

Abstract
Oculomotor neurons (CN3s) and trochlear neurons (CN4s) exhibit remarkable resistance to degenerative motor neuron diseases such as amyotrophic lateral sclerosis (ALS) when compared to spinal motor neurons (SMNs). The ability to isolate and culture primary mouse CN3s, CN4s and SMNs would provide an approach to study mechanisms underlying this selective vulnerability. To date, most protocols have been for heterogeneous cell cultures, which can confound interpretation of experimental outcomes. To minimize the problems associated with mixed-cell populations, pure cultures are indispensable. Here, we describe in detail the first protocol to efficiently purify and cultivate CN3s/CN4s alongside SMNs counterparts from the same embryos using embryonic day 11.5 (E11.5) IslMN:GFP transgenic mouse embryos. We provide details on tissue dissection and dissociation, FACS-based cell isolation, and in vitro cultivation of cells from CN3/CN4 and SMN nuclei. This protocol adds a novel in vitro CN3/CN4 culture system to existing protocols and simultaneously provides a pure species- and age-matched SMN culture for comparison. Analyses focusing on the morphological, cellular, molecular, and electrophysiological characteristics of motor neurons are feasible in this culture system. We hope this protocol will enable research into the mechanisms that define motor neuron development, selective vulnerability, and disease.
Keywords: Motor neuron, oculomotor neuron, trochlear neuron, primary culture, mouse embryonic motor neuron culture, FACS, IslMN:GFP transgenic mouse, cell purification, cell isolation
SUMMARY
Here, we present a protocol to yield homogeneous cell cultures of primary oculomotor, trochlear, and spinal motor neurons. These cultures can be used for comparative analyses of the morphological, cellular, molecular, and electrophysiological characteristics of ocular and spinal motor neurons.
INTRODUCTION
The culture of primary motor neurons is a powerful tool which enables the study of neuronal development, function, and susceptibility to exogenous stressors. Motor neuron cultures are particularly useful for the study of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS)1,2, whose disease mechanisms are incompletely understood. Interestingly, despite significant cell death of spinal motor neurons (SMNs) in both ALS patients and ALS model mice, oculomotor neurons (CN3s) and trochlear neurons (CN4s) are relatively spared1,3–9. Therefore, comparative analyses of pure cultures of CN3s/CN4s and SMNs could give important clues about mechanisms underlying relative vulnerability. Unfortunately, a major barrier to such analyses has been the inability to grow purified cultures of these motor neurons.
Many protocols have been described for the purification of SMNs from animal models. Most of these protocols use density gradient centrifugation10–12 and/or p75NTR-antibody-based cell-sorting panning techniques13–16. Density gradient centrifugation exploits the larger size of SMNs relative to other spinal cells, whereas p75NTR is an extracellular protein expressed exclusively by SMNs in the spinal cord. Nearly 100% pure SMN cultures have been generated by one or both of these protocols11,12,14. These protocols have not been successful for generating CN3/CN4 cultures because CN3s/CN4s do not express p75NTR, are smaller than SMNs and therefore more difficult to isolate based on size, and other specific CN3/CN4 markers have not been identified. Instead, in vitro studies of CN3s or CN4s have relied on dissociated17–21, explant17,22–26, and slice27,28 cultures, which are composed of heterogeneous cell types, and no protocols have existed for isolation and culture of primary CN3s or CN4s.
Here, we describe a protocol for the visualization, isolation, purification, and cultivation of CN3, CN4, and spinal motor neurons from the same embryonic day 11.5 (E11.5) IslMN:GFP transgenic mice29 (Figures 1 and 2A). IslMN:GFP specifically labels motor neurons with a farnesylated GFP that localizes to the cell membrane. This protocol enables species- and age-matched comparison of multiple types of motor neurons in order to elucidate pathological mechanisms in motor neuron disease.
Figure 1: Scheme for preparation of mouse embryonic motor neurons.

The schematic illustrates the steps involved in the isolation and culture of mouse embryonic motor neurons and the approximate time in hours or days for each step. The order of the dissection procedure for CN3/CN4 and for SMN are each labelled sequentially 1 through 4. Abbreviations: CN3/CN4: oculomotor neuron/trochlear neuron; SMN: spinal motor neuron; FACS: fluorescence-activated cell sorting; h: hour; d: day.
Figure 2: Dissection of the ventral midbrain and the cervical (C1) - lumbar (L2-L3) portion of the ventral spinal cord.

A) Lateral (a) and dorsal (b) views of GFP-positive motor neurons in an E11.5 IslMN:GFP transgenic mouse embryo under fluorescein isothiocyanate (FITC) illumination. A whole mount E11.5 embryo was prepared as previously described32 in order to make the embryo transparent. Subsequently, the embryo was analyzed by immunofluorescence labeling with anti-GFP staining (green). Images were captured under a confocal microscope. Scale bars: 200 μm (lateral view) and 400 μm (dorsal view). Abbreviations: S: superior; I: inferior; V: ventral; D: dorsal. B-H) Dissection steps highlighted on images of E11.5 ventral midbrain and ventral spinal cord tissues taken with an equipped camera under bright light (Bb) or FITC illumination using a fluorescence dissection stereomicroscope. Scale bars: 200 μm (D) and 1 mm (A-C, E-H). B) a) Remove the face and tail of the embryo by cutting along the red lines. b) Embryo positioned for dissection. Positioning of the front of the microscope is indicated by *. C) Cut along the solid red line in order to slit open the roof of the fourth ventricle [a) lateral view and b) dorsal view]. Use this opening to cut along the surface of the embryo dorsal to the brain (trajectory indicated by dashed red arrow). This will expose the tissue containing mesenchyme, CN3, and CN4, which can be lifted out of the cranium. For SMN dissection, insert forceps into the same opening between the fourth ventricle and its roof, then cut toward the caudal side of the embryo (trajectory indicated by dashed yellow arrow). D) Final view of the ventral midbrain containing bilateral GFP-positive CN3 and CN4 nuclei. The edges of the tissue are highlighted by a red rectangle. Cut along yellow dotted line to collect CN3 and CN4 nuclei separately, if desired. E) After opening the rest of the hindbrain and spinal cord, pinch off flapping dorsal tissues above the red lines on both sides with tweezers [a) before and b) after]. F) Bilaterally remove excess tissue ventral to the spinal cord along the red line [a) before and b) after]. G) Cut the ventral spinal cord at the two locations indicated by the red lines. On the rostral side, cut the floating ventral spinal cord transversely above C1 where the first GFP-positive anterior horn projects. Cut the caudal end of the spinal cord transversely at the upper boundary of the lower limb. Once these cuts are made, the cervical (C1) through lumbar (L2-L3) portion of the ventral spinal cord can be dissected away. H) Final view of the ventral spinal cord containing GFP-positive SMN columns.
PROTOCOL
All experiments utilizing laboratory animals were performed in accordance with NIH guidelines for the care and use of laboratory animals, and with approval of the Animal Care and Use Committee of Boston Children’s Hospital.
1. Setting up Timed Matings Prior to Dissection
-
1.1.
To generate prenatal embryonic mice for motor neuron harvest, set up timed matings between adult IslMN:GFP transgenic mice 11.5 days prior to the day of neuron isolation. Before setting up timed matings, weigh each female mouse. For the purpose of developing this protocol, 129S1/C57BL/6J IslMN:GFP mice aged 2–9 months were used and timed matings were set up in the evening.
-
1.2.
Examine female mice for vaginal plugs the following morning. The date on which the plug is identified is embryonic day 0.5.
