

Abstract
The efficient delivery of antisense oligonucleotides (ASOs) to the targeted cells and organs remains a challenge, in particular, in vivo. Here, we investigated the ability of a library of biodegradable lipid nanoparticles (LNPs) in delivering ASO to both cultured human cells and animal models. We first identified three top-performing lipids through in vitro screening using GFP-expressing HEK293 cells. Next, we explored these three candidates for delivering ASO to target proprotein convertase subtilisin/kexin type 9 (PCSK9) mRNA in mice. We found that lipid 306-O12B-3 showed efficiency with the median effective dose (ED50) as low as 0.034 mg·kg-1, which is a notable improvement over the efficiency reported in the literature. No liver or kidney toxicity was observed with a dose up to 5 mg·kg-1 of this ASO/LNP formulation. The biodegradable LNPs are efficient and safe in the delivery of ASO and pave the way for clinical translation.
Keywords: antisense oligonucleotide, biodegradable lipid nanoparticle, PCSK9
Graphical Abstract
Introduction
Antisense oligonucleotides (ASOs) are single-strand DNAs serving as effective drugs to reduce the messenger RNA level in cells. ASOs are usually 16–25 bases long and are designed to hybridize with mRNA through Watson-Crick base-pairing specificity.1, 2, 3, 4 This DNA-RNA hybrid recruits and activates the endonuclease RNase H, which cleaves the RNA strand, leading to the degradation of the mRNA and halting protein translation.5, 6, 7 Moreover, ASO can redirect the mRNA splicing machinery to include or delete specific exons by complementary binding to RNA sequences at the early transcriptional stage.8,9 ASO affiliating to mRNA can also sterically inhibit the formation of the ribosomal complex, resulting in the hindrance of protein translation.10,11
The use of synthetic ASO to regulate gene expression has been developed for many years. Within the past few years, the US Food and Drug Administration (FDA) approved two ASO-mediated therapies for the treatment of Duchenne muscular dystrophy and spinal muscular atrophy, respectively.12,13 However, optimization of ASO delivery is still urgently needed for the further advancement of ASO in the clinic. An optimal delivery system needs to be cell specific, controllable, and able to protect the nucleic acids from nuclease degradation.14
Substantial progress has been made in developing lipid-based and polymer-based nanocarriers to facilitate nucleic acid delivery.15, 16, 17, 18, 19, 20, 21 Among them, cationic lipid-like nanoparticles are most advanced in terms of clinical translation, as demonstrated by the FDA approval of patisiran, a lipid nanoparticle (LNP)-formulated RNAi-based therapy for the treatment of the hereditary transthyretin amyloidosis.22
Recently, we have used the combinatorial library strategy to synthesize lipid-like materials for delivering various biologics, including proteins and nucleic acids, both in vitro and in vivo.23,24 The subset of synthetic lipids, which contain disulfide bonds, has been demonstrated to be biodegradable and has shown excellent biocompatibility.25, 26, 27, 28 The high concentration of glutathione (GSH) in cytoplasm tends to intracellularly cleave and degrade LNPs through disulfide bond exchange once the nanoparticles reach the reductive intracellular environment.29,30
In this study, we investigated the potential application of this novel class of synthetic lipids for ASO delivery both in vitro and in vivo. For in vivo study, we chose proprotein convertase subtilisin/kexin type 9 (PCSK9) as a therapeutic target, because it plays an essential role in cholesterol metabolism by regulating low-density lipoprotein (LDL) receptor degradation. The antibody against PCSK9 has been developed and approved by the FDA for the treatment of hyperlipidemia.31 ASO or RNAi may be a better strategy to manage the hyperlipidemia than antibody therapeutics, due to the potential longer-lasting effect and thus less-frequent dosing of ASOs and RNAi compared with antibody. However, this strategy relies on a robust, targeted delivery method that specifically delivers the ASO to the liver.
Herein, we explored the capability of the bioreducible lipids to deliver ASO for efficient mRNA silencing both in vitro and in vivo (Figure 1A). We conducted a screening of a small library of the bioreducible lipids by delivering GFP silencing ASO in GFP-expressing cells and identified three lipids, 113-O14B-3, 113-O16B-3, and 306-O12B-3, showing high efficiency in gene knockdown. They all showed superior delivery efficacy than Lipofectamine 2000 (LPF 2000), a commercially available transfection reagent. These lipid nanoparticles also exhibit lower toxicity than that of LPF 2000 in vitro. Further, we evaluated the capability of these synthetic lipids in delivering PCSK9 silencing ASO in mouse liver for efficient PCSK9 mRNA knockdown. The top-performing lipid was found to be 306-O12B-3, with a median effective dose (ED50) around 0.034 mg·kg-1. The knockdown of the PCSK9 mRNA level by ASO/LNP complexes was also demonstrated to effectively reduce the total PCSK9 protein and serum cholesterol level in mice, demonstrating a functional knockdown. Meanwhile, no hepatotoxicity or nephrotoxicity was observed. This study showed that the bioreducible lipids can be good candidates for ASO delivery and may pave the way for the advancement of ASO-based therapeutics.
Figure 1.

Design of Bioreducible Lipids for Effective ASO Delivery
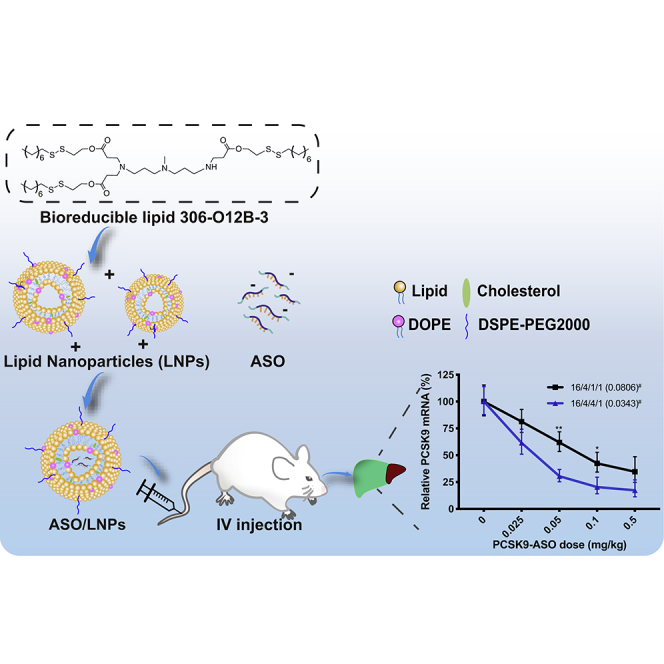
(A) Schematic illustration of the mechanism of LNPs for ASO delivery both in vitro and in vivo. For in vitro study, cultured HEK cell lines stably expressing GFP were treated with the LNPs packing with ASO. The ASO sequences were able to target and silence GFP mRNA, followed by the fade in GFP intensity. On the other hand, the optimal lipids screened from in vitro study were applied to PCSK9 mRNA knockdown in vivo. BALB/c mice were intravenously administrated with ASO-loaded LNPs. Once the ASO/LNP complexes arrived at hepatocytes, the LNPs were destabilized and degraded at the reductive intracellular environments, triggering the release of ASO. The PCSK9 targeting ASO then specifically hybridized with mRNA by Watson-Crick base pairing, followed by the nonspecific activation of endonuclease RNase H, which cleaved the RNA strand only in the form of a DNA-RNA hybrid. This mechanism led to the degradation of mRNA and the halt of PCSK9 protein translation. (B) Synthetic route (I) and the chemical structure of lipid 306-O12B-3 (II). (C) The chemical structures of amine head (R) groups and acrylate tails (R′).
