

SUMMARY
Biological membranes are complex barriers in which membrane proteins and thousands of lipidic species participate in structural and functional interactions. Developing a strategic approach that allows uniform labeling of membrane proteins while maintaining a lipidic environment that retains functional interactions is highly desirable for in vitro fluorescence studies. Herein, we focus on complementing current methods by integrating the powerful processes of unnatural amino acid mutagenesis, bioorthogonal labeling and the detergent-free membrane protein solubilization based on the amphiphilic styrene-maleic acid (SMA) polymer. Importantly, the SMA polymer preserves a thermodynamically stable shell of phospholipids. The approach that we present is both rapid and generalizable providing a population of uniquely-labeled membrane proteins in lipid nanoparticles for quantitative fluorescence-based studies.
Keywords: membrane protein, non-canonical amino acid, styrene maleic acid copolymer, bioorthogonal labeling, single-molecule imaging
Graphical Abstract
eTOC
Membrane proteins are challenging targets for detailed biophysical analysis. Here, Swiecicki et al. apply an integrated approach involving unnatural amino acid mutagenesis, bioorthogonal labeling and detergent-free membrane protein solubilization based on an amphipathic polymer. When combined, these technologies deliver uniquely-labeled membrane proteins in a native-like lipid bilayer for fluorescence-based studies.
INTRODUCTION
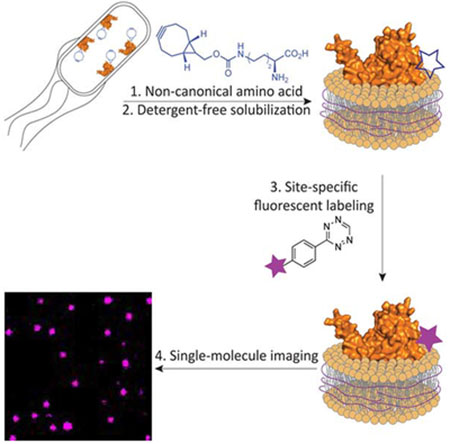
Single-molecule fluorescence imaging of nucleic acids and soluble proteins is providing an unprecedented level of detail regarding the organization and dynamics of supra molecular complexes, and has provided new insight into important cellular processes (Petrov et al., 2012; Ticau et al., 2017; Wang and Greene, 2011). However, although approximately 25% of human proteins are predicted to be α-helical transmembrane proteins, fluorescence-based studies of membrane proteins still demand more versatile approaches (Almen et al., 2009). Such a need is further underscored by the fact that membrane proteins constitute 60% of approved drug targets (Overington et al., 2006). Current approaches for studying membrane proteins in vitro is a daunting task as they often require isolation protocols that are challenging to optimize and are non-generalizable. State-of-the-art strategies for preparing samples for fluorescence studies of membrane proteins involve the solubilization of the target membrane protein with detergent, site-selective labeling and finally reconstitution into liposomes or nanodiscs (McLean et al., 2018; Nath et al., 2010; Whorton et al., 2007). To be successful, these methods require considerable handling and optimization at each step. A major issue is that the introduction of surfactants disrupts native interactions and is not suitable for investigating weak intra- and intermolecular interactions (Etzkorn et al., 2013). Moreover, detergents solubilize by replacing native membrane components and fall far short of capturing the subtle, but essential, roles of the membrane environment. Herein, we present a general strategic approach that addresses these current technical hurdles and affords, in a few days, fluorescently-labeled membrane proteins embedded in soluble lipoparticles. This approach integrates three recently introduced methods: (i) genetic code expansion in Escherichia coli using the pyrrolysyl-tRNA synthetase/pyrrolysyl-tRNA pair for the incorporation of a non-canonical amino acid (ncAA), (ii) efficient bioorthogonal labeling and (iii) detergent-free membrane protein solubilization that preserves a native-like membrane environment (Figure 1). All of these methods are established, but they have not previously been incorporated into a single streamlined work flow that may be pivoted towards any membrane protein.
Figure 1. General approach for solubilization and labeling of a target membrane protein for fluorescence-based applications.

The addition of BCN within the native protein sequence is performed using the M. mazei pyrrolysyl ncAA system. Once solubilized using SMA polymer, the protein is labeled using the rapid cycloaddition with a tetrazine-linked fluorophore (Knowles et al., 2009; Lang et al., 2012).
A broad palette of methods has been developed for fluorescence labeling of proteins. In cell protein fluorescent labeling can be conveniently performed using intrinsically fluorescent tags (e.g. GFP) or self-labeling tags (e.g. SNAP- or CLIP-tags) Gautier et al., 2008; Keppler et al., 2003; Phillips, 2001). Unfortunately, these tags are large and not suited for single-molecule methods that require the label to be placed at defined positions. Site-specific labeling of protein can alternatively be performed in vitro, through modification of cysteine (Chen and Wu, 2016). However, these strategies can be low yielding or even prohibitive if the native protein sequence contains cysteines. Alternatively, genetic-code expansion methods enable the incorporation of ncAAs with intrinsic fluorescent properties or with side chains for efficient orthogonal conjugation as “minimalist” tags for labeling (Chen and Wu, 2016). ncAAs can be introduced by repurposing a non-sense codon, most commonly the amber stop codon (TAG). This expansion of the genetic code, termed amber suppression, requires the addition of an engineered tRNA synthetase/tRNATAG pair. tRNA synthetase/tRNATAG pairs have been successfully engineered for the incorporation of small fluorescent ncAAs, such as acridon-2-ylalanine (Acd) and 3-(6-acetylnaphthalen-2-ylamino)-2-aminopropanoic acid (Anap), which have been applied in the biophysical characterization of membrane proteins (Kalstrup and Blunck, 2013; Padmanarayana et al., 2014). Although powerful for bulk fluorescence studies of proteins, intrinsically fluorescent ncAAs cannot be exploited for single-molecule imaging applications because of limited brightness. We therefore chose to implement the two-step strategy: (i) insertion of a ncAA followed by (ii) biorthogonal labeling with a fluorescent dye compatible with single-molecule studies.
