

Abstract
Understanding molecular mechanisms underlying the complexity of allosteric regulation in proteins has attracted considerable attention in drug discovery due to the benefits and versatility of allosteric modulators in providing desirable selectivity against protein targets while minimizing toxicity and other side effects. The proliferation of novel computational approaches for predicting ligand–protein interactions and binding using dynamic and network-centric perspectives has led to new insights into allosteric mechanisms and facilitated computer-based discovery of allosteric drugs. Although no absolute method of experimental and in silico allosteric drug/site discovery exists, current methods are still being improved. As such, the critical analysis and integration of established approaches into robust, reproducible, and customizable computational pipelines with experimental feedback could make allosteric drug discovery more efficient and reliable. In this article, we review computational approaches for allosteric drug discovery and discuss how these tools can be utilized to develop consensus workflows for in silico identification of allosteric sites and modulators with some applications to pathogen resistance and precision medicine. The emerging realization that allosteric modulators can exploit distinct regulatory mechanisms and can provide access to targeted modulation of protein activities could open opportunities for probing biological processes and in silico design of drug combinations with improved therapeutic indices and a broad range of activities.
Keywords: Allostery, allosteric modulators, network analysis, MD-TASK, drug resistance, precision medicine
1. Introduction
Allosteric regulation is often a mechanism of choice for proteins and biomolecular assemblies to operate in complex signalling cascades and to modulate their activity levels, adapting to binding partners in the cellular environment during signal transduction, catalysis, and gene regulation [1,2,3,4,5]. The advances in X-ray crystallography, Nuclear Magnetic Resonance (NMR), and biophysical techniques have enabled numerous detailed investigations of large protein systems and conformational dynamic processes at atomic resolution [6,7,8,9,10,11,12,13,14,15,16,17,18,19]. These developments have facilitated the integration of computational and experimental studies of allosteric regulation, eventually leading to new conceptual outlooks and attempts to develop a unified theory of this allosteric phenomenon. The thermodynamics-based conformational selection model of allosteric regulation has been particularly fruitful in explaining a wide range of experiments by assuming that a statistical ensemble of preexisting conformational states and communication pathways is inherent to any protein system and can be modulated through allosteric ligand perturbations [20,21,22,23,24,25,26]. While great leaps have been made in the field of molecular modelling, NMR spectroscopy, and X-ray crystallography, it should be noted that no single method can provide allostery information for all cases due to the complexity and incomplete understanding of allosteric phenomena.
Understanding molecular mechanisms of allosteric regulation in proteins has attracted considerable attention in both academia and industry owing to the importance of discovering allosteric modulators of therapeutically important targets [27]. These efforts are motivated by fundamental differences in structural and evolutionary diversity between active and allosteric sites even among structurally similar proteins of the same family. While active sites for structurally related proteins and protein families are often highly conserved and present a formidable challenge for design of selective modulators, allosteric binding is typically more dynamic and structurally and evolutionarily diverse, thereby often alleviating conceptual difficulties in the design of target-specific therapies and addressing lingering problems of toxicity and side effects [28]. Another important incentive for the development of allosteric drugs is that, while traditional orthosteric drugs usually inhibit protein activity, allosteric modulators may not only inhibit but also increase protein activity (allosteric activators) [29]. In the last decade, drug discovery has been shifting its focus toward targeting allosteric sites in order to improve compound selectivity [28,29,30,31,32,33]. Allosteric drugs also feature distinct physicochemical properties, adding further freedom for discovery of novel active compounds, and can often be combined with orthosteric drugs into synergistic drug cocktails to modulate and improve enzyme activities, specificity, and pharmacological profiles.
While orthostery-based therapies have enhanced the quality of life for patients, they have brought forth many daunting challenges for which allostery may provide new solutions. Drug discovery against more diverse protein targets can result in less toxic and more specific therapies. The incorporation of dynamic and network analysis tools has proven their effectiveness in drug discovery studies of several target proteins [32,33,34,35] and offer a promising direction for the analysis of large datasets [36]. With the maturation of open-source projects, the availability of cheaper computation, and large datasets, in silico simulations are a very attractive venture for early-stage drug discovery as they offer cost-effective drug development. The integration of such approaches into robust, reproducible, and customizable workflows should make in silico allosteric drug discovery more efficient and reliable. In this review article, we discuss how the integration of state-of-the-art structural, dynamic, and network-based approaches for simulation of ligand–protein binding can provide a comprehensive methodological framework for advancing computer-aided discovery of allosteric sites and allosteric modulators of protein functions and mechanisms.
2. Part I: Overview of Allostery and Allosteric Drugs
2.1. What Is Allostery?
Allostery is generally defined as a reversible functional and conformational modulation at one site resulting from a remote perturbation in a protein [27,37,38]. These remote events can be instantiated from both covalent (residue mutations and chemical reactions) and non-covalent (intermolecular interactions) events and are well-summarized by Nussinov and Tsai [27]. The phenomenon is also extended to include entropic changes, which may prevail even when no conformational change may be apparent [39,40,41]. Allostery-driven conformational changes are not expressed as two discrete conformational transitions but rather exist as an equilibrium comprising a population of various conformations [42]. More generally, allostery is an inherent property of biomacromolecules [43] and its dysfunction is linked to the cause of several diseases [27]. Two examples of inherent allosteric mechanisms include the protein binding by a hormone to drive conformational changes that affect protein–protein interactions in signalling cascades and small molecules regulating catalytic activity by binding at loci far from an active site [44].
2.2. Understanding Allosteric Mechanisms Using Existing Approaches
Allosteric effects are not easily detectable by any single method as they can take many forms, and a diverse pool of conformational samples is often needed to expose these rare events [41,45,46,47]. Current understanding of the details of allosteric mechanisms is still fragmentary [48]. However, the recent experimental breakthroughs in NMR technologies have enabled structural studies of large protein systems and conformational dynamic processes at atomic resolution, providing unique insights into allosteric mechanisms [12,14,15,17,18]. While studies of allosteric regulation often emphasize thermodynamic aspects of the mechanism and incorporate a population-shift conformational selection paradigm, the critical role of conformational dynamics delineated in NMR studies led to the development of the “dynamics-driven” framework of allosteric phenomenon. In dynamics-driven allostery, effector ligands can induce allosteric effects through global redistribution of protein fluctuations and can propagate signals through dynamic modulation of functional motions even in the absence of visible structural changes [5,13,49,50,51,52]. Recent time-resolved infrared spectroscopy experiments have indicated that allosteric transitions occur on multiple timescales. A time-dependent view of allosteric communication revealed that allostery can be manifested by hierarchical propagation of structural and dynamical changes, suggesting a high degree of conformational heterogeneity of the ensemble of communication routes in proteins [53,54]. Relaxation dispersion NMR methods have also enabled the detection of rapid conformational exchanges between ground and excited states occurring on the s–ms timescale that facilitated characterization of hidden excited states that play a significant role in dynamic modulation of protein function and allosteric mechanisms [14,15]. Structural identification and characterization of lowly populated states by high-pressure NMR can allow for detection of reversible transitions under thermodynamic equilibrium conditions that are functionally relevant for allosteric mechanisms [55,56]. Pressure-dependent chemical shifts may also measure redistributions in conformational entropy and specify dynamic allosteric mechanisms, offering an exciting experimental platform for design of allosteric modulators specifically targeting lowly populated functional states [55,56,57].
Allosteric interactions and communications can be conveniently described and characterized by dynamic networks of interactions between components of biological systems. The organization and evolution of dynamic residue interaction networks in proteins allows for formation of ensembles of pathways that transmit signals by propagating conformational fluctuations and functional motions between distant sites. Recent years have witnessed the development of various approaches that investigate NMR chemical-shift perturbations to identify allosteric networks in proteins [58,59,60,61,62,63].
The ensembles of allosteric communications and protein residues involved in signal transmission via population-shift or dynamics-based allostery can be experimentally examined by NMR spectroscopy [64,65,66]. NMR chemical shift responses to bound ligands are commonly employed as diagnostic tools for identifying coupled networks within allosteric proteins that could quantify potential communication pathways [14,50,67].
NMR chemical exchange saturation transfer (CEST) experiments [68,69,70] can identify invisible hidden states and characterize slow-to-intermediate conformational exchanges. NMR chemical-shift covariance (CHESCA) and projection (CHESPA) analyzes can identify residue interaction networks that show correlated changes in chemical shifts due to allosteric perturbations caused by ligand binding or mutations [71,72,73]. NMR chemical-shift perturbations have also been combined with Markov modelling and network analysis to reveal the dynamic flow of communication between allosteric communities in proteins [74].
There is increasing evidence that dynamic allostery may be a common feature in many protein interactions, and as a result, allosteric mechanisms are no longer viewed as being mediated solely through structural transitions that select specific stable conformational states.
Even though NMR technologies opened the doors to us to greatly understand the mechanisms of allostery, over 80% of the structures available from the Protein Data Bank (PDB) comprise macromolecules solved by X-ray crystallography [75]. These are a representation of the most frequent conformations adopted by proteins from their respective pools of conformation ensembles [76]. As allosteric sites are rarely observed in crystallographic structures, a large amount of conformational sampling may be needed to uncover such high-energy states. For instance, cryptic (or hidden) allosteric sites sporadically appear during conformational transitions of a protein in the presence of a bound ligand. They are a recently discovered form of allostery whereby ligands can bind to pockets that are not present in crystal structures [77]. These “hidden” allosteric sites are essentially invisible in crystal structures, apart from some chanceful detection due to the stabilization of the rarer, higher-energy conformation by certain compounds [78]. Fortunately, an abundance of computational tools have been designed over the years to examine allosteric mechanisms in protein systems [36]. In silico conformation sampling techniques allow us to unveil such rarely observed protein conformations [78] by simulating protein motion, thus facilitating the discovery of potentially druggable sites [79]. Exhaustive sampling is impossible, and highly precise quantum mechanics (QM) simulations are limited to small systems or portions of larger systems. Coarse-grained molecular dynamics (MD), markov state models (MSMs), and elastic network models (ENM) offer speed improvements by reducing complexity of all-atom protein representation while maintaining topological and network integrity of protein systems that drive allosteric mechanisms and global regulatory functions. The explored approaches have included normal mode analysis (NMA) [80,81,82,83], MD [84], and machine learning [85]. Brown and coworkers successfully coupled full-atom MD simulations to dynamic residue network (DRN) analysis to study allosteric effects in disease-causing variants [86]. These computational approaches will be further discussed in the second part of this review article.