Results
Construction of Lipid Library
The bioreducible lipids were synthesized by the Michael addition between the primary or secondary amines and acrylate containing a disulfide bond (Figure 1BI), according to our published procedure.26 Bioreducible lipids were named “R-O[x]B-n” using the following method: the amine head is indicated by a two- or three-digit serial number (R). The letter “O” indicates an acrylate linker between the amine head and hydrophobic tail. The number [x] indicates the number of carbon atoms in the hydrophobic tail of the acrylate. The “B” refers to a bioreducible lipid, i.e., containing a disulfide bond in the tail. The last number “n” indicates the total number of hydrophobic tails of the synthetic lipids and is usually either 3 or 4. For example, 306-O12B-3 indicates the lipid prepared by reacting amine 306 with acrylate O12B, resulting in a lipid with 3 tails. Figure 1BII shows the chemical structure of the lipid, termed “306-O12B-3.”
Chemical Modification of ASO Sequence
For in vitro screening of the lipids, the HEK cell line stably expressing GFP (GFP-HEK cells) was used as a model to evaluate the efficiency of the ASO delivery using these bioreducible lipids. Because the activity of ASO depends on many factors, such as chemical linkages, guanine and cytosine (GC) content, and secondary structure of mRNA targeting, it is necessary to optimize the sequences and chemistry pattern of ASO to achieve optimal gene silence of a target gene. We first designed five ASO sequences targeting the GFP gene, each 20 nucleotides in length (identified as G-ASO-1 to G-ASO-5) with phosphodiester linkages between each nucleotide (Figure S1A). We screened the delivery of these ASOs to the GFP-HEK cells using the commercial transfection reagent LPF 2000. As shown in Figure S1B, the #5 ASO showed higher GFP silencing efficiency than other ASOs, so we chose the #5 ASO for further study. This sequence was optimized as described in literature using chemically modified phosphorothioate bonds and 2′-O-methyl (2-OME)-modified ribose (Figure S1C). The modified 5th ASO showed a 55.6% improvement in GFP mRNA repression, demonstrating that the chemical modification of ASO played a crucial role in improving the capacity and stability in ASO (Figure S1D). The chemically modified scrambled ASO, delivered by LPF 2000, showed no effect in GFP silencing, indicating ASO sequence-specific gene silence. Thus, all ASOs are phosphorothioate bonds and 2-OME modified in the following experiments.
Formulation and Characterization of LNP
For in vitro ASO delivery, we used the pure lipids, referred as “nonformulated LNPs,” in which no other helper lipids were added to the formulation. The ASO and the lipid sample were complexed at a 1/15 (w/w) ratio. The particle size and zeta potential were measured using dynamic light scattering (DLS). As shown in Figure S2A, the blank, nonformulated LNPs ranged from 90 to 300 nm effective diameter, whereas the sizes increased to 150 nm to 500 nm after complexing with ASO. The size increase is due to the complexation between the lipid and ASO. The zeta potential of the nanocomplex (Figure S2B) showed an obvious surface-charge decrease from positive (0.2 to 28 mV) before complexation with ASO to negative (−31.7 to −12.2 mV) after ASO complexation. This demonstrates that the nanocomplexation is driven by the electrostatic interaction between the negative-charged ASO and positive-charged LNPs.
For in vivo ASO delivery, in addition to the active bioreducible lipid, we also added helper lipids, including cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and polyethylene glycol (PEG)ylated lipid in the formulation. The LNPs produced with colipids were referred as “formulated LNPs.” The ASO and the formulated LNPs were mixed at a 1/15 (w/w) ratio of ASO to bioreducible lipid. The nanocomplex size and surface charge were determined using DLS, and the morphology was visualized using transmission electronic microscopy (TEM). The sizes of the ASO/LNP nanocomplexes are in the range of 90 to 150 nm. Figures S2C–S2F showed the typical morphology of the LNP formulation with bioreducible lipid (306-O12B-3), before and after complexing with ASO. Both showed spherical shape, and the particle size is consistent with the DLS analysis. The ASO encapsulation efficiency using formulated LNPs was measured by the Quant-iT OliGreen ssDNA assay kit, and all of the lipid formulations exhibited high ASO encapsulation efficiency above 97% (Figure S3).
Screening the Bioreducible Lipids for ASO Delivery In Vitro
For in vitro study, lipid nanoparticles were fabricated via directly dissolving the pure lipids in sodium acetate buffer (25 mM, pH 5.2).
For the in vitro screening, we fixed the weight ratio of synthetic lipids to GFP-ASO at 15/1 and delivered the GFP-ASO to the GFP-HEK cells at a concentration of 0.1 μg/mL. The GFP expression level of the HEK cells was analyzed by flow cytometry after 16 h of post-transfection. The ASO delivery efficacy was determined by the ratio of the mean fluorescent intensity of treated cells to that of untreated ones. As shown in Figure 2A, GFP-ASO-alone treatment exhibits negligible GFP suppression, similar to PBS vehicle control treatment, indicating the inability to penetrate the cell membranes by ASO itself. The cells treated with the ASO/LNP complexes showed different GFP silencing levels. We found that 113-O16B-3 and 306-O16B-3 showed higher GFP silencing than LPF 2000, a commercial in vitro gene transfection reagent, with 53.5% of GFP knockdown efficacy.
Figure 2.

