

Abstract
Most human cancers arise from epithelial tissues, which are apical-basally polarized and possess intercellular adhesive junctions. Epithelial cells grow to characteristic densities, often from proliferative progenitors, which arrest as they mature. Homeostatic mechanisms can maintain this characteristic density if it is exceeded (crowding) or is too low (e.g., in response to wounding). During tumor initiation and progression this homeostatic mechanism is lost. Some aspects of cell polarity are also lost, although many carcinomas retain intercellular junctions and even apical domains. In other cases, and particularly in recurrent tumors, however, the cells become predominantly mesenchymal. A major question, still only incompletely answered, is whether the proteins that determine cell polarity function as tumor suppressors or tumor promoters. Here we discuss recent advances in understanding the role of polarity proteins and homeostasis in cancer.
Keywords: Epithelia, homeostasis, tumor suppressor, tumor promoter, Hippo signalling
Introduction
Epithelial cells either in culture or in situ can continue to proliferate even when attached to neighboring cells by intercellular junctions, but they arrest when they have achieved a characteristic density (Figure 1). Squamous epithelial cells usually arrest at a lower density than columnar epithelia, for example. In some cases, as in the intestine, proliferating stem cells generate transit amplifying cells that stop dividing when they differentiate, while in other tissues such as the kidney there is no clear hierarchy of stem to progenitor to mature cells, and during development the epithelial cells proliferate as needed to expand the surface area of the organ [1–3].
Figure 1. Apico-basal polarity proteins play a critical role in homeostatic control of cell density.
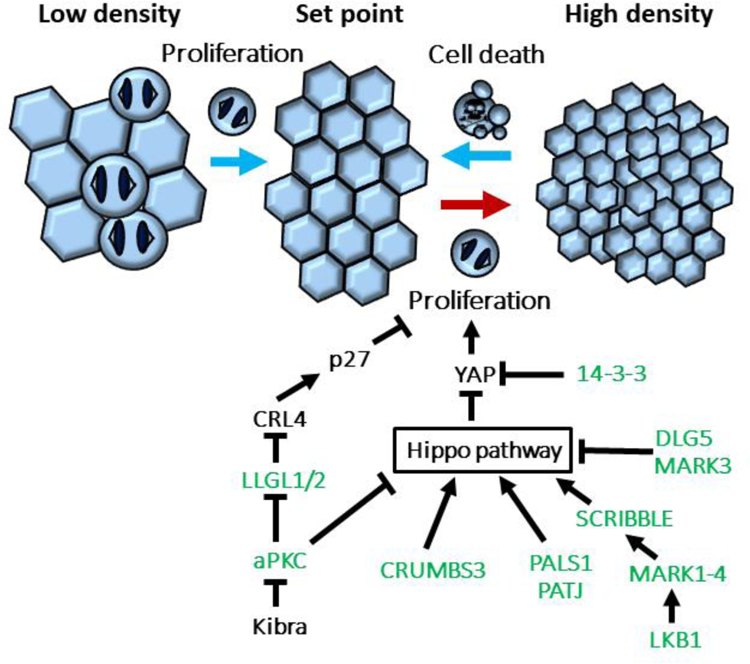
If cell density is too low or too high, epithelial cells return to homeostatic density by proliferation or by cell extrusion and death, respectively (depicted by light blue arrows). If cells fail to respond to density signals, they continue to proliferate and cause hyperplasia (red arrow). Polarity proteins (shown in green) control density-dependent proliferation via different mechanisms, including Hippo signaling pathway and CRL4 ubiquitin E3 ligase complex.
Mechanical forces play a key role in determining epithelial cell density. So, for instance, stretching an epithelial sheet can trigger cells to enter mitosis [4]; while compression can induce cell extrusion and apoptosis [5] (Figure 1). An early step in cancer initiation is a loss of homeostatic control, so that cells no longer respond to density signals and continue to proliferate. Hyperplasia within the epithelial sheet can inappropriately generate multi-layered structures through extrusion induced by over-crowding, or by cell migration, or misorientation of the division plane. One can conceive of apoptosis (or anoikis) of extruded cells as a fail-safe mechanism to eliminate epithelial cells that are not correctly positioned and have escaped the organizational constraints of the epithelial sheet. If apoptosis is inhibited, however, then cells that are extruded can survive to form disorganized tissue masses. In tubular organs such as the breast this can result in occlusion of the lumens, as occurs with ductal carcinoma in situ [6,7].