-
1.3.
Weigh female mice and examine for pups using ultrasound (see Table of Materials) between E8.5–11. In addition to recognition of vaginal plugs, successful mating can be confirmed by detection of weight gain in female mice (usually >1.5g on E9.5, if there are more than 5–6 embryos) and visual confirmation of embryos under ultrasound. Embryos are easily detectable by ultrasound after E9.5. Females that gain weight as described above are more often pregnant than those that do not gain weight, so ultrasounds are conducted only on females that have gained weight. However, female mice can gain weight for reasons other than pregnancy, so weight gain alone is not a reliable indicator of pregnancy. Ultrasound confirmation prevents unnecessary sacrifice of females that are not pregnant, but is not crucial if unavailable.
Table of Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| Alexa Fluor 2488-conjugated goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | A-11001 | 1:400 |
| Alexa Fluor 594-conjugated F(ab’)2 goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | A-11072 | 1:400 |
| B27 Supplement (50X), serum free | Thermo Fisher Scientific | 17504-044 | |
| BD FACSAria llu SORP Flow Cytometer | BD Bioscience | - | This has 4 laser system equipped with 405, 488, 594, and 640 nm lasers. |
| BD Falcon 70μm Nylon Cell Strainers | CORNING | 352350 | For filtering the dissociating cells before FACS. |
| BD Falcon Round Bottom Test Tubes With Snap Cap | CORNING | 352054 | |
| BDNF Human | ProSpec-Tany TechnoGene, Ltd. | CYT-207 | |
| Cell Culture microplate, 96 well, PS, F-bottom (Chimney Well) | Greiner Bio-One International | 655090 | We tried multiple 96-well dishes and this was the best one for culture and analyses after ICC |
| Circular Cover Glasses for microscopy | Karl Hecht & Assistent | 1001/14 | We used thie coverslip since the area was large (diamater: 14 mm). |
| CNTF Human | ProSpec-Tany TechnoGene, Ltd. | CYT-272 | |
| Cyclopiazonic acid from Penicillium cyclopium | Sigma-Aldrich | C1530 | CPA. One of ER stressors. |
| 4′,6-diamidino-2-phenylinodole (DAPI) | Thermo Fisher Scientific | D1306 | |
| Dimethyl sulfoxide | Sigma-Aldrich | D2650 | DMSO |
| Dumont #5 Forceps Inox Tip Size .05 × .01 mm Biologie Tips | Roboz Surgical Instrument | RS-5015 | |
| Forskolin | Thermo Fisher Scientific | BP25205 | |
| GDNF Human | ProSpec-Tany TechnoGene, Ltd. | CYT-305 | |
| GlutaMAX supplement | Thermo Fisher Scientific | 35050-061 | |
| Hanks’ Balanced Salt Solution (HBSS) | Thermo Fisher Scientific | 14175-095 | |
| Hibernate E | BrainBits | HE | |
| Hibernate E low fluorescence | BrainBits | HELF | Fluorescence which hinders observation of embryo’s GFP expressions should be low. |
| Horse serum, heat inactivated, New Zealand origin | Thermo Fisher Scientific | 26050-070 | |
| IBMX | Tocris Cookson | 2845 | Isobutylmethylxanthine |
| Laminin | Thermo Fisher Scientific | 23017-015 | |
| Leibovitz’s L15 medium | Thermo Fisher Scientific | 11415064 | |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250 | |
| Micro Dissecting Scissors | Roboz Surgical Instrument | RS-5913 | |
| Micro Knife 4.75” 1.7 × 27 mm blade | Roboz Surgical Instrument | RS-6272 | |
| Moria Mini Perforated Spoon | Fine Science Tools | 10370-19 | |
| mouse monoclonal antibody to neuronal class III β-tubulin (TUBB3) | BioLegend | 801202 | 1:500, TUJ1 |
| Nikon Perfect Focus Eclipse Ti live cell fluorescence microscope and Elements software | Nikon | - | Differential interference contrast images and immunocytochemistry images of the cell cultures were captured with these equipments |
| Nitric Acid 90%, Fuming (Certified ACS) | Fisher Scientific | A202-212 | For rinsing coverslips |
| Olympus 1.7ml Microtubes, Clear | Genesee Scientific | 22-281 | These are the tubes that we discribed ‟1.7 mL microcentrifuge tubes” in the context. |
| Papain Dissociation System | Worthington Biochemical Corp | LK003150 | Papain solution and alubumin-ovomucoid inhibitor solution are prepared from this kit. |
| Penicillin-streptomycin (10,000 U/ml) | Thermo Fisher Scientific | 15140-122 | |
| Phosphate buffered saline (PBS) | Thermo Fisher Scientific | 10010-023 | |
| Poly D-lysin (PDL) | MilliporeSigma | A-003-E | |
| rabbit monoclonal antibody to Islet1 | Abcam | ab109517 | 1:200 |
| SMZ18 and SMZ1500 zoom stereomicroscopes with DS-Ri1 camera | Nikon | - | Dissection was performed and images of dissected embryos and tissues are captured under these fluorescence microscopes. |
| Sylgard 170 Black Silicone Encapsulant - A+B 0.9 Kg kit | Dow Corning | 1696157 | We make dissection dishes using this kit. |
| TC treated Dishes, 100 × 20 mm | Genesee Scientific | 25-202 | We make dissection dishes using this dish. |
| Thum Dressing Forceps 4.5” Serrated 2.2 mm Tip Width | Roboz Surgical Instrument | RS-8100 | |
| Transducer for LOGOQ e VET | GE Healthcare | L8-18i-RS | For ultrasound on female mice |
| Veterinary ultrasound machine | GE Healthcare | LOGOQ e VET | For ultrasound on female mice |
| Zeiss LSM 700 series laser scanning confocal microscope and Zen Software | Carl Zeiss | - | Confocal image of the embryo was captured with these equipments |
2. Dissection Conditions and Preparation of Instruments
-
2.1.
Perform all coating (except acid-cleaning of coverslips), media preparation, tissue dissociation (except centrifugation and incubation), and culture work in a laminar flow hood to ensure the sterility of media and embryonic motor neurons.
-
2.2.
With careful attention to sterile technique, tissue dissection can be conducted outside of a laminar flow hood with minimal risk of contamination.
-
2.3.
Sterilize one dissection plate, one pair of micro-dissecting scissors, one pair of thumb dressing forceps, two pairs of Dumont #5 tweezers, one micro dissecting knife, and one Moria mini perforated spoon by immersing in 70% ethanol prior to use.
3. PDL/Laminin Coating of Dishes/Coverslips
Note 1. Culture dissociated primary motor neurons in 96-well or 24-well plates, depending on the number of cells required for the application. Cells can be imaged directly in the tissue culture plate without the use of coverslips if the wells are optically transparent and of thicknesses compatible with imaging.
-
3.1.
For applications requiring coverslips, prepare acid-cleaned, sterilized, and air-dried coverslips at least two days prior to neuron isolation as previously described30. Batches of coverslips can be prepared in this manner well in advance of experiments and can be stored up to 6 months without impact on experimental quality.
-
3.2.Prepare a working solution of 20 μg/ml poly D-lysine (PDL) in phosphate buffered saline (PBS) two days prior to neuron isolation.
-
3.2.1.Aliquot PDL (1mg/ml) in advance and store at −20 °C as a stock solution.
-
3.2.1.
-
3.3.
Cover the surface of each coverslip or well of the tissue culture plate with sufficient PDL solution working solution (e.g. 100 μl per well on 96-well plates or 500 μl per well on 24-well plates with or without coverslips). Incubate overnight at 37 °C.