In vitro Screening of Lipids for ASO Delivery
(A) HEK cells stably expressing GFP were treated with or without ASO/LNP complexes. ASO can specifically target GFP mRNA sequences (GFP-ASO). With fixed acrylate tail length at O16B, lipids of ten different heads in two to four tail numbers were screened. The LNPs were mixed with GFP-ASO at the ratio of 15/1 (w/w); *p < 0.01 versus all other samples. (B) The three tail numbers of lipids were further investigated in different tail lengths (O12B-O18B) with top-head groups of 113 and 306; *p < 0.01 versus all other samples. (C) Optimization of the N/P ratio of the three best-performing lipids 113-O14B-3, 113-O16B-3, and 306-O12B-3. The ASO/LNP complexes were prepared by mixing GFP-ASO with fixed concentration at 0.1 μg/mL, and differing LNP concentrations ranged from 0.2 μg/mL to 2 μg/mL; *p < 0.01 versus all other samples at the same lipid group. (D) The comparison of the ASO transfection efficiency mediated by bioreducible lipids or LPF 2000. The GFP-HEK cells were treated with ASO/LNP complexes with GFP-ASO concentration increasing from 0.3 to 30 nM, with the fixed N/P ratio of 15/1. (E) The cell viability of GFP-HEK cells treated with bioreducible LNPs or LPF 2000 in a dose-dependent manner; same conditions as (D). For all negative control groups, GFP-HEK cells were treated with 16.7 nM of formulated, scrambled GFP-ASO; naked GFP-ASO; or PBS; *p < 0.01 versus all other samples at the same concentration group. For all of the in vitro experiments, the LNPs were fabricated by directly dissolving in sodium acetate buffer (25 mM, pH 5.2), followed by a brief sonication and dialysis. The GFP intensity was then analyzed by flow cytometry after 16 h of postdelivery. Data are presented as mean ± SD (n ≥ 3).
The amine heads (113 and 306 shown in Figure 1C), used to synthesize these lipids, share some structural similarities. Both amine heads contained three amino groups and two of them evenly distributed at the end of the structures with the potential to induce different numbers of carbon tails. Further, the synthetic lipids showed the most efficient in vitro ASO delivery when the lipids were synthesized with three branches of carbon tails instead of two or four tails. To investigate whether the carbon tail length also affected the ASO delivery, we synthesized lipids derived from amines 113 and 306 with three tails using different tail lengths (O12B, O14B, O16B, and O18B, shown in Figure 1C). As shown in Figure 2B, three of eight synthetic lipids (113-O14B-3, 113-O16B-3, and 306-O12B-3) in the expanded library showed higher delivery efficiency than LPF 2000 in vitro. In particular, 306-O12B-3 nanoparticles exhibited the highest GFP silencing efficiency, with 81.5% of GFP-negative cells observed. Also, those lipids showed high cell viability compared to that of LPF 2000, demonstrating low cell toxicity of those different tail lengths (Figure S4).
We further investigated the influence of the ratio of the lipid to ASO (N/P ratio). The N/P ratio is the molar ratio of the nitrogen in synthetic lipid to the phosphate groups in the ASO. The top three performing lipids from the screening (113-O14B-3, 113-O16B-3, and 306-O12B-3) were chosen for this study. The N/P ratio of lipid to ASO was varied from 2.5 to 20. As shown in Figure 2C, when N/P ratios increased from 2.5 to 15, the GFP silencing efficiency was improved from 10.9% to 79.5%, whereas the increase of N/P ratios above 15 did not augment ASO delivery efficiency. Hence, we chose the N/P ratio of 15/1 for following experiments to achieve sufficient delivery efficacy with good biocompatibility. Interestingly, while N/P ratio is defined as a molar ratio, given the exact molecular weights of our particular lipids and ASOs, a 15/1 N/P ratio was found to correlate to the 15/1 weight ratio as well. Thus, for the remainder of this paper, we report delivery parameters of our synthetic lipids as a weight/weight ratio for practical purposes.
We next checked the in vitro cytotoxicity and gene-knockdown efficiency of the LNP/ASO nanocomplexes in a dose-dependent matter. Commercially available transfection reagent LPF 2000 is used as control for comparison. The N/P ratio of LNP/ASO is fixed at 15/1, and the dose of GFP-ASO varying from 0.3 to 30 nM was used to treat the GFP-HEK cells. The GFP gene-knockdown efficiency was evaluated using a flow cytometer, and the cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As Figure 2D shows, all three lipids showed higher GFP silencing efficiency than that of LPF 2000 at each dose of GFP-ASO, except for the 0.3-nM concentration, which is too low to show significant gene silencing in all lipids. To eliminate the potential of unspecific targeting, we also formulated the scrambled GFP-ASO sequence with our LNPs as a negative. No GFP silencing effect was observed, indicating the specificity of silencing. Figure 2E showed the dose-dependent cell-viability results from the three synthetic lipids and control LPF 2000. The three top candidates showed the similar cell viability trend as the GFP-ASO doses increased. Even at the highest LNP/ASO complex concentration (30 nM), the cell viability from all three bioreducible lipid-treated cells maintained above 76.4%, whereas the cells treated with LPF 2000 at the same dose reduced to 47.9%. These results suggested that our lipids were not only highly effective but also more biocompatible and less cytotoxic than LPF 2000 for ASO delivery.
Biodistribution of ASO-Loaded LNPs In Vivo
For in vivo delivery, in addition to the active synthetic lipid, excipients, including cholesterol, DOPE, and PEGylated lipid (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000 [DSPE-PEG2000]), were used to formulate the LNPs. ASO labeled with near-infrared fluorescent dye (ASO-Alexa 750) was used as a cargo to form nanocomplexes with LNPs.
The top three lipids (113-O14B-3, 113-O16B-3, and 306-O12B-3) identified from the in vitro study were chosen for in vivo delivery of ASO-Alexa 750. The active lipid to the Alexa 750-labeled ASO ratio was fixed at 15/1 (w/w). At 1 h of postinjection, major organs were collected and then visualized using IVIS (in vivo imaging system). As shown in Figure 3, all of the ASO-LNP-treated mice exhibited most fluorescence signals in the liver, indicating the LNP/ASO complexes mainly targeted and accumulated to this organ. It is noticeable that the ASO-Alexa 750s complexed with 306-O12B-3 showed the highest fluorescence intensity in the liver, which is about 2-fold higher than that from mice injected with ASO/113-O14B-3 complexes (Table S1). Besides liver, we also observed the fluorescent signals in the organs, such as spleen, kidney, and lung, although much less than liver. As expected, no fluorescence was detected from the PBS-injected control mice.
Figure 3.

Biodistribution of ASO/LNP Complexes in Major Organs (Brain, Heart, Lung, Liver, Spleen, and Kidney)
The three best-performing lipids, 113-O14B-3, 113-O16B-3, and 306-O12B-3, chosen from the in vitro screening studies were formulated in the following formulation: lipids/cholesterol/DOPE/DSPE-PEG2000 at the weight ratio 16/4/1/1. The Alexa 750-labeled ASOs were mixed with formulated LNPs at the weight ratio of 1/15, followed by the intravenous injection to mice. The results showed that all of the ASO-loaded LNPs exhibited most fluorescence signals in the liver, indicating the complexes mainly accumulating in this organ. Also, the ASO-Alexa 750s delivered by 306-O12B-3 exhibited the strongest fluorescence intensity in the liver.