We decided to employ the Methanosarcina mazei pyrrolysyl-tRNA synthetase (PylRS)/pyrrolysyl-tRNATAG system due to its efficiency and versatility for amber suppression (Wan et al., 2014). In particular, the PylRS Y306A Y384F double mutant (PylRSAF) was used to incorporate the bicyclononyne-lysine (BCN) ncAA. BCN is stable in biological media and reacts rapidly with fluorescent tetrazine derivatives via a high-yielding strain-promoted inverse-electron-demand Diels-Alder cycloaddition (Figure 1) Borrmann et al., 2012; Lang et al., 2012; Plass et al., 2011).
One limitation of the amber suppression methodology is the competition between the tRNATAG and the endogenous release factor 1 (RF1) that can induce early termination of translation. To overcome this, we performed heterologous expression in the genomically-recoded organism (GRO) Escherichia coli strain C321.ΔprfA. The GRO is deleted in RF1 and all amber stop codons are replaced by synonymous ochre stop codons (Lajoie et al., 2013). Very importantly, the scope of this method could also be expanded to the insertion of two orthogonal ncAAs for dual labeling of membrane proteins for Forster resonance energy transfer (FRET) experiments (Willis and Chin, 2018).
To complement ncAA mutagenesis and labeling, detergent-free solubilization of membrane proteins is achieved using the unique properties of the styrene-maleic acid (SMA) copolymer (Figure 1)Dorr et al., 2014; Knowles et al., 2009; Lee et al., 2016). The SMA polymer generates discoidal lipoparticles that are 10-20 nm in diameter. One of the major advantages of this solubilization method is that the SMA polymer preserves a thermodynamically-stable phospholipid shell around the target protein (Dorr et al., 2014; Hazell et al., 2016). Applied together, ncAA mutagenesis, bioorthogonal labeling and detergent-free solubilization have the potential to deliver labeled membrane proteins into lipoparticles for fluorescence-based analyses.
RESULTS
The protein N-linked glycosylation (pgl) pathway of the human pathogen Campylobacter jejuni is the target of this study (Figure 2A)Larkin and Imperiali, 2011; Szymanski et al., 1999). The pgl pathway involves nine membrane-associated steps, which have been biochemically characterized (Hartley et al., 2013; Larkin and Imperiali, 2011; Schwarz and Aebi, 2011). PglC and PglA have different membrane topologies and as such are good candidates for establishing the proof-of-concept for the integrated approach; PglA is membrane-associated and PglC is a monotopic membrane protein (Entova et al., 2018; Glover et al., 2005; Ray et al., 2018). Moreover, the purification of PglC is challenging because the hydrophobic domain that is critical for structure makes it a recalcitrant target for detailed biophysical studies (Lukose et al., 2015).
Figure 2. Detergent-free solubilization of membrane proteins of the N-linked protein glycosylation (pgl) pathway in C. jejuni.

(A) The pgl pathway in C. jejuni Larkin and Imperiali, 2011). PglC is a phosphoglycosyl transferase that catalyzes the formation of a phosphodiester bond between phospho-N,N′-diacetylbacillosamine (diNAcBac) from UDP-N,N′-diacetylbacillosamine and undecaprenyl-phosphate (Und-P) Ray et al., 2018). PglA is a glycosyltransferase that catalyzes transfer of N-acetylgalactosamine (GalNAc) from UDP-GalNAc to form Und-P-P-diNAcBac-GalNAc (Glover et al., 2005).
(B) SDS-PAGE analysis of the elution fractions following Ni2+NTA-affinity purification of PglC (24 kDa) and PglA (45 kDa) in SMALP (Coomassie staining). See Table S1 for information about commercially available SMA polymers. See also Figure S1.
(C) Activity of PglC in SMALP evaluated using the UMP-Glo assay (Promega), which allows quantification of UMP release from the PglC-catalyzed reaction (Das et al., 2016). Data are represented as mean ± SEM, n = 3; replicates were performed on distinct aliquots taken from a common protein purification using SMA for solubilization.
A cornerstone of the approach is the detergent-free solubilization of membrane proteins. Several SMA polymers are currently available (Table S1). Therefore, the efficiencies of three SMAs with different styrene to maleic acid ratios (3:1, 2.3:1 and 1.2:1; from Polyscope) were compared. The selection criteria used for the polymer are: (i) purification yield, (ii) protein purity and (iii) homogeneity of the SMA-lipoparticles (SMALPs). PglC and PglA were heterologously expressed in the GRO. The addition of a SUMO tag at the N-terminus of PglC has been shown to improve the detergent-mediated purification of this membrane protein (Entova et al., 2018; Lukose et al., 2015). Therefore, we used both the His6-SUMO-tagged and PglC-His8 constructs to challenge the SMA-mediated solubilization. His6-SUMO-PglC, PglC-His8 and PglA-His6 were solubilized directly from crude cell-envelope fractions (CEFs) using the three SMA polymers and isolated through a Ni2+-affinity purification. While the most hydrophobic SMA (3:1 styrene to maleic acid) did not solubilize the proteins, the 2.3:1 and 1.2:1 SMAs efficiently solubilized all three constructs (Figure 2B and Figure S1). Interestingly, these polymers solubilized SUMO-PglC and PglC in comparable amounts, allowing omission of the extra tag that had previously been absolutely required for the detergent-mediated solubilization and purification. Particle sizing further demonstrated that the direct solubilization of PglC and PglA by the 2.3:1 SMA yielded membrane protein in particles of homogeneous size (Figure S1). This polymer was used for the rest of this study. Importantly, PglC was found to be catalytically active in SMALP (Figure 2C).
