

Abstract
The field of genetic epidemiology is relatively young and brings together genetics, epidemiology, and biostatistics to identify and implement the best study designs and statistical analyses for identifying genes controlling risk for complex and heterogeneous diseases (i.e., those where genes and environmental risk factors both contribute to etiology). The field has moved quickly over the past 40 years partly because the technology of genotyping and sequencing has forced it to adapt while adhering to the fundamental principles of genetics. In the last two decades, the available tools for genetic epidemiology have expanded from a genetic focus (considering 1 gene at a time) to a genomic focus (considering the entire genome), and now they must further expand to integrate information from other “-omics” (e.g., epigenomics, transcriptomics as measured by RNA expression) at both the individual and the population levels. Additionally, we can now also evaluate gene and environment interactions across populations to better understand exposure and the heterogeneity in disease risk. The future challenges facing genetic epidemiology are considerable both in scale and techniques, but the importance of the field will not diminish because by design it ties scientific goals with public health applications.
Keywords: genetic epidemiology, genome-wide study designs, genomics, -omics, public health genetics
The emergence of modern genetics is generally attributed to Gregor Mendel, the Austrian monk who used peas to explain the principles of inheritance. His work laid the foundation for genetics and defined the basic ideas of genes as the functional unit of inheritance where different alleles of a gene control an observable phenotype (1). After the discovery of the molecular structure of DNA in 1953, this led to the central dogma of genetics where information encoded in DNA is transcribed into RNA and then translated into protein, which ultimately results in a phenotype.
However, the field of genetic epidemiology is much younger, and was first described by Neel and Schull in 1954 as “epidemiologic genetics” where the focus was on trying to understand the role of genetics in determining risk to chronic diseases, where known nongenetic factors also influence risk (2, 3). The mixing of distinct disciplines (genetics and epidemiology) required individuals to think about how genes and nongenetic factors might control familial aggregation of a phenotype (a disease or a quantitative trait) where multiple risk factors are in play. In 1967, Morton, Chung, and Mi coined the term “genetic epidemiology” and defined it as “a science that deals with etiology, distribution, and control of disease in groups of relatives and with inherited causes of disease in populations” (3, p. 1). From this definition, the new field of genetic epidemiology evolved, drawing methods and tools from epidemiology, biostatistics, and genetics to analyze the rapidly expanding forms of genetic data from families and population-based samples to address essential questions of genetic aggregation and susceptibility and to identify causal genomic variants (Figure 1).
Figure 1.
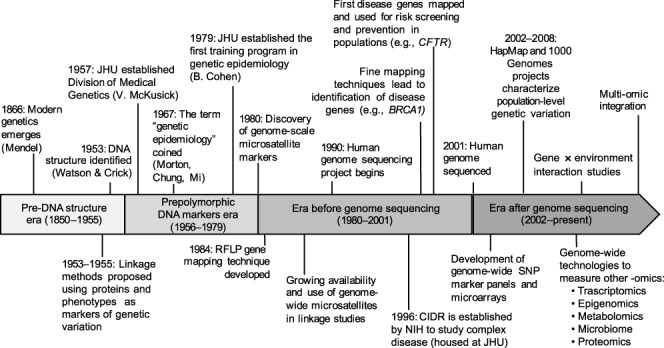
Timeline of the development of genetic epidemiology. BRCA1, Breast cancer type 1 susceptibility protein; CFTR, cystic fibrosis transmembrane conductance regulator; CIDR, Center for Inherited Disease Research; JHU, Johns Hopkins University; NIH, National Institutes of Health; RFLP, restriction fragment length polymorphism; SNP, single nucleotide polymorphism.
As we celebrate the centennial of the Department of Epidemiology at Johns Hopkins University, we also celebrate 40 years of research and teaching in genetic epidemiology. At Johns Hopkins University there is a rich history of both medical genetics, started by the pioneering work of Victor McKusick and Barton Childs in the 1950s, and epidemiology, led by Wade Hampton Frost in the 1920s. Thus, it is not surprising that in 1979, Bernice Cohen, a human geneticist, and Abraham Lilienfeld, a physician and Chair of Epidemiology, partnered with McKusick to establish the first graduate training program in genetic epidemiology in the world. They were active in integrating these two scientific disciplines and introducing students in both the School of Medicine and School of Public Health to research methods suitable for defining the role genetics could play in health and disease. In 1978, P. C. Huang, Bernice Cohen, and Abraham Lilienfeld edited the first textbook focused on genetics from a public health perspective (4). This was followed by the textbook by Khoury, Cohen, and Beaty (1993), Fundamentals of Genetic Epidemiology, which laid out the principles of the emerging scientific discipline (5).