The central role of apical/basal polarity in epithelia raises the question of whether homeostatic control of cell density is linked to the polarity machinery, and if polarity proteins function as tumor suppressors. One key mechanism of density control is the Hippo signaling pathway, which controls the YAP/TAZ transcriptional co-activators and is intimately linked to the cell polarity machinery [8]. There are also examples where loss of polarity proteins such as PAR3 clearly promote tumor growth and metastasis, but other examples in which the data are ambiguous, or where polarity proteins can promote tumorigenesis [9].
This review will discuss recent progress in untangling these issues, and identify areas where further studies are required.
Polarity proteins and Hippo signaling in epithelial homeostasis
The Hippo signaling pathway involves a protein kinase cascade, the downstream effectors of which are the YAP/TAZ transcriptional co-activators. When the Hippo kinase cascade is activated, YAP/TAZ are phosphorylated and retained in the cytoplasm, promoting their degradation and inhibiting function. Inactivation of Hippo signaling permits nuclear accumulation of YAP/TAZ and stimulates cell proliferation. Hippo signaling has many inputs, and YAP/TAZ localization can be determined not just by phosphorylation but also by sequestration to the cell cortex [8]. Perhaps unexpectedly, the polarity machinery plays a central role in Hippo regulation, with multiple levels of interaction between the two pathways, and it is likely that there is still much to learn about the intricacies and meaning of these interactions.
If one considers the polarity machinery in epithelial cells to be divided into four parts, comprising an apical complex (CRUMBS, PALS1, PATJ), the PAR complex (PAR3, PAR6, aPKC), a lateral cluster (SCRIB, LLGL, DLG), and a fourth, partially cytoplasmic group (PAR4/LKB1, PAR1/MARK, PAR5/14–3-3) it is surprising to realize that all of them associate with components of the Hippo pathway in one way or another (Figure 1).
In Drosophila, the apical polarity protein Crumbs associates with Expanded, a member of the FERM domain protein family distantly related to NF2 and other family members. Expanded in turn binds directly to the fly version of YAP (called Yorkie), recruiting it to the apical membrane and blocking nuclear accumulation [10]. Loss of Crumbs or Expanded results in tissue hyperproliferation in Drosophila [11]. However, mammals do not have a clear homolog of Expanded, and Crumbs3 loss is not associated with overgrowth in mammalian epithelia. Nonetheless, Crumbs3 expression during proximal airway development results in a similar recruitment of YAP to the apical cortex as occurs in Drosophila, and this YAP retention mechanism is required for normal airway cell differentiation [12]. The PALS1 polarity protein, which associates with PATJ at tight junctions, interacts with another Hippo regulator, NF2 (Merlin), that forms a complex with Angiomotin (AMOT) and YAP. At least in over-expression studies using HEK293 cells, this complex can regulate activity of the small GTPase RAC1 [13]; and phosphorylation of AMOT at Ser176 retains the complex at tight junctions, inhibiting YAP function, while dephosphorylation permits nuclear accumulation of AMOT-YAP to promote cell proliferation [14]. Notably, NF2 is a known tumor suppressor the loss of which is closely linked to neurofibromatosis, sporadic meningiomas, ependymomas, schwannomas, and pleural mesotheliomas [15].
Within the PAR complex, activation of the apical polarity protein aPKCζ induces YAP activation and subsequent over-proliferation and multilayering in MDCK cells [16]. KIBRA, which in many cell types is an upstream activator of Hippo signaling, binds to and inhibits aPKCζ, displacing PAR3, but the biological function of this inhibition remains unclear [17].
Another polarity protein that affects Hippo signaling is DLG5 (Discs Large 5), which localizes to lateral membranes. DLG5 serves as a scaffold for interaction of upstream Hippo kinases MST1/2 and the MARK3 protein, which inhibits MST1/2 kinase activity, and DLG5 KO mice have increased Hippo signaling activity [18]. Why this specific isoform of DLG and not others is linked to Hippo remains unclear. A second lateral polarity protein is SCRIB (Scribble), which has also been implicated in Hippo regulation, and has been reported to bind to TAZ in an inhibitory complex with the LATS and MST kinases [19]. Interestingly, yet another polarity protein, LKB1 (PAR4) kinase, acts through one of its substrates, the PAR1/MARK polarity kinase, to regulate Scribble localization and the activity of the Hippo pathway [20]. A key question for the future is how all these interactions work together to regulate epithelial Hippo signaling in a coherent manner. It will also be important to determine how Hippo signaling regulates apical-basal cell polarity.