2.3. Understanding the Allosteric Effects of Disease and Drug-Resistant/Sensitive Mutations—Precision Medicine
Human diseases and traits have been associated with single nucleotide polymorphisms (SNPs) through the use of genome-wide association studies (GWAS) [87,88]. There is even considerable genetic variation between just two random individuals, where 10,000 non-synonymous single nucleotide polymorphisms (nsSNPs) were found to exist between their exomes [89]. These mutations have the potential to alter distal functional sites of enzymes by means of allosteric signalling—where the dynamics of the entire structure is transformed as a result of the mutation [90,91,92]. Fortunately, progress in allosteric research has made it possible to determine allosteric molecular mechanisms in a myriad of allosteric systems in much detail [93]. For example, network analysis is usually performed to determine the pathways that connect the mutation and the active site and aids in the study of allosteric communication [86,94,95]. A recent publication studied six validated non-synonymous single nucleotide variations (nsSNVs) to identify underlying mechanisms responsible for CA-II deficiencies resulting in the phenotype of osteopetrosis with renal tubular acidosis and cerebral calcification [96]. In this study, Sanyanga et al. combined MD and DRN [97] analysis and showed that nsSNVs have indirect/allosteric effects, providing greater insights into SNV mechanism of action. Hence, the study proposed taking steps towards the treatment of CA-II deficiencies. Further, the authors highlighted the importance of studying missense mutation effects in proteins with combined approaches and, hence, in a broader sense, precision medicine-related research. This computational approach is detailed in SubSection 3.3.
Precision medicine combines pharmacology and genomics to exploit genetic variation in the human population in order to deliver innocuous but powerful drugs to certain groups of individuals. This is based on the idea that an individual’s genetic makeup determines their reaction to a drug, good or bad [88,98]. For instance, mutations can be linked to altered drug sensitivities in patients [99,100,101]. This opens the door to personalized medicine, where knowledge of drug-resistant and drug-sensitive SNPs can assist in the development of effective biomarkers [99] and allow treatments to be tailored to individual patients [102,103]. Furthermore, understanding structural changes caused by nsSNPs would enable the design of novel drugs to target these mutations and, thus, is key in advancing precision/personalized medicine [104,105]. One example can be given from stroke and stroke-related medications. Clopidogrel and Warfarin, antiplatelet/anticoagulant drugs, are widely used for primary and secondary prevention of stroke. It has been shown that genetic polymorphisms resulting in reduced function of cytochrome P450 2C19 (CYP2C19) were associated with increased cardiovascular risk and mortality in coronary artery disease patients on clopidogrel treatment. A study of clopidogrel response in the Amish population indicated that the CYP2C19*2 variant accounts for 12% of the variation in platelet aggregation after clopidogrel treatment for 7 days [106]. A recent review identified that, among patients with ischemic stroke and being treated with clopidogrel, carriers of CYP2C19 loss-of-function alleles (*2, *3, and *8) have increased risk of recurrent stroke and composite vascular events compared to noncarriers [107]. Another example can be taken from efavirenz, an antiretroviral medicine, metabolized by cytochrome P450 2B6 (CYP2B6), UDP-glucuronosyltransferase 2B7 (UGT2B7), and CYP2A6 [108]. In this study, the authors investigated SNP associations with plasma drug levels in Zimbabwean HIV-positive patients and observed elevated plasma levels of efavirenz, which may lead to toxicity in patients due to CYP2B6*6 and CYP2B6*18 mutations. In both cases, the molecular mechanism is not known. The integrated protocol suggested in Part II would be a good starting point to elucidate the mechanism of the variations in drug sensitivity and to understand the allosteric effects of the mutations.
Mutations have also been associated with drug resistance in numerous pathogenic diseases such as influenza [109], tuberculosis [110], malaria [111], and HIV/AIDS [112]. A detailed understanding of pathogenicity as well as drug-resistance mechanism(s) would be essential for designing novel therapies. Sequencing the genome of pathogens can identify the drug-resistant and drug-sensitive mutations and leads to a deeper understanding of the cognate molecular mechanisms. One example can be given from a recent article by Sheik Amamuddy and colleagues [113]. The authors looked at eight Food and Drug Administration (FDA)-approved drugs and the mutations of HIV protease due to drug usage. As shown in Figure 1, the investigated mutations were interspersed within the protein (observed from collections of variants) and were not limited to the orthosteric site. While not explicitly designed to extract out the effects of specific allosteric mutations in the study, the method was able to expose underlying conserved signals buried within noisy dynamic data generated from the combined effect of orthosteric and allosteric mutations. The signals comprised a conserved and practically symmetrical lateral expansion coupled to an inward contraction captured from an ensemble of simulations. This method can easily be adapted to investigate allosteric events by designing an experiment-comprising allosteric loci, which in the case of HIV protease, for instance, could be accessory DRMs (resistance mutations not in contact with the antiretroviral drug) distal to the active site. Their approach combining MD, network analysis, and statistical calculations indicated that, regardless of the drug used, these mutations induce a common allosteric behavior in the protein in the presence of the drug, probably to dislodge the latter from the binding site. Hence, understanding the allosteric behavior of the drug targets due to mutations and identifying allosteric sites as alternative drug targeting sites would help in the design of alternative modulatory molecules. Allosteric modulators, as explained in the following section, would have many benefits over orthosteric drugs.
Figure 1.

Three-dimensional mapping of variation positions for the 8 FDA-approved HIV protease inhibitors (atazanavir (ATV), darunavir (DRV), fosamprenavir (FPV), indinavir (IDV), lopinavir (LPV), nelfinavir (NFV), saquinavir (SQV), and tipranavir (TPV)) used to investigate the effects of drug resistance: Coloured cartoon representations depict the fulcrum, elbow, flap, cantilever, and interface, while the variation loci are shown as red spheres. Even though single positions are shown, some positions comprise multiple residue variations, some of which are validated drug resistance mutations (DRMs) (as per the 2017 update [114]). Figure obtained from Reference [115].
2.4. Orthosteric versus Allosteric Drugs
Orthosteric and allosteric drugs are distinguished from each other by their molecular mechanism of action. The former generally binds to the active site and is in competition with biological cofactors or substrates. Alternatively, allosteric drugs and modulators can modify or obstruct the active site when bound to an allosteric site. As allosteric binding sites are significantly less conserved compared to orthosteric sites, the principal advantage of allosteric drugs lie in their higher specificities and thus lower risks of toxic side effects [116,117,118,119]. In contrast, orthosteric site conservation allows an orthosteric drug to bind not only to the intended protein but also to unintended homologous protein family members, contributing to adverse side effects. Off-target toxicities are widespread amongst anticancer drugs in clinical trials [120]. Allosteric sites are also highly enriched in hydrophobic residues, whereas orthosteric sites are enriched with polar residues [43]. A study by Smith and colleagues hinted at the higher rigidity and aromaticity of allosteric modulators [121], thus providing an additional criterion for selecting modulators that are more likely to bind to an allosteric site. Allosteric modulators induce conformational changes and distal effects on the orthosteric site residues [122]. Understanding the structural and molecular mechanism of the induced effects is important for the rational design of allosteric modulators and site identification.
Orthosteric drugs have been successfully used in therapy, for instance, to reduce viral loads in HIV patients to undetectable levels [123]. Unfortunately, the selective pressures of drug treatment have brought forth associated transmissible drug resistance mutations (DRMs), which render these same drugs less effective over time, thus constraining future treatment strategies [124]. Some critically important examples of drug resistance concerns include the emergence of extensively drug resistant tuberculosis [125], where typical treatment options cease to work, and the recent surge of multidrug drug-resistant HIV across Africa, America, and Asia [126]. The last report issued by the Centers for Disease Control and Prevention listed 18 antibiotic resistant pathogens, including those resistant to many commonly prescribed antibiotics [127]. The discovery and use of novel allosteric drugs may therefore open the door for a wider array of future therapies as their more diverse loci could alleviate selection pressures from the currently low-performing orthosteric drugs.
Targeting allosteric sites of functional proteins in Mycobacterium tuberculosis (Mtb) has played a key role in research towards alleviation of TB. The protein pyruvate kinase was observed to exhibit some allosteric mechanism and deemed an attractive target of Mtb therapy [128]. Similarly, an allosteric inhibitor was identified for the Mtb enzyme ornithine acetyltransferase [129] and yet another for the enzyme tryptophan synthase [130]. Bacterial and viral infection agents can be targeted by looking at proteins common to them. Rab II is a protein from the Rab family of GTPases and has been identified as important in a number of disease-causing agents, including influenza A virus; pneumonia, caused by Chlamydia pneumoniae; and Chlamydia trachomatis, which causes a number of sexually transmitted infections by associating with other proteins to facilitate transport processes in the cell [131,132,133]. This protein has been shown by Kumar et al. to possess allosteric-binding sites, and its allosteric inhibition would be an effective remedy option towards eradication [134]. Another good example showcasing the relevance of allosteric research in disease alleviation is the case of the American trypanosomiasis (Chagas disease), where pockets with allosteric potential were identified and characterized in a cysteine protease (cruzain) in efforts to inhibit the causative agent Trypanosoma cruzi [135].
2.5. FDA-Approved Allosteric Drugs
Protein allostery research has been ongoing for years, and our understanding of protein allosteric modulation is strengthening [43]. Publicly available data concerning protein allosteric sites and their modulators is steadily increasing—the AlloSteric Database (ASD) now holds over 1900 protein targets and more than 82,000 allosteric modulators [136]. Despite continually growing investment in allosteric research, only 19 of these modulators are approved drugs compared to the current total of more than 3700 approved drugs [136,137], almost all of which bind to an orthosteric site [138]. This comparison exposes the difficulty of allosteric drug discovery. The paucity of allosteric modulators most likely results from a mixture of their reduced binding affinity, their relatively higher hydrophobicity compared to orthosteric ligands [139], and their frequently adverse structure–activity relationships [30]. Furthermore, the discovery of initial hit compounds is impeded by the difficulty in detecting allosteric binding sites as well as the shortage of knowledge of allosteric interactions and their consequences for protein modulation [43]. To date, only one approved allosteric drug has been discovered solely using in silico methods, namely enasidenib (Table 1). Overall, there is a substantial need for the elucidation of protein–modulator interactions in allosteric binding.