PCSK9 Gene Knockdown Using LNP/PCSK9-ASO Complexes in Mice
The PCSK9 expression is involved in hypercholesterolemia and premature atherosclerotic cardiovascular disease (ASCVD), and antibody against PCSK9 has gained FDA approval for the management of hypercholesterolemia.31,32 Here, we evaluated whether our lipid formulation can deliver PCSK9-targeted ASO (PCSK9-ASO) for efficient PCSK9 knockdown in mice. The top three synthetic lipids (113-O14B-3, 113-O16B-3, and 306-O12B-3) identified from in vitro study were used to form LNPs for PCSK9-ASO delivery in vivo. To increase the serum stability and circulation time of the LNP/ASO complexes, we formulated LNPs with other helper lipids. The helper lipids cholesterol, DOPE, and DSPE-PEG2000 were used in the LNP formulation. In a typical formulation, the synthetic lipid, cholesterol, DOPE, and DSPE-PEG2000 were set at the weight ratio of 16/4/1/1. The synthetic lipid to PCSK9-ASO ratio was set as 15/1 (w/w). The PCSK9-ASO/LNP complexes were injected in mice through the tail vein at the PCSK9-ASO doses of 0.05, 0.1, 0.5, 1, and 1.5 mg·kg-1. A control group was injected with saline via the tail vein and another control group with scrambled ASO encapsulated in the same lipid formulations. Livers were harvested after 72 h after administration for PCSK9 mRNA quantization using RT-PCR. As shown in Figure 4, the PCSK9 mRNA level in mice liver was found to be ASO dose dependent. In general, the PCSK9 mRNA level decreased as the injected PCSK9-ASO dose increased for all three lipid formulations. Among them, 306-O12B-3 LNP formulation showed the best silencing effect with an ED50 of 0.088 mg·kg-1 in the mRNA level. Thus, we chose 306-O12B-3 as the leading lipid for further study, including formulation optimization and in vivo biocompatibility.
Figure 4.

In Vivo Evaluation of the Three Best-Performing Lipids (113-O14B-3, 113-O16B-3, and 306-O12B-3) for PCSK9-ASO Delivery
The “#” represents the ED50 (mg·kg-1) for different lipids in PCSK9-ASO delivery in vivo. All of the LNPs were composed of cationic lipid, cholesterol, DOPE, and DSPE-PEG2000 (16/4/1/1 w/w) with a PCSK9-ASO-to-lipid ratio of 1/15 (w/w). Animals were treated with single-dose intravenous administration at varying ASO doses (mg·kg-1). Mice were sacrificed after 72 h of postinjection. PCSK9 mRNA levels relative to β-actin mRNA levels were determined in liver samples. Data points are expressed as a percentage of saline control animals and represent group mean ± SD (n = 3).
The Test of Two Different DOPE Fractions In Vivo
It has been reported that the formulation ratios of the helper lipids may influence the delivery efficiency of the nanoparticle system. Thus, we investigated whether increasing the amount of the DOPE in the LNP formulation can improve the ASO delivery in vivo. In the study shown in Figure 4, the weight ratio of cationic lipid/cholesterol/DOPE/DSPE-PEG2000 was set as 16/4/1/1. To evaluate the influence of the DOPE on the in vivo delivery, we kept the ratio of cationic lipid (306-O12B-3), cholesterol, and DSPE-PEG2000 not changed, whereas we increased the DOPE. The final ratio of cationic lipid/cholesterol/DOPE/DSPE-PEG2000 was set as 16/4/4/1 in the formulation. We kept the active cationic lipid-to-ASO ratio the same at 15/1 (w/w) in both cases. The formulations were injected into mice via tail vein at PCSK9-ASO doses of 0.025, 0.05, 0.1, or 0.5 mg·kg-1. After 72 h of postinjection, livers were harvested for PCSK9 mRNA determination, and the blood was collected for serum PCSK9 protein and total cholesterol measurement. As shown in Figure 5A, we found that increasing the DOPE fraction in the LNP improved the in vivo ASO delivery efficiency. For 16/4/4/1 formulation (cationic lipid/cholesterol/DOPE/DSPE-PEG2000), an ED50 of 0.034 mg·kg-1 PCSK9-ASO was obtained, compared with an ED50 of 0.088 mg·kg-1 in the 16/4/1/1 formulation for the PCSK9 mRNA knockdown in the mouse liver. Both PCSK9 protein in serum and serum cholesterol level were significantly decreased after the LNP/ASO treatment in a dose-dependent manner (Figures 5B and 5C). Meanwhile, no PCSK9 silencing effect was observed in the scrambled PCSK9-ASO (S-PCSK9-ASO) delivered by the 306-O12B-3 in either 16/4/4/1 or 16/4/1/1 formulation (Figure S5). This result demonstrated the specific binding of functional PCSK9-ASO to the corresponding mRNA sequence. At the dose of 0.1 mg·kg-1 of ASO, the PCSK9 protein and serum cholesterol level also decrease to 50% of the normal level. We also tested some other lipid composition and ratios (Figure S6), but none of them exhibited higher PCSK9 silencing efficiency than the 16/4/4/1 formulation.
Figure 5.

Improved Efficacy of the Optimized Formulation for the Lipid 306-O12B-3 Compared to the Initial Formulation in PCSK9-ASO Delivery In Vivo
The “#” represents the ED50 (mg·kg-1) for different lipids in PCSK9-ASO delivery in vivo. (A) The in vivo PCSK9 mRNA silencing level mediated by the optimized formulation (cationic lipid/cholesterol/DOPE/DSPE-PEG2000 at the ratio of 16/4/4/1, w/w) was compared to that of the initial screening formulation (16/4/1/1, w/w). (B) The optimized formulation evaluated by the serum PCSK9 protein level. (C) The optimized formulation assessed by the serum total cholesterol level. The lipid and PCSK9-ASO were combined at a ratio of 15/1 (w/w); *p < 0.01 or **p < 0.01 versus all other samples at the same dose group. Animals were treated with single-dose intravenous administration at varying ASO dose levels. The control group was injected with saline. Mice were sacrificed after 72 h of postinjection. PCSK9 mRNA levels relative to β-actin mRNA levels were determined in liver samples. Data points are expressed as a percentage of saline control animals and represent group mean ± SD (n = 3).