Hippo-independent cell density control through polarity proteins
Despite the apparent ubiquity of Hippo signaling in epithelial growth control there are several mechanisms that appear to function through the polarity machinery independently of YAP/TAZ or their upstream regulators. For example, loss of the latero-basal polarity proteins Llgl1 and Llgl2 results in the over-proliferation of epithelial cells at high density, but not at low density. These two proteins appear to act redundantly in this homeostatic process, but not through Hippo. Instead, Llgl1/2 inhibit the multimeric CRL4 E3-ligase complex, by sequestering VprBP away from this complex. The CRL4 complex is necessary for degradation of the cell cycle kinase inhibitor p27, so loss of Llgl1/2 results in VprBP binding to and activating CRL4, which degrades p27 and allows the cell cycle to proceed even at high cell densities (Figure 1). Intriguingly, Llgl1/2 binding of VprBP increases with increasing cell density, suggesting a regulated mechanism. However, the molecular basis for this mechanism remains to be elucidated. Although phosphorylation of Llgl2 by aPKC decreases Llgl1/2 binding of VprBP and increases proliferation, this phosphorylation is independent of cell density [21].
Interestingly, CRL4DCAF1 (DDB1- and CUL4-associated factor 1) has been reported to ubiquitylate and inhibit the Hippo-pathway Lats1/2 kinases in the nucleus, and NF2 binds to and suppresses CRL4 function [22]. The Cullin that forms the platform for this multimeric E3 ligase is amplified in many types of solid tumors. It seems possible, therefore, that this pathway is somehow linked to Llgl1/2 to control cell density-dependent proliferation.
Polarity proteins as tumor suppressors
Perturbation of cell polarity is thought to be an early event in the progression of tumorigenesis, and apical–basal polarity has been described as a barrier to carcinogenesis [9,23]. Conversely, loss of cell polarity is often considered a ‘precondition’ and a hallmark for cancer [24]. However, there are different definitions of cell polarity, which often confuses this issue. For example, if the PAR3 polarity protein is silenced in MDCK cells aPKC becomes mislocalized, but apical proteins such as podocalyxin, and tight junction proteins are unperturbed [25], so one could conclude that these cells either retain cell polarity or have lost it, depending on the chosen markers. In many solid cancers some polarity proteins are mislocalized or show reduced expression, but these correlations do not necessarily signify loss of polarized cell morphology, and do not address causality. Moreover, carcinomas frequently retain epithelial characteristics and continue to express polarity proteins, so it remains in many cases unclear if loss of any aspect of polarity is a cause or a consequence of tumorigenesis.
Polarity proteins were first implicated as tumor suppressors when early studies in Drosophila larvae revealed massive tissue overgrowth in response to loss-of-function mutations in polarity genes Discs large (dlg), Lethal Giant larvae (llgl) and Scribble (scrib) [26–29]. Deletion of several Hippo components gives a similar phenotype, but it is unclear whether Llgl, Dlg or Scrib work through this pathway in Drosophila. Recently, Scrib knockdown in wing imaginal discs was reported to induce overgrowth as a result of mitochondrial dysfunction and fission, through Drp-1 upregulation [30]; and Llgl can activate Notch through JNK signaling [31].
Despite the early identification of these tumor suppressor genes in flies, there seemed to be no functional correspondence with the mammalian homologs, and until recently it was controversial as to whether polarity genes were of any importance in human cancer. Nonetheless, a few cases demonstrated that some polarity proteins have a strong impact on cancer progression (Figure 2). PAR3 depletion in the murine mammary gland promotes tumor growth and metastasis in multiple oncogene models, for example [32,33]. More recently, loss of PAR3 in keratinocytes was found to promote malignant melanoma through a cell non-autonomous mechanism that creates a permissive niche for melanocyte transformation [34]. Additionally, loss of PAR3 in the prostate gland causes high grade intraepithelial neoplasia [35]. The underlying mechanism appears to involve Hippo signaling – disassembly of a PAR3/NF2/LATS1 complex prevents phosphorylation of LATS1, leading to YAP/TAZ activation. However, the same authors report elsewhere that PAR3 also acts as a prostatic tumor promoter, through a PAR3/aPKC/KIBR complex that inactivates Hippo signaling [36]. Clearly, further work is required to resolve these apparent contradictions.
Figure 2: The mechanism of Mammalian polarity proteins in tumorigenesis.