Table 1.
List of currently approved allosteric drugs [136] in alphabetical order.
| Drug/Code Name | Medical Condition | Mechanism | Enzyme Target | Discovery Method | 2D Structure |
|---|---|---|---|---|---|
| Carglumic Acid | Acute hyper- ammonaemia | Activator | Carbamoyl phosphate synthetase 1 | Experiments in rats, both in vivo and in vitro [140] |
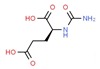
|
| Cinacalcet | Hyper- parathyroidism | Activator | G protein- coupled receptor | Functional responses of cells regulated by calcium receptor activity: PTH secretion by parathyroid cells, calcitonin secretion by C-cells, and bone resorption by osteoclasts. [141] |
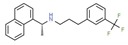
|
| Clonazepam | Epilepsy | Activator | -amino- butyric acid (GABA) | Perifused frog neuro- intermediate lobes [142] |
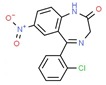
|
| Cobimetinib | Melanoma | Inhibitor | MAPK1, MEK1 & MEK2 | Structural insight—manipulation of previously known MEK inhibitors’ structure. Ligand- binding affinity assays [143] |
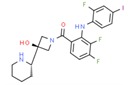
|
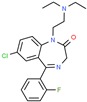
| Cyclothiazide | Hypertension | Activator | AMPA Receptor | AMPA- and KA-induced [3H]NE release from slices of rat hippocampus [144] |
|
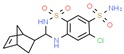
| Drotaverine | Irritable bowel syndrome | Inhibitor | L-type Ca2+ channel | Saturation studies. Dissociation kinetics [145] |
|
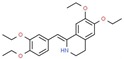
| Enasidenib | Acute myeloid leukemia | Inhibitor | IDH2 | In silico: Binding free energy, conformational change [146] |
|
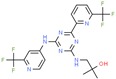
| Flurazepam | Insomnia | Activator | GABA-A receptor | Site-directed mutagenesis. Concentration-response analysis [147] |
|
| Ivermectin | Parasite infestations | Activator | Alpha7 neuronal nicotinic acetylcholine receptor | Mutagenesis. Cell line, culture, and recordings [148] |
|
| Ketazolam | Anxiety disorder | Activator | GABA-A receptor | Increase of GABA level in cat spinal cord and in the total brain of mice and rats [149] |
|
| Lorazepam | Anxiety disorder | Activator | -adrenergic receptor | Transfection. Ligand-binding affinity assays [150] |
|
| Maraviroc | HIV | Inhibitor | C-C chemokine receptor type 5 | Displacement binding assays. Dissociation kinetics [151] |
|
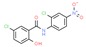
| Niclosamide | Neuropathic pain | Inhibitor | Group 1 metabotropic glutamate receptor | Calcium mobilization assays. Cross-receptor selectivity experiments. Computati- onal molecular modeling analysis. NP-evoked mechanical hyperalgesia model in rats [152] |
|
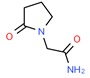
| Piracetam | Dementia, vertigo, cortical myoclonus, dyslexia, and sickle cell anemia | Activator | AMPA Receptor | Enzyme crystallization. Crystal structure determination. Structure analysis [153] |
|
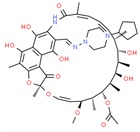
| Rifapentine | Tuberculosis | Inhibitor | DNA- dependent RNA polymerase | Site-directed mutagenesis. In vitro transcription. RFP binding assays [154] |
|
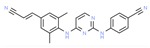
| Rilpivirine | HIV | Inhibitor | HIV-1 reverse transcriptase | X-ray crystallo- graphy. Molecular modeling. Optimizing lead compounds [155] |
|
| Sirolimus | Immuno- suppressive | Inhibitor | FK Binding Protein-12 | Site-directed mutagenesis. FKBP12- Rapamycin (Sirolimus) binding assays [156] |
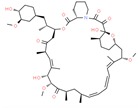
|
| Ticagrelor | Stroke; Acute coronary syndrome undergoing percutaneous coronary intervention | Inhibitor | G protein- coupled receptor | ATP analogue production. Platelet inhibition and patient outcome (PLATO) trial [157] |
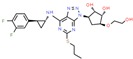
|
| Trametinib | Melanoma | Inhibitor | MEK1 & MEK2 | Enzymatic and cellular studies. Pharmacokinetic analysis [158] |
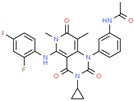
|
3. Part II: An Integrated In Silico Approach for Allosteric Drug Discovery
3.1. The Main Workflow
Central to this work, we propose an integration of in silico approaches that can be used for allosteric drug discovery (Figure 2 and Figure 3). The proposed workflow has been developed as a result of our research over the last few years, as indicated below and in the sections that follow. The workflow diagram gives a complete picture of all techniques, but in a particular case, only some of the approaches would actually be used. Our holistic approach starts with the acquisition of a high-quality drug target and goes through a series of approaches for allosteric site prediction before finally proceeding to the identification of allosteric modulators. We emphasize that the structural reliability of the chosen target is of utmost importance for the success of the subsequent steps. As done for most in silico allostery detection approaches, we start by finding putative allosteric binding sites before zooming onto them to determine and characterize possible binders by analyzing stabilities from conformational sampling processes. As many of the approaches and methods are common between allosteric site and allosteric modulator identification, we will follow the order given in Figure 2A while introducing them. Some of the approaches were shown to be in agreement with each other (Figure 2B). These will be indicated where necessary.
Figure 2.

(A) Our proposed integrated workflow for allosteric site identification, which starts with the acquisition of a drug target and (B) different concepts and techniques from molecular simulation that can provide correlating information to discover and characterize allosteric events in proteins.
Figure 3.

Our proposed integrated workflow for identifying allosteric modulators.
Some examples of the use of the proposed workflow to investigate allosteric phenomena in proteins of medical significance are as follows: (1) Perturbation response scanning (PRS) was combined with all-atom MD and DRN to investigate the allosteric potential of remote residues to effect conformational changes [35] and has been supplemented with docking to investigate the distal effects of ligand binding [29] in human Hsp protein; (2) homology modelling, docking, MD, essential dynamics, and DRN were jointly used to identify allosteric modulators of malarial Hsp, after which coarse-grained conformational sampling simulations coupled to residue network analysis showed corroborating results, obtained from a fraction of the computation time required compared to the all-atom simulations [33]; and (3) allosteric effects associated to alpha carbonic anhydrase SNVs were discovered by coupling all-atom MD to DRN—further characterization was achieved using essential dynamics and binding free energy landscape analysis [96]. As can be observed from these examples, one has a better chance of discovering and characterizing allosteric events by combining multiple techniques, among which simulating protein motion and coupling to DRN appear to be very effective.
3.2. Allosteric Site and Modulator Prediction
3.3. Drug Target Acquisition
As a first step for in silico allosteric modulator identification, one requires a target protein. Three-dimensional structures for the investigated target may already be available fully or in part from the PDB [159]. In the absence of an experimental structure, a computational modelling approach, such as homology modelling, can be used to model an entire protein. Further homology modelling may be useful to insert non-synonymous residue variations using experimental structures as templates, but it can also be used to impute missing ones from incomplete experimentally determined structures [86]. Over the years, the Research Unit in Bioinformatics group developed detailed homology modelling approaches, and the methodology is given in a number of group articles [32,33,35,160,161,162].
There is a long list of user-friendly web servers available for modelling protein 3D structures, some examples of which include I-TASSER [163], ModWeb [164], Phyre2 [165], PRIMO [166], RaptorX [167], Robetta [168], HHPred [169], and SWISS-MODEL [170]. MODELLER [171] can be used as a stand-alone tool by the more technically inclined users or when more customised solutions are needed, such as loop refinement or when large volumes of modelling jobs are to be calculated. As a last resort for target domains with no detectable homologs, ab initio modelling may be an option [172,173]. However, compared to the aforementioned target acquisition approaches (threading and homology modelling), the latter is accurate only for small proteins [174,175]. More recently, a command-line protein structure prediction tool based on a recurrent neural network has been developed, showing promising accuracies and producing models in a fraction of the time using only a position-specific scoring matrix and the target sequence as inputs [176]. Finally, before the structure can be used in the subsequent steps, one should validate the quality of the models via local and global metrics. ANOLEA [177], QMEAN [178], ProSA [179], Verify3D [180], and ModFOLD [181] are a few examples used to determine local quality scores, while z-DOPE [182], resolution, and QMEAN are a few examples of global metrics. Using the MODELLER tool, a per-residue profile of the z-DOPE can also be obtained for pinpointing poorly modelled regions [183]. As no single evaluation criterion is perfect, one should investigate multiple metrics to ascertain the validity of the model. As a general rule in homology modelling, (1) the template resolution should be as high as possible, while the R-free and R-value should be minimal, as they are the residuals between experimental diffraction data and the predicted crystallographic model, and (2) the environmental conditions should closely match those that are to be experimented on [184], such as the presence of a co-crystallized ligand and the receptor protonation states. Unfortunately the presence of certain molecular entities may require some prior planning due to the incomplete transferability across certain force fields [185] for use in downstream approaches (such as MD). Finally, given that differences in receptor conformation specify different topologies and alter the ligand-accessible surfaces, it is clear that this plays a major role in experimental designs aimed at discovering allosteric sites.
3.3.1. Mining Literature and Databases for Allostery Information
Before attempting to identify an allosteric site of a protein, it is good practice to determine whether the literature already describes it from previous work. The AlloSteric Database (ASD), which hosts a collection of known allosteric targets and their modulators, is a major source of relevant literature and annotations pertaining to the concept of allostery [136]. The database has grown to become a central resource for the storage, retrieval, and analysis of allosteric protein datasets [136,186,187]. It organizes information about allosteric regulation by receptor target and modulator (activator, inhibitor, or regulator), and entries incorporate additional annotations such as the associated interactions, sites, pathways, functions, and any linked disorders [136]. A good example of the use of the ASD in allostery research can be drawn from work done by Astl and Verkhivker [188]. Using information retrieved from the database, they combined coarse-grained MD and residue interaction network to demonstrate the differential effects of allosteric inhibitors and activators on the global dynamics and network organizations within protein systems, comprising 300 diverse proteins and complexes. In the case of insufficient information characterizing allostery within the chosen target protein, one can proceed to cavity-finding approaches for the discovery and characterization of de novo allosteric sites.