-
3.4.The following day, wash ×3 with sterilized water.
-
3.4.1.Parafilm-sealed and washed PDL-coated plates can be stored at 4 °C for up to 1 month if they are dried completely after the final wash.
-
3.4.1.
-
3.5.Prepare a working solution with 10 μl laminin (1.1–1.2mg/ml) in 1.2 ml PBS.
-
3.5.1.Aliquot laminin stocks (1.1–1.2mg/ml) in advance and store at −80 °C.
-
3.5.1.
-
3.6.
Cover the surface of each coverslip or well with sufficient laminin solution to evenly coat the surface. Incubate for at least 2 h at 37 °C prior to use. PDL/laminin-coated plates and coverslips can be stored up to 1 week at 37 °C, but freshly coated laminin is preferred. Remove laminin directly before plating.30 Laminin should not be allowed to dry out. If the plates are to be stored for more than several hours, they should be wrapped in parafilm.
4. Preparation of Dissection and Motor Neuron Culture Medium and Dissociation Solutions
-
4.1.Prepare dissection medium.
-
4.1.1.Thaw heat-inactivated horse serum overnight at 4 °C, prepare 1 ml aliquots, and store at −20 °C. Thaw aliquots at RT directly before use.
-
4.1.2.Store B27-supplement (50X) in 1 ml aliquots at −20 °C. Thaw aliquots at RT immediately before use. Avoid freeze/thaw cycles.
-
4.1.3.Store GlutaMAX supplement (100X) at 4 °C or −20 °C. Divide into 0.5 ml aliquots if storing at −20 °C.
-
4.1.4.Store penicillin-streptomycin (10,000 U/ml) in 1 ml aliquots at −20 °C. Thaw aliquots at RT immediately before use.
-
4.1.5.To make dissection medium, mix 9.4 ml Hibernate E with 200 μl horse serum (2%), 200 μl of 50X B27 supplement (1X), 100 μl of 100X GlutaMAX supplement (1X), and 100 μl of 10,000 U/ml penicillin-streptomycin (100 U/ml). All parentheses indicate final concentrations of each reagent. Use this medium for collecting dissected tissues and making the final suspension of dissociated cells.
-
4.1.6.Add 500 μl dissection medium to individual 1.7 mL microcentrifuge tubes for tissue collection the day before neuron isolation. Prepare one tube for each tissue type that will be collected (e.g. positive control, negative control, CN3/CN4, SMN) and store at 4 °C prior to use.
-
4.1.7.Combine 49 ml Hibernate E low fluorescence media with 1 ml B27 supplement (1X final concentration) and fill a 24-well plate with this medium (2 ml/well) the day before neuron isolation. This dish will be used for collecting mouse embryos. Store at 4 °C prior to use.
-
4.1.1.
-
4.2.Prepare motor neuron culture medium.
-
4.2.1.Prepare 250 μl aliquots of 25 mM 2-mercaptoethanol in Leibovitz’s L15 medium. Store at −20 °C.
-
4.2.2.Generate 5 μl aliquots of 100 μg/ml solutions of BDNF, CNTF, and GDNF diluted in sterilized water. Store at −80 °C and thaw aliquots at RT immediately before use.
-
4.2.3.Prepare 10 mM forskolin solution by adding DMSO (64 μl) to 5 mg (1.0670 μmol) of forskolin and vortex well to dissolve completely. Then add sterilized water (1.003 ml) to the DMSO solution and vortex well. Store 12 μl aliquots of 10 mM forskolin at −20 °C and thaw aliquots at RT immediately before use.
-
4.2.4.Prepare 12 μl aliquots of 100 mM isobutylmethylxanthine (IBMX) diluted in DMSO. Store at −20 °C and thaw aliquots at RT immediately before use.
-
4.2.5.To make motor neuron culture medium, mix 9.4 ml neurobasal medium with 200 μl horse serum (2%), 200 μl of 50X B27 supplement (1X), 100 μl of 100X GlutaMAX supplement (1X), 100 μl of 10,000 U/ml penicillin-streptomycin (100 U/ml), and 20 μl of 25 mM 2-mercaptoethanol (50 μM), preferably directly before use. This step can be performed up to 1 day before neuron isolation. All parentheses indicate final concentrations of each reagent.
-
4.2.6.Just before use, add 1 μl each of 100 μg/ml BDNF (10 ng/ml), CNTF (10 ng/ml), and GDNF (10 ng/ml); and 10 μl of 10 mM forskolin (10 μM); and 10 μl of 100 mM IBMX (100 μM) to motor neuron culture medium. Pre-warm medium to 37 °C. All parentheses indicate final concentrations of each reagent.
-
4.2.1.
-
4.3.Prepare dissociation solutions.
-
4.3.1.Prepare a papain solution (20 units/ml papain and 0.005% DNase) and an albumin-ovomucoid inhibitor solution (1mg/ml ovomucoid inhibitor, 1mg/ml albumin, and 0.005% DNase) following manufacturer’s instruction.
-
4.3.2.Prepare 500 μl aliquots of each of ovomucoid inhibitor and papain solutions and store at −80 °C. Thaw aliquots at 37 °C immediately before use.
-
4.3.1.
5. Ventral Midbrain and Spinal Cord dissection
Note: Perform all of the following except for steps 5.1.1–5.1.3 and 5.1.5–5.1.6 under a fluorescence dissection stereomicroscope. Total dissection time per experiment is typically 3–5 h, depending on the proficiency of dissection technique and the number of motor neurons required for each experiment.
-
5.1.Ventral midbrain dissection
-
5.1.1.Euthanize a pregnant mouse approximately 11.5 days post-fertilization by carbon dioxide gas and cervical dislocation.
-
5.1.2.Spray the abdomen thoroughly with ethanol and remove the uterus using sterile micro-dissecting scissors and thumb dressing forceps.
-
5.1.3.Wash the uterus briefly in sterile PBS, then transfer to the dissection plate filled with pre-chilled sterile PBS.
-
5.1.4.Remove the IslMN:GFP-positive embryos carefully from the uterus using sterile micro-dissecting scissors, thumb dressing forceps, and Dumont #5 tweezers in ice-cold sterile PBS under the bright light of the microscope.
-
5.1.5.Using a sterile Moria mini-perforated spoon, transfer each embryo to a separate well of a 24-well plate filled with pre-chilled Hibernate-E low fluorescence medium supplemented with 1× B27. Keep the 24-well plate on ice.
-
5.1.6.Transfer one embryo to a sterile dissection plate and cover completely with ice-cold sterile Hank’s balanced salt solution (HBSS).
-
5.1.7.All of the following dissection steps are performed under fluorescein isothiocyanate (FITC) illumination of the microscope. Using tweezers, remove the tail and face of the embryo without damaging midbrain (Figure 2Ba).
-
5.1.8.Place the embryo prone with limbs straddled underneath and tail pointing toward the front of the microscope (toward the dissector which is indicated by *) (Figures 2Bb).
-
5.1.9.Using tweezers, slit open the roof of the fourth ventricle in order to generate a small opening. Use this opening to hook tweezers into the space created between the fourth ventricle and its roof, and dissect along the dorsal surface of the embryo rostral to the cortex and lateral to the floor plate and motor column (Figure 2Ca and b).
-
5.1.10.Open the dissected tissue in an open-book manner to reveal the GFP-positive CN3 and CN4 nuclei. A small piece of tissue from the ventral midbrain containing mesenchyme, CN3, and CN4 will now be exposed.