Forty years ago, we were just beginning to consider how to integrate and effectively use the tools of two distinct scientific disciplines to answer fundamental questions about how genes influence risk of major chronic diseases such as type 2 diabetes, cardiovascular diseases, and common cancers. At the time, there was no reference human genome sequenced, and there were no public repositories of common haplotypes or single nucleotide polymorphic (SNP) markers, and there were no genome-wide association studies (GWAS). Here, we highlight the role of genetic epidemiology within the broader realm of epidemiology, and we discuss the strides made in our understanding the genetic control of complex disease that arose through critical advances in technology during this time frame. We then briefly discuss directions and challenges to be pursued in the future.
THE EARLY YEARS
Statistical, computational, and technological innovations in genetic epidemiology have set standards for reporting results and synthesizing knowledge not only for the field of epidemiology but also for other scientific fields. As genetic epidemiologists, we develop and apply statistically sound methods to identify genetic factors associated with disease, gather knowledge about underlying causal mechanisms using several different kinds of genetic data, and use this knowledge for improving public health, whether through prevention or intervention in controlling human diseases. Genetic epidemiology is still epidemiology—the study designs must still protect from recognized confounders or biases such as sampling, information, and ascertainment biases; population stratification (i.e., confounding by ancestry); and sample or specimen errors, and there must be transparent reporting of methods and analysis, as well as replication/validation to confirm findings (6).
The field of genetic epidemiology works with traditional population-based study designs as well as family-based designs to ask a wide range of scientific questions. It applies a broad range of methodological designs, from the purely descriptive to the highly analytical (7). Early genetic epidemiologic studies were focused on trying to assess the role of family history on the risk of disease and to quantify familial aggregation, whether it results from genetic or environmental causes (8). Sometimes evidence for familial aggregation can be based solely on population prevalence rates (Kpop) and the observed risk among relatives of cases (stratified by the degree of relationship). The ratio of these prevalence rates (λ = Krel/Kpop) is a direct measure of familial aggregation and determines statistical power under specialized study designs such as affected sib-pair studies that are used to test for consequences of genetic linkage. Other measures of familial aggregation include assessing risk of disease if there is any family history of the disease, including family members in the study, and through adoption and migration studies. Beyond these simple risk ratios, the most common summary statistic to measure any potential degree of genetic control is “heritability.” Heritability dates back to R. A. Fisher (1918), who developed the theory underlying “polygenic” models, where an unspecified number of unobserved, independent, autosomal genes control a quantitative phenotype (9), and extends to our current definition of “narrow sense heritability” attributed to J. L. Lush (10, 11). This linear model predicts an observed phenotype as the simple sum of effects due to genetic and nongenetic factors, and it allows partitioning the observed total phenotypic variance (σ2) into corresponding genetic and nongenetic components of variance.
The early years of genetic epidemiology were about characterizing disease aggregation in families and in the population and then estimating an overall heritability. The aim was to consider the contribution of genes to disease risk using what was observed (vs. unobserved genetics), and these methods continue to be used today to summarize the importance of genes. However, after the discovery of DNA, the attention of the field rapidly turned towards mapping genes associated with disease phenotypes (See Figure 2).
Figure 2.
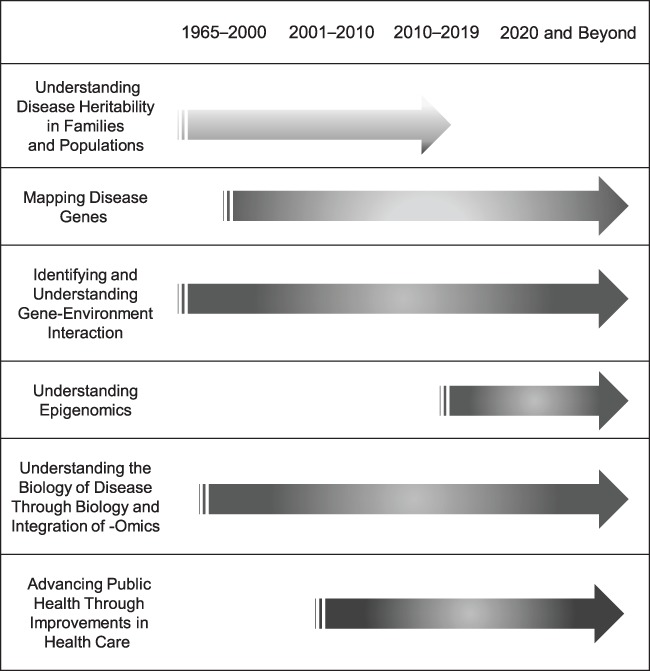
Overview of genetic epidemiology contributions to public health.