We also investigated the tolerability of the optimized 306-O12B-3 formulation in mice. Animals were intravenously (i.v.) injected with a PCSK9-ASO dose up to 5 mg·kg-1. The LNPs comprising 306-O12B-3 were well tolerated at all dose levels and observed no clinically significant change in mouse behavior, spleen size, and key hepatotoxic or nephrotoxic parameters (Table 1).
Table 1.
Clinical Chemistry and Hematology Parameters for ASO/LNP Complex-Treated Mice
| Groups | ALT (U/L) | AST (U/L) | Cr (mg/dL) | BUN (mg/dL) | Spleen (mg) |
|---|---|---|---|---|---|
| 0 mg·kg-1 | 20.24 ± 2.78 | 129.14 ± 17.89 | 0.35 ± 0.09 | 20.28 ± 2.39 | 76.8 ± 3.6 |
| 1 mg·kg-1 | 20.53 ± 2.03 | 131.76 ± 15.88 | 0.35 ± 0.09 | 20.11 ± 3.69 | 77.3 ± 3.1 |
| 3 mg·kg-1 | 17.88 ± 3.38 | 129.95 ± 20.21 | 0.36 ± 0.07 | 20.9 ± 2.51 | 79.7 ± 1.6 |
| 5 mg·kg-1 | 19.84 ± 3.5 | 133.34 ± 15.08 | 0.37 ± 0.1 | 21.99 ± 2.34 | 80.9 ± 2.2 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Cr, creatinine. Data points are expressed as a percentage of saline control animals and represent group mean ± SD (n = 3).
Discussion
The therapeutic application of oligonucleotides, including ASOs and small interfering RNA (siRNA), has been recognized as potent tools against myriad diseases. Due to the stability and the low off-target effect of ASO, some ASO-mediated gene silencing has achieved a successfully clinical translation.33 As polyanionic and hydrophilic molecules, however, naked ASO is incapable of self-penetrating across the negatively charged cell membranes to reach the intracellular space where the target mRNA resides. Further, their anionic and small-sized characteristics would trigger the nonspecific combination with serum nucleases, followed by the rapid renal filtration.34 Also, there is potential for off-target effects and immunogenicity.35 As such, the sophisticated delivery of ASO remains as a vital challenge to the advance of ASO-based therapeutics.
Currently, the cationic lipid nanoparticles were developed as one of the most efficient systems for a broad spectrum of clinical applications. The LNPs are capable of protecting encapsulated ASOs from nuclease degradation, as well as localizing macromolecules to target organs. To achieve a specific pharmacological effect, the LNP platforms can achieve the same or better therapeutic index with a lower ASO dose comparing with the naked chemical-modified ASO.
The bioreducible lipid-like materials developed by our group showed superior effects in nucleic acids and protein delivery across the mammalian cells. Here, we evaluated whether these bioreducible LNPs can also achieve a good performance in encapsulating and then delivering ASO to intracellular sites of action, as well as to targeting organs (Figure 1A). Different from other traditional amino lipids, both ester bonds and disulfide bonds are integrated into the hydrocarbon tails of our biodegradable materials. On the one hand, the acrylate tails can be degraded through ester hydrolysis into hydrophilic metabolites, followed by a rapid removal from plasma and tissues, thus reducing the cytotoxicity. On the other hand, once LNPs reach the reductive intracellular environment, the incorporated disulfide bond can also be cleaved and degraded by short peptide GSH inside cells through the disulfide bond exchange. This mechanism facilitates oligonucleotide release from LNPs, specifically upon delivery to the intracellular target, and simultaneously eliminates cytotoxicity arising from the sustained loading of LNPs. These structural designs explain the higher ASO delivery efficacy and biocompatibility of our bioreducible lipids compared to the widely used commercial agent, Lipofectamine 2000 (Figures 2D and 2E).
The combinatorial synthesis and high-throughput screening have also been utilized in the cationic lipid library production and lipid screening. The concept allows for rapid synthesis of a large number of lipid structures based on structure-activity relationships.24 The subsequent lipid screening facilities the identification of top-performing lipids and formulations, which target different organs. Notably, the lipid materials screened in vitro and in vivo, which were found to have the best performance, shared some similarities: (1) more than two hydrophobic carbon tails, and (2) one tail fewer than a full substitution of the amine head—i.e., an amine head that could accommodate 4 tails instead containing 3 tails (Figure 1C). Based on the structural advantages and in vitro screening outcomes, the biodistribution of the leading lipids was analyzed via systemic administration, indicating that our ASO/LNP delivery systems have excellent access to the liver (Figure 3). The success in passively targeting hepatocytes may ascribe, in part, to the discontinuous and fenestrated endothelial cells that allow the circulating nanoparticles extravasating out of the bloodstream.36 We also observed that those lipids could target other organs, like lung, spleen, and kidney, which offers potentials for other therapeutic applications. In the future, the active lipid and helper formulations may be optimized for specific targeting to these other organs.
As one of the primary drug-accumulating organs, livers have gained much attention from the oligonucleotide therapies. For example, a subset of patients with hypercholesterolemia has been found to have an overexpression of PCSK9 genes in the liver. In response, pharmaceuticals based around antibody- and siRNA-mediated knockdown of PCSK9 expression have been explored. The AMG145 (by Amgen), a humanized monoclonal antibody, specifically binds to the catalytic domain of PCSK9 on the surface of hepatic cells, leading to a decrease of circulating LDL cholesterol levels above 50%. However, anti-PCSK9 antibodies can only extracellularly block the function of PCSK9.37 The ionizable amino lipid, DLin-MC3-DMA, developed by Anylam, was used to deliver siRNA targeting PCSK9 mRNA intracellularly, resulting in gene knockdown with an ED50 of 0.07 mg·kg-1.17,18,32,38, 39, 40, 41, 42 When our bioreducible lipids are employed to achieve ASO-mediated PCSK9 mRNA silencing in mice, the best-performing lipid, 306-O12B-3, generated an ED50 of 0.088 mg·kg-1. Subsequently, optimization of the DOPE ratios in the lipid formulation renders an enhancement of the ED50 of our lipid delivery down to 0.034 mg·kg-1 (Figure 4). This improvement of the ASO delivery efficacy is also validated by the decrease of the PCSK9 protein and serum cholesterol level (Figure 5). Based on the literature, it is believed that moderately increasing the amount of DOPE promotes LNPs transforming to an inverted hexagonal structure, which more readily destabilizes the anionic endosome membranes at the acidic cell environment. This disruption leads to a faster release of the encapsulated ASOs in the cytoplasm.43,44 Our results, although not focused on the mechanisms, appear to confirm this belief, as a moderate increase in DOPE in the formulation improved the delivery efficacy. The increased efficiency allows for a reduction in the dose of LNP needed for the delivery, reducing the undesired toxic accumulation. This was corroborated by the toxicity studies in mice. No clinically significant increase was observed in crucial hepatotoxic or nephrotoxic parameters for the optimized formulation of the 306-O12B-3 lipid.