3.3.2. Cavity-Finding Approaches
Pockets/cavities present in proteins might be functionally important and have been recognized as conventional sites for ligand binding [189]. For those targets that are not well characterized for such pockets, the identification of these sites is thus a starting point for potential allosteric-site prediction and structure-based drug design, with the effects of ligand binding confirming allostery [190]. There is a wide array of freely available cavity-finding tools, mostly in the form of web servers which use a variety of approaches to provide potential starting points for the discovery allosteric sites. The methods employed include, amongst others, the use of NMA, energy evaluations, machine learning, and MD simulations. As features characteristic to allostery are not completely understood, no single predictive method can be exhaustive and it is best to utilize a combination of approaches to increase the level of support for the potential sites. Some of the most recent cavity-finding approaches are summarized in Table 2.
Table 2.
Web servers for the prediction of allosteric sites.
| Web Server and URL | Functionality | Input | Output |
|---|---|---|---|
| AlloDriver [220] | Identifies potential driver mutations implicated in cancer and maps them to binding sites. | A list of annotated cancer-related mutations. | Returns a list of ranked driver mutations annotated by residue loci, scores and binding site (allosteric and orthosteric), amongst many other features. |
| AlloFinder [221] | AlloFinder identifies possible allosteric sites via dynamic perturbations and algorithms present in Allosite. It also screens for possible binders against the identified sites. Protein-ligand complexes are then scored using Alloscore algorithms. | The receptor PDB file and a ligand library. | Displays protein-ligand complex for docked ligands within the putative allosteric site. Further, a table reports the volume of the predicted allosteric site, the perturbation score, the drug-like score, the allosteric site score and the AlloScore score. Additionally, the top 100 potential allosteric ligands are ranked according to their Alloscore. Finally, the predicted site and the predicted ligands are mapped using allosterome data. |
| AlloPred [82] | Uses NMA to identify potential allosteric pockets. | The receptor PDB file and active site residues. | Displays protein structure and a list of pockets with Allopred and Fpocket rankings as well as NMA effect per residue. |
| Alloscore [222] | Uses a linear combination of non-bonded interaction terms, a deformation term and geometric features to predict the binding affinities of protein-ligand interactions. | The receptor PDB file and a pre-docked ligand MOL2 file. | File with potential ligands and their allosteric interactions (hydrogen bonds, van der Waals, hydrophobic interactions and Alloscore values). |
| AlloSigMA [223] | Calculates energetics of allosteric signalling resulting from ligand binding, mutations or a combination of the two. | The receptor PDB file. | The allosteric free energy profile, colouring residues according to difference in free energy between the ligand bound and the apo-protein. |
| Allosite 2.0 [85] | Predicts allosteric sites by means of pocket-based analysis and support vector machine (SVM) classifier algorithms. | The receptor PDB file. | Window showing the structure and identified potential allosteric sites. Pockets can be viewed on the displayed protein structure. Properties of the pocket include: (i) Its volume, (ii) Total solvent-accessible surface area (SASA), (iii) Polar SASA and (iv) Druggability score |
| AllosMod [84] | Makes use of MD simulations and energy landscapes to identify allosteric conformational changes. | The receptor PDB file and its sequence. | Returns a zipped file of further input files to be MD-run by the user via MODELLER and analysed using a provided Python script. |
| Cavity (Submodule of CavityPlus) [190] | Identifies cavities and provides their respective drug scores. | The receptor PDB file. | Displays the structure, potential cavities and constituting residues with their respective drug scores, which determine cavity druggability. |
| CorrSite (Submodule of CavityPlus) [190] | Identifies possible allosteric sites from those picked up by CavityPlus on the basis of correlated motion between allosteric and orthosteric cavities. | PDB file of a proposed orthosteric site or predetermined cavities obtained from the Cavity tool. | Displays the structure with mapped orthosteric and allosteric sites. Cavities are labelled with their corresponding correlation scores to the orthosteric site. |
| CovCys (Submodule of CavityPlus) [190] | Identifies druggable cysteine residues for covalent allosteric ligand design. | Cavities identified by the Cavity web server. | Maps any of the selected sites onto the protein structure and displays a table of Cys residues labelled by cavity ID, targetability, pKa value, exposure and their pocket binding affinity. |
| DynOmics ENM [83] | Predicts allosteric communication using ENM. | The receptor PDB file. | (i) JSmol window showing structure color-coded by the size of motions driven by the slowest two modes, lowest mobility (blue) to highest mobility (red) regions, (ii) Molecular motions animation, (iii) Mapped RMSF, (iv) 3D and 2D display of selected modes, (v) Cross correlations between residue fluctuations, and (vi) Inter-residue contact maps |
| MCPath [224] | Identifies regions in a protein structure which may function in allosteric communication using a Monte Carlo-based approach. | The receptor PDB file and pathway data (initial residue index, length and number of paths). | List of all pathways ranked according to their probabilities and populated pathways. 3D structure onto which the top three populated pathways and their residues are mapped. |
| PARS [81] | Uses NMA to identify possible allosteric pockets which, upon binding of a ligand, cause a regulatory effect in the protein. | The receptor PDB file and its sequence. | Table with identified pockets ranked according to their potential as allosteric sites. |
| SPACER [80] | Combines ENM and docking to predict allosteric communication. | The receptor PDB file. | List of ligand binding sites, for which the following can be explored: (i) Local closeness - the output structure is colored according to surface local closeness values, (ii) Binding leverage - quantifies the cost of the binding site deformation in the presence of a ligand, and (iii) Characteristics of the communication strength between a putative allosteric site and another binding site. |
| STRESS [225] | Identifies allosteric hotspot residues which result in large protein conformational changes when bound by a small ligand. | The receptor PDB file. | Ranked list of predicted sites each with an index of the binding site obtained from Monte Carlo simulations, a binding leverage score and their respective residues. |
3.3.3. Blind Docking
Small-molecule docking to a target protein can give us an indication as to whether and how strongly a ligand might bind to a certain surface, but limiting the search space to a primary binding site (i.e., targeted docking) can be oblivious to allosteric pockets. In blind docking (BD), the whole protein surface is scanned for putative binding sites and is especially useful when one has not been determined a priori [191,192,193]. Several studies have used BD for allosteric site identification, for which we give some examples in this section. Iorga and colleagues have used BD of three known allosteric modulators to reveal putative binding sites from the acetylcholine-binding protein and in homology-modelled human nicotinic receptors [194]. Grant and coworkers identified potential allosteric pockets on the Ras protein catalytic domain via BD of 267 putative ligands from PubChem BioAssay database [195]. Pavlovicz and colleagues used BD to identify negative allosteric sites on the numan and neuronal nicotinic acetylcholine receptors [196]. Jin and colleagues used BD with tyrosine phosphatase 1B to reveal a hydrophobic and less conserved allosteric site in contrast to its negatively charged but highly conserved active site [197]. Based on this study, an alternative drug-design strategy was proposed. Lastly, a study by Chen and colleagues employed BD to explore a specific allosteric site for non-peptidic inhibitors located behind the catalytic triad of the dengue virus-2 NS2B-NS3 protease [198].
Due to the increased search space, stochastic strategies are typically used to make the exploration of the larger target surface computationally tractable [199]. One way to overcome this limitation is to divide the search surface [200] and/or to increase the search exhaustiveness.
In their pioneering paper introducing BD using AutoDock, Hetényi and van der Spoel recommended over 100 independent docking runs with flexible ligands [192], which would be analogous to the exhaustiveness parameter in AutoDock Vina, for example. Further, in a comprehensive study evaluating ten common docking programs [201,202,203,204,205,206,207,208,209,210], Wang and colleagues showed that these tools correctly predicted the ligand poses even though the binding energies could not be estimated accurately [211]. As BD involves a larger volume to scan, the docking process can be limited by efficiencies of the sampling algorithms and complexity of the scoring functions [193]. A more accurate estimation of the ligand-binding energy was achieved via quantum mechanical (QM) and semi-empirical QM-based rescoring schemes [212,213]. This rescoring scheme was successfully applied to HIV-1 protease (with 22 ligands) [214], cyclin-dependent kinase 2 (with 31 ligands) [215], and casein kinase 2 (with 23 halogenated ligands) [216].
Key regions important for the stability of subtilisin (an industrially important serine protease) were determined using PRS, in which the covariance matrix was obtained from equilibrated portions of MD trajectories [217]. PRS was, for the first time, applied to large, flexible proteins by Penkler et al. (2017), determining key residues involved in allosteric control in the 70 kDa heat shock protein (Hsp70) [218] using an implementation of the PRS algorithm available from the MD-TASK package [97]. More recently, the same approach was coupled with DRN to determine potential effector residue loci promoting allosterically driven conformation interconversions in human Hsp90 [35] before targeting the same sites (at the C-terminus) by high-throughput virtual screening for druggable pockets [29]. After Penkler’s work showcased the application of PRS to highly dynamic proteins, Amusengeri and Tastan Bishop employed a similar workflow in their work in Hsp72 and Hsc70 to search for allosteric modulators [32] from the South African natural compounds database (SANCDB) [219]. While linearly impacting a protein allows us to promptly assess the likelihood of transitioning from a static starting state to a target state, measuring and summarising topological changes from conformational sampling processes opens up a new avenue for the detection and characterization of allosteric effects, which we describe in the next section focussed on the application network analysis in assessing protein dynamics.
3.3.4. Perturbation Response Scanning
By factoring out the bound small molecule, to instead perturb single residues from a receptor and to record the linear response, is a rapid way of approximating biological perturbations in proteins. These perturbation events can be instantiated by various intrinsic and extrinsic factors, which may in turn have far-reaching effects [27,36]. In perturbation response scanning (PRS), a series of uniformly distributed forces are sequentially applied to each residue of an equilibrated protein conformation to estimate the agreement with a desired target state [226]. The perturbation is obtained from the dot product between a covariance matrix and a force vector. The covariance matrix can be obtained from various models of interatomic potential such as those used in MD [29] or by inverting a Hessian matrix [227] as typically done in ENM [226]. The uniform force vector impacts a given residue multiple times, and a correlation consensus is obtained for that residue’s overall effect against a targeted conformation. The algorithm is sequentially applied to each residue of the protein to yield a correlation value in each case. The PRS algorithm has been successfully used to uncover mechanisms of allostery in various experiments. For instance, Gerek and Ozkan used PRS to compare allosteric transitions in two PDZ domain proteins (proteins linked to cancer and to Alzheimer’s and Parkinson’s diseases) by perturbing inverted Hessian matrices obtained from ENM models [228].