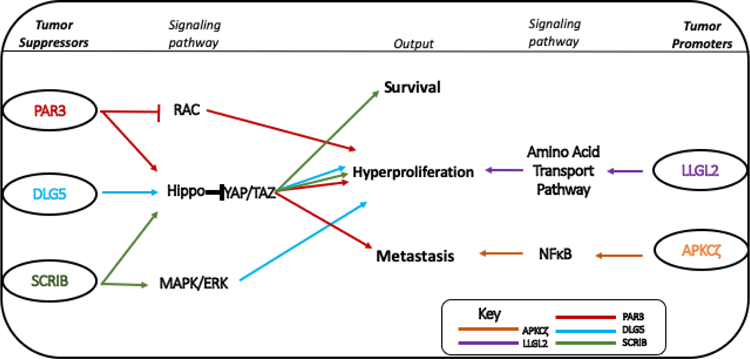
PAR3 (red), DLG5 (blue) and SCRIB (green) usually function as tumor suppressors, preventing tumorigenesis via regulation of the RAC, HIPPO and MAPK/ERK pathways. LLG2 (purple) and APKCζ (orange) are tumor promoters. LLG2 promotes hyperproliferation via the amino acid transport pathway and aPKCζ can promote metastasis via NFκB signaling.
As described above, loss of DLG5 also results in the activation of YAP/TAZ. YAP is also known to be a driver of spindle cell carcinoma, an aggressive subtype of squamous cell carcinoma, and synergizes with loss of epithelial cell polarity [37]. SCRB deficiency has been reported to enhance liver tumor growth in vivo, and over-expression can suppress growth of Hepatocellular carcinoma cells in culture [38]. A mechanism was proposed in which Scrib disrupts a positive feed-back loop between YAP1 and c-MYC in HCC cells, and simultaneously regulates the MAPK/ERK and Hippo signaling pathways [38]. In the mammary gland, depletion of SCRB results in luminal filling, decreased apoptosis and over-proliferation of mammary epithelial cells [39].
Polarity proteins as tumor promoters
Despite the links described above, the relationship of polarity proteins to cancer is not straightforward, because in several instances these proteins seem able to promote rather than suppress tumorigenesis. In Drosophila, for example, aPKC can behave as an oncogene, causing neoplastic growth in eye imaginal disc epithelia by disrupting the Hippo signaling pathway [40], and depletion of mammalian PKCζ inhibits invasion and metastasis of breast cancer cells in mice [41]. On the other hand, PAR3 seems able to function in both pro-oncogenic and tumor repressor capacities. Using a mouse skin tumorigenesis model, Iden and colleagues demonstrated that loss of PAR3 reduced papilloma formation and growth, suggesting a tumor promoting activity; but the PAR3-deficient mice were also more susceptible to keratoacanthomas [42].
Most interestingly, Llgl2 was recently demonstrated to be a tumor promoter in breast cancers [43]. Llgl2 (but not a second isoform, Llgl1) is over-expressed in the majority of ER+ breast cancers and high expression is associated with poor prognosis and resistance to endocrine treatment. Llgl2 is induced in response to estradiol, and Llgl2 depletion reduced cell proliferation. Remarkably, Llgl2 forms a trimeric complex with a leucine transporter, SLC7A5, and with the SNARE protein YKT6, a regulator of membrane fusion, to increase surface levels of SLC7A5 and promote leucine uptake. The increased availability of leucine stimulates cell proliferation and confers resistance to anti-estrogen treatment [43].
Summary and outlook
Recent years have dramatically expanded our understanding of the numerous links between the polarity machinery and Hippo signaling, and their involvement in epithelia homeostasis. It has also become increasingly clear that failure to regulate homeostasis is a key early attribute of carcinoma. However, the relationship of cell polarity to cancer is much more complicated than was initially appreciated. Polarity proteins can function as either tumor suppressors or tumor promoters depending on context, and closely related isoforms can display quite different functions.