In summary, we synthesized and examined a library of bioreducible lipids in order to explore the potency and toxicity of these lipid-like materials in ASO delivery. For in vitro studies, we identified three best-performing lipids presenting powerful ASO delivery capability. Simultaneously, these three lipids attained higher efficacy and lower toxicity compared to the widely used transfection agent Lipofectamine 2000 in a dose-dependent manner. When applied on in vivo investigation, lipid 306-O12B-3 outperformed other screened lipids at mRNA silencing in mice after i.v. administration. One of the leading formulations showed no significant increase in toxicology parameters with a dose up to 5 mg·kg-1. The promising capacity and safety indicated that the optimized 306-O12B-3 formulation was a potent lipid material for ASO delivery to the body system and offered another potential for clinical applications.
Materials and Methods
General
All chemicals used for lipid synthesis were purchased from Sigma-Aldrich. The bioreducible lipids were synthesized using approaches as previously reported. All ASOs, including the chemical-modified ones, and DNA fragments were synthesized by Integrated DNA Technologies (IDT). GFP-HEK cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) complemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin-streptomycin (Gibco). The fluorescent intensity for GFP-HEK cells was analyzed by the flow cytometer (BD FACSCalibur; BD Biosciences, CA, USA). Female BALB/c mice (age 6–8 weeks, weight 18–20 g) were ordered from Charles River. The Institutional Animal Care approved the animal protocol used in this study. The Institutional Animal Care and Use Committee (IACUC) of Tufts University (B2018-96) and all in vivo experiments were executed under approved animal-care guidelines.
Preparation and Formulation of the Bioreducible LNPs
For the in vitro study, we directly dissolved the pure lipids in sodium acetate buffer (25 mM, pH 5.2) to obtain a 1-mg/mL solution and directly used them for ASO delivery. The lipid samples produced using this process were referred to as nonformulated LNPs, in which no other helper lipids were added in the formulation. To formulate ASO using nonformulated LNPs for the in vitro study, we mixed the ASO and the lipid sample at a 1/15 (w/w) ratio and incubated them for 15 min at room temperature.
For in vivo study, in addition to the synthetic bioreducible lipid, other helper lipids, including cholesterol, DOPE, and PEGylated lipid, are also included in the LNP formulation. The LNPs produced with colipids were referred to as formulated LNPs. To produce formulated LNPs, the synthesized lipid, cholesterol, DOPE, and DSPE-PEG2000 were first dissolved in ethanol and mixed in a weight ratio of 16/4/1/1 (or 16/4/4/1). 100 μL solution was then added dropwise to 800 μL of sodium acetate buffer (25 mM, pH 5.2), and the resulting solution was mixed using brief vortex. Finally, we removed the ethanol in the formulation by dialysis (molecular weight cutoff [MWCO] 3.5 kDa) for 2 h. We mixed the formulated LNP solution with ASO at a 15/1 (w/w) ratio of bioreducible lipid to ASO and incubated it at room temperature before tail-vein injection in mice.
Characterization of LNPs
Hydrodynamic size and zeta potential for LNPs were recorded on a Zeta-PALS particle size analyzer (Brookhaven Instruments). The ASO loading efficiency was determined by the Quant-iT OliGreen ssDNA Assay Kit (Life Technologies). TEM images were captured by an FEI Technai Spirit Transmission Electron Microscope.
Intracellular Delivery of ASO/LNP Complexes
GFP-HEK cells were seeded in a 48-well plate at a density of 20,000 cells per well for 24 h of incubation time before the experiment. Starting with LNPs to ASO at a weight ratio of 15/1, 16.7 nM ASO was thoroughly mixed with LNPs in Opti-MEM medium (Thermo Fisher Scientific), followed by 15 min of incubation at room temperature. The ASO/LNP complex solutions were then added to the cells, and the cell plate was incubated at 37°C for 16 h before flow cytometry analysis. The weight ratio of ASO to LNPs was adjusted in a gradient according to the different purpose of investigation. For N/P ratio study, the LNP concentrations for each well ranged from 0.2 μg/mL to 2 μg/mL, with a fixed ASO concentration at 0.1 μg/mL. Meanwhile, in ASO dose-dependent experiments, different concentrations of ASO and LNPs were mixed in vials ranging from 0.002 μg/mL to 0.2 μg/mL and 0.033 μg/mL to 3.3 μg/mL (all concentrations referred to final ASO and LNP concentrations to cells). The LNPs and ASO were mixed at an N/P ratio of 15/1. The GFP silencing efficiency was calculated by dividing the mean fluorescence intensity of ASO-treated cells by that of untreated controls. The functional GFP-ASO sequence (#5 ASO) was as follows: 5′-TTGCCGGTGGTGCAGATAAA-3′. The scrambled GFP-ASO sequence was as follows: 5′-GGAGTACACTATATCGGTGG-3′.
Cytotoxicity Assay of ASO/LNP Complexes
The cytotoxicity of LNPs against GFP-HEK cells was examined by the MTT assay. GFP-HEK cells were seeded in a 96-well plate at the density of 5,000 cells per well for 24 h before the experiment. Cells were treated with ASO/LNP complexes in a fixed weight ratio of 15/1 containing a raised ASO concentration ranging from 0.002 μg/mL to 0.2 μg/mL. After 16 h of incubation at 37°C, the cell plate was treated with 5 mg/mL MTT solution (in PBS) for each well, and the cells were incubated for another 4 h at 37°C. The mixed solution in each well was then removed, followed by adding 200 μL dimethyl sulfoxide (DMSO) to dissolve the colored crystal on the bottom of each well. After gently mixing by a gyratory shaker for 5 min, the absorbance for each well was read by a microplate reader (SpectraMax M2; Molecular Devices) at a wavelength of 570 nm.
Biodistribution of ASO-Loaded LNPs in Different Organs
We separately prepared LNPs in the following formulation: lipid/cholesterol/DOPE/DSPE-PEG2000 at the weight ratio 16/4/1/1. After dialysis, the formulated LNPs were mixed with ASOs conjugating with a far-red fluorescent dye (ASO-Alexa 750) at the weight ratio of 15/1. Then the ASO-Alexa 750/LNP complexes were intravenously injected into BALB/c mice (female, 6 weeks of age). After 1 h, mice were anesthetized and perfused with saline containing 10% formalin solution. Afterward, major organs (brain, heart, lung, liver, spleen, and kidney) were collected. The fluorescent signal distribution was visualized using the Spectrum CT Biophotonic Imager (PerkinElmer, Boston, MA, USA).
