrains inflammatory immune responses (green). In contrast, inhibition or deficiency of Wnt3a (pink) promotes degradation of β-catenin, lessens the constraint and elevates NF-κB activity (pink), leading to enhancement of pro-inflammatory cytokine levels and neutrophil infiltration, consequently exacerbated tissue damage in mice endotoxemia model.
Wnt proteins have been assigned with both pro- and anti- inflammation. For Wnt3a-mediated β-catenin signaling, studies by Neumann et al reported that wnt3a promotes anti-inflammatory responses and is constitutively expressed by bronchial epithelial cells [14, 22]. Moreover, numerous studies have shown that Wnt3a-mediated β-catenin accumulation suppresses the activity of NF-κB through different mechanisms [40, 43]. In contrast, several recent studies reported the opposite effects of Wnt3a in LPS-stimulated epithelial cells and septic mice model, showing Wnt3a inhibition reduces the magnitude of inflammation in vitro and in vivo [24, 37]. In our hands, we found that Wnt3a is promptly induced upon the challenge of LPS and play an anti-inflammatory role in primary human monocytes and endotoxemia mice model, which consolidates the anti-inflammatory function of Wnt3a and indicates that inducible Wnt3a might be an alternative anti-inflammatory mechanism to avoid the subsequent overwhelming inflammatory response. The underlying reasons for the discrepant function of Wnt3a and their expression model are currently unknown. However, other studies reporting more controversial results about the function and kinetic expression of Wnt proteins [44–46] suggest the important influences of different contexts such as cell type, concentration and specificity of inhibitors, animal facilities, along with other unknown factors on the results of experiments. A recent report highlights this point, which showed that only subtle changes to the context of delivery of the Wnt3a signal, arginine methylation of a single component of the Wnt3a signalosome, is sufficient to dramatically alter downstream canonical signals [47]. Thus, more careful, independent investigations of Wnt-mediated cellular responses are required to validate the role of Wnt3a-Dvl3 signaling in TLR-mediated inflammatory responses.
Although Wnt3a or Dvl3 silence indeed reduce the expression of β-catenin in our study, we observed that LPS stimulation enhances β-catenin expression is earlier than that of Wnt3a and Dvl3, indicting there are other mechanisms other than Wnt3a may be also involved in the β-catenin expression. GSK3β compartmentalization has been identified with distinct influences in PI3K-Akt or Wnt3a signaling circumstance [35, 36]. Our previous studies have demonstrated LPS-mediated PI3K activation enhances phosphorylation of GSK3β and accumulation of β-catenin [11, 48]. Therefore, it is possible that TLR4-mediated β-catenin accumulation resulted from both PI3K-Akt and Wnt3a-Dvl3 signaling. On the other hand, increase of β-catenin is concurrently with the decreases of IκB and wane quickly after the new synthesized IκB playing back their locking function to limited the activity of NF-κB. The hierarchy in the expression of these suppressive proteins strongly suggests Wnt3a-mediated anti-inflammatory signaling could be an alternative earlier anti-inflammatory signaling mechanism to restrain the potential excessive transcription of inflammatory mediators. More deep studies to validate the function of Wnt3a-β-catenin signaling axis and elucidate the hierarchy of these anti-inflammatory mechanisms will aid to develop more interventional targets for the control of inflammatory diseases. In addition, Wnt3a-β-catenin signaling has been shown to be highly oncogenic when dysregulated [49, 50], and to be involved in the development of degenerative diseases, including systemic sclerosis [50–52], inflammatory bowel disease [53] and asthma [54]. Therefore, our findings about the anti-inflammatory function of Dvl3-mediated GSK3β-β-catenin may have relevance beyond the manipulation of host-pathogen interactions.