Acknowledgements
This work was supported by grant CA197571 and CA197571-S1 (to IGM) from the National Cancer Institute, NIH
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
- 1.Gehart H, Clevers H: Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol 2019, 16:19–34. [DOI] [PubMed] [Google Scholar]
- 2.Combes AN, Phipson B, Lawlor KT, Dorison A, Patrick R, Zappia L, Harvey RP, Oshlack A, Little MH: Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development 2019, 146:dev174177. [DOI] [PubMed] [Google Scholar]
- 3.Marcheque J, Bussolati B, Csete M, Perin L: Concise Reviews: Stem Cells and Kidney Regeneration: An Update. Stem Cells Transl Med 2019, 8:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S: A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154:1047–1059. [DOI] [PubMed] [Google Scholar]
- 5.Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J: Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484:546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danes CG, Wyszomierski SL, Lu J, Neal CL, Yang W, Yu D: 14–3-3 zeta down-regulates p53 in mammary epithelial cells and confers luminal filling. Cancer Res 2008, 68:1760–1767. [DOI] [PubMed] [Google Scholar]
- 7.Oudenaarden CRL, van de Ven RAH, Derksen PWB: Re-inforcing the cell death army in the fight against breast cancer. J Cell Sci 2018, 131:jcs.212563. [DOI] [PubMed] [Google Scholar]
- 8.Ma S, Meng Z, Chen R, Guan KL: The Hippo Pathway: Biology and Pathophysiology. Annu Rev Biochem 2018, 88:577–604. [DOI] [PubMed] [Google Scholar]
- 9.Saito Y, Desai RR, Muthuswamy SK: Reinterpreting polarity and cancer: The changing landscape from tumor suppression to tumor promotion. Biochim Biophys Acta Rev Cancer 2018, 1869:103–116. [DOI] [PubMed] [Google Scholar]
- 10.Badouel C, Gardano L, Amin N, Garg A, Rosenfeld R, Le Bihan T, McNeill H: The FERM-domain protein Expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev Cell 2009, 16:411–420. [DOI] [PubMed] [Google Scholar]
- 11.Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y, Wu S, Pan D: The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci U S A 2010, 107:10532–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szymaniak AD, Mahoney JE, Cardoso WV, Varelas X: Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev Cell 2015, 34:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R, et al. : A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 2011, 19:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **14.Moleirinho S, Hoxha S, Mandati V, Curtale G, Troutman S, Ehmer U, Kissil JL: Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. Elife 2017, 6:e23966.Moleirinho et al. show that Angiomotin (Amot) phosphorylation leads to its association with E-cadherin and polarity proteins Pals1/PATJ. Dephosphorylation of Amot shifts its localization from tight junctions to the nucleus, where it stimulates YAP-TEAD interaction and subsequent transcription of YAP target genes.
- 15.Petrilli AM, Fernandez-Valle C: Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archibald A, Al-Masri M, Liew-Spilger A, McCaffrey L: Atypical protein kinase C induces cell transformation by disrupting Hippo/Yap signaling. Mol Biol Cell 2015, 26:3578–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshihama Y, Chida K, Ohno S: The KIBRA-aPKC connection: A potential regulator of membrane trafficking and cell polarity. Commun Integr Biol 2012, 5:146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwan J, Sczaniecka A, Heidary Arash E, Nguyen L, Chen CC, Ratkovic S, Klezovitch O, Attisano L, McNeill H, Emili A, et al. : DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genes Dev 2016, 30:2696–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al. : The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147:759–772. [DOI] [PubMed] [Google Scholar]
- 20.Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, et al. : A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 2014, 16:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita K, Ide M, Furukawa KT, Suzuki A, Hirano H, Ohno S: Tumor suppressor protein Lgl mediates G1 cell cycle arrest at high cell density by forming an Lgl-VprBP-DDB1 complex. Mol Biol Cell 2015, 26:2426–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P, et al. : Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halaoui R, McCaffrey L: Rewiring cell polarity signaling in cancer. Oncogene 2015, 34:939–950. [DOI] [PubMed] [Google Scholar]
- 24.Ellenbroek SI, Iden S, Collard JG: Cell polarity proteins and cancer. Semin Cancer Biol 2012, 22:208–215. [DOI] [PubMed] [Google Scholar]
- 25.McCaffrey LM, Macara IG: The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev 2009, 23:1450–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bilder D: Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev 2004, 18:1909–1925. [DOI] [PubMed] [Google Scholar]
- 27.Martin-Belmonte F, Perez-Moreno M: Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 2011, 12:23–38. [DOI] [PubMed] [Google Scholar]
- 28.Wodarz A, Nathke I: Cell polarity in development and cancer. Nat Cell Biol 2007, 9:1016–1024. [DOI] [PubMed] [Google Scholar]
- 29.Levayer R, Moreno E: Mechanisms of cell competition: themes and variations. J Cell Biol 2013, 200:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yadav AK, Srikrishna S: scribble (scrib) knockdown induces tumorigenesis by modulating Drp1-Parkin mediated mitochondrial dynamics in the wing imaginal tissues of Drosophila. Mitochondrion 2019, 44:103–110. [DOI] [PubMed] [Google Scholar]
- *31.Paul MS, Singh A, Dutta D, Mutsuddi M, Mukherjee A: Notch signals modulate lgl mediated tumorigenesis by the activation of JNK signaling. BMC Res Notes 2018, 11:247.Paul et al. report that co-expression of Nact and lgl-IR in Drosophila eye disc results in overgrowth, loss of positional clues and upregulation of MMP1 expression. They propose that the lgl loss-of-function wing phenotype is dependent on elevated Notch signaling, consistent with previous studies.