DISCOVERY OF DNA
Rapid development of cost-effective technologies and large-scale, global, collaborative scientific efforts have led to an explosion of gene discovery over the past few decades. These technical advances, coupled with large-scale, global efforts to map risk genes, have resulted in empirical identification of many risk variants but still build upon many concepts and theories about the genetic contribution to disease developed early in the 20th century.
In 1909, Wilhelm Johannsen first introduced the term “gene” to define the functional unit of heredity. In the following decades, there was considerable debate over and searches for the molecular basis of inheritance. During this time, genetic studies relied on the use of protein biomarkers (e.g., blood types and serum proteins) to map causal genes and test for cosegregation with disease in families. It was not until 1953 that DNA was understood by Watson and Crick (with unacknowledged contributions from Rosalind Franklin) as having all the properties necessary to act as the mechanism for genetic inheritance (12). In the 1960s, certain genetic conditions, such as phenylketonuria (PKU), were screened for among babies, with subsequent management providing direct impact (via dietary changes) on a genetic disease. In the late 1970s, the Sanger sequencing method was developed and created the ability to “read” DNA nucleotides sequentially (13). Over the next decade, rapid advances in laboratory techniques and automated processing (e.g., polymerase chain reaction, restriction fragment length polymorphisms, yeast and bacterial artificial chromosomes) laid the groundwork needed to discover the first disease-causing genes through genetic mapping and then to sequence the entire human genome. Development of these techniques in the 1980s led to the identification of the first causal genes for Mendelian diseases (i.e., those conforming to autosomal or X-linked recessive or dominant modes of inheritance). For example, mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR) result in cystic fibrosis among individuals who are homozygotes (14–16). This discovery was the first Mendelian disease mapped in humans, and it resulted in effective screening and treatment in a public health context. The growing availability, during the 1980s and 1990s, of restriction fragment length polymorphism and SNP panels led to the discovery of several other disease genes (e.g., breast cancer type 1 (BCRA1), Dystrophin).
USING GENETIC EPIDEMIOLOGY TO MAP GENES
In the early days of gene mapping, multiplex families (i.e., those with 2 or more affected relatives) were the main focus of research because they provided a way to follow or track mutations within a family. While there were gene mapping successes using laboratory methods developed in the 1970s to 1980s, there was consensus among the scientific community that more detailed gene maps were needed. In 1990, the Human Genome Project was launched with the goal of providing a physical map for all 3.2 billion nucleotides in the human genome (17, 18). A working draft human genome was completed in 2001, followed by the completed version in 2003 (19–21). The Human Genome Project provided the blueprint for future research to identify specific genes controlling risk to human diseases.
At the turn of the 21st century, the cost to sequence 1 genome was tens of millions of dollars, and thus, measuring the DNA sequence for many individuals to use in genetic epidemiology was not feasible. At about the same time, single base extension chemistry and DNA microarrays were developed, which enabled researchers to measure a large number of positions in the genome in a cost-effective way. In 2002, the International HapMap Project was initiated to define patterns of common genetic variants and haplotype structure in humans; it included 270 individuals from Africa, Asia, and Europe (22). The development of “next generation” or “massively parallel” sequencing methods, in the mid 2000s, led to a substantial drop in genome sequencing costs and helped launch the 1000 Genomes Project in 2008, which sought to discover all variants with a minor allele frequency of >1%, in an expanded set of 2,504 individuals from diverse populations (23–26). These laboratory advances and foundational concepts enabled countless descriptive studies of the genetic variation in different populations and the history of human migration over time. The results of these projects have also enabled genome-wide mapping studies for complex diseases using both common and rare variants.
Genetic epidemiology needed to adapt methods to accommodate these new types of genetic data. Similarly, ideas and hypotheses generated long before DNA markers were available could now be tested with empirical data. Family-based methods such as linkage analysis, transmission disequilibrium tests, and family-based association tests successfully mapped genes for a number of human diseases (27–29). In addition, utilization of existing case-control and cohort studies led to the identification of thousands of genomic risk factors across a wide range of complex common diseases using both candidate gene and GWAS designs (30, 31).