-
5.1.11.Carefully separate the ventral midbrain from the embryo and remove meningeal tissue using tweezers and a micro-dissecting knife.
-
5.1.12.Dissect the bilateral GFP-positive CN3 and CN4 nuclei away from the floor plate and other GFP-negative surrounding tissue using tweezers and a micro-dissecting knife (Figure 2D). Maximize GFP-positive motor neurons in the excised tissue but take care not to damage them (avoid touching).
-
5.1.13.If collection of separate CN3 and CN4 nuclei is desired, cut along the midline of these two nuclei (yellow dotted line in Figure 2D).
-
5.1.14.Using a P1000 pipette, collect the dissected ventral midbrain tissue with minimal HBSS and place in the CN3/CN4-labelled 1.7 mL microcentrifuge tube filled with Dissection Media (4.1.5.). Store on ice until dissociation.
-
5.1.15.Continue pooling ventral midbrains from additional embryos in the same tube until the total number meets the experimental requirement (refer to “8. Culture of Purified Primary Motor Neurons” for ideal cell numbers). Since tissues are subject to stress during dissociation and sorting, pooled collection of at least 10 ventral midbrains yielding approximately 1×104 CN3/CN4 motor neurons is recommended.
-
5.1.1.
-
5.2.
Ventral spinal cord dissection
-
5.2.1.
Keep the embryo prone with the head facing the front of the microscope (toward the dissector). Hold the embryo with one pair of tweezers and insert the tip of the other pair of tweezers into the unopened caudal part of the fourth ventricle.
-
5.2.2.
Open the rest of the hindbrain and spinal cord dorsally over the whole rostro-caudal extent of the embryo. Open by cutting dorsal tissue, starting from the fourth ventricle and working toward the central canal of the caudal spinal cord using forceps as scissors (Figure 2Ca and b). Take care not to damage the ventral spinal cord (avoid contact) during this procedure.
-
5.2.3.
Hold the embryo with one pair of tweezers and pinch off the flap of dorsal tissue on each side with the other pair of tweezers (Figures 2Ea and b).
Note: Excised dorsal tissues contain dorsal skin, mesenchyme, dorsal root ganglia (DRGs), dorsal hindbrain, and spinal cord. Remove as much of these tissues as possible without damaging SMN nuclei, since they are adhesive and can trap SMNs during filtering or cause clogging during FACS sorting.
-
5.2.4.
Remove the ventral spinal cord by using the micro-dissection knife to pierce directly below the GFP-positive SMN. Lift up the ventral spinal cord with saw-like movements on both sides (Figures 2Fa and b).
-
5.2.5.
Cut the floating ventral spinal cord transversely directly above C1 where the first GFP-positive anterior horn projects (Figure 2G). Also cut transversely at the upper boundary of the lower limb (Figure 2G). Remove the cervical (C1) - lumbar (L2-L3) portion of the ventral spinal cord after this procedure.
-
5.2.6.
Place the ventral spinal cord dorsal side up and hold by pressing the GFP-negative tissue between the GFP-positive SMN columns with one pair of tweezers. Remove the remaining attached mesenchyme, DRGs, and dorsal spinal cord by trimming both sides of the GFP-positive SMN column with the micro-dissection knife (Figure 2H). Take care to maximize GFP-positive motor neurons without damaging them.
-
5.2.7.
Using a P1000 pipette, collect the dissected ventral spinal cord tissue with minimal HBSS and place in the SMN-labelled 1.7 mL microcentrifuge tube filled with Dissection Media. Store on ice until dissociation.
-
5.2.8.
Continue pooling ventral spinal cords from additional embryos in the same tube until the total number meets the experimental requirements. Since tissues are subject to stress during dissociation and sorting, pooled collection of at least 3 ventral spinal cords yielding approximately 2.1×104 SMN is recommended.
-
5.2.9.
Collect facial motor neurons and extremities of IslMN:GFP mouse embryos as GFP-positive and -negative controls for fluorescence-activated cell sorting (FACS), respectively. Extremities are GFP-negative because the GFP-positive axons of the spinal motor neurons have not yet extended into the extremities at this embryonic age.
6. Tissue Dissociation
Note: Total dissociation time is typically 1.5 h per experiment.
-
6.1.
Warm papain and albumin-ovomucoid inhibitor solution aliquots to 37 °C 30 min prior to dissociation.
-
6.2.
Briefly spin down microdissected tissues at a low speed.
-
6.3.
Using a P100 pipette, carefully remove as much Hibernate E as possible without aspirating tissues.
Note: Be sure to remove all residual Hibernate E after this step to avoid reducing the efficacy of papain dissociation in the next step.
-
6.4.
Add the appropriate volume of papain solution (Table 1) to each of the 1.7 mL microcentrifuge tubes containing microdissected tissue samples. The appropriate volume of papain for dissociation was determined in order to maximize effective dissociation while minimizing stress on the cells.
-
6.5.
Gently triturate ×8 with a P200 pipette. Perform all trituration steps gently to preserve motor neuron viability.
-
6.6.
Incubate the tubes containing the tissues for 30 min at 37 °C, agitating by finger flicking ×10 every 10 min.
-
6.7.
Gently triturate each suspension ×8 with a P200 pipette after incubation.
-
6.8.
Spin down the cells at 300 ×g for 5 min.
-
6.9.
To ensure the efficacy of ovomucoid inhibition in the next step, use a P1000 pipette to remove and discard as much supernatant as possible without aspirating the tissues.
-
6.10.
Resuspend pellets in the appropriate volume of albumin-ovomucoid inhibitor solution (Table 1) by gently triturating ×8 with a P200 pipette.
-
6.11.
Wait 2 min to allow any remaining pieces of undissociated tissue to settle to the bottom of the tube.
-
6.12.
Collect as much supernatant as possible without aspirating undissociated tissues using a P200 pipette. Transfer the supernatant to fresh 1.7 mL microcentrifuge tubes.
-
6.13.
Typically, some chunks of tissue will remain undissociated after step 6.12. To maximize the final yield of dissociated cells while minimizing stress on the cells dissociated previously (contained in the supernatant of step 6.12), repeat steps 6.10–6.12 for the undissociated tissues that remain in the original 1.7 mL microcentrifuge tubes.
-
6.14.
Spin down the cells at 300 ×g for 5 min.
-
6.15.
Carefully remove and discard the supernatant using a P1000 pipette.
-
6.16.
Resuspend the pellet in the appropriate volume of Dissection Media (Table 1) by pipetting ×8 using a P1000 pipette. The appropriate volume of final suspension was determined so that cell density does not exceed 107 cells/ml, which can block the stream of the flow cytometry machine, but also so that cells are not excessively dilute, which results in a slowed sorting speed.
-
6.17.
Filter the suspensions through 70 μm cell strainers to eliminate any large clumps or undigested tissue. Transfer the suspensions into 5 ml round bottom polystyrene test tubes and store on ice until required.
Table 1: Appropriate volumes of papain, albumin-ovomucoid, and final suspension (Hibernate E) used in dissociation steps.