Sites for amber suppression were rationally selected using sequence homology analyses and the predicted structures of PglC and PglA from C. jejuni (Figure S2). As expected, the amber suppression efficiency strongly depends on the position of the ncAA (Figure 3A–B, Table S2, S3). For some variants, the suppression efficiency was higher than 50 %, reaching 80 % for PglAW102TAG. Differences in suppression efficiency are likely to be due to a combination of factors, including the codon context of the amber codon in the mRNA and the position of the suppression site in the protein tertiary structure (Miller and Albertini, 1983). The suppression efficiency is difficult to predict, and as Hostetler et al. recommended for soluble proteins, testing a large number of positions for the installation of ncAA in a new membrane protein target is necessary (Hostetler et al., 2018). We also noted that in the absence of the BCN ncAA, the expression of full-length protein is possible. This might be due to the loading of a canonical amino acid at the amber stop codon, which is enhanced by the absence of RF1 (Lajoie et al., 2013). In summary, positions located within different structural elements and on different faces of PglC and PglA were successfully substituted with BCN, offering the opportunity to evaluate the labeling efficiency in different physico-chemical environments.
Figure 3. Incorporation on the BCN non-canonical amino acid into membrane proteins.

(A) Expression yield of PglC (left) or PglA (right) variants normalized to the expression of the corresponding WT protein, in the presence or absence of BCN. Expression was quantified by anti-His western blot.
(B) Heatmap representing the amber suppression efficiency at individual sites on PglC (left) and PglA (right) (see Figure S2 for details on the structure prediction). The structure of PglC from C. jejuni was predicted with the online server iTasser using the PglC crystal structure (PDB: 5W7L) from C. concisus as template (Ray et al., 2018; Roy et al., 2010; Yang et al., 2015; Zhang, 2008). The position of PglC with respect to the membrane plane was determined using the server “Orientations of Proteins in Membranes” Lomize et al., 2012). Positions selected for the insertions of non-canonical amino acids were selected based on their position within this predicted structure (surfaced exposed and in a diverse physico-chemical environment) and also based on the alignment of 15,000 sequences (non-conserved residues) Lukose et al., 2015). The structure of PglA from C. jejuni was predicted with the online server iTasser using the structure of the glycosyl transferases WbnH (PDB: 4XYW) from E. coli as the template (Martinez-Fleites et al., 2006; Roy et al., 2010; Yang et al., 2015; Zhang, 2008). Position of PglA with respect to the membrane plane was also determined using the server “Orientations of Proteins in Membranes”. Amber suppression efficiencies were calculated as the difference between the quantity of variant expressed in the presence and absence of BCN, normalized by the quantity of expressed WT protein. See also Tables S2 and S3, and Figure S2.
Several PglC and PglA variants were further expressed for labeling. All of the target protein variants were successfully solubilized into SMALP (Figure S2). The labeling of PglC and PglA reaches up to 95 % conversion, depending on the position of the BCN in the sequence (Figure 4A–B, Tables S2 and S3 and Figure S3). We observed that the labeling efficiency was systematically higher if BCN was located close to, or embedded in, the membrane (Figure 4C). This result may find its origin in the high local concentration of the hydrophobic Cy3 fluorophore within the membrane promoting the reaction. A similar observation has been previously reported for membrane proteins solubilized in detergent (Tian et al., 2015). In order to increase the conversion of only partially labeled residues, the concentration of the tetrazine derivative, the temperature and/or incubation time were increased. However, these attempts did not afford additional conversion. We also attempted to use hydrophilic tetrazine dye derivatives, such as sulfo-Cy3 methyltetrazine. In this case the labeling of PglC was less efficient, probably because of the lower reactivity of methyltetrazine in comparison to the tetrazine moiety (Figure S3) Chen and Wu, 2016). If positions located within the soluble domain of a membrane protein are the targets for labeling, the more reactive bicyclic trans-cyclooctene-lysine (sTCO) ncAA would be the preferred alternative (Lang et al., 2012);Plass et al., 2012). To challenge the generality of this strategic approach for the solubilization and labeling of membrane proteins, we applied it to two other proteins, LpxM and WbaP. LpxM is a fatty acid acyltransferase involved in the biosynthesis of lipid A in E. coli. This protein, which is unrelated to PglC and PglA, exhibits a reentrant membrane topology (Entova et al., 2018). WbaP is a phosphoglycosyltransferase, which catalyzes the transfer of galactose-1-phosphate onto undecaprenyl phosphate and participate to O-antigen biosynthesis in Salmonella enterica. In contrast to PglC and LpxM, WbaP is a polytopic membrane protein. The N-terminal domain comprises four transmembrane helices while the C-terminal domain is homologous to PglC and include a rerentrant helix (Furlong et al., 2015). The method was applied to LpxM and WbaP to generate membrane proteins in SMALPs. In brief, variants of LpxM-His6 and WbaP-His8 were expressed in GRO and the amber suppression was evaluated (Figure S4). Three LpxM and two WbaP variants were selected for expression and purification using the 2.3:1 SMA (Table S1). Reaction with Cy5-tetrazine yielded LpxM and WbaP proteins labeled in SMALP, suitable for single-molecule studies (Figure S4).
Figure 4. Incorporation on the BCN non-canonical amino acid into membrane proteins and labeling with a tetrazine-linked Cy3 fluorophore derivative.

(A) Imaging of a western blot membrane showing Cy3 fluorescence (top). Anti-His western blot of the same SDS-PAGE showing that the labeling of PglC can be quantitative (bottom). Lanes containing the unreacted and reacted PglC in SMALP are placed side-by-side for clarity. PglC variants are 24 kDa, labeled PglC-Cy3 variants are 25 kDa. See Figure S2 for the details on the purification of PglC and PglA variants in SMALP.
(B) Quantitative evaluation of the efficiency of labeling in PglC. Data are represented as mean ± SEM, n = 3; replicates were performed on distinct CEF aliquots.
(C) Heatmap representing the labeling efficiency in PglC (left) and PglA (right). See also Tables S2 and S3 and Figure S3 and S4.