The identification of these genes associated with many complex diseases has not always yielded obvious causal variants in part because most SNPs are in noncoding regions and are merely in linkage disequilibrium (LD) with some unobserved causal variant. Furthermore, these genome-wide significant SNPs combined did not account for the estimated heritability of most common diseases and quantitative phenotypes (32), creating the so-called “missing heritability” issue. Yet the collective implications of GWAS findings (cataloged in https://www.ebi.ac.uk/gwas/) clearly emphasize 2 broad points: 1) most complex phenotypes are influenced by multiple genes, and 2) many genes influence multiple phenotypes. Neither of these broad conclusions is surprising given the biological complexity underlying the pathogenesis of most common, complex diseases. Although the scientific insights provided by large-scale GWAS are indisputable (33, 34), it remains difficult to translate these genetic risk factors to the level of the individual. Implications for medicine and public health are varied and eventually will include improving diagnostics, screening, and risk prediction, as well as identification of multiple genetic risk factors and biomarkers useful in better understanding the underlying biology and possible improvement of treatment and intervention. However, identifying causal alleles remains a major goal of genetic research, and this will require different biological, laboratory, and statistical approaches.
Availability of cost-efficient measurement tools and confirmation of the theory that numerous common variants with small effect sizes contribute to common complex disease risk (35–38) led to relying on ever-larger sample sizes, facilitated by large scientific consortiums. The success of these disease-focused consortia efforts resulted in the emergence of megacohorts, consortiums, and biobanks (UK Biobank, All of Us Cohort, TOPMed Consortium). This has led to more “team science” with large-scale international collaboration of epidemiologists, geneticists, biostatisticians, and clinicians all coming together to share data, results, and standardized tools to identify genes and genetic variants associated with diseases (39).
OTHER -OMICS
The rapid technological advances extend beyond the DNA sequence to other -omics, including epigenomics, transcriptomics, metabolomics, and the microbiome (40). Advances in computational speed have aided our ability to look across the genome to integrate the different ‑omics. They also create the opportunity to test hypotheses about genomics at the tissue and even single-cell level, which moves from the biology of populations to the tissue and even the cellular level while building medical inferences, which must in turn be applied back to individuals and populations.
One of the major challenges since sequencing of the genome has been trying to understand how genes function. The Encyclopedia of DNA Elements (ENCODE) Consortium was designed to enable understanding of the way the genome functions and identified biologically active regions of the genome outside of canonical coding regions (41). Measuring which genes are turned on or off has moved from in situ hybridization to reverse-transcriptase quantitative polymerase chain reaction to microarray to RNA-sequencing measurement methods. Our ability to measure gene expression levels across the genome has led to identifying molecular profiles unique to tumor cells, which are now routinely used for diagnosis, prognosis, and treatment of some cancers. Other commonly measured genome-wide epigenetic marks include DNA methylation, chromatin structure, and histone tail modifications. The RoadMap Epigenomics Project and International Human Epigenome Consortium are currently working towards developing a reference map of the epigenome across a large number of cell types (42) to determine interindividual epigenetic variation possibly related to disease. Epigenetic alterations have already been linked to a diverse set of diseases, including cancer and autoimmune, psychiatric, and metabolic diseases. DNA methylation measures at hundreds of loci were found to be related to the biological aging process and led to the development of “epigenetic clock” tools for studying aging and health in populations (43, 44). These clocks have been shown to be associated with adverse health outcomes, including all-cause mortality, cancer, infection with human immunodeficiency virus-1, menopause, and cardiovascular disease (45–49).
The Genotype-Tissue Expression (GTEx) project is a public reference data set to study tissue-specific expression patterns across 53 tissues (50). Integrating multiple variant annotations or other -omics data and use of pathway-based enrichment (51–53) and functional SNP prioritization tools (54–56) might help to find causal genetic variants and elucidate underlying biological mechanisms (57–59). Different statistical approaches to integrate these ‑omics are now used (60–63). For instance, PrediXcan application to a GWAS involves estimating the transcriptome using weights derived from reference transcriptome data sets (such as GTEx) and then implementing a combined gene-based test of association for estimated gene expression with any disease outcome (61). Other approaches use statistical methods, -omics databases, and data from functional laboratory experiments to prioritize and/or predict biological consequences of genetic variants identified via GWAS (64–73). For example, a combination of genetic sequence data, chromatin state enrichment, and DNA methylation maps helped to pinpoint likely causal genetic variants and provided novel insights into the pathogenesis of type 2 diabetes (74, 75). These integrated methods will be critical to the future of epidemiology, as we more completely understand mechanism and causality and can eventually translate that to public health action.
FUTURE EPIDEMIOLOGIC DIRECTIONS
Two areas that will continue to have a large influence on the broader field of epidemiology are the environment and causal inference. We anticipate that these areas and the still-evolving novel methods necessary to address them will continue to grow and begin to define a new era in genetic epidemiology.