PCSK9-ASO/LNP Delivery In Vivo
For the ASO dose-dependent study in vivo, fifty-four female BALB/c mice were randomly separated into three groups (n = 18 for each group). Each group received different lipid nanoparticles, 113-O14B-3, 113-O16B-3, and 306-O12B-3, via tail-vein injection. In each group, mice were further randomly divided into six subgroups (n = 3), and each group was given a single-dose injection at 1.5 mg, 1 mg, 0.5 mg, 0.1 mg, and 0.05 mg ASO kg-1, respectively. The control group was injected with saline. After 72 h of postinjection, mice were sacrificed by CO2 inhalation, and livers were collected. Liver tissues (100 mg) were lysed and homogenized in 1 mL TRIzol (Life Technologies) by a bead bug tissue homogenizer (2 min, 3,500 rpm). Total tissue RNA was extracted with TRIzol, according to the manufacturer’s instructions. Reverse transcription was executed by ImProm-II reverse transcriptase (Promega) and 250 ng oligo(dT)12-18 primers (Thermo Fisher Scientific) at 42°C for 60 min with 1 μg total RNA. The resulting complementary DNA (cDNA) was analyzed by quantitative PCR (qPCR) for 40 cycles and 58–60°C annealing temperature on a StepOne Plus thermocycler (Applied Biosystems, Foster City, CA). PCSK9 mRNA levels were normalized to β-actin mRNA and presented relative to saline controls. The murine PCSK9-ASO sequence was as follows: 5′-GGGCTCATAGCACATTATCC-3′. The scrambled PCSK9-ASO sequence was as follows: 5′-ATCCGGTCCTATACACTGAG-3′. The primer sequences were as follows: total PCSK9: forward 5′-TCAGTTCTGCACACCTCCAG-3′, reverse 5′-GGGTAAGGTGCGGTAAGTCC-3′; for murine β-actin: forward 5′-AGTGTGACGTTGACATCCGT-3′, reverse 5′-GCAGCTCAGTAACAGTCCGC-3′.
In the different DOPE fraction investigations, thirty female BALB/c mice were randomly partitioned into two groups (n = 15 for each group). Each group of mice was intravenously injected with different LNP formulations. Each group was further randomly divided into five subgroups (n = 3) and rendered a single-dose injection at 0.5 mg, 0.1 mg, 0.05 mg, or 0.025 mg ASO kg-1, respectively. The control group was injected with saline. The mice were sacrificed after 72 h of postinjection by CO2 inhalation. The liver tissues and blood (about 0.2 mL) were then collected. The PCSK9 mRNA expression level was measured by qPCR as described above. The blood coagulated at room temperature for 30 min and then centrifuged for 10 min at 10,000 rpm to obtain the serum. Serum PCSK9 protein content was examined with Cirulex human PCSK9 ELISA kits, according to the manufacturer’s instructions (MBL, Woburn, MA, USA). Simultaneously, the cholesterol level in blood serum was analyzed by Wako Diagnostics total cholesterol E kits following the manufacturer’s instructions (FUJIFILM, Richmond, CA, USA).
Examination of Hepatotoxicity and Nephrotoxicity
Twelve female BALB/c mice were randomly partitioned into four groups (n = 3). Each group was intravenously injected with ASO/LNP complexes with the optimal formulation (lipid/ cholesterol/ DOPE/DSPE-PEG2000 at a weight ratio of 16/4/4/1) at a single dose of 5 mg, 3 mg, or 1 mg·kg-1, respectively. The control group was injected with saline. Mice behaviors were observed daily, and the spleen size and some hematology parameters were evaluated in the 72 h of postinjection. Mice were sacrificed by CO2 inhalation. The blood samples (about 0.2 mL) were then collected by mandibular vein puncture. To obtain blood serum, the blood was allowed to coagulate at room temperature for 30 min and then centrifuged for 10 min at 10,000 rpm. The hepatotoxicity was elevated by serum biochemical parameters, alanine aminotransferase (ALT) and aspartate aminotransferase (AST). ALT and AST were measured by ALT activity kits and AST activity kits (Sigma-Aldrich) following the manufacturer’s guidelines. Meanwhile, serum biochemical parameters, creatinine (Cr) and blood urea nitrogen (BUN), were allowed to assess the nephrotoxicity, which was determined by Cr activity kits and BUN activity kits (Sigma-Aldrich), according to the manufacturer’s instructions.
Statistical Analysis
All data were expressed as mean ± standard deviation (SD). Statistical analysis was performed using two-way analysis of variance (ANOVA), followed by the Turkey-Kramer multiple comparison test for more than two groups. Student’s t test was used for comparing two groups using Prism (v.8; GraphPad Software, La Jolla, CA, USA). Values of p <0.05 were considered as significant.
Author Contributions
L.Y. designed and conducted the experiments, analyzed data, and wrote the manuscript. F.M. helped conduct the LNP characterization and revised the manuscript. F.L. helped with the i.v. injection and the hepatotoxicity and nephrotoxicity examination. J.C. conducted the TEM imaging. X.Z. offered some suggestions for in vitro delivery. Q.X. designed the entire study, interpreted and analyzed data, as well as critically revised and finalized the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We acknowledge support from NIH (R01 EB027170-01 and 1R21EB024041-01).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.01.018.