The detergent-free solubilization of membrane proteins into SMALPs is an empirical process; therefore, it is important to determine the number of target protein monomers located within a particle. The fluorescent properties of the label can be advantageously used to determine this number by counting photobleaching steps (Das et al., 2007). PglC and PglA variants were labeled in SMALP using a tetrazine-linked Cy5 fluorophore (Cy5 is prone to photobleaching) and purified away from excess dye (Figure S5). First, the activity of Cy5-labeled PglC variants in SMALP was evaluated using the UMP-Glo assay. Each displayed reduced, but significant, activity with respect to PglC WT (19 to 32 % depending on the variant; Figure S5). Individually-labeled protein variants in SMALP were imaged using single-molecule total internal reflection fluorescence microscopy (smTIRF) (Figure 5A–B). For all protein variants, approximately 90% of the visible fluorescent spots exhibited a single-step photobleaching behavior. The residual ~10% exhibited a double-step behavior (Figure 5B–C), which can be attributed either to a single SMALP loaded with two proteins, or to two adjacent SMALPs loaded with a single protein. In conclusion, the analysis of photobleaching behavior confirmed that most SMALPs were loaded with individual membrane proteins.
Figure 5. Determination of the loading of PglC and PglA SMA-lipoparticles.

(A) Representative smTIRF image Cy5-PglAK321TAG in SMALP. A total of 50 frames were averaged for clarity.
(B) Example of 1- or 2-step photobleaching traces corresponding to the fluorescent dots that are circled on the image (A).
(C) Frequency of single, double or triple photobleaching steps observed for Cy5-PglC (left) or Cy5-PglA (right) variants in SMALP. Over 100 fluorescent traces were analyzed for each variant. See also Figure S5.
DISCUSSION
Single-molecule fluorescence imaging of membrane proteins can contribute to a much deeper understanding of the intramolecular dynamics of membrane proteins and the intermolecular organization and dynamics of labile membrane complexes (e.g. intermolecular FRET studies). Despite recent successes, labeling membrane proteins for in vitro fluorescence studies remains challenging to generalize. The presented approach combines generalizable methods that can rapidly and efficiently deliver stable populations of labeled proteins in a native-like membrane environment for fluorescence-based studies. Essentially, it takes just two days to go from cell lysis to video acquisition: one day for the cell lysis, isolation of the cell envelope fraction and SMALP; one day for the purification, labeling and imaging. We are confident that the approach can be further applied to the study of large complexes of membrane proteins, as demonstrated by the recently reported structure of a 464 kDa supercomplex in SMALP (Sun et al., 2018). We also anticipate that this method to be applicable to mammalian proteins heterologously expressed in E. coli, as well as to mammalian proteins expressed in mammalian cells. Indeed, the SMA-mediated solubilization of proteins located at the plasma membrane of mammalian cells has also been successfully applied to the human adenosine A2A receptor, a GPCR, expressed in HEK293T cell or to the human Kv channels expressed in COS-1 cells (Jamshad et al., 2015; Karlova et al., 2019). Furthermore, the genetic code expansion of mammalian cells is well-documented and the pyrrolysyl-tRNA synthetase/pyrrolysyl-tRNA pair that was use in this study is also orthogonal to the endogenous mammalian translation machinery, making it a system of choice (Italia et al., 2017). One limitation is the unavailability of a mammalian equivalent of GRO, i.e. a mammalian cell line depleted in the release factor competing with the suppressor tRNA. Strict depletion of the eukaryotic release factor 1 (eRF1) is not feasible as eRF1 recognizes all three stop codons and there is no eukaryotic equivalent to RF2. Efforts have been successfully focusing on engineering eRF1 to specifically reduce the termination at the amber stop codon while preserving termination property at ochre and opal stop codon (Schmied et al., 2014). Combined with an optimized pyrrolysyl-tRNA synthetase/pyrrolysyl-tRNA pair for expression in mammalian cells, it significantly increased amber suppression. In conclusion, we believe that our strategic approach that integrates the (i) genetic code expansion in the pyrrolysyl-tRNA synthetase/pyrrolysyl-tRNA pair for the incorporation of a ncAA, (ii) efficient bioorthogonal labeling and (iii) detergent-free membrane protein solubilization can be translated to mammalian proteins expressed in mammalian cells. Finally, the study of interactions amongst transmembrane domains or between transmembrane domains and membrane constituents is an actively emerging research field that aims to understand how the membrane environment modulates protein function (Yin and Flynn, 2016). Since this method yields membrane proteins in a thermodynamically-stable shell of phospholipids without exposure to detergents, it will also empower the study of the multiple roles of the membrane environment on protein-protein interactions. We thus anticipate this approach to be broadly applicable to many diverse biologically-relevant membrane-associated phenomena.