Despite activation of Dvls was involved in multiple signaling pathways such as PI3K, PKC, and Rac-1, which are all important regulators of innate immune responses [55, 56], there is a surprising lack of direct evidence of immunomodulatory function of Dvl proteins in innate immunity. Recent studies have reported Dvl proteins could shuttle between the cytoplasm and the nuclear [57, 58]. Moreover, disrupted immigration through mutation of the nuclear localization sequence substantially impairs the activity of β-catenin pathway [57, 58], indicating there might be different pools of Dvls with unidentified functions. In our study, we demonstrated for the first time that Dvl3 is inducible by LPS and critical for anti-inflammatory role of Wnt3a in primary human monocytes. Moreover, we found knockdown of Dvl3 not only blocks Wnt3a signaling but also impairs the downstream activity of GSK3β-mediated β-catenin, which highlighted the key function of Dvl3 in Wnt3a signaling. Our findings about Wnt3a-Dvl3 function as an anti-inflammatory regulator is also consistent with the mainstream opinion of this signaling in other experimental contexts [30, 59, 60]. While we addressed the function of Dvl3, we can’t exclude other two isoforms also function in Wnt3a-mediated canonical signaling. Knockdown of Dvl3 leading to only part of alteration of GSK3β-β-catenin activity suggests the influences of Dvl3 might be compromised by other isoforms of Dvl proteins. Thus, more deeply studies to separate the function of other individual Dvl proteins in TLR-mediated immune response will be required to characterize the role of Dvl proteins and delineate the signaling anti-inflammatory function of Wnt3a.
In our study, we demonstrated for the first time that TLR4 activation enhances Wnt3a expression at the early stage of LPS challenge. Since Wnt3a is also a critical player in every subsequent stage of tissue healing process [12], our results suggest possible dual interrelated roles of de novo wnt3a: Quenching the flame of inflammation and initiate the subsequent tissue regeneration upon the injury. This kind of interaction between TLR4 and Wnt3a signaling well addresses the orchestra and integrity of signaling networks upon the challenge of injury or infections pathogens. On the other hand, recent studies have demonstrated that Wnt3a and Dvl proteins degradation are regulated by ubiquitination system [61, 62]. Our data showed LPS stimulation fails to induce a significant increase of Wnt3 or Dvl3 mRNA. Thus, it is very possible that LPS stimulation enhances the expression of Wnt3a and Dvl3 is through suppression of their ubiquitination, especially when we considered a plethora of ubiquitination E3 ligase could be activated by LPS in innate cells [63]. Our unpublished data also showed that LPS leads to the phosphorylation of Nedd4–2 (Neural precursor cell Expressed Developmentally Down-regulated 4–2), a HECT (Homologous to the E6-AP C Terminus) domain E3 ubiquitin ligase, in monocytes and THP1, which could be a pathway to modify the activation of Wnt3a-Dvl3 signaling. Future studies focusing on the dynamic expression of individual Wnt3a signaling molecules and the function of ubiquitination will validate our conclusion and provide more insights in inflammation and subsequent growth signaling.
In summary, we have demonstrated the anti-inflammatory function of Wnt3a-Dvl3 signaling in TLR4-mediated inflammatory responses. Moreover, TLR4 activation enhances Wnt3a and Dvl3 expression, leads to inactivation of GSK3β and subsequent accumulation of β-catenin, which will ultimately suppress the activity of NF-κB in human monocytes. Our results indicate that TLR4-mediated Wnt3a-Dvl3 signaling axis acts as a rheostat to restrain the intensity of inflammation in innate immune cells. Combined with our in vivo data showing the protective effects of Wnt3a in endotoxemia murine model, our findings suggested that Wnt3-Dvl3 signaling could be a potential interventional target to manipulate the direction and intensity of inflammatory responses.
Highlights.
TLR4 activation enhances the expression of Wnt3a and Dvl3 in innate immune cells.
Wnt3a-Dvl3 reduces pro-inflammatory cytokine production via β-catenin/NF-κB pathway.
Wnt3a restrains inflammation intensity and protects mice from lethal endotoxemia
Wnt3a signaling could be a target to manipulate the intensity of inflammation.