Environmental accommodation
The study of gene-environment interactions (i.e., genotype-specific phenotypic responses to different environmental risk factors) remains an active area of research that started with candidate gene studies and evolved to genome-wide and gene-environment-wide interaction studies (7, 76, 77). These interaction analyses are challenging because of the large number of variables available for investigation—tens of millions of genetic variants and potentially thousands of environmental risk factors. Therefore, statistical methods with improved statistical power have been developed and continue to emerge (77). Alternative strategies to address the large number of SNPs available for testing include some methods for prioritizing genomic variants to be tested for potential gene × environment interaction based on a priori biological knowledge (78–80). Studies to examine genomic risk burden and environmental factor interactions using polygenic risk scores are emerging (81, 82). The issue of breadth of studies (sample size) versus depth (repeated, longitudinal measurements) are both important, and we hope that with continued technologic advances the ability to measure the environment (e.g., physical environment, nature, or other nongenetic factors) continues to expand with reduced costs. Large sample sizes with unified genetic and environmental data will be needed to understand risk and how disease processes can be modified (77).
Using genetics as an instrumental variable in epidemiology
Genetic epidemiology has also been useful for traditional epidemiologic studies through Mendelian randomization analysis, where causal effects of one trait on another (even in the presence of unmeasured confounding) are estimated using genetic markers as instruments in an instrumental variable framework. With some assumptions, the instrumental variable approach mimics the random assignment of treatments in a randomized controlled trial (RCT) in public health. Genetic epidemiology provides easily measured instrumental variables: genetic markers, where risk alleles are randomly assigned at conception if the null hypothesis of independence is actually true. Mendelian randomization studies, thought of as intermediate between observational studies and RCTs, have the ability to inform RCTs in the absence of reliable evidence to prioritize drug targets (83). For instance, Mendelian randomization studies have consistently demonstrated C-reactive protein to be a simple marker rather than a causal risk factor for cardiometabolic diseases (83), thus potentially saving time and money by avoiding an RCT destined to fail (84). Consistent with RCT results on the drug ezetimibe (84), Mendelian randomization study (85) implicated lowering of LDL cholesterol via inhibition of the Niemann-Pick C1-like intracellular cholesterol transporter 1 gene (NPC1L1) as causing reduced risk of coronary heart disease (84). Future studies that seek to accumulate evidence to support exposure effects on health outcomes will likely continue to use genetic markers as instrumental variables, particularly as megacohorts with genome-wide marker panels continue to emerge.
CHALLENGES
Despite the advances we have made in genetics over the past four decades, there are important challenges we face in genetic epidemiology that we must address going forward. These include diversity of research populations, privacy, and communication.
Diversity of research populations
Epidemiology understands the need for heterogeneous study groups, so data can be evaluated across different populations that vary in exposures, risks, and environments. For genetic studies especially, ancestry represents the history of populations, and this is reflected in differences in allele frequencies and underlying haplotype block structures. However, the GWAS Catalog (https://www.ebi.ac.uk/gwas/) shows nearly 80% of all current participants as being of European ancestry even though they only represent 16% of the global population (86). Furthermore, the fraction of non-European GWAS has not improved since 2014 (86). It is especially important to draw subjects from non-European populations into new genetic studies. As we consider methods for risk prediction, therapeutic development, and diagnostic scores based on genetic studies, this work will yield limited or no benefits for non-European populations if study populations are constructed only from European-ancestry populations. This is not only about inclusion in research; for genetics it will also determine the downstream clinical and public health benefits. We need to take an active role as epidemiologists to augment diversity in research study populations.
Privacy
Deidentified genetic research data are now shared through several national and international databases (Database of Genotypes and Phenotypes, European Genome-Phenome Archive), making these data available to a broad range of researchers. There are concerns about sharing genomic data with additional metadata because this can lead to direct or indirect identification of individuals despite being “deidentified” (87). This is not unique to genetics; the combination of birthdate, sex, and 5-digit zip code can uniquely identify 87% of all US residents (88). However, with the increased use of direct-to-consumer testing, new concerns are raised about access to genetic information by law enforcement, recently underscored by identification of a cold-case serial killer using a relative match in a public genealogy database (89). These databases have also uncovered nonpaternity, marital affairs, and biological parents of closed adoptions, situations that have created social and familial unrest for people who thought that information would never be unveiled. A study using a genealogy database company, MyHeritage, including 1.28 million people genotyped using direct-to-consumer testing, predicted the identification of a third cousin or closer relation for 60% of individuals of European ancestry (90). And as the database size grows to 3 million US individuals of European descent, the prediction would match 99% of people to a third cousin or closer relation. This study also showed that the inclusion of geography, age, and sex would narrow the identification of an individual after finding a relative that matched. The balance of what we can learn about individuals and the common good of broadly sharing genetic data must be considered carefully. Although these data were in the public domain, the same concerns exist in the research domain. Understanding the necessary protections for research participants is critical and will remain important for all epidemiologic studies that include genetic data from an individual or their family members. Epidemiologists, unlike other data end-users, are often responsible for consent and enrollment; thus, a full understanding of all current and future risks is important for researchers and for those outside of scientific research.