3.3.5. Interaction Networks in Proteins Dynamics
The Usefulness of Network Theory in Investigating Protein Dynamics and Allostery
As reviewed recently by Liang and coworkers, the last decade has produced an array of various tools that bridge multiple interdisciplinary concepts for the study of protein dynamics and allostery regulatory mechanisms [36]. More specifically, in allosteric signalling, the perception and long-distance relay of the trigger signal is associated with the rewiring of an intricately connected network of non-covalently interacting protein residues [36]. This ultimately leads to directly observable conformational changes and/or to entropic changes, where no conformational change is seen. Simplifying the protein topology as a network of nodes connected by edges to represent the residues and their interaction (or strength of interaction), respectively [23,229,230], allows for the investigation at various levels of the allosteric effects, which are otherwise convoluted with protein entropy. The increasingly popular integration of network theory for protein dynamic analysis thus plays a major role in robustly deconvoluting complex relational behaviours into a more interpretable and quantifiable form [230,231,232,233,234,235,236,237,238,239,240]. Several studies have pointed out the tremendous usefulness of modelling and analyzing proteins as dynamic entities oscillating between various allosteric states in order to extract meaning from the complexity of allosteric regulation [29,80,81,94,224,241,242,243,244,245].
Dynamic Residue Network Analysis
Networks are typically represented by either an adjacency matrix or an edge list [36]. In static networks, a binary contact is inferred between each protein residue pair (defined by C-alpha or C-beta atoms) using a defined cut-off distance (i.e., 6.7Å). The DRN approach extends this idea by computing the time-averaged version of static contact networks using MD simulation data [97]. Different variations of time-averaged networks have been used to investigate allosteric effects in various proteins, for instance, in characterizing (1) the dynamics of catalysis in Cyclophilin A variants [95], (2) in characterizing the effects of damaging non-synonymous SNVs of the renin–angiotensinogen complex [86] and carbonic anhydrase 2 [96], and (3) in investigating cross-domain allostery in human heat shock proteins [29,32]. The MD-TASK package was the first downloadable tool packaging a set of nonconventional methods meant to analyze MD simulations. These comprise a set of freely available Python scripts aimed at computing DRN metrics in addition to other scripts used for carrying out MD analysis [97]. More specifically, these network metrics comprise the time-averaged versions of both betweenness centrality (BC) and averaged shortest path (L), calculated for each protein residue (node) using MD trajectory data. While BC measures the number of geodesics (shortest paths between two nodes) going through an intervening node [246], L averages the number of geodesics inbound to a node. Both metrics gain robustness in predicting medium-to-long-distance relational information from averaged information across conformational ensembles. By computing the average geodesics between every residue pair, L would effectively be maximized when a structure is compact and minimized otherwise. As BC is evaluated for intervening residues linking pairs of residues, it gives a good indication of the importance of that residue for information flow within the protein. Penkler et al. for the first time showed that there is correlation between BC and 1/RMSF (root mean square fluctuation) as well as between BC and 1/L [35]. Interestingly, the article also highlighted a high correlation between PRS hot spot residues and residues with high BC values. This correlation, later, was also identified in other studies [32]. More recently, Kimuda coupled DRN to the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) analysis and principal components analysis (PCA) to identify 18 novel potential inhibitors of pteridine reductase 1 in Trypanosoma brucei—the causative agent of Human African Trypanosomiasis—five of which inhibited the pathogen’s growth in vitro [247].
In a new approach developed by Sheik Amamuddy and coworkers, a statistically guided network was able to show two coupled conformational changes associated with antiretroviral (ARV) drug resistance, using one-tailed t-tests on collections of residue pairwise distances determined from ensembles of protease structures sampled by MD [113]. Despite the presence of a multitude of nsSNVs and selected DRMs, situated at multiple loci within the viral drug target, they were able to reproducibly detect these motions in HIV proteases with a high degree of conservation across 8 FDA-approved inhibitors despite the chaotic nature of protein dynamics. This technique, which combines t-tests and the degree centrality, may hold a lot of promise for the analysis of alternate phenotypes (such as in the study of drug resistance) and may be invaluable in the characterization of allosteric effects. The recently published statistically guided network construction technique proved highly sensitive in detecting distinct motions from ensembles comprising hundreds of drug-resistant and drug-sensitive HIV protease variants, whereby present nsSNVs were dispersed within the protein structures [113]. The calculation of the degree centrality in this approach is very attractive for making inferences from batches of comparable protein variants, with potential applications in comparing allosteric effects between alternate pairs of a given phenotype.
Coevolution and Residue Interaction Networks
A functional site that mediates communication pathways and determines organization of the residue interaction networks often coincides or tightly couples with coevolving residues. Statistical coupling analysis (SCA), mutual information (MI) model, and covariance-based approaches have employed sequence-based analysis of residue coevolution in homologous families to show that functional residues in residue networks are connected via strong evolutionary relationships [248,249,250,251,252,253,254,255] Coevolution of protein residues can reflect correlated functional dynamics of these sites in mediating residue–residue contacts [256], protein folding transitions [257], and allosteric signalling in protein complexes [258]. Coevolving residues could also form direct communication paths in the interaction networks with connections weighted according to dynamic couplings and coevolutionary interaction strengths between nodes [259,260,261]. Structurally stable and quasi-independent modules of physically interacting coevolving residues (protein sectors) appeared to play a key role in mediating protein stability and long-range interactions [250,251]. Several computational methods have been developed to evaluate the extent of mutual information and coevolutionary dependencies between residue pairs [252,253,262,263,264]. Computational analysis of residue interaction networks and community analysis have shown that local dynamic modules anchored around functional residues can serve as building blocks to connect distant functional regions and to mediate allosteric conformational transitions [94,252,265,266,267,268]. Dynamic and coevolutionary residue correlations may also act as synchronizing forces that determine modular organization of allosteric interaction networks and enable efficient allosteric regulation [94,269,270]. These results have motivated the development of novel community-based methods for modelling ensembles of allosteric communication pathways in protein structures [94,269,270]. Using this computational framework, it was found that efficient allosteric communications in various signalling proteins could be controlled by structurally stable functional centers that exploit dynamically coupled residues in their local communities to propagate cooperative structural changes.
After having scanned the structure of interest using the allosteric site prediction approaches to obtain various sources of support for putative ligand binding sites, a consensus can be drawn before proceeding to the identification of allosteric modulators.
3.3.6. Conformational Sampling
Molecular Dynamics
MD is an invaluable tool in the hands of protein allostery researchers [44]. MD experiments simulate biological protein movement at various levels of theory, for which molecular mechanical approximations (coupled to Newton’s second law of motion) [271] are the most common for relatively large solvated protein systems. At the junction between the quantum and coarse-grained atomic models, all-atom MD simulations provide a good trade-off between accuracy and speed for conformational sampling, producing quality spatiotemporal data associated to protein action. To investigate allostery, a researcher would typically dock a prospective binder against a protein of interest before characterizing any ligand or protein changes. For such work, conventional methods of analysis can be supplemented by DRN (Part II, Section 3.3.5). These methods can also be used to study the allosteric effects of protein variants. Utilizing sequence data, it is possible to elucidate the mechanisms associated with disease-causing sequence variants using protein dynamics and allosteric regulations [36,272]. All-atom MD may not be able to sample enough conformations under reasonable computation time in the case of rare events or for changes that occur over longer periods. Coarse-graining simulations and accelerated sampling approaches are the next best approaches and are discussed in the next section.
Coarse-Grained Simulations and Stochastic Markov State Models
Coarse-grained models are computationally effective and enable simulations of long timescales for large allosteric systems and assemblies, thereby allowing for observation of allosteric structural and dynamic changes [33]. Functional and large-scale flexibility changes in allosteric systems can be predicted using several popular coarse-grained methods such as CABS-flex [273,274,275,276], NMSim [277], and FlexServ [278]. CABS-flex uses a coarse-grained model in combination with an efficient search protocol [274,275] and allows the generation of trajectories that can accurately recapitulate the all-atom MD simulations for long-time processes [274,275,279]. NMSim is a computationally efficient alternative to all-atom MD simulations and can be employed for sampling of large conformational space and pathway generation [277]. FlexServ method and server provides access to three coarse-grained algorithms for simulations of protein flexibility: discrete dynamics, NMA, and Brownian Dynamics [278]. These coarse-grained approaches provide robust and efficient means for simulation and analysis of large conformational changes and allosteric transitions in protein systems that otherwise are difficult to observe in all-atom MD simulations. ENM are a type of coarse-grained NMA, which can substantially reduce the computational demands to efficiently explore protein dynamics around a single energy minimum by simplifying interatomic interactions as spring-connected nodes (protein C and/or C atoms) [227,280,281,282,283,284,284]. Several studies involving this approach have verified its success in identifying functionally relevant protein conformations [175,285,286,287,288,289,290,291,292]. Further, coarse-grained NMA [293,294,295] and ENM integrated with the information-based Markovian theory of signal propagation [296,297,298,299,300,301,302,303] have provided a generalized formalism of allosteric communication pathways in proteins.