Acknowledgments
This research was supported by Grants DE023633 and DE026727 (to H.W.), DE 024509 (to S. M. U.), and DE025388 (to S.L.) from the U.S. National Institutes of Health, National Institute of Dental and Craniofacial Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- [1].Nathan C, Points of control in inflammation, Nature, 420 (2002) 846–852. [DOI] [PubMed] [Google Scholar]
- [2].Rothschild DE, McDaniel DK, Ringel-Scaia VM, Allen IC, Modulating inflammation through the negative regulation of NF-B signaling, Journal of Leukocyte Biology, 103 (2018) 1131–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lawrence T, Willoughby DA, Gilroy DW, Anti-inflammatory lipid mediators and insights into the resolution of inflammation, Nature Reviews Immunology, 2 (2002) 787–795. [DOI] [PubMed] [Google Scholar]
- [4].Murray PJ, Smale ST, Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways, Nat Immunol, 13 (2012) 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martin M, Rehani K, Jope RS, Michalek SM, Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3, Nature Immunology, 6 (2005) 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chi HB, Flavell RA, Acetylation of MKP-1 and the Control of Inflammation, Science Signaling, 1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lopez-Vazquez A, Garcia-Banuelos JJ, Gonzalez-Garibay AS, Urzua-Lozano PE, Del Toro-Arreola S, Bueno-Topete MR, Sanchez-Enriquez S, Munoz-Valle JF, Jave-Suarez LF, Armendariz-Borunda J, Bastidas-Ramirez BE, IRS-1 pY612 and Akt-1/PKB pT308 Phosphorylation and Antiinflammatory Effect of Diindolylmethane in Adipocytes Cocultured with Macrophages, Med Chem, 13 (2017) 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ohlsson C, Hammarstedt A, Vandenput L, Saarinen N, Ryberg H, Windahl SH, Farman HH, Jansson JO, Moverare-Skrtic S, Smith U, Zhang FP, Poutanen M, Hedjazifar S, Sjogren K, Increased adipose tissue aromatase activity improves insulin sensitivity and reduces adipose tissue inflammation in male mice, Am J Physiol Endocrinol Metab, 313 (2017) E450–e462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fan H, Bitto A, Zingarelli B, Luttrell LM, Borg K, Halushka PV, Cook JA, Beta-arrestin 2 negatively regulates sepsis-induced inflammation, Immunology, 130 (2010) 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou H, Gao S, Duan X, Liang S, Scott DA, Lamont RJ, Wang H, Inhibition of serum- and glucocorticoid-inducible kinase 1 enhances TLR-mediated inflammation and promotes endotoxin-driven organ failure, Faseb j, 29 (2015) 3737–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang H, Brown J, Gao S, Liang S, Jotwani R, Zhou H, Suttles J, Scott DA, Lamont RJ, The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells, J Immunol, 191 (2013) 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bastakoty D, Young PP, Wnt/beta-catenin pathway in tissue injury: roles in pathology and therapeutic opportunities for regeneration, Faseb j, 30 (2016) 3271–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Whyte JL, Smith AA, Helms JA, Wnt Signaling and Injury Repair, Cold Spring Harbor Perspectives in Biology, 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schaale K, Neumann J, Schneider D, Ehlers S, Reiling N, Wnt signaling in macrophages: Augmenting and inhibiting mycobacteria-induced inflammatory responses, European Journal of Cell Biology, 90 (2011) 553559. [DOI] [PubMed] [Google Scholar]
- [15].Willert K, Jones KA, Wnt signaling: is the party in the nucleus?, Genes Dev, 20 (2006) 1394–1404. [DOI] [PubMed] [Google Scholar]
- [16].Bradley EW, Drissi MH, WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-kappaB pathways, Mol Endocrinol, 24 (2010) 1581–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT, The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape, Trends Genet, 16 (2000) 279–283. [DOI] [PubMed] [Google Scholar]
- [18].Dejmek J, Safholm A, Kamp Nielsen C, Andersson T, Leandersson K, Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1alpha signaling in human mammary epithelial cells, Mol Cell Biol, 26 (2006) 6024–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lai SL, Chien AJ, Moon RT, Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis, Cell Res, 19 (2009) 532–545. [DOI] [PubMed] [Google Scholar]
- [20].Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, Trumper L, Binder C, Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines, Proc Natl Acad Sci U S A, 103 (2006) 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chae WJ, Bothwell ALM, Canonical and Non-Canonical Wnt Signaling in Immune Cells, Trends Immunol, 39 (2018) 830–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neumann J, Schaale K, Farhat K, Endermann T, Ulmer AJ, Ehlers S, Reiling N, Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages, Faseb j, 24 (2010) 4599–4612. [DOI] [PubMed] [Google Scholar]