Communication
Communication between scientists and the broader community is also critical in all parts of epidemiology. Explaining concepts of risk, prediction, and confounding can be a challenge. For genetic epidemiology, where genetic risk might be altered by penetrance of specific alleles or gene expression, or modified by environmental exposure, it becomes especially complicated. Historically, genetic medicine has had the benefit of genetic counselors who can explain these concepts to families or individuals carrying risk alleles for Mendelian disorders. However, the increased availability of genetic information and the direct marketing of sequencing information to consumers has not been accompanied by an increase in available trained individuals to explain the allelic and genetic heterogeneity controlling risk to complex diseases. Additionally, clinicians are not being prepared in their medical curricula to address individual genetic risks summarized as either polygenic risk scores or sequencing studies. Translating causal findings from genetic studies also remains a challenge. We must find opportunities and mechanisms for continued education, so individuals (personalized medicine) and populations (personalized public health) are given the knowledge—not only the data—for informed health decisions.
CONCLUSIONS
For a relatively young field, genetic epidemiology has made rapid progress over the past 40 years. Much of this progress has been led by advances in laboratory and computational technologies. Methods enabling high throughput and cost-effective ways to measure genetic variants in populations, rapid generation and public dissemination of genetic resources, and large-scale team-based science have expanded tools for research, and this information will need to be incorporated into public health. In less than 20 years, this field has seen costs drop from $2.7 billion per genome sequence to $1,000 per genome sequence and has made substantial contributions to biomedical and population sciences. As sequencing costs continue to drop and new technologies (e.g., nanopores) emerge, we advocate for a larger number, and more diverse, set of individuals to be sequenced and for this data to be merged with a unified collection of multi-omic and environmental exposure data. Additionally, new laboratory methods such as clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 (91, 92), which can alter genomic sequence at the single base-pair level, hold considerable promise for correcting mutations in the DNA sequences that produce disease, but these also create ethical concerns that must be considered carefully. In the decades to come, it will remain a challenge to decipher the large amount of data produced to understand how the molecular pieces fit together and, in the context of external environment risk factors, to influence health outcomes and work out how to best communicate and use that information to improve public health in an ethically responsible way. Forty years ago at Johns Hopkins, we began to incorporate genetic ideas and family history information into epidemiologic studies, and since then we have mapped genetic variants, predicted risk based on genetic profiles, and tested for potential interactions between environment and genes for a multitude of complex diseases, such as chronic obstructive pulmonary disease, autism, birth defects, infection with human immunodeficiency virus, other viral infections, diabetes, cardiovascular disease, schizophrenia, inflammatory bowel disease, enteric infections, and more. We look forward to this next era, when some of these genetic discoveries will transition from “findings” to results with real impact on public health and policy.
ACKNOWLEDGMENTS
In recognition of the Johns Hopkins Department of Epidemiology Centennial, the Genetic Epidemiology program would like to recognize the professorial faculty who participated in this program from its inception to present day (1979–2019): Dr. Bernice Cohen (Program Director, deceased), Dr. Terri H. Beaty (Program Director), Dr. Muin Khoury, Dr. Emily Harris, Dr. Gloria Petersen, Dr. W.H. Linda Kao (deceased), Dr. M. Daniele Fallin (Program Director), Dr. Yin Yao, Dr. Priya Duggal (Program Director), Dr. Christine Ladd-Acosta, Dr. Robert Wojiechowski, Dr. Debashree Ray, and Dr. Genevieve Wojcik. With heartfelt thanks to the numerous genetic epidemiology students and trainees who have enriched this academic program.
Coflict of interest: none declared.