Given the complexity of thermodynamic and kinetic factors underlying allosteric regulatory events, stochastic MSMs [304,305,306,307,308,309,310] have become increasingly useful states-and-rates network models with the software infrastructure [311,312,313,314] for describing the probability of transitions between functional states during allosteric events [77,315,316]. Combined with MD simulations, MSM approaches can provide detailed network connectivity maps of states on the free-energy landscape and can estimate the effect of allosteric perturbations on the conformational equilibrium and kinetics of allosteric transitions. Rare transition events between long-lived states are a key feature of allosteric proteins that are difficult to observe in direct MD simulations because of the long simulation timescales needed. To circumvent these limitations, MSMs can be built from multiple shorter simulations yet describe long timescale dynamics accurately. While protein structure network models describe allosteric interactions between residue nodes, MSM network maps are markedly different by representing discrete protein states as the system nodes. The connections between Markovian state modeling and network-centric analysis of protein structure have never been rigorously explored for understanding of allosteric processes, leaving a significant conceptual void in the current knowledge and the existing repertoire of computational approaches. The integration of the experiment-informed Markovian modeling of protein dynamics and the information-theoretical description of dynamic flows and entropy transfer in the networks of protein states represents an interesting and promising avenue for further exploration of allostery. Flow-based model methods [317,318,319,320,321,322,323,324,325] operate through a stochastic walk on the dynamics of the network rather than on its topological structure, where communities consist of dynamically interconverting conformations among which the dynamic flow can persist for a long time and define functionally significant states. The apparatus of the map equation can reveal modular patterns in the dynamic flows of allosteric proteins, allowing to map conformational transformations between allosteric states and to reconstruct regulatory mechanisms.
In the following section, we describe some analysis strategies that have shown their efficacy in investigation of allosteric effects using data obtained from conformational sampling.
3.3.7. Trajectory Analysis
After performing preliminary quality checks on the simulated protein dynamics data, conventional metrics such as the root mean square deviation (RMSD), residue root mean square fluctuation (RMSF), radius of gyration (Rg), and geometry calculations (distances and angles) can be performed to infer the stabilities (or instabilities) within protein–ligand complexes for the previously docked compounds [29,32,233,326,327]. Distal effects of interfacial residue variations within the renin–angiotensinogen complex were uncovered by Brown and colleagues using RMSF alongside BC [86]. Dynamic cross correlation (DCC) is an additional option for characterizing the residue motions sampled from MD simulations, an example of which can be found from work done by Bowerman and Wereszczynski to characterize the allosteric effects of a thrombin antagonist [328]. An implementation of a DCC algorithm is available from MD-TASK. For a bird’s eye view of the distribution and clustering of sampled conformations, essential dynamics (ED) can be supplemented with energies in free-energy landscape (FEL) calculations to analyze the stability of protein complexes under various conditions [329,330]. More specifically, it has been used by Amusengeri and Tastan Bishop to investigate the effects of allosteric ligand binding in human heat shock proteins [32].
As each of the aforementioned methods highlights different facets of simulated protein dynamics, we provide some details about their principles and the types of information that can be retrieved using such techniques. In each case, the proteins or complexes are assumed to be free from periodic boundaries and corrected from rotational and translational effects. In the following section, we describe several approaches that can be used for analyzing results obtained from simulated protein dynamics.
RMSD
The root mean square deviation evaluated between a target and a given reference structure gives an idea of the overall deviation of all atoms of the structure from those of the reference. The RMSD evaluated at time is calculated as shown below, where N is the number of atoms, m is the atomic mass, x is the generalized coordinate, and is the frame used as reference, being generally the frame at :
| (1) |
For an MD simulation, it is important that the structures are properly aligned such that correct distances are evaluated. As it is often the case that trajectory-specific topologies are used in independent simulations, care should be applied when interpreting and comparing pairs of such systems, for instance, in an allosteric modulator-bound and a modulator-free protein or in a wild-type and a variant, as each protein may have a slightly different starting conformation as reference. Work by Penkler and Tastan Bishop comparing allosteric effects from ligand-bound and ligand-free Hsp90 clearly suggests the presence of multiple receptor conformations, as seen from the multimodal distribution of RMSD data [29].
RMSF
While RMSD calculates an average obtained from a collection of atom coordinates, an RMSF value is computed for each atom as the standard deviation of atomic fluctuations recorded over time. For each atom, the RMSF gives an estimate of its variability around its time-averaged value, once more from aligned conformations. Comparing RMSF values for homologous residue positions across two systems thus allows for the local inspection of differential residue flexibility, given an allosteric perturbation.
| (2) |
As an example of the use of RMSF for the detection and partial characterization of allosteric effects, Brown and coworkers observed increased rigidities across several deleterious variants of the renin-angiotensinogen complex with respect to the wild-type complex at sites distal to the dimer interface where known nsSNVs had been introduced [86].
Radius of Gyration
In the calculation of Rg, radii are first computed between each atomic () center of mass (COM) and the molecular COM (), before computing the RMSD to give an indication of the degree of compactness for a protein at a given frame i.
| (3) |
A higher averaged radius hints at a less compact structure. For instance, a residue mutation situated at the interface between two interacting protein surfaces may destabilize them and increase the Rg value. Unless the protein conformational changes are large enough, one may not always find significantly different Rg distributions. In such cases, one may focus on more localized regions, such as an individual domain for example. Sheik Amamuddy and coworkers used Rg distributions as one of their methods to investigate drug-resistance-related changes from ensembles of drug-resistant and drug-susceptible HIV protease structures of the B subtype [113].
Dynamic Cross Correlation
In essence, DCC performs pairwise correlations of atomic motion, generally by normalizing the covariance matrix over the product of the respective standard deviations of atomic fluctuations recorded over time for each atom pair. Provided the simulations are not too short, this allows one to compare motions that trend apart or together within a single system. As for the other metrics, it can be used to compare analogous systems that have experienced a given allosteric perturbation to determine correlative properties from the experiment.
Geometry Calculations
Geometry calculations comprise the calculations of distances, bond angles, and dihedrals. For example, one may compare the domain–domain distance in a multi-domain protein or the inter-residue distances resulting from the introduction of a non-synonymous residue mutation or the binding of a ligand at a site away from the active site. In an experiment investigating allostery-related effects in the redox-dependent conformational dynamics in the multidomain human protein disulphide isomerase (hDPI), Karamzadeh and coworkers computed several geometric features, such as the inter-domain angles and distances, torsion angles, and pairwise residue distances in order to build machine-learning classifiers able to discriminate between the redox states [331].
Essential Dynamics and Free-Energy Landscape
The application of ED to analyze protein behaviour mainly focuses on the dominant modes of motion, which are obtained by decomposing the covariance matrix and by ranking the eigenvectors (principal components) in descending order of the corresponding eigenvalues [292,332]. By plotting the first two or three principal components against each other, we obtain a general idea of the distribution for all generated conformations obtained from a conformational sampling experiment, as exemplified from ED analyzes for various proteins [333,334,335,336]. On the resulting figure, one may obtain conformationally distinct clusters of the simulated proteins or complexes. When supplemented with energy levels, the 3D FEL plot effectively highlights areas of low and high energies, which typically correspond to the metastable and transitory states respectively. According to the principle of minimal frustration, troughs in the landscape are generally populated by the more native conformations [337]. Given an allosteric signal, the population of protein conformations can be shifted to an alternate state against an otherwise higher energy barrier. The FEL is thus a very important tool in investigating the allosteric effect, when conformational changes are present, as exemplified by a case study showcasing the application of computational methods to discover allosteric modulators in malarial proteins [33], and in the characterization of Discorhabdin N as potential allosteric modulators of anticancer drug targets [32].
4. Conclusions
While we have gained important insights into the function of allosteric proteins, the quantitative characterization of these highly dynamic and often elusive processes continues to present formidable technical and conceptual challenges. Allosteric events in biological systems occur on different spatial and temporal scales and involve a complex interplay of thermodynamic and dynamic changes that are difficult to observe, simulate, and interpret.
Experimental studies of protein systems indicate that allosteric regulation may involve a combination of the classical models of allostery, i.e., conformational selection, dynamic allostery, and induced fit. An understanding, at the structural level, of the relationships between protein robustness, allosteric drug binding, and disease may be of use in the development of theoretical and experimental approaches bridging structure-based network analysis of protein targets with modelling of protein interaction networks and pathways. The complexity and diversity of these processes require innovative theoretical and data-driven approaches that can bridge advances in structural and quantitative biology in transformative yet practical ways. The development of novel integrated research strategies should address these challenges by strengthening and advancing the interface between molecular biophysics, network biology, and data science. We argue that the next breakthrough in the discovery of allosteric drugs may require such integration of traditional biophysical approaches with systems biology and experiment-guided machine-learning tools to bridge a detailed microscopic analysis with macroscopic modelling of allosteric phenomena in cellular networks and signalling pathways.
Author Contributions
Conceptualization, G.M.V. and Ö.T.B.; supervision, G.M.V., Ö.T.B.; writing—original draft, O.S.A., W.V., C.M., A.K., S.A., O.O., G.M.V., and Ö.T.B.; writing—review and editing, O.S.A., G.M.V., and Ö.T.B. All authors have read and agreed to the published version of the manuscript.