The appropriate volumes of papain and albumin-ovomucoid to be used with various numbers of ventral midbrain and ventral spinal cord tissues were modified from the manufacturer’s instruction after several rounds of optimization. Since tissues are subject to stress during dissociation and sorting, pooled collection of more than 10 ventral midbrains and more than 3 ventral spinal cords is recommended. The volume of papain was determined by considering the balance between effective dissociation and the stress of this procedure. The volume of albumin-ovomucoid inhibitor solution is half of that of papain. The appropriate volume of Hibernate E final suspension was determined such that cell density does not exceed 107 cells/ml, but also such that the cells do not become excessively diluted.
| Number of midbrains (X) | Papain | Albumin-ovomucoid | Hibernate E |
|---|---|---|---|
| 10≦X≦20 | 200 μl | 100 μl | 600 μl |
| 20<X≦30 | 300 μl | 150 μl | 700 μl |
| 30<X≦40 | 400 μl | 200 μl | 800 μl |
| Number of spinal cords (Y) | Papain | Albumin-ovomucoid | Hibernate E |
| 3≦Y≦5 | 200 μl | 100 μl | 500 μl |
| 5<Y≦10 | 400 μl | 200 μl | 800 μl |
| 10<Y≦15 | 600 μl | 300 μl | 1200 μl |
7. Fluorescence-activated Cell Sorting (FACS)
Note 1: This protocol was optimized using a FACS sorter equipped with a 15 mw 405 nm violet laser, a 100 mw 488 nm blue laser, a 75 mw 594 nm orange laser, and a 40 mw 640 nm red laser. Cells were sorted as sheath fluid in sterile PBS under aseptic conditions through a 100 μm nozzle. In order to minimize cell stress, the flow rate was set to a sample pressure of 1–3, such that a maximum of 1000–4000 events per second were acquired.
Note 2: Total FACS time is typically 1–2 h per experiment.
-
7.1.
Set up voltages for forward and side scatter so that the cell population can be visualized properly. Setting up appropriate voltages for cell sorting is complex and requires an experienced FACS operator.
-
7.2.
First plot cells based on size [Forward Scatter Area (FSC-A)] versus internal complexity [Side Scatter Area (SSC-A)] to distinguish different cell populations. Draw a gate around live cells as indicated in Figure 3Aa and Ba to exclude debris and dead cells. Group the cells within the gated region as population 1 (P1).
-
7.3.
To exclude cell clumps and doublets, plot P1 cells next based on Side Scatter Width (SSC-W) versus SSC-A. Gate the population of single cells as population 2 (P2) (Figure 3Ab and Bb).
-
7.4.
Plot P2 cells based on Forward Scatter Width (FSC-W) versus FSC-A and gate the population of single cells as population 3 (P3) (Figure 3Ac and Bc).
Note: Using two consecutive gates in 7.3 and 7.4 excludes cell clumps and doublets (high FSC-W and high FSC-A).
-
7.5.
Gate P3 cells based on GFP versus allophycocyanin (APC). The APC channel detects auto-fluorescence and gating on this channel avoids capturing auto-fluorescent cells. Use GFP-negative cells to adjust the voltage for FITC/GFP fluorescent channels. Ideally, position gates for these cell populations around 102. Select gate thresholds for GFP-positive population 4 (P4) individually for each type of motor neuron (Figure 3Ad and Bd).
Note: Set the GFP gate much higher for SMNs than for CN3s/CN4s in order to obtain a pure culture (Figure 3Ad and Bd). A lower GFP gate for SMN cultures leads to contamination of the cultures by glia and non-motor neurons. This is likely because there is low-level GFP expression in some glia and non-motor neurons due to a leaky promoter. The percentage of GFP-positive cells as compared to total cells is typically 0.5–1.5% for CN3s/CN4s and 1.5–2.5% for SMNs. If dissection was successful, these numbers can be used as a benchmark to determine the appropriate position for the GFP-positive gate (Figures 3Ae and Be).
-
7.6.
Perform FACS according to manufacturer’s protocol. Collect P4 cells into fresh 1.7 mL microcentrifuge tubes filled with 500 μl motor neuron culture medium. Store on ice until plating.
Note: Although cells can be sorted directly into wells, this results in an uneven number of cells per well. Sort the cells into 1.75 mL microcentrifuge tubes and then plate manually in order to achieve more even plating distribution.
Figure 3: Representative sort plots of ventral midbrains (A) and ventral spinal cords (B).

(Aa and Ba). Forward Scatter Area (FSC-A) versus Side Scatter Area (SSC-A) sort plot before exclusion of debris and dead cells. Ab, Bb, Ac, Bc) Sort plots for exclusion of cell clumps (b) and doublets (c) based on Width (SSC-W) versus SSC-A and Forward Scatter Width (FSC-W) versus FSC-A, respectively. Ad and Bd) Sort plots to isolate IslMN:GFP -positive motor neurons. Set the GFP gate higher for SMNs (Bd) than for CN3s/CN4s (Ad) in order to obtain a pure culture. Ae and Be) Percentages of cells gated for collection by FACS sorting. %Parent represents the percentage of cells in the current gated population relative to the number of cells in the previous gated cell population, whereas %Total represents the percentage of gated cells relative to total cells. Expected percentages of GFP-positive cells as compared to total cells (boxed in red) are 0.5–1.5 % for CN3/CN4 and 1.5–2.5% for SMN. If dissection was performed successfully, these percentages can be used as a benchmark to set up the GFP-positive gate in (Ad and Bd).
8. Culture of Purified Primary Motor Neurons
-
8.1.
Dilute FACS-isolated CN3/CN4 and SMN suspensions with prewarmed (37 °C) motor neuron culture medium to densities of 5×103 and 1×104 cells/ml, respectively.
Note: One E11.5 embryo yields approximately 1×103 CN3/CN4 and 7×103 SMN. However, these yields rely heavily on the purity of dissected tissues, the thoroughness of cell dissociation, and the appropriate thresholding of GFP gates during FACS.
-
8.2.
Transfer 96-well plates precoated with PDL and laminin from the 37 °C tissue culture incubator to the laminar flow hood and aspirate laminin from each well. Use plates and coverslips immediately without washing.
-
8.3.
Add 200 μl of diluted CN3/CN4 and SMN suspensions into the each well of PDL/laminin-coated 96-well plates. Final cell densities should be 1×103 and 2×103 cells/well for CN3/CN4 and SMN, respectively.
Note: Initial plating density of SMNs in 96-well plates (2×103 cells/well) is double that of CN3s/CN4s (1×103 cells/well) in order to obtain similar final motor neuron numbers and densities at 2 and 9 DIV (4–6×102 and 2–4×102 cells per well, respectively).
-
8.4.
Culture neurons in a 37 °C, 5% CO2 incubator.
-
8.5.
Feed neurons every 5 days by removing half of the old media (100 μl) and replacing with the same volume of fresh motor neuron culture medium. Neuronal processes should become visible on 1 DIV and become thicker and longer by 14 DIV (Figure 4). Neuronal cell bodies become enlarged and tend to aggregate in long-term cultures, particularly for SMNs (Figure 4).
Note: Perform all medium and solution changes by leaving half of the original medium volume in order to avoid detaching cultured cells. This includes fixation and immunocytochemistry (ICC) steps. If all media is removed, regardless of how gently, most of the cells will detach and be washed away.
Figure 4: Phase-contrast images of primary CN3/CN4 and SMN monocultures at 2, 7, and 14 DIV.