Abbreviations
- GWAS
genome-wide association study
- RCT
randomized controlled trial
- SNP
single nucleotide polymorphism
References
- 1. Mendel G. Experiments Concerning Plant Hybrids [in German]. Proceedings of the Natural History Society of Brünn (IV). 1865. [Google Scholar]
- 2. Neel J, Schull W. Human Heredity. Chicago, IL: University of Chicago Press; 1954. [Google Scholar]
- 3. Morton N. Outline of Genetic Epidemiology. Basel, Switzerland: Karger; 1982. [Google Scholar]
- 4. Huang PC, Cohen BH, Lilienfeld AM, eds. Genetic Issues in Public Health and Medicine. Springfield, IL: Charles C. Thomas; 1978. [Google Scholar]
- 5. Khoury MJ, Beaty TH, Cohen BH. Fundamentals of Genetic Epidemiology. New ed. Oxford, United Kingdom: Oxford University Press; 1993. [Google Scholar]
- 6. Ioannidis JPA, Boffetta P, Little J, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. 2008;37(1):120–132. [DOI] [PubMed] [Google Scholar]
- 7. Khoury MJ, Wacholder S. Invited commentary: from genome-wide association studies to gene-environment-wide interaction studies—challenges and opportunities. Am J Epidemiol. 2009;169(2):227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lilienfeld AM. Formal discussion of: genetic factors in the etiology of cancer: an epidemiologic view. Cancer Res. 1965;25(8):1330–1335. [PubMed] [Google Scholar]
- 9. Fisher RA. XV. The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinburgh. 1918;52:399–433. [Google Scholar]
- 10. Lush J. Genetic aspects of the Danish system of progeny-testing swine. Iowa Agric Home Econ Exp Station. 1936;18(204):108–195. [Google Scholar]
- 11. Bell AE. Heritability in retrospect. J Hered. 1977;68(5):297–300. [DOI] [PubMed] [Google Scholar]
- 12. Watson JD, Crick FHC. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. [DOI] [PubMed] [Google Scholar]
- 13. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rommens JM, Iannuzzi MC, Kerem BS, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–1065. [DOI] [PubMed] [Google Scholar]
- 15. Riordan JR, Rommens JM, Kerem BS, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. [DOI] [PubMed] [Google Scholar]
- 16. Kerem BS, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. [DOI] [PubMed] [Google Scholar]
- 17. Collins F. Galas D. a new five-year plan for the U.S. human genome project. Science. 1993;262(5130):43–46. [DOI] [PubMed] [Google Scholar]
- 18. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998–2003. Science. 1998;282(5389):682–689. [DOI] [PubMed] [Google Scholar]
- 19. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. [DOI] [PubMed] [Google Scholar]
- 20. International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931–945. [DOI] [PubMed] [Google Scholar]
- 21. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. [DOI] [PubMed] [Google Scholar]
- 22. International Hapmap Consortium , Gibbs RA, Belmont JW, et al. The International HapMap Project. Nature .2003;426(6968):789–796. [DOI] [PubMed] [Google Scholar]
- 23. Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. 1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ott J. Analysis of Human Genetic Linkage. Baltimore, MD: Johns Hopkins University Press; 1999. [Google Scholar]
- 28. Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet. 1993;52(3):506–516. [PMC free article] [PubMed] [Google Scholar]
- 29. Gauderman WJ, Witte JS, Thomas DC. Family-based association studies. J Natl Cancer Inst Monogr. 1999;(26):31–37. [DOI] [PubMed] [Google Scholar]
- 30. Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273(5281):1516–1517. [DOI] [PubMed] [Google Scholar]
- 32. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pritchard JK. The allelic architecture of human disease genes: common disease-common variant... Or not? Hum Mol Genet. 2002;11(20):2417–2423. [DOI] [PubMed] [Google Scholar]
- 36. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40(6):695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83(3):311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saint PA, Génin E. How important are rare variants in common disease? Brief Funct Genomics. 2014;13(5):353–361. [DOI] [PubMed] [Google Scholar]
- 39. Thun MJ, Hoover RN, Hunter DJ. Bigger, better, sooner—scaling up for success. Cancer Epidemiol Biomarkers Prev. 2012;21(4):571–575. [DOI] [PubMed] [Google Scholar]
- 40. Kidd BA, Peters LA, Schadt EE, et al. Unifying immunology with informatics and multiscale biology. Nat Immunol. 2014;15(2):118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. ENCODE Project overview. https://www.encodeproject.org/help/project-overview/.
- 42. Project RE Reference epigenome mapping centers. http://www.roadmapepigenomics.org/overview/mapping-centers.
- 43. Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perna L, Zhang Y, Mons U, et al. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marioni RE, Shah S, McRae AF, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levine ME, Lu AT, Chen BH, et al. Menopause accelerates biological aging. Proc Natl Acad Sci. 2016;113(33):9327–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kresovich J, Xu Z, O’Brien K, et al. Methylation-based biological age and breast cancer risk. J Natl Cancer Inst. 2019;111(10):1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015;212(10):1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. GTEx Project GTEx Portal. https://gtexportal.org/home/. Accessed October 3, 2019.