Funding
O.S.A. is funded as a postdoctoral fellow by H3ABioNet, which is supported by the National Institutes of Health Common Fund grant number (U41HG006941). W.V. is supported by the National Research Foundation (NRF) South Africa (Grant Number 105267) for a PhD fellowship. C.M. is funded by the CSIR DST Interbursary Support (IBS) for PhD bursary. A.K. is funded as a PhD student by the Organization for Women in Science for the Developing World (OWSD) and by the Swedish International Development Cooperation Agency (SIDA) fund reservation No. 3240287275. G.M.V. acknowledges support by the Kay Family Foundation Grant A20-0032. Ö.T.B. acknowledges Grand Challenges Africa program (GCA/DD/rnd3/023). Grand Challenges Africa is a program of the African Academy of Sciences (AAS) implemented through the Alliance for Accelerating Excellence in Science in Africa (AESA) platform, an initiative of the AAS and the African Union Development Agency (AUDA-NEPAD). GC Africa is supported by the Bill & Melinda Gates Foundation (BMGF), Swedish International Development Cooperation Agency (SIDA), German Federal Ministry of Education and Research (BMBF), Medicines for Malaria Venture (MMV), and Drug Discovery and Development Centre of University of Cape Town (H3D). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965 doi: 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 2.Koshland D.E., Nemethy J.G., Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966 doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 3.Changeux J.P. Allostery and the Monod-Wyman-Changeux Model After 50 Years. Annu. Rev. Biophys. 2012;41:103–133. doi: 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- 4.Changeux J.P., Edelstein S.J. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 5.Popovych N., Sun S., Ebright R.H., Kalodimos C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006 doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boehr D.D., Dyson H.J., Wright P.E. An NMR Perspective on Enzyme Dynamics. Chem. Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 7.Jarymowycz V.A., Stone M.J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006 doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 8.Mittermaier A. New Tools Provide New Insights in NMR Studies of Protein Dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 9.Sprangers R., Velyvis A., Kay L.E. Solution NMR of supramolecular complexes: Providing new insights into function. Nat. Methods. 2007;4:697–703. doi: 10.1038/nmeth1080. [DOI] [PubMed] [Google Scholar]
- 10.Mittermaier A.K., Kay L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Kay L.E. NMR studies of protein structure and dynamics—A look backwards and forwards. J. Magn. Reson. 2011;213:492–494. doi: 10.1016/j.jmr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Korzhnev D.M., Kay L.E. Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res. 2008;41:442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 13.Kalodimos C.G. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011;20:773–782. doi: 10.1002/pro.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenzweig R., Kay L.E. Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem. 2014;83:291–315. doi: 10.1146/annurev-biochem-060713-035829. [DOI] [PubMed] [Google Scholar]
- 15.Kay L.E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol. 2016;428:323–331. doi: 10.1016/j.jmb.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 16.Lisi G.P., Loria J.P. Solution NMR Spectroscopy for the Study of Enzyme Allostery. Chem. Rev. 2016;116:6323–6369. doi: 10.1021/acs.chemrev.5b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C., Kalodimos C.G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017;46:317–336. doi: 10.1146/annurev-biophys-070816-033701. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Y., Kalodimos C.G. NMR Studies of Large Proteins. J. Mol. Biol. 2017;429:2667–2676. doi: 10.1016/j.jmb.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Lisi G.P., Loria J.P. Allostery in enzyme catalysis. Curr. Opin. Struct. Biol. 2017;47:123–130. doi: 10.1016/j.sbi.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Gunasekaran K., Ma B., Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins: Struct. Funct. Genet. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 21.Liu J., Nussinov R. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc. Natl. Acad. Sci. USA. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai C.J., del Sol A., Nussinov R. Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. J. Mol. Biol. 2008 doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai C.J., del Sol A., Nussinov R. Protein allostery, signal transmission and dynamics: A classification scheme of allosteric mechanisms. Mol. BioSyst. 2009;5:207. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Sol A., Tsai C.J., Ma B., Nussinov R. The Origin of Allosteric Functional Modulation: Multiple Pre-existing Pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhuravlev P.I., Papoian G.A. Protein functional landscapes, dynamics, allostery: A tortuous path towards a universal theoretical framework. Q. Rev. Biophys. 2010;43:295–332. doi: 10.1017/S0033583510000119. [DOI] [PubMed] [Google Scholar]
- 26.Blacklock K., Verkhivker G.M. Computational Modeling of Allosteric Regulation in the Hsp90 Chaperones: A Statistical Ensemble Analysis of Protein Structure Networks and Allosteric Communications. PLoS Comput. Biol. 2014;10:e1003679. doi: 10.1371/journal.pcbi.1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nussinov R., Tsai C.J. Allostery in Disease and in Drug Discovery. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 28.Nussinov R., Tsai C.J., Csermely P. Allo-network drugs: Harnessing allostery in cellular networks. Trends Pharmacol. Sci. 2011 doi: 10.1016/j.tips.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penkler D.L., Tastan Bishop Ö. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Sci. Rep. 2019;9:1600. doi: 10.1038/s41598-018-35835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenthur C.J., Gentry P.R., Mathews T.P., Lindsley C.W. Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 2014;54:165–184. doi: 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szilagyi A., Nussinov R., Csermely P. Allo-Network Drugs: Extension of the Allosteric Drug Concept to Protein- Protein Interaction and Signaling Networks. Curr. Top. Med. Chem. 2013;13:64–77. doi: 10.2174/1568026611313010007. [DOI] [PubMed] [Google Scholar]
- 32.Amusengeri A., Taştan Bishop Ö. Discorhabdin N, a South African natural compound, for Hsp72 and Hsc70 allosteric modulation: Combined study of molecular modeling and dynamic residue network analysis. Molecules. 2019;24:188. doi: 10.3390/molecules24010188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amusengeri A., Astl L., Lobb K.A., Verkhivker G.M., Taştan Bishop Ö. Establishing computational approaches towards identifying malarial allosteric modulators: A case study of Plasmodium falciparum Hsp70s. Int. J. Mol. Sci. 2019;20:5574. doi: 10.3390/ijms20225574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verkhivker G.M. Dynamics-based community analysis and perturbation response scanning of allosteric interaction networks in the TRAP1 chaperone structures dissect molecular linkage between conformational asymmetry and sequential ATP hydrolysis. Biochim. Biophys. Acta Proteins Proteom. 2018;1866:899–912. doi: 10.1016/j.bbapap.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Penkler D.L., Atilgan C., Taştan Bishop Ö. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018;58:383–404. doi: 10.1021/acs.jcim.7b00630. [DOI] [PubMed] [Google Scholar]
- 36.Liang Z., Verkhivker G.M., Hu G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief. Bioinform. 2019 doi: 10.1093/bib/bbz029. [DOI] [PubMed] [Google Scholar]
- 37.Changeux J.P., Christopoulos A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes. Metab. 2017;19:4–21. doi: 10.1111/dom.12959. [DOI] [PubMed] [Google Scholar]
- 38.Greener J.G., Sternberg M.J. Structure-based prediction of protein allostery. Curr. Opin. Struct. Biol. 2018;50:1–8. doi: 10.1016/j.sbi.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Motlagh H.N., Wrabl J.O., Li J., Hilser V.J. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nussinov R., Tsai C.J. Allostery without a conformational change? Revisiting the paradigm. Curr. Opin. Struct. Biol. 2015;30:17–24. doi: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Guo J., Zhou H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016;116:6503–6515. doi: 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner J.R., Lee C.T., Durrant J.D., Malmstrom R.D., Feher V.A., Amaro R.E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016;116:6370–6390. doi: 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu S., He X., Ni D., Zhang J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019;62:6405–6421. doi: 10.1021/acs.jmedchem.8b01749. [DOI] [PubMed] [Google Scholar]
- 44.Hertig S., Latorraca N.R., Dror R.O. Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations. PLoS Comput. Biol. 2016;12:1–16. doi: 10.1371/journal.pcbi.1004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardy J.A., Lam J., Nguyen J.T., O’Brien T., Wells J.A. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. USA. 2004 doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu S., Huang W., Zhang J. Recent computational advances in the identification of allosteric sites in proteins. Drug Discov. Today. 2014;19:1595–1600. doi: 10.1016/j.drudis.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 47.Song K., Liu X., Huang W., Lu S., Shen Q., Zhang L., Zhang J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017;57:2358–2363. doi: 10.1021/acs.jcim.7b00014. [DOI] [PubMed] [Google Scholar]
- 48.Wodak S.J., Paci E., Dokholyan N.V., Berezovsky I.N., Horovitz A., Li J., Hilser V.J., Bahar I., Karanicolas J., Stock G., et al. Allostery in Its Many Disguises: From Theory to Applications. Structure. 2019;27:566–578. doi: 10.1016/j.str.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tzeng S.R., Kalodimos C.G. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- 50.Tzeng S.R., Kalodimos C.G. Protein dynamics and allostery: An NMR view. Curr. Opin. Struct. Biol. 2011;21:62–67. doi: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Tzeng S.R., Kalodimos C.G. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 52.Kalodimos C.G. Protein function and allostery: A dynamic relationship. Ann. N. Y. Acad. Sci. 2012;1260:81–86. doi: 10.1111/j.1749-6632.2011.06319.x. [DOI] [PubMed] [Google Scholar]
- 53.Buchenberg S., Sittel F., Stock G. Time-resolved observation of protein allosteric communication. Proc. Natl. Acad. Sci. USA. 2017;114:E6804–E6811. doi: 10.1073/pnas.1707694114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stock G., Hamm P. A non-equilibrium approach to allosteric communication. Philos. Trans. R. Soc. B Biol. Sci. 2018;373:20170187. doi: 10.1098/rstb.2017.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalbitzer H.R., Rosnizeck I.C., Munte C.E., Narayanan S.P., Kropf V., Spoerner M. Intrinsic allosteric inhibition of signaling proteins by targeting rare interaction states detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. 2013;52:14242–14246. doi: 10.1002/anie.201305741. [DOI] [PubMed] [Google Scholar]
- 56.Williamson M.P., Kitahara R. Characterization of low-lying excited states of proteins by high-pressure NMR. Biochim. Biophys. Acta Proteins Proteom. 2019;1867:350–358. doi: 10.1016/j.bbapap.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 57.Munte C.E., Beck-Erlach M., Kremer W., Koehler J., Kalbitzer H.R. Distinct conformational states of the alzheimer β-amyloid peptide can be detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. 2013;52:8943–8947. doi: 10.1002/anie.201301537. [DOI] [PubMed] [Google Scholar]