SIGNIFICANCE
Membrane proteins are ubiquitous and essential in all living systems. They impact functions as diverse and essential as nutrient transport, energy conversion and storage, as well as signal transduction. While fluorescence-based techniques have provided crucial insight into intra- and intermolecular dynamics and interactions of soluble proteins, similar in vitro studies with membrane proteins are challenging to perform. A major issue is that each membrane protein must be extracted from the membrane using detergents, which disrupt native interactions and deplete the native lipid environment. Herein we present an integrated strategic approach that enables the site-specific incorporation of fluorescent labels into proteins in lipid nanoparticles that feature a native-like membrane bilayer. The method combines unnatural amino-acid mutagenesis and biorthogonal conjugation for site-specific labeling of membrane proteins. It takes advantage of the pyrrolysine tRNA/aminoacyl tRNA synthase system for the introduction of a strained cycloalkyne that displays excellent reactivity with commercially-available tetrazine-tagged fluorescent dyes. Solubilization of target membrane proteins together with the native lipid environment is achieved using an amphipathic styrene-maleic acid (SMA) copolymer. The SMA solubilizes membrane proteins directly from native membranes, bypassing the use of detergent. We establish proof of principle using structurally-distinct enzymes including PglC and PglA from the N-linked protein glycosylation (pgl) pathway of the human bacterial pathogen Campylobacter jejuni. Once solubilized and labeled, we demonstrate that membrane proteins can be directly used for single-molecule fluorescence studies in a similar fashion to biopolymers including nucleic acids and soluble proteins, thus transforming membrane proteins into tractable targets for biophysical analyses. The approach is rapid, practical and provides samples of labeled membrane proteins suitable for biophysical studies with minimum handling. We believe that this integrated strategic approach represents a major advance towards the lexicon of methods for the study of membrane proteins.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILBILITY
Further information and requests for resources and reagents should be directed to the Lead Contact, Prof Barbara Imperiali (imper@mit.edu). All unique/stable reagents generated in this study will be made available by the Lead Contact on request but we may require a completed Materials Transfer Agreement
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For transformation or protein expression, E. coli C321.ΔprfA cells were grown in 5 mL LB-L (Lysogeny broth-Lennox: 10 g/L tryptone, 5 g/L yeast extract and 5 g/L NaCl) with supplemented with carbenicillin (50 μg/mL) at 34 °C overnight under agitation (170 rpm) Lajoie et al., 2013). If needed, temperature was varied during protein expression (decreased to 25 °C for the expression of WbaP).
METHOD DETAILS
Transformations of Escherichia coli C321.ΔprfA cells with the pEvol plasmid and pZE21 plasmids
Cells were sequentially transformed with (i) the pEvol plasmid that encodes the pyrrolysine tRNA synthetase double mutant PylRSAF and the cognate pyrrolysine tRNA as well as with (ii) the pZE21 plasmid that encodes PglC, PglA, LpxM or WbaP variants (Borrmann et al., 2012; Elowitz and Leibler, 2000; Plass et al., 2011; Young et al., 2010). Both transformations were performed in the same way: E. coli C321.ΔprfA cells grown overnight were harvested at 4 °C and washed with 10% glycerol in distilled water three times and finally resuspended in 200 μL 10% glycerol in distilled water. 50 μL of resuspended cells were transformed with DNA using 100 ng of plasmid by electroporation using an E. coli pulser at 2.5 kV. Transformed cells were then incubated for 1 h in 1 mL Super Optimal broth with Catabolite repression (SOC) medium at 34 °C and plated on selective LB-L agar plates (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl and 15 g of agar) supplemented with carbenicillin (50 μg/mL), chloramphenicol (30 μg/mL) and incubated overnight at 30 °C. Kanamycin (30 μg/mL) was the third antibiotic used for selection after transformation with the pZE21 plasmid containing the target membrane protein for expression.
Plasmid Constructs
Campylobacter jejuni PglC and PglA, Escherichia coli LpxM and Salmonella enterica WbaP were subcloned into the pZE21 vector for protein expression in E. coli C321.ΔprfA. PglC, PglA, LpxM and WbaP mutagenesis was performed by QuikChange site-directed mutagenesis. All variants were confirmed by sequencing.
Membrane protein expression in C321.ΔprfA cells
Transformed E. coli C321.ΔprfA cells were incubated overnight in 5 mL LB-L containing carbenicillin, chloramphenicol and kanamycin. 50 mL of a LB-L broth shaking culture (200 rpm; 34 °C) supplemented with chloramphenicol, carbenicillin and kanamycin for selection were inoculated with 250 μL of the overnight culture (dilution x200). When the culture reached an OD of 0.2, a stock of 80 mM bicyclononyne-lysine non-canonical amino acid (BCN, Sirius Fine Chemicals) in 0.2 M NaOH, 15% DMSO, was prepared, diluted to 16 mM in 1 M HEPES buffer pH 7.5. Cells were induced with 0.05 % L-arabinose (from a 20 % stock in water) and BCN was finally added to a 1 mM final concentration. The culture was further incubated for 40 min at 34 °C. Cells were finally induced with anhydrotetracyclin (final concentration of 50 ng/mL) and incubated overnight at 34 °C. Cells expressing WbaP were incubated overnight at 25 °C because expression at low temperature yielded the highest amount of proteins, as evaluated by Western blot on whole cells. Finally, cells were harvested by centrifugation, flash frozen and stored at −80 °C.
Isolation of the cell-envelope fraction (CEF)
The CEF preparation was carried out strictly at 4 °C. Cell pellets from 50 mL culture were resuspended in 20 ml buffer A (50 mM HEPES, pH 7.5, 150 mM NaCl) containing lysozyme (5 mg), protease inhibitor cocktail (20 μl) and DNAse I (10 μl). Cells were sonicated (50 % amplitude, 1 sec ON/2 sec OFF, 2x2 min). Cells were always kept on ice during sonication. The resultant lysate was centrifuged at 9,000 g for 45 min at 4 °C. The cloudy supernatant was collected in a new centrifuge tube and further centrifuged at 140,000 g for 65 min at 4 °C. A membrane pellet (cell envelope fraction, CEF) was produced. The CEF was re-suspended in the appropriate amount of buffer B (50 mM HEPES, pH 8.0, 150 mM NaCl) to achieve a 50-100 mg/mL membrane concentration.
SMA-mediated solubilization of membrane proteins
The CEF (50-100 mg/mL, 0.5 mL) was thawed on ice and the same amount of 5 % w/v SMA in buffer B was added. The mixture was gently rocked for 2 h at room temperature and the remaining insoluble material was pelleted at 100,000 g for 45 min. The supernatant was incubated overnight at 4 °C together with 0.3 mL of Ni2+-NTA pre-equilibrated with buffer B supplemented with 10 mM imidazole. The Ni2+-NTA resin was washed with 10 column volumes (CVs) of buffer B supplemented with 20 mM and the target protein in SMALP was eluted with 2 CVs of buffer B supplemented with 500 mM imidazole.