- [23].Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J, beta-Catenin activity negatively regulates bacteria-induced inflammation, Lab Invest, 87 (2007) 613–624. [DOI] [PubMed] [Google Scholar]
- [24].Jang J, Jung Y, Kim Y, Jho EH, Yoon Y, LPS-induced inflammatory response is suppressed by Wnt inhibitors, Dickkopf-1 and LGK974, Sci Rep, 7 (2017) 41612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gao C, Chen YG, Dishevelled: The hub of Wnt signaling, Cell Signal, 22 (2010) 717–727. [DOI] [PubMed] [Google Scholar]
- [26].Lee YN, Gao Y, Wang HY, Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, −2, and −3, Cell Signal, 20 (2008) 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang J, Yang J, Han D, Zhao X, Ma J, Ban B, Zhu X, Yang Y, Cao D, Qiu X, Dvl3 polymorphism interacts with life events and pro-inflammatory cytokines to influence major depressive disorder susceptibility, Sci Rep, 8 (2018) 14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pan W, Choi SC, Wang H, Qin Y, Volpicelli-Daley L, Swan L, Lucast L, Khoo C, Zhang X, Li L, Abrams CS, Sokol SY, Wu D, Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation, Science, 321 (2008) 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qin Y, Li L, Pan W, Wu D, Regulation of phosphatidylinositol kinases and metabolism by Wnt3a and Dvl, J Biol Chem, 284 (2009) 22544–22548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brown J, Wang H, Hajishengallis GN, Martin M, TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk, J Dent Res, 90 (2011) 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yin H, Zhou H, Kang Y, Zhang X, Duan X, Alnabhan R, Liang S, Scott DA, Lamont RJ, Shang J, Wang H, Syk negatively regulates TLR4-mediated IFNbeta and IL-10 production and promotes inflammatory responses in dendritic cells, Biochim Biophys Acta, 1860 (2016) 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang H, Brown J, Gu Z, Garcia CA, Liang R, Alard P, Beurel E, Jope RS, Greenway T, Martin M, Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-beta-signaling pathways regulates the innate inflammatory response, J Immunol, 186 (2011) 5217–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hermida MA, Dinesh Kumar J, Leslie NR, GSK3 and its interactions with the PI3K/AKT/mTOR signalling network, Adv Biol Regul, 65 (2017) 5–15. [DOI] [PubMed] [Google Scholar]
- [34].McCubrey JA, Rakus D, Gizak A, Steelman LS, Abrams SL, Lertpiriyapong K, Fitzgerald TL, Yang LV, Montalto G, Cervello M, Libra M, Nicoletti F, Scalisi A, Torino F, Fenga C, Neri LM, Marmiroli S, Cocco L, Martelli AM, Effects of mutations in Wnt/beta-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity-Diverse effects on cell growth, metabolism and cancer, Biochim Biophys Acta, 1863 (2016) 2942–2976. [DOI] [PubMed] [Google Scholar]
- [35].Voskas D, Ling LS, Woodgett JR, Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling?, F1000 Biol Rep, 2 (2010) 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McNeill H, Woodgett JR, When pathways collide: collaboration and connivance among signalling proteins in development, Nat Rev Mol Cell Biol, 11 (2010) 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gatica-Andrades M, Vagenas D, Kling J, Nguyen TTK, Benham H, Thomas R, Korner H, Venkatesh B, Cohen J, Blumenthal A, WNT ligands contribute to the immune response during septic shock and amplify endotoxemia-driven inflammation in mice, Blood Adv, 1 (2017) 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang H, Brown J, Martin M, Glycogen synthase kinase 3: a point of convergence for the host inflammatory response, Cytokine, 53 (2011) 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kabiri Z, Greicius G, Zaribafzadeh H, Hemmerich A, Counter CM, Virshup DM, Wnt signaling suppresses MAPK-driven proliferation of intestinal stem cells, J Clin Invest, 128 (2018) 3806–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma B, Fey M, Hottiger MO, WNT/beta-catenin signaling inhibits CBP-mediated RelA acetylation and expression of proinflammatory NF-kappaB target genes, J Cell Sci, 128 (2015) 2430–2436. [DOI] [PubMed] [Google Scholar]