- 58.Cembran A., Kim J., Gao J., Veglia G. NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 2014;16:6508–6518. doi: 10.1039/C4CP00110A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robustelli P., Stafford K.A., Palmer A.G. Interpreting Protein Structural Dynamics from NMR Chemical Shifts. J. Am. Chem. Soc. 2012;134:6365–6374. doi: 10.1021/ja300265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Selvaratnam R., Chowdhury S., VanSchouwena B., Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. USA. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selvaratnam R., VanSchouwen B., Fogolari F., Mazhab-Jafari M.T., Das R., Melacini G. The Projection Analysis of NMR Chemical Shifts Reveals Extended EPAC Autoinhibition Determinants. Biophys. J. 2012;102:630–639. doi: 10.1016/j.bpj.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boulton S., Akimoto M., Selvaratnam R., Bashiri A., Melacini G. A Tool Set to Map Allosteric Networks through the NMR Chemical Shift Covariance Analysis. Sci. Rep. 2015;4:7306. doi: 10.1038/srep07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Narayanan C., Bafna K., Roux L.D., Agarwal P.K., Doucet N. Applications of NMR and computational methodologies to study protein dynamics. Arch. Biochem. Biophys. 2017;628:71–80. doi: 10.1016/j.abb.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smock R.G., Gierasch L.M. Sending Signals Dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swain J., Gierasch L. The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 66.Grutsch S., Brüschweiler S., Tollinger M. NMR Methods to Study Dynamic Allostery. PLoS Comput. Biol. 2016;12:e1004620. doi: 10.1371/journal.pcbi.1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi L., Kay L.E. Tracing an allosteric pathway regulating the activity of the HslV protease. Proc. Natl. Acad. Sci. USA. 2014;111:2140–2145. doi: 10.1073/pnas.1318476111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Long D., Bouvignies G., Kay L.E. Measuring hydrogen exchange rates in invisible protein excited states. Proc. Natl. Acad. Sci. USA. 2014;111:8820–8825. doi: 10.1073/pnas.1405011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anthis N.J., Clore G.M. Visualizing transient dark states by NMR spectroscopy. Q. Rev. Biophys. 2015;48:35–116. doi: 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuwen T., Sekhar A., Kay L.E. Separating Dipolar and Chemical Exchange Magnetization Transfer Processes in 1H-CEST. Angew. Chem. Int. Ed. 2017;56:6122–6125. doi: 10.1002/anie.201610759. [DOI] [PubMed] [Google Scholar]
- 71.Boulton S., Melacini G. Advances in NMR Methods To Map Allosteric Sites: From Models to Translation. Chem. Rev. 2016;116:6267–6304. doi: 10.1021/acs.chemrev.5b00718. [DOI] [PubMed] [Google Scholar]
- 72.Boulton S., Selvaratnam R., Ahmed R., Melacini G. Implementation of the NMR CHEmical Shift Covariance Analysis (CHESCA): A Chemical Biologist’s Approach to Allostery. In: Ghose R., editor. Methods in Molecular Biology. Springer; New York, NY, USA: 2018. pp. 391–405. [DOI] [PubMed] [Google Scholar]
- 73.Xu Y., Zhang D., Rogawski R., Nimigean C.M., McDermott A.E. Identifying coupled clusters of allostery participants through chemical shift perturbations. Proc. Natl. Acad. Sci. USA. 2019;116:2078–2085. doi: 10.1073/pnas.1811168116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aoto P.C., Martin B.T., Wright P.E. NMR Characterization of Information Flow and Allosteric Communities in the MAP Kinase p38γ. Sci. Rep. 2016;6:28655. doi: 10.1038/srep28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burley S.K., Berman H.M., Bhikadiya C., Bi C., Chen L., Di Costanzo L., Christie C., Dalenberg K., Duarte J.M., Dutta S., et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47:D464–D474. doi: 10.1093/nar/gky1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu S., Ji M., Ni D., Zhang J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today. 2018;23:359–365. doi: 10.1016/j.drudis.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 77.Bowman G.R., Bolin E.R., Hart K.M., Maguire B.C., Marqusee S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. USA. 2015;112:2734–2739. doi: 10.1073/pnas.1417811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oleinikovas V., Saladino G., Cossins B.P., Gervasio F.L. Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations. J. Am. Chem. Soc. 2016;138:14257–14263. doi: 10.1021/jacs.6b05425. [DOI] [PubMed] [Google Scholar]
- 79.Vajda S., Beglov D., Wakefield A.E., Egbert M., Whitty A. Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018;44:1–8. doi: 10.1016/j.cbpa.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goncearenco A., Mitternacht S., Yong T., Eisenhaber B., Eisenhaber F., Berezovsky I.N. SPACER: Server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013;41:W266–W272. doi: 10.1093/nar/gkt460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Panjkovich A., Daura X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics. 2014;30:1314–1315. doi: 10.1093/bioinformatics/btu002. [DOI] [PubMed] [Google Scholar]
- 82.Greener J.G., Sternberg M.J. AlloPred: Prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinform. 2015;16:1–7. doi: 10.1186/s12859-015-0771-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H., Chang Y.Y., Lee J.Y., Bahar I., Yang L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017;45:W374–W380. doi: 10.1093/nar/gkx385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weinkam P., Pons J., Sali A. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. USA. 2012;109:4875–4880. doi: 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang W., Lu S., Huang Z., Liu X., Mou L., Luo Y., Zhao Y., Liu Y., Chen Z., Hou T., et al. Allosite: A method for predicting allosteric sites. Bioinformatics. 2013;29:2357–2359. doi: 10.1093/bioinformatics/btt399. [DOI] [PubMed] [Google Scholar]
- 86.Brown D.K., Sheik Amamuddy O., Tastan Bishop Ö. Structure-Based Analysis of Single Nucleotide Variants in the Renin-Angiotensinogen Complex. Glob. Heart. 2017 doi: 10.1016/j.gheart.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gallagher M.D., Chen-Plotkin A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018;102:717–730. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brown D.K., Tastan Bishop Ö. Role of Structural Bioinformatics in Drug Discovery by Computational SNP Analysis: Analyzing Variation at the Protein Level. Glob. Heart. 2017;12:151–161. doi: 10.1016/j.gheart.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ng P.C., Levy S., Huang J., Stockwell T.B., Walenz B.P., Li K., Axelrod N., Busam D.A., Strausberg R.L., Venter J.C. Genetic variation in an individual human exome. PLoS Genet. 2008 doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guarnera E., Berezovsky I.N. On the perturbation nature of allostery: Sites, mutations, and signal modulation. Curr. Opin. Struct. Biol. 2019;56:18–27. doi: 10.1016/j.sbi.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 91.Kurochkin I.V., Guarnera E., Wong J.H., Eisenhaber F., Berezovsky I.N. Toward allosterically increased catalytic activity of insulin-degrading enzyme against amyloid peptides. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b00783. [DOI] [PubMed] [Google Scholar]
- 92.Guarnera E., Berezovsky I.N. Toward Comprehensive Allosteric Control over Protein Activity. Structure. 2019;27:866–878.e1. doi: 10.1016/j.str.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 93.Tee W.V., Guarnera E., Berezovsky I.N. On the Allosteric Effect of nsSNPs and the Emerging Importance of Allosteric Polymorphism. J. Mol. Biol. 2019;431:3933–3942. doi: 10.1016/j.jmb.2019.07.012. [DOI] [PubMed] [Google Scholar]
- 94.Stetz G., Verkhivker G.M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017;13:e1005299. doi: 10.1371/journal.pcbi.1005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Doshi U., Holliday M.J., Eisenmesser E.Z., Hamelberg D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc. Natl. Acad. Sci. USA. 2016;113:4735–4740. doi: 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sanyanga T.A., Nizami B., Taştan Bishop Ö. Mechanism of action of non-synonymous single nucleotide variations associated with α-carbonic anhydrase II deficiency. Molecules. 2019;24:3987. doi: 10.3390/molecules24213987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown D.K., Penkler D.L., Sheik Amamuddy O., Ross C., Atilgan A.R., Atilgan C., Tastan Bishop Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics. 2017;33:2768–2771. doi: 10.1093/bioinformatics/btx349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nussinov R., Jang H., Tsai C.J., Cheng F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019 doi: 10.1371/journal.pcbi.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J., et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H., et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keshava N., Toh T.S., Yuan H., Yang B., Menden M.P., Wang D. Defining subpopulations of differential drug response to reveal novel target populations. NPJ Syst. Biol. Appl. 2019;5:36. doi: 10.1038/s41540-019-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumar R.D., Chang L.W., Ellis M.J., Bose R. Prioritizing Potentially Druggable Mutations with dGene: An Annotation Tool for Cancer Genome Sequencing Data. PLoS ONE. 2013;8:e67980. doi: 10.1371/journal.pone.0067980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Niu B., Scott A.D., Sengupta S., Bailey M.H., Batra P., Ning J., Wyczalkowski M.A., Liang W.W., Zhang Q., McLellan M.D., et al. Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 2016;48:827–837. doi: 10.1038/ng.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang J.O., Oh S., Ko G., Park S.J., Kim W.Y., Lee B., Lee S. VnD: A structure-centric database of disease-related SNPs and drugs. Nucleic Acids Res. 2011;39:D939–D944. doi: 10.1093/nar/gkq957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brown D.K., Taştan Bishop Ö. HUMA: A platform for the analysis of genetic variation in humans. Hum. Mutat. 2018;39:40–51. doi: 10.1002/humu.23334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shuldiner A.R. Association of Cytochrome P450 2C19 Genotype With the Antiplatelet Effect and Clinical Efficacy of Clopidogrel Therapy. JAMA. 2009;302:849. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan Y., Chen W., Xu Y., Yi X., Han Y., Yang Q., Li X., Huang L., Johnston S.C., Zhao X., et al. Genetic Polymorphisms and Clopidogrel Efficacy for Acute Ischemic Stroke or Transient Ischemic Attack. Circulation. 2017;135:21–33. doi: 10.1161/CIRCULATIONAHA.116.024913. [DOI] [PubMed] [Google Scholar]
- 108.Maimbo M., Kiyotani K., Mushiroda T., Masimirembwa C., Nakamura Y. CYP2B6 genotype is a strong predictor of systemic exposure to efavirenz in HIV-infected Zimbabweans. Eur. J. Clin. Pharmacol. 2012;68:267–271. doi: 10.1007/s00228-011-1118-0. [DOI] [PubMed] [Google Scholar]
- 109.Hussain M., Galvin H., Haw T.Y., Nutsford A., Husain M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017;10:121–134. doi: 10.2147/IDR.S105473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dookie N., Rambaran S., Padayatchi N., Mahomed S., Naidoo K. Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 2018;73:1138–1151. doi: 10.1093/jac/dkx506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Menard D., Dondorp A. Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. 2017 doi: 10.1101/cshperspect.a025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koigi P., Ngayo M., Khamadi S., Ngugi C., Nyamache A. HIV type 1 drug resistance patterns among patients failing first and second line antiretroviral therapy in Nairobi, Kenya. BMC Res. Notes. 2014;7:890. doi: 10.1186/1756-0500-7-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sheik Amamuddy O., Bishop N.T., Taştan Bishop Ö. Characterizing early drug resistance-related events using geometric ensembles from HIV protease dynamics. Sci. Rep. 2018;8:17938. doi: 10.1038/s41598-018-36041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wensing A., Calvez V., Gunthard H., Johnson V., Paredes R., Pillay D., Shafer R., Richman D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2017;24:132–133. [PMC free article] [PubMed] [Google Scholar]
- 115.Sheik Amamuddy O.S.A. Ph.D. Thesis. Rhodes University; Makhanda, South Africa: 2019. Application of Machine Learning, Molecular Modelling and Structural Data Mining against Antiretroviral Drug Resistance in HIV-1. [Google Scholar]
- 116.Yang J.S., Seo S.W., Jang S., Jung