Labeling membrane proteins in SMALP
100 μL of the eluted volume was mixed together with Cy3- or Cy5-tetrazine (1-10 μM, Jena Bioscience). The solution was gently rocked at room temperature for 1 h. The membrane protein solubilized in SMALP was separated from the free dye using a Zeba Spin Desalting Column following the manufacturer’s instructions (7 kDa molecular weight cut-off; Thermo Scientific).
SDS-PAGE and western blot analysis
CEF and Ni2+-NTA fractions, and labeling efficiencies were analyzed by SDS-PAGE followed by western blot. 12 % acrylamide gels were used for SUMO-PglC and PglA, 15 % acrylamide gels were used for PglC and 12 % or 8-16 % gradient gels were used for LpxM and WbaP. Gel shift assays were run at 180 V for 4 h at 4 °C, while the standard SDS-PAGE gels were run at 180 V for 60 min at room temperature. Western blots were run at 100 V for 1 h at 4 °C. For the detection of the His-tag, mouse anti-His antibody (LifeTein) was used followed by a detection using the IRDye 680RD secondary goat anti-mouse antibody (LI-COR Biosciences). Due to the spectral overlap between IRDye 680RD and Cy5, only the labeling by Cy3-tetrazine was evaluated by this method.
Size-exclusion chromatography
Size exclusion chromatography was performed on a GE ÄKTAprime plus FPLC system applying with a Superdex 200 10/300 GL column at a 0.5 mL/min flow rate. Detection was at 280 nm and 0.25 mL fractions were collected.
Particle sizing
Dynamic light scattering (DLS) was performed on a DynaPro Titan Instrument (Wyatt Technology) using disposable UVettes (Eppendorf). The administered laser power was 50-70 % and 10 scans (5-10 s) at 25 °C were accumulated to generate the data points. The data were processed using the globular protein model.
PglC activity assay
Activity assays were performed as described previously using the UMP-Glo assay (Promega) Das et al., 2016). In brief, 50 nM enzyme and 20 μM of both UDP-N,N’-diacetylbacillosamine and Und-P substrates were reacted in assay buffer (50 mM HEPES, 100 mM NaCl, 3 mM MgCl2, pH 7.5). The assay was not supplemented with detergent. The reaction was quenched using the UMP-Glo reagent. Luminescence was read in a 96-well plate using a SynergyH1 multimode plate reader (Biotek) and the corresponding UMP concentration was deduced by comparison with a standard curve.
smTIRF imaging
A flow cell was assembled using cleaned glass slides and Ni2+-NTA-coated coverslips (low loading coverslips from MicroSurfaces). A solution of labeled protein was introduced by capillary action into the channels (30 μL, 100 pM of labeled protein, as determined by absorption measurement of stock solutions). Single-molecule experiments were performed using a multi-wavelength total internal reflection fluorescence (TIRF) microscopy setup. The setup included an Eclipse Ti microscope (Nikon) equipped with a 60× Apo-TIRF oil immersion objective lens (NA 1.49; Nikon) placed on a vibration cancellation table (TMC). Labeled proteins in the evanescent field were excited at 642 nm and fluorescence was imaged on an ImagEM X2 EM-CCD cameras (Hamamatsu). Images were acquired using MetaMorph and processed using ImageJ or a custom-made software written in MATLAB. Over 100 fluorescence time-course traces were visually inspected for each labeled variants for the step photobleaching analysis. A fluorescence trace was considered as “productive” if a fluorescence signal was observed for at least three frames and yielded to a complete extinction of fluorescence within 50 s (duration of the acquisition). “Unproductive” fluorescence time-course traces are those for which the fluorescence did not bleach completely or the signal-to-noise ratio was too low to observe steps (in particular auto-fluorescence of the glass coverslip), or the fluorescence signal was too brief to be considered as significant. “Unproductive time-course traces were excluded from the analysis and account for 10-35 % of the total fluorescent spots detected by the software written in MATLAB.
QUANTIFICANTION AND STATISTICAL ANALYSIS
The statistical details of experiments can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti His monoclonal antibody | LifeTein | Cat#LT0426 |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat#926-32210 |
| IRDye® 680LT Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat#926-68020 |
| Bacterial Strains | ||
| Escherichia coli DH5α competent cells | Maintained in our lab | N/A |
| Escherichia coli C321.ΔprfA (GRO) | Lajoie et al., 2013 | Addgene bacterial strain #48998 |
| Chemicals | ||
| SMA 3:1 | Polyscope | SL 25010 |
| SMA 2.3:1 | Polyscope | SL 30010 |
| SMA 1.2:1 | Polyscope | SL 40005 |
| SMA 2000 | Cray Valley | SMA 2000 |
| DIBMA | BASF | Sokalan CP9 |
| Cy3-tetrazine | Jena Bioscience | Cat#CLK-014-05 |
| Cy5-tetrazine | Jena Bioscience | Cat#CLK-015-05 |
| Sulfo-Cy3-methyltetrazine | Jena Bioscience | Cat#CLK-1018-1 |
| TAMRA-tetrazine | Jena Bioscience | Cat#CLK-017-05 |
| BCN | Sirius Fine Chemicals | Cat#SC-8016 |
| Critical Commercial Assays | ||
| UMP/CMP-Glo glycosyltransferase assay | Promega | Cat#VA1132 |
| Recombinant DNA | ||
| pZE21-GFPaav | Elowitz and Leibler, 2000 | Addgene plasmid #26643 |
| pEvol plasmid that encodes the pyrrolysine tRNA synthetase double mutant PylRSAF and the cognate pyrrolysine tRNA | Profs. Carsten Schultz and Edward Lemke | N/A |
| Campylobacter jejuni PglC | Hartley et al., 2013 | N/A |
| Campylobacter jejuni PglA | Hartley et al., 2013 | N/A |
| Escherichia coli LpxM | Entova et al., 2018 | N/A |
| Salmonella enterica WbaP, sequence optimized for expression in E. coli | Genewiz | N/A |
| Software and Algorithms | ||
| ChemDraw 18.0 | PerkinElmer Informatics | https://www.perkinelmer.com/category/chemdraw |
| Illustrator CC 2019 | Adobe | https://www.adobe.com |
| ImageJ | National Institutes of Health | https://imagej.net/Fiji/Downloads |
| Matlab R2018b | MathWorks | https://www.mathworks.com/products/matlab.html |
| PyMOL | Schrödinger LLC | https://pymol.org/2/ |
| Other | ||
| Zeba spin desalting column, 7K MWCO | ThermoFisher Scientific | Cat#89882 |
| Ni-NTA glass coverslip | MicroSurfaces Inc | Cat#Ni_01 |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and Virus Strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; and Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
HIGHLIGHTS.