- 32.Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK: Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat Cell Biol 2013, 15:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCaffrey LM, Montalbano J, Mihai C, Macara IG: Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 2012, 22:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mescher M, Jeong P, Knapp SK, Rubsam M, Saynisch M, Kranen M, Landsberg J, Schlaak M, Mauch C, Tuting T, et al. : The epidermal polarity protein Par3 is a non-cell autonomous suppressor of malignant melanoma. J Exp Med 2017, 214:339–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *35.Zhou PJ, Wang X, An N, Wei L, Zhang L, Huang X, Zhu HH, Fang YX, Gao WQ: Loss of Par3 promotes prostatic tumorigenesis by enhancing cell growth and changing cell division modes. Oncogene 2019, 38:2192–2205.Zhou et al. report that co-deletion of Par3 and Lats1 results in prostate tumor initiation, but co-deletion of Par3 and YAP reverses the phenotype to normal.
- 36.Zhou PJ, Xue W, Peng J, Wang Y, Wei L, Yang Z, Zhu HH, Fang YX, Gao WQ: Elevated expression of Par3 promotes prostate cancer metastasis by forming a Par3/aPKC/KIBRA complex and inactivating the hippo pathway. J Exp Clin Cancer Res 2017, 36:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **37.Vincent-Mistiaen Z, Elbediwy A, Vanyai H, Cotton J, Stamp G, Nye E, Spencer-Dene B, Thomas GJ, Mao J, Thompson: YAP drives cutaneous squamous cell carcinoma formation and progression. Elife 2018, 7:e33304.Vincent-Mistiaen et al. show that in human spindle cell carcinoma Yap is localized to the nucleus. They also show that in mice activated Yap and K5 can induce formation of squamous cell carcinoma and spindle cell carcinoma. Upon scratch wounding, spindle cell carcinomas develop and the expression of ZEB1, the EMT transcription factor, is induced. This revealed a synergistic relationship between wound healing and YAP to induce progression of squamous cell carcinoma to spindle cell carcinoma.
- 38.Kapil S, Sharma BK, Patil M, Elattar S, Yuan J, Hou SX, Kolhe R, Satyanarayana A: The cell polarity protein Scrib functions as a tumor suppressor in liver cancer. Oncotarget 2017, 8:26515–26531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russ A, Louderbough JM, Zarnescu D, Schroeder JA: Hugl1 and Hugl2 in mammary epithelial cells: polarity, proliferation, and differentiation. PLoS One 2012, 7:e47734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archibald A, Mihai C, Macara IG, McCaffrey L: Oncogenic suppression of apoptosis uncovers a Rac1/JNK proliferation pathway activated by loss of Par3. Oncogene 2015, 34:3199–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paul A, Danley M, Saha B, Tawfik O, Paul S: PKCzeta Promotes Breast Cancer Invasion by Regulating Expression of E-cadherin and Zonula Occludens-1 (ZO-1) via NFkappaB-p65. Sci Rep 2015, 5:12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iden S, van Riel WE, Schafer R, Song JY, Hirose T, Ohno S, Collard JG: Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer Cell 2012, 22:389–403. [DOI] [PubMed] [Google Scholar]
- **43.Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, Asara JM, Brown M, Muthuswamy SK: LLGL2 rescues nutrient stress by promoting leucine uptake in ER(+) breast cancer. Nature 2019, 569:275–279.Saito et al. found that increased LLGL2 (but not LLGL1) expression in ER+ breast cancer associates with poor survival. They demonstrated that LLGL2 is required for cell proliferation under nutrient stress conditions. LLGL2 localizes the leucine transporter SLC7A5 to the cell surface, which is necessary for cell proliferation. Importantly, the authors found that estrogen treatment increased cell proliferation via induction of LLGL2 expression. Hence, estrogen mediates adaptation to nutrient stress through expression of LLGL2, which supports leucine transport.