- 51. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet. 2007;81(6):1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luo W, Friedman MS, Shedden K, et al. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Edwards SL, Beesley J, French JD, et al. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93(5):779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kichaev G, Yang WY, Lindstrom S, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10(10):e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chung D, Yang C, Li C, et al. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS Genet. 2014;10(11):e1004787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiong Q, Ancona N, Hauser ER, et al. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res. 2012;22(2):386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang YT, Vanderweele TJ, Lin X. Joint analysis of SNP and gene expression data in genetic association studies of complex diseases. Ann Appl Stat. 2014;8(1):352–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li L, Kabesch M, Bouzigon E, et al. Using eQTL weights to improve power for genome-wide association studies: a genetic study of childhood asthma. Front Genet. 2013;4:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet. 2014;94(4):559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gamazon ER, Wheeler HE, Shah KP, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang J, Fritsche LG, Zhou X, et al. A scalable Bayesian method for integrating functional information in genome-wide association studies. Am J Hum Genet. 2017;101(3):404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hao X, Zeng P, Zhang S, et al. Identifying and exploiting trait-relevant tissues with multiple functional annotations in genome-wide association studies. PLoS Genet. 2018;14(1):e1007186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spain SL, Barrett JC. Strategies for fine-mapping complex traits. Hum Mol Genet .2015;24:R111–R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics. 2010;26(16):2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9. [DOI] [PubMed] [Google Scholar]
- 70. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86(1):6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thompson JR, Gögele M, Weichenberger CX, et al. SNP prioritization using a Bayesian probability of association. Genet Epidemiol. 2013;37(2):214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Minelli C, De Grandi A, Weichenberger CX, et al. Importance of different types of prior knowledge in selecting genome-wide findings for follow-up. Genet Epidemiol. 2013;37(2):205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thurner M, van de Bunt M, Torres JM, et al. Integration of human pancreatic islet genomic data refines regulatory mechanisms at type 2 diabetes susceptibility loci. Elife. 2018;7:e31977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thomas D. Gene-environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11(4):259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gauderman WJ, Mukherjee B, Aschard H, et al. Update on the state of the science for analytical methods for gene-environment interactions. Am J Epidemiol. 2017;186(7):762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu L, Li Y, Tollefsbol TO. Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol. 2008;10(1–2):25–36. [PMC free article] [PubMed] [Google Scholar]
- 79. Ladd-Acosta C, Fallin MD. The role of epigenetics in genetic and environmental epidemiology. Epigenomics. 2016;8(2):271–283. [DOI] [PubMed] [Google Scholar]
- 80. Ritchie MD, Davis JR, Aschard H, et al. Incorporation of biological knowledge into the study of gene-environment interactions. Am J Epidemiol. 2017;186(7):771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peyrot WJ, Milaneschi Y, Abdellaoui A, et al. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry. 2014;205(2):113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pasman J, Verweij K, Vink J. Systematic review of polygenic gene-environment interaction in tobacco, alcohol, and cannabis use. Behav Genet. 2019;49(4):349–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pingault JB, O’Reilly PF, Schoeler T, et al. Using genetic data to strengthen causal inference in observational research. Nat Rev Genet. 2018;19(9):566–580. [DOI] [PubMed] [Google Scholar]
- 84. Zheng J, Baird D, Borges M-C, et al. Recent developments in Mendelian randomization studies. Curr Epidemiol Reports. 2017;4(4):330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ference BA, Majeed F, Penumetcha R, et al. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Martin AR, Kanai M, Kamatani Y, et al. Current clinical use of polygenic scores will risk exacerbating health disparities. bioRxiv; 2018. (doi: 10.1101/441261). Accessed October 3, 2019. [DOI] [Google Scholar]
- 87. Shi X, Wu X. An overview of human genetic privacy. Ann N Y Acad Sci. 2017;1387(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sweeney L. Simple Demographics Often Identify People Uniquely. Carnegie Mellon University, Data Privacy Working Paper 3 2000. https://dataprivacylab.org/projects/identifiability/paper1.pdf. Accessed October 3, 2019.
- 89. Maron DF. Cold cases heat up as law enforcement uses genetics to solve past crimes. Scientific American. July 2, 2018. https://www.scientificamerican.com/article/cold-cases-heat-up-as-law-enforcement-uses-genetics-to-solve-past-crimes/. Accessed October 3, 2019 [Google Scholar]
- 90. Erlich Y, Shor T, Pe’er I, et al. Identity inference of genomic data using long-range familial searches. Science. 2018;362(6415):690–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. O’Connell MR, Oakes BL, Sternberg SH, et al. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516(7530):263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]