Membrane proteins can be efficiently solubilized by styrene-maleic acid co-polymers
Incorporation of unnatural amino acids into membrane proteins is position dependent
Labeling with tetrazine-linked dyes is most efficient at or near the membrane
The combined technologies enable powerful single-molecule fluorescence studies
ACKNOWLEDGEMENTS
The pEvol plasmid containing the pyrrolysine tRNA and tRNA synthetase double mutant genes was generously provided by Profs. Carsten Schultz and Edward Lemke. The pZE21-GFPaav vector was a gift from Prof. Michael Elowitz (Addgene plasmid # 26643; http://n2t.net/addgene:26643; RRID: Addgene_26643). C321.ΔA (GRO) was a gift from Prof. George Church (Addgene plasmid # 48998). We are grateful to Cray Valley, Polyscope and BASF for generously providing samples of copolymers and Dr. Stefan Scheidelaar as well as Dr. Thomas Laursen for facilitating these gifts. We thank Dr. Caroline Koehrer for helpful discussions in the implementation of the incorporation of ncAA. We are grateful to Profs. Steve Bell and Ibrahim Cissé for access to their TIRF microscopes as well as to Dr. Wonki Cho and to Takuma Inoue for technical advices. We acknowledge the MIT Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems. This investigation has been aided by a fellowship from The Jane Coffin Childs Memorial Fund for Medical Research, by a grant from The Philippe Foundation and by the National Institutes of Health (NIH GM-039334).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Almen MS, Nordstrom KJ, Fredriksson R, and Schioth HB (2009). Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann A, Milles S, Plass T, Dommerholt J, Verkade JM, Wiessler M, Schultz C, van Hest JC, van Delft FL, and Lemke EA (2012). Genetic encoding of a bicyclo[6.1.0]nonyne-charged amino acid enables fast cellular protein imaging by metal-free ligation. Chembiochem 13, 2094–2099. [DOI] [PubMed] [Google Scholar]
- Chen X, and Wu YW (2016). Selective chemical labeling of proteins. Org Biomol Chem 14, 5417–5439. [DOI] [PubMed] [Google Scholar]
- Das D, Walvoort MT, Lukose V, and Imperiali B (2016). A Rapid and Efficient Luminescence-based Method for Assaying Phosphoglycosyltransferase Enzymes. Sci Rep 6, 33412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Darshi M, Cheley S, Wallace MI, and Bayley H (2007). Membrane protein stoichiometry determined from the step-wise photobleaching of dye-labelled subunits. Chembiochem 8, 994–999. [DOI] [PubMed] [Google Scholar]
- Dorr JM, Koorengevel MC, Schafer M, Prokofyev AV, Scheidelaar S, van der Cruijsen EA, Dafforn TR, Baldus M, and Killian JA (2014). Detergent-free isolation, characterization, and functional reconstitution of a tetrameric K+ channel: the power of native nanodiscs. Proc Natl Acad Sci U S A 111, 18607–18612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, and Leibler S (2000). A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338. [DOI] [PubMed] [Google Scholar]
- Entova S, Billod JM, Swiecicki JM, Martin-Santamaria S, and Imperiali B (2018). Insights into the key determinants of membrane protein topology enable the identification of new monotopic folds. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, and Wagner G (2013). Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure 21, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong SE, Ford A, Albarnez-Rodriguez L, and Valvano MA (2015). Topological analysis of the Escherichia coli WcaJ protein reveals a new conserved configuration for the polyisoprenyl-phosphate hexose-1-phosphate transferase family. Scientific Reports 5, 9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Correa IR Jr., Kindermann M, Beaufils F, and Johnsson K (2008). An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15,128–136. [DOI] [PubMed] [Google Scholar]
- Glover KJ, Weerapana E, and Imperiali B (2005). In vitro assembly of the undecaprenylpyrophosphate-linked heptasaccharide for prokaryotic N-linked glycosylation. Proc Natl Acad Sci U S A 102, 14255–14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MD, Schneggenburger PE, and Imperiali B (2013). Lipid bilayer nanodisc platform for investigating polyprenol-dependent enzyme interactions and activities. Proc Natl Acad Sci U S A 110, 20863–20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell G, Arnold T, Barker RD, Clifton LA, Steinke NJ, Tognoloni C, and Edler KJ (2016). Evidence of Lipid Exchange in Styrene Maleic Acid Lipid Particle (SMALP) Nanodisc Systems. Langmuir 32, 11845–11853. [DOI] [PubMed] [Google Scholar]
- Hostetler ZM, Ferrie JJ, Bornstein MR, Sungwienwong I, Petersson EJ, and Kohli RM (2018). Systematic Evaluation of Soluble Protein Expression Using a Fluorescent Unnatural Amino Acid Reveals No Reliable Predictors of Tolerability. ACS chemical biology 13, 2855–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italia JS, Zheng Y, Kelemen RE, Erickson SB, Addy PS, and Chatterjee A (2017). Expanding the genetic code of mammalian cells. Biochemical Society transactions 45, 555–562. [DOI] [PubMed] [Google Scholar]
- Jamshad M, Charlton J, Lin Y-P, Routledge SJ, Bawa Z, Knowles TJ, Overduin M, Dekker N, Dafforn TR, Bill RM, et al. (2015). G-protein coupled receptor solubilization and purification for biophysical analysis and functional studies, in the total absence of detergent. Biosci Rep 35, e00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalstrup T, and Blunck R (2013). Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proc Natl Acad Sci U S A 110, 8272–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlova MG, Voskoboynikova N, Gluhov GS, Abramochkin D, Malak OA, Mulkidzhanyan A, Loussouarn G, Steinhoff HJ, Shaitan KV, and Sokolova OS (2019). Detergent-free solubilization of human Kv channels expressed in mammalian cells. Chemistry and physics of lipids 219, 50–57. [DOI] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, and Johnsson K (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Knowles TJ, Finka R, Smith C, Lin YP, Dafforn T, and Overduin M (2009). Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J Am Chem Soc 131, 7484–7485. [DOI] [PubMed] [Google Scholar]
- Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, et al. (2013). Genomically recoded organisms expand biological functions. Science 342, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, and Chin JW (2012). Genetic Encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J Am Chem Soc 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin A, and Imperiali B (2011). The Expanding Horizons of Asparagine-Linked Glycosylation. Biochemistry 50, 4411–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Knowles TJ, Postis VL, Jamshad M, Parslow RA, Lin YP, Goldman A, Sridhar P, Overduin M, Muench SP, et al. (2016). A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat Protoc 11, 1149–1162. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, and Lomize AL (2012). OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40, D370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukose V, Luo L, Kozakov D, Vajda S, Allen KN, and Imperiali B (2015). Conservation and Covariance in Small Bacterial Phosphoglycosyltransferases Identify the Functional Catalytic Core. Biochemistry 54, 7326–7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Fleites C, Proctor M, Roberts S, Bolam DN, Gilbert HJ, and Davies GJ (2006). Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem Biol 13, 1143–1152. [DOI] [PubMed] [Google Scholar]
- McLean MA, Gregory MC, and Sligar SG (2018). Nanodiscs: A Controlled Bilayer Surface for the Study of Membrane Proteins. Annu Rev Biophys, 10.1146/annurev-biophys-070816-033620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH, and Albertini AM (1983). Effects of surrounding sequence on the suppression of nonsense codons. J Mol Biol 164, 59–71. [DOI] [PubMed] [Google Scholar]
- Nath A, Trexler AJ, Koo P, Miranker AD, Atkins WM, and Rhoades E (2010). Single-molecule fluorescence spectroscopy using phospholipid bilayer nanodiscs. Methods Enzymol 472, 89–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, and Hopkins AL (2006). How many drug targets are there? Nat Rev Drug Discov 5, 993–996. [DOI] [PubMed] [Google Scholar]
- Padmanarayana M, Hams N, Speight LC, Petersson EJ, Mehl RA, and Johnson CP (2014). Characterization of the Lipid Binding Properties of Otoferlin Reveals Specific Interactions between PI(4,5)P2 and the C2C and C2F Domains. Biochemistry 53, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A, Chen J, O’Leary S, Tsai A, and Puglisi JD (2012). Single-molecule analysis of translational dynamics. Cold Spring Harb Perspect Biol 4, a011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GJ (2001). Green fluorescent protein--a bright idea for the study of bacterial protein localization. FEMS microbiology letters 204, 9–18. [DOI] [PubMed] [Google Scholar]
- Plass T, Milles S, Koehler C, Schultz C, and Lemke EA (2011). Genetically encoded copper-free click chemistry. Angew Chem Int Ed Engl 50, 3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass T, Milles S, Koehler C, Szymanski J, Mueller R, Wiessler M, Schultz C, and Lemke EA (2012). Amino acids for Diels-Alder reactions in living cells. Angew Chem Int Ed Engl 51, 4166–4170. [DOI] [PubMed] [Google Scholar]
- Ray LC, Das D, Entova S, Lukose V, Lynch AJ, Imperiali B, and Allen KN (2018). Membrane association of monotopic phosphoglycosyl transferase underpins function. Nat Chem Biol 14, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, and Zhang Y (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmied WH, Elsässer SJ, Uttamapinant C, and Chin JW (2014). Efficient Multisite Unnatural Amino Acid Incorporation in Mammalian Cells via Optimized Pyrrolysyl tRNA Synthetase/tRNA Expression and Engineered eRF1. Journal of the American Chemical Society 136, 15577–15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz F, and Aebi M (2011). Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol 21, 576–582. [DOI] [PubMed] [Google Scholar]
- Sun C, Benlekbir S, Venkatakrishnan P, Wang Y, Hong S, Hosler J, Tajkhorshid E, Rubinstein JL, and Gennis RB (2018). Structure of the alternative complex III in a supercomplex with cytochrome oxidase. Nature 557, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski CM, Yao R, Ewing CP, Trust TJ, and Guerry P (1999). Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol 32, 1022–1030. [DOI] [PubMed] [Google Scholar]
- Tian H, Sakmar TP, and Huber T (2015). Micelle-Enhanced Bioorthogonal Labeling of Genetically Encoded Azido Groups on the Lipid-Embedded Surface of a GPCR. Chembiochem 16, 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticau S, Friedman LJ, Champasa K, Correa IR Jr., Gelles J, and Bell SP (2017). Mechanism and timing of Mcm2-7 ring closure during DNA replication origin licensing. Nat Struct Mol Biol 24, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W, Tharp JM, and Liu WR (2014). Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim Biophys Acta 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, and Greene EC (2011). Single-molecule studies of transcription: from one RNA polymerase at a time to the gene expression profile of a cell. J Mol Biol 412, 814–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, and Sunahara RK (2007). A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A 104, 7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis JCW, and Chin JW (2018). Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs. Nat Chem 10, 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015). The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, and Flynn AD (2016). Drugging Membrane Protein Interactions. Annu Rev Biomed Eng 18, 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TS, Ahmad I, Yin JA, and Schultz PG (2010). An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Zhang Y (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.