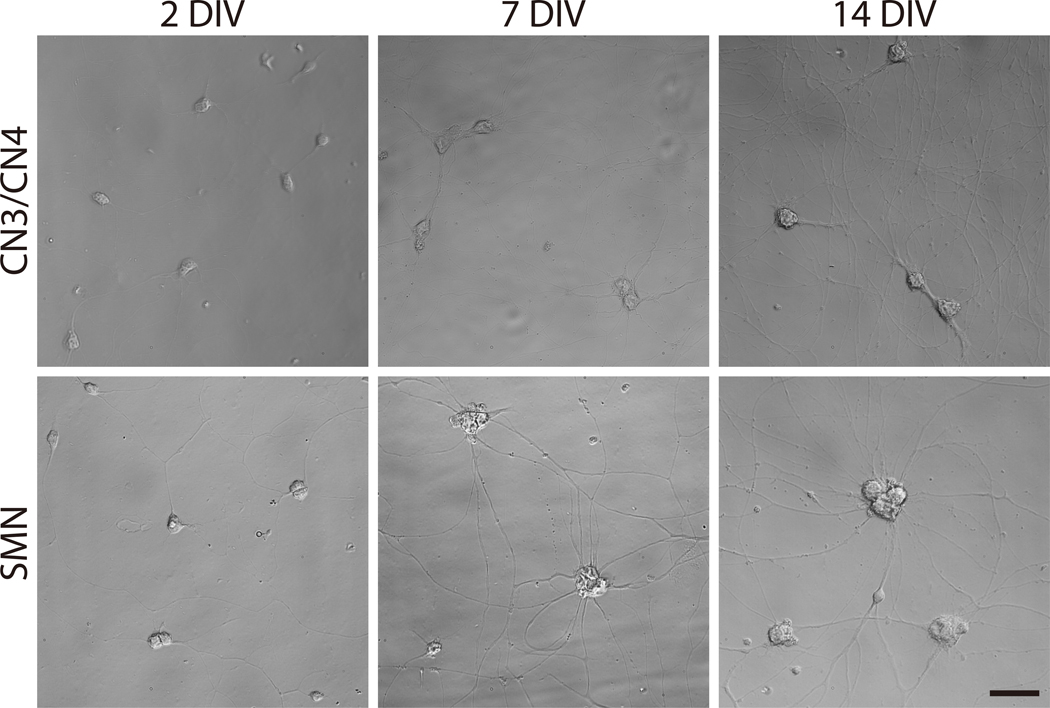
Representative differential interference contrast images of primary CN3/CN4 and SMN cultures were captured at 2, 7, and 14 DIV with inverted fluorescence microscope using corresponding image acquisition and processing software and 40× objectives. Neuronal processes become thicker and longer by 14 DIV. Neuronal cell body sizes become enlarged and tend to aggregate in long-term cultures, especially for SMNs. Both cultures can be maintained at least 14 DIV. Scale bar: 50 μm.
REPRESENTATIVE RESULTS
The aim of this protocol was to highly purify and culture both primary CN3s/CN4s and SMNs long-term to enable comparative analyses of the mechanisms underlying motor neuron disorders (see Figures 1 and 2 for overview).
Once neurons have been successfully isolated and grown in culture, nearly pure primary CN3/CN4 and SMN cultures can be obtained (Figure 5A and B) and maintained for at least 14 DIV (Figures 4 and 6). Purities of CN3/CN4 and SMN cultures at 2 DIV were 93.5 ± 2.2% and 86.7 ± 4.7%, respectively, when assessed by ICC using the motor neuron marker Islet1 and neuronal marker TUJ1 (Figure 5B). However, these high purities rely heavily on the age of the embryos and on setting appropriate thresholds for GFP gates during FACS (Figure 3). Dissection of embryos at E10.5 is more difficult than dissection at E11.5 due to increased softness and adhesiveness of tissues, resulting in decreased motor neuron yields. However, the purities of E10.5 CN3s/CN4s and SMNs are comparable to those for E11.5 embryos (92.8% and 82.2% at 2 DIV, respectively; data obtained from a single experiment). However, the purities of CN3s/CN4s and SMNs dramatically decrease when E13.5 embryos are used, even if only the highest GFP-positive population is collected (20.7% and 7.4% at 2 DIV, respectively; data obtained from a single experiment), probably due to the expression of GFP in non-motor neurons (Figure 7). This same tendency also holds for E12.5 cultures, although it is much less dramatic. Therefore, embryos at E12.5 or older are inappropriate for use in the purification of motor neurons using this protocol.
Figure 5: Characterizations of isolated E11.5 mouse CN3/CN4 and SMN cultures.

A) Representative immunocytochemistry images of E11.5 mouse CN3s/CN4s (top) and SMNs (bottom) cultured for 2 DIV. Neurons were analyzed by immunofluorescence labeling with the neuronal marker TUJ1 (green) and the motor neuron marker Islet1 (red), and nuclei were counterstained with DAPI (blue). Almost all of the cultured cells were motor neurons (TUJ1+, Islet1+). Images were captured with inverted fluorescence microscope using corresponding image acquisition and processing software and 20× objectives. Samples were imaged and processed to achieve maximum signal intensity without saturated pixels. All of the microscopic work and image processing in the following figures were performed in these conditions unless otherwise specified. Scale bar: 100 μm. B) The purities of E11.5 mouse CN3/CN4 and SMN cultures at 2 DIV. The purities of CN3/CN4 and SMN cultures were 93.5 ± 2.2% and 86.7 ± 4.7%, respectively. Dead neuronal cell bodies were assessed by screening for pyknotic nuclear morphology and membrane swelling. Neuronal processes that showed signs of beading and swelling were classified as degenerating processes. Cells with neither cell body death nor degenerating processes were counted as viable non-motor neurons (TUJ1+, Islet1-) or viable motor neurons (TUJ1+, Islet1+).33 The purities of motor neuron cultures were calculated as the number of viable motor neurons divided by the total number of viable non-motor neurons plus viable motor neurons. Values represent the mean ± SEM of 3 separate experiments. Not significant (P > 0.05) by Student’s t test. Cell counting was performed manually under 20× magnification.
Figure 6: Representative immunocytochemistry of primary CN3/CN4 and SMN monocultures at 2, 7, and 14 DIV.

Primary CN3/CN4 and SMN cultures were analyzed at 2, 7, 14 DIV by immunofluorescence labeling with TUJ1 (green) and nuclei were counterstained with DAPI (blue). Neuronal processes become thicker and longer by 14 DIV. Neuronal cell body sizes become enlarged and tend to aggregate in long-term cultures, particularly for SMNs. Both CN3/CN4 and SMN cultures can be maintained at least 14 DIV. Images were captured under 10× magnification. Scale bar: 200 μm.
Figure 7: Characterization of E13.5 mouse CN3/CN4 and SMN isolated cultures.

E13.5 CN3/CN4 and SMN were isolated and cultured using this protocol and analyzed at 2 DIV by immunofluorescence labeling with TUJ1 (green) and Islet1 (red), and nuclei were counterstained with DAPI (blue). Many non-motor-neuronal cells (TUJ1+, Islet1-), as indicated by arrows, were present, resulting in a drastic decrease in both CN3/CN4 and SMN purity, with the decrease more pronounced in SMN cultures. Images were captured under 20× magnification. Scale bar: 100 μm.
Pure motor neuron cultures are valuable for understanding isolated growth patterns, behaviors, and vulnerabilities of motor neurons. This example demonstrates how these cultures can be used to test motor neuron responses to chemical treatment. To determine if primary CN3s/CN4s and SMNs show differential responses to endoplasmic reticulum (ER) stressors, primary monocultures of CN3s/CN4s and SMNs were obtained using this protocol and treated with varying concentrations of an ER stressor, cyclopiazonic acid (CPA). Neurons were treated with cyclopiazonic acid (CPA; 5, 10, 15, 20, 25, or 30 μM) or vehicle control [dimethyl sulfoxide (DMSO)] at 2 DIV and fixed 3 days later for ICC to evaluate survival ratios (Figure 8A). The number of viable neurons in each sample was counted and survival ratios were calculated as the number of viable cells in drug-treated wells divided by the number of viable cells in the wells treated with DMSO. CN3/CN4 monocultures were significantly more resistant to CPA treatment (10–25 μM) as compared to SMN monocultures (Figure 9 and Figure 8B)31.