Supplemental Information
References
- 1.Raouane M., Desmaële D., Urbinati G., Massaad-Massade L., Couvreur P. Lipid conjugated oligonucleotides: a useful strategy for delivery. Bioconjug. Chem. 2012;23:1091–1104. doi: 10.1021/bc200422w. [DOI] [PubMed] [Google Scholar]
- 2.Crooke S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen X., Corey D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46:1584–1600. doi: 10.1093/nar/gkx1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crooke S.T. Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta. 1999;1489:31–44. doi: 10.1016/s0167-4781(99)00148-7. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura H., Oda Y., Iwai S., Inoue H., Ohtsuka E., Kanaya S., Kimura S., Katsuda C., Katayanagi K., Morikawa K. How does RNase H recognize a DNA.RNA hybrid? Proc. Natl. Acad. Sci. USA. 1991;88:11535–11539. doi: 10.1073/pnas.88.24.11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017;35:230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 7.Lima W.F., Vickers T.A., Nichols J., Li C., Crooke S.T. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS ONE. 2014;9:e101752. doi: 10.1371/journal.pone.0101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Juliano R.L., Ming X., Nakagawa O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 2012;45:1067–1076. doi: 10.1021/ar2002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vickers T.A., Crooke S.T. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015;43:8955–8963. doi: 10.1093/nar/gkv920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mercatante D.R., Kole R. Control of alternative splicing by antisense oligonucleotides as a potential chemotherapy: effects on gene expression. Biochim. Biophys. Acta. 2002;1587:126–132. doi: 10.1016/s0925-4439(02)00075-3. [DOI] [PubMed] [Google Scholar]
- 11.Chery J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016;4:35–50. doi: 10.14304/surya.jpr.v4n7.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein C.A. Eteplirsen Approved for Duchenne Muscular Dystrophy: The FDA Faces a Difficult Choice. Mol. Ther. 2016;24:1884–1885. doi: 10.1038/mt.2016.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.US Food and Drug Administration FDA approves first drug for spinal muscular atrophy. Press Anouncement. 2016 https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy December 23, 2016. [Google Scholar]
- 14.Rinaldi C., Wood M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018;14:9–21. doi: 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- 15.Falzarano M.S., Passarelli C., Ferlini A. Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther. 2014;24:87–100. doi: 10.1089/nat.2013.0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akinc A., Zumbuehl A., Goldberg M., Leshchiner E.S., Busini V., Hossain N., Bacallado S.A., Nguyen D.N., Fuller J., Alvarez R. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 18.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love K.T., Mahon K.P., Levins C.G., Whitehead K.A., Querbes W., Dorkin J.R., Qin J., Cantley W., Qin L.L., Racie T. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 21.Wang M., Glass Z.A., Xu Q. Non-viral delivery of genome-editing nucleases for gene therapy. Gene Ther. 2017;24:144–150. doi: 10.1038/gt.2016.72. [DOI] [PubMed] [Google Scholar]
- 22.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 23.Chang J., Chen X., Glass Z., Gao F., Mao L., Wang M., Xu Q. Integrating Combinatorial Lipid Nanoparticle and Chemically Modified Protein for Intracellular Delivery and Genome Editing. Acc. Chem. Res. 2019;52:665–675. doi: 10.1021/acs.accounts.8b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altınoglu S., Wang M., Xu Q. Combinatorial library strategies for synthesis of cationic lipid-like nanoparticles and their potential medical applications. Nanomedicine (Lond.) 2015;10:643–657. doi: 10.2217/nnm.14.192. [DOI] [PubMed] [Google Scholar]
- 25.Takeda Y.S., Wang M., Deng P., Xu Q. Synthetic bioreducible lipid-based nanoparticles for miRNA delivery to mesenchymal stem cells to induce neuronal differentiation. Bioeng. Transl. Med. 2016;1:160–167. doi: 10.1002/btm2.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M., Alberti K., Varone A., Pouli D., Georgakoudi I., Xu Q. Enhanced intracellular siRNA delivery using bioreducible lipid-like nanoparticles. Adv. Healthc. Mater. 2014;3:1398–1403. doi: 10.1002/adhm.201400039. [DOI] [PubMed] [Google Scholar]
- 27.Sun S., Wang M., Alberti K.A., Choy A., Xu Q. DOPE facilitates quaternized lipidoids (QLDs) for in vitro DNA delivery. Nanomedicine (Lond.) 2013;9:849–854. doi: 10.1016/j.nano.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Wang M., Zuris J.A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. USA. 2016;113:2868–2873. doi: 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirazi R.S., Ewert K.K., Leal C., Majzoub R.N., Bouxsein N.F., Safinya C.R. Synthesis and characterization of degradable multivalent cationic lipids with disulfide-bond spacers for gene delivery. Biochim. Biophys. Acta. 2011;1808:2156–2166. doi: 10.1016/j.bbamem.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang M., Alberti K., Sun S., Arellano C.L., Xu Q. Combinatorially designed lipid-like nanoparticles for intracellular delivery of cytotoxic protein for cancer therapy. Angew. Chem. Int. Ed. Engl. 2014;53:2893–2898. doi: 10.1002/anie.201311245. [DOI] [PubMed] [Google Scholar]
- 31.Schmitz J., Gouni-Berthold I. Anti-PCSK9 Antibodies: A New Era in the Treatment of Dyslipidemia. Curr. Pharm. Des. 2017;23:1484–1494. doi: 10.2174/1381612823666170130155036. [DOI] [PubMed] [Google Scholar]
- 32.Fitzgerald K., Frank-Kamenetsky M., Shulga-Morskaya S., Liebow A., Bettencourt B.R., Sutherland J.E., Hutabarat R.M., Clausen V.A., Karsten V., Cehelsky J. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porensky P.N., Burghes A.H. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum. Gene Ther. 2013;24:489–498. doi: 10.1089/hum.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juliano R., Bauman J., Kang H., Ming X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009;6:686–695. doi: 10.1021/mp900093r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deleavey G.F., Damha M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Shi B., Keough E., Matter A., Leander K., Young S., Carlini E., Sachs A.B., Tao W., Abrams M., Howell B., Sepp-Lorenzino L. Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 2011;59:727–740. doi: 10.1369/0022155411410885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaparro-Riggers J., Liang H., DeVay R.M., Bai L., Sutton J.E., Chen W., Geng T., Lindquist K., Casas M.G., Boustany L.M. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J. Biol. Chem. 2012;287:11090–11097. doi: 10.1074/jbc.M111.319764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basha G., Novobrantseva T.I., Rosin N., Tam Y.Y., Hafez I.M., Wong M.K., Sugo T., Ruda V.M., Qin J., Klebanov B. Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol. Ther. 2011;19:2186–2200. doi: 10.1038/mt.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cullis P.R., Hope M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017;25:1467–1475. doi: 10.1016/j.ymthe.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.FitzGerald K. Alnylam Pharmaceuticals; 2010. PCSK9 RNAi Therapeutics; pp. 1–21.https://www.alnylam.com/web/Files/Presentations/ALNY-PCSK9Conference-March2010.pdf [Google Scholar]
- 42.Maier M.A., Jayaraman M., Matsuda S., Liu J., Barros S., Querbes W., Tam Y.K., Ansell S.M., Kumar V., Qin J. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du Z., Munye M.M., Tagalakis A.D., Manunta M.D., Hart S.L. The role of the helper lipid on the DNA transfection efficiency of lipopolyplex formulations. Sci. Rep. 2014;4:7107. doi: 10.1038/srep07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mochizuki S., Kanegae N., Nishina K., Kamikawa Y., Koiwai K., Masunaga H., Sakurai K. The role of the helper lipid dioleoylphosphatidylethanolamine (DOPE) for DNA transfection cooperating with a cationic lipid bearing ethylenediamine. Biochim. Biophys. Acta. 2013;1828:412–418. doi: 10.1016/j.bbamem.2012.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.