- [41].Schulte W, Bernhagen J, Bucala R, Cytokines in sepsis: potent immunoregulators and potential therapeutic targets--an updated view, Mediators Inflamm, 2013 (2013) 165974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wagner JG, Roth RA, Neutrophil migration during endotoxemia, J Leukoc Biol, 66 (1999) 10–24. [DOI] [PubMed] [Google Scholar]
- [43].Ma B, Hottiger MO, Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation, Front Immunol, 7 (2016) 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hii HP, Liao MH, Chen SJ, Wu CC, Shih CC, Distinct Patterns of Wnt3a and Wnt5a Signaling Pathway in the Lung from Rats with Endotoxic Shock, PLoS One, 10 (2015) e0134492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sharma A, Yang WL, Ochani M, Wang P, Mitigation of sepsis-induced inflammatory responses and organ injury through targeting Wnt/beta-catenin signaling, Sci Rep, 7 (2017) 9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu CH, Nguyen TT, Irvine KM, Sweet MJ, Frazer IH, Blumenthal A, Recombinant Wnt3a and Wnt5a elicit macrophage cytokine production and tolerization to microbial stimulation via Toll-like receptor 4, Eur J Immunol, 44 (2014) 1480–1490. [DOI] [PubMed] [Google Scholar]
- [47].Bikkavilli RK, Malbon CC, Wnt3a-stimulated LRP6 phosphorylation is dependent upon arginine methylation of G3BP2, J Cell Sci, 125 (2012) 2446–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang H, Garcia CA, Rehani K, Cekic C, Alard P, Kinane DF, Mitchell T, Martin M, IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-beta, J Immunol, 181 (2008) 6797–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Clevers H, Wnt/beta-catenin signaling in development and disease, Cell, 127 (2006) 469–480. [DOI] [PubMed] [Google Scholar]
- [50].Nakashima N, Liu D, Huang CL, Ueno M, Zhang X, Yokomise H, Wnt3 gene expression promotes tumor progression in non-small cell lung cancer, Lung Cancer, 76 (2012) 228–234. [DOI] [PubMed] [Google Scholar]
- [51].Chen Y, Guan Y, Liu H, Wu X, Yu L, Wang S, Zhao C, Du H, Wang X, Activation of the Wnt/beta-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice, Biochem Biophys Res Commun, 420 (2012) 397–403. [DOI] [PubMed] [Google Scholar]
- [52].Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, Gottardi CJ, Atit R, Whitfield ML, Varga J, Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells, Arthritis Rheum, 64 (2012) 2734–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].You J, Nguyen AV, Albers CG, Lin F, Holcombe RF, Wnt pathway-related gene expression in inflammatory bowel disease, Dig Dis Sci, 53 (2008) 1013–1019. [DOI] [PubMed] [Google Scholar]
- [54].Lee H, Bae S, Choi BW, Yoon Y, WNT/beta-catenin pathway is modulated in asthma patients and LPS-stimulated RAW264.7 macrophage cell line, Immunopharmacol Immunotoxicol, 34 (2012) 56–65. [DOI] [PubMed] [Google Scholar]
- [55].Ziemba BP, Falke JJ , A PKC-MARCKS-PI3K regulatory module links Ca2+ and PIP3 signals at the leading edge of polarized macrophages, PLoS One, 13 (2018) e0196678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li G, Zeng L, Cheng H, Han J, Zhang X, Xie H, Acupuncture Administration Improves Cognitive Functions and Alleviates Inflammation and Nuclear Damage by Regulating Phosphatidylinositol 3 Kinase (PI3K)/Phosphoinositol-Dependent Kinase 1 (PDK1)/Novel Protein Kinase C (nPKC)/Rac 1 Signaling Pathway in Senescence-Accelerated Prone 8 (SAM-P8) Mice, Med Sci Monit, 25 (2019) 4082–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Itoh K, Brott BK, Bae GU, Ratcliffe MJ, Sokol SY, Nuclear localization is required for Dishevelled function in Wnt/beta-catenin signaling, J Biol, 4 (2005) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gan XQ, Wang JY, Xi Y, Wu ZL, Li YP, Li L, Nuclear Dvl, c-Jun, beta-catenin, and TCF form a complex leading to stabilization of beta-catenin-TCF interaction, J Cell Biol, 180 (2008) 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S, PI3K-mediated negative feedback regulation of IL-12 production in DCs, Nat Immunol, 3 (2002) 875–881. [DOI] [PubMed] [Google Scholar]
- [60].Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B, Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine, Science, 329 (2010) 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ, Moon RT, The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation, Nat Cell Biol, 8 (2006) 348–357. [DOI] [PubMed] [Google Scholar]
- [62].Jiang X, Charlat O, Zamponi R, Yang Y, Cong F, Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases, Mol Cell, 58 (2015) 522–533. [DOI] [PubMed] [Google Scholar]
- [63].Skaug B, Jiang X, Chen ZJ, The role of ubiquitin in NF-kappaB regulatory pathways, Annu Rev Biochem, 78 (2009) 769–796. [DOI] [PubMed] [Google Scholar]