Figure 8: Representative application of primary motor neuron culture demonstrating that CN3s/CN4s are selectively resistant to ER stress induced by CPA.

A) Experimental outline: primary CN3/CN4 and SMN monocultures were treated with CPA or vehicle control (DMSO) at 2 DIV and cell viabilities were evaluated through immunocytochemistry analysis after 3 days of treatment. This outline has been modified from published work31. B) Quantification of survival ratios of CN3s/CN4s and SMNs treated with 5–30 μM CPA for 3 days from 2 DIV. Neurons were analyzed by immunofluorescent labeling of cells with TUJ1, and nuclei were counterstained with DAPI. Survival ratios were calculated as the number of viable cells (refer to Figure 5B legend) in drug-treated wells divided by the number of viable cells in wells containing vehicle alone (DMSO). Cell counting was performed manually under 20× magnification. Values represent the mean ± SEM of 4 separate experiments. *P < 0.05; ***P < 0.005 by Student’s t test. This figure has been modified from published work31.
Figure 9. Representative immunocytochemistry of primary CN3/CN4 and SMN monocultures after 3-day exposure to increasing concentrations of CPA beginning at 2 DIV.

Neurons were analyzed by immunofluorescent labeling of cells with TUJ1 (green) and nuclei were counterstained with DAPI (blue). Primary CN3s/CN4s were more resistant to CPA treatment than primary SMNs. Images were captured under 10× magnification. Scale bar: 200 μm.
In conclusion, this protocol allows for the generation of highly purified primary mouse embryonic CN3/CN4 and SMN cultures that provide a powerful and reliable system for the investigation of neuronal behavior.
DISCUSSION
Historically, in vitro studies of CN3 and/or CN4 motor neurons have relied on heterogeneous cultures such as dissociated17–21, explant17,22–26, and slice27,28 cultures because these cells cannot be distinguished from surrounding cells based on size, and specific markers for these cells have not been reported. The present protocol is a comprehensive method for the isolation and culture of primary E11.5 murine CN3s/CN4s and SMN from the same embryos and confirm the high purity of the cultures. By generating pure SMN and CN3/CN4 cultures from the same mouse embryos, the protocol enables well controlled comparisons of the in vitro behaviors of CN3s/CN4s versus SMNs isolated from wildtype as well as mutant embryos.
The pure cultures of CN3s/CN4s and SMNs generated by this protocol allow comparative studies of morphological, cellular, molecular, and electrophysiological characteristics of these motor neurons. In theory, because other cranial motor neuron populations can be visualized and dissected from this IslMN:GFP transgenic mouse line (including abducens, motor trigeminal, facial, and hypoglossal), this protocol could be expanded for their isolation and culture as well, provided that FACS GFP gates are adjusted appropriately. Finally, the FACS-sorted motor neurons derived from this protocol can be subjected to genomic [eg. Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)] and/or transcriptomic (eg. RNA sequence40) analyses to study normal development and the selective vulnerability of specific motor neuron subtypes in neurodegenerative disorders.31
There are multiple steps in this protocol that are critical to maximize the number of pure, healthy motor neurons in the isolated culture system. (1) During the dissection, the GFP-negative tissues (eg. mesenchyme and DRGs) should be maximally removed without damaging the motor neurons, since these tissues are adhesive and can trap SMNs during filtering or cause clogging during FACS sorting. (2) During tissue dissociation, the minimal essential volume of papain should be used and the cells must be treated gently with minimal but sufficient trituration. Papain was used for the tissue dissociation step in this protocol, since preliminary data indicated that it is less destructive than trypsin to both CN3/CN4 and SMN. Survival ratios based on plated numbers of CN3s/CN4s and SMNs at 2 days in vitro (DIV) increased from 39.5% to 52.7% and from 52.3% to 58.4%, respectively, when papain was used instead of trypsin (0.25%, 4 min incubation). Although these numbers are derived from a single experiment performed before full optimization, additional reports also suggest that trypsin is suboptimal for cell extraction from nervous system tissues12,34–36. (3) During FACS, fluorescent vital dyes (propidium iodide and calcein blue) and small sorting nozzles (eg. 70 μm) should not be used since they are deleterious to motor neuron survival. Use of large sorting nozzles (100 μm or larger) is highly recommended since SMN cell death increases significantly when a 70 μm nozzle is used. Setting the appropriate gating thresholds for GFP-positive cells in FACS is a critical step in order to obtain pure cultures. (4) Motor neuron cultures are supplemented with forskolin, IBMX, and growth factors (BDNF, CNTF, and GDNF). Forskolin and IBMX have been reported to additively promote SMN survival37,38. Preliminary data from the present studies suggest that forskolin and IBMX also additively increase CN3/CN4 survival. Survival ratio based on plated numbers of CN3s/CN4s at 2 DIV increased from 17.5% to 26.9%, 31.9%, and 37.0% when IBMX, forskolin, and IBMX+forskolin were added, respectively (numbers are based on a single experiment performed prior to full optimization of cell culture conditions). Perform all medium changes and washes of cultured cells by leaving half of the original volume to avoid detaching cultured cells. (5) Finally, reduce the time spent between dissection and plating of motor neurons (e.g. by shortening dissection time using multiple dissectors) to improve the viability of the cultures.
There are four major potential problems that may arise when following this protocol. (1) Low yield of motor neurons after FACS. Potential causes include using young embryos (e.g. E10.5), which have fewer motor neurons; insufficient removal of adhesive GFP-negative tissues during dissection (e.g. mesenchyme and DRGs), which can trap motor neurons and lead to their removal during filtration; insufficient papainization/trituration during dissociation; and/or setting the GFP-positive gate too high during FACS. (2) Low purity of the motor neuron cultures, which most likely arises from the use of older embryos (eg. E12.5) and/or from setting the GFP-positive gate too low during FACS. (3) Low number of attached motor neurons in culture. Potential causes for this include inappropriate FACS sorting and/or inadequate PDL/laminin coating of plates/coverslips. (4) Low viability of cultured motor neurons. Potential causes include rough and/or prolonged dissection; excessive papainization/trituration during dissociation; inappropriate handling of cells throughout the protocol (e.g. rough pipetting of cells, failure to place cells on ice, failure to pre-chill PBS and HBSS); and/or excessive time between euthanization of pregnant mice and final plating of the cells. Use of reagents which are not fresh and/or inappropriate concentrations can also impair experimental outcomes.
There are three major limitations of this protocol. (1) IslMN:GFP transgenic mice and FACS sorting are both fairly expensive. They are, however, crucial for this protocol as there is currently no alternative method capable of generating highly purified CN3s/CN4s in a more economical fashion. (2) The small E10.5-E12.5 age window for the embryonic mice, and the difficulty of confirming that appropriately aged embryos are present, especially if an ultrasound machine is not available. If only pure SMNs are required, they can be derived from E12.5–15.0 mouse embryos using methods such as gradient centrifugation10–12 and/or p75NTR-antibody-based cell-sorting panning techniques13–16. (3) Protein-based assays that require a large amount of starting material (e.g. western blot analysis) thus are not feasible from these cultures due to the small yield of motor neurons (especially CN3s/CN4s). Stem cell-derived motor neurons31,39, which can be generated limitlessly, could in theory be substituted for this purpose.
ACKNOWLEDGEMENTS
We thank Brigitte Pettmann (Biogen, Cambridge, MA, USA) for instruction in SMN dissection techniques; the Dana Farber Cancer Institute Flow Cytometry Facility, the Immunology Division Flow Cytometry Facility of Harvard Medical School, The Joslin Diabetes Center Flow Cytometry Core, Brigham and Women’s Hospital Flow Cytometry Core, and Boston Children’s Hospital Flow Cytometry Research Facility for FACS isolation of primary motor neurons; A.A. Nugent, A.P. Tenney, A.S. Lee, E.H. Nguyen, M.F. Rose, additional Engle laboratory members, and Project ALS consortium members for technical assistance and thoughtful discussion. This study was supported by Project ALS. In addition, R.F. was funded by the Japan Heart Foundation / Bayer Yakuhin Research Grant Abroad and NIH Training grant in Genetics T32 GM007748; J.J. was supported by the NIH/NEI training program in the Molecular Bases of Eye Diseases (5T32EY007145-16) through Schepens Eye Research Institute and by the Developmental Neurology Training Program Postdoctoral Fellowship (5T32NS007473-19) through Boston Children’s Hospital; M.C.W was supported by NEI (5K08EY027850) and Children’s Hospital Ophthalmology Foundation (Faculty Discovery Award); and E.C.E. is a Howard Hughes Medical Institute Investigator.
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
REFERENCE
- 1.Kiernan MC et al. Amyotrophic lateral sclerosis. Lancet. 377 (9769), 942–55 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Wood-Allum C, Shaw PJ Motor neurone disease: a practical update on diagnosis and management. Clinical medicine (London, England). 10 (3), 252–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nijssen J, Comley LH, Hedlund E Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta neuropathologica. 133 (6), 863–85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gizzi M, DiRocco A. Sivak M, Cohen B Ocular motor function in motor neuron disease. Neurology. 42 (5), 1037–46 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Kanning KC, Kaplan A, Henderson CE Motor neuron diversity in development and disease. Annual review of neuroscience. 33, 409–40 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Nimchinsky EA et al. Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. The Journal of comparative neurology. 416 (1), 112–25 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Angenstein F et al. Age-dependent changes in MRI of motor brain stem nuclei in a mouse model of ALS. Neuroreport. 15 (14), 2271–4 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Niessen HG et al. In vivo quantification of spinal and bulbar motor neuron degeneration in the G93A-SOD1 transgenic mouse model of ALS by T2 relaxation time and apparent diffusion coefficient. Experimental neurology. 201 (2), 293–300 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Spiller KJ et al. Selective Motor Neuron Resistance and Recovery in a New Inducible Mouse Model of TDP-43 Proteinopathy. The Journal of neuroscience. 36 (29) 7707–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham JM Isolation of a mouse motoneuron-enriched fraction from mouse spinal cord on a density barrier. ScientificWorldJournal. 2, 1544–6 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gingras M, Gagnon V, Minotti S, Durham HD, Berthod F Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. Journal of neuroscience methods. 163 (1), 111–8 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Beaudet MJ et al. High yield extraction of pure spinal motor neurons, astrocytes and microglia from single embryo and adult mouse spinal cord. Scientific reports. 5, 16763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camu W, Henderson CE. Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. Journal of neuroscience methods. 44 (1), 59–70 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Arce V et al. Cardiotrophin-1 requires LIFRbeta to promote survival of mouse motoneurons purified by a novel technique. Journal of neuroscience research. 55 (1), 119–26 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Wiese S et al. Isolation and enrichment of embryonic mouse motoneurons from the lumbar spinal cord of individual mouse embryos. Nature protocols. 5 (1), 31–8 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Conrad R et al. Lectin-based isolation and culture of mouse embryonic motoneurons. Journal of visualized experiments. (55), doi: 10.3791/3200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerner O et al. Stromal cell-derived factor-1 and hepatocyte growth factor guide axon projections to the extraocular muscles. Developmental neurobiology. 70 (8), 549–64 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Ferrario JE et al. Axon guidance in the developing ocular motor system and Duane retraction syndrome depends on Semaphorin signaling via alpha2-chimaerin. Proceedings of theNational Academy of Sciences of the United States of America. 109 (36), 14669–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark C, Austen O, Poparic I, Guthrie S α2-Chimaerin regulates a key axon guidance transition during development of the oculomotor projection. The Journal of neuroscience. 33 (42), 16540–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theofilopoulos S et al. Cholestenoic acids regulate motor neuron survival via liver X receptors. The Journal of clinical investigation. 124 (11), 4829–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montague K, Guthrie S, Poparic I In Vivo and In Vitro Knockdown Approaches in the Avian Embryo as a Means to Study Semaphorin Signaling. Methods in molecular biology. 1493, 403–16 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Porter JD, Hauser KF Survival of extraocular muscle in long-term organotypic culture: differential influence of appropriate and inappropriate motoneurons. Developmental biology. 160 (1), 39–50 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Serafini T et al. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 87 (6), 1001–14 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Varela-Echavarría A Tucker, A. Püschel, A.W. Guthrie, S. Motor axon subpopulations respond differentially to the chemorepellents netrin-1 and semaphorin D. Neuron. 18 (2), 193–207 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Irving C, Malhas A, Guthrie S, Mason I Establishing the trochlear motor axon trajectory: role of the isthmic organiser and Fgf8. Development. 129 (23), 5389–98 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Butowt R, Rind HB, von Bartheld CS GDNF increases the survival of developing oculomotor neurons through a target-derived mechanism. Molecular and cellular neurosciences.24 (1), 41–56 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Whitman MC, et al. Loss of CXCR4/CXCL12 Signaling Causes Oculomotor Nerve Misrouting and Development of Motor Trigeminal to Oculomotor Synkinesis. Investigative ophthalmology & visual science. 59, (12), 5201–5209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitman MC, Bell JL, Nguyen EH, Engle EC Ex Vivo Oculomotor Slice Culture from Embryonic GFP-Expressing Mice for Time-Lapse Imaging of Oculomotor Nerve Outgrowth. Journal of visualized experiments. (Pending Publication), e59911, In-press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewcock JW, Genoud N, Lettieri K, Pfaff SL The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 56: 604–20 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Bibel M, Richter J, Lacroix E, Barde YA, Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nature protocols. 2 (5), 1034–43 (2007). [DOI] [PubMed] [Google Scholar]
- 31.An D. Fujiki R,et al. Stem cell-derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons. eLife. 8:e44423 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber AB, et al. Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48 (6), 949–64 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Plachta N, et al. Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nature neuroscience. 10 (6), 712–9 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Eide L, McMurray CT Culture of adult mouse neurons. BioTechniques. 38 (1), 99–104 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Brewer GJ, Torricelli JR Isolation and culture of adult neurons and neurospheres. Nature protocols. 2 (6), 1490–8 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Seibenhener ML, Wotten MW Isolation and culture of hippocampal neurons from prenatal mice. Journal of visualized experiments. (65), doi: 10.3791/3634 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanson MG Jr., Shen S, Wiemelt AP, McMorris FA, Barres BA Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. The Journal of neuroscience. 18 (18), 7361–71 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamas NJ, et al. Neurotrophic requirements of human motor neurons defined using amplified and purified stem cell-derived cultures. Plos one. 9 (10), doi: 10.1371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazzoni EO, et al. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nature neuroscience 16 (9), 1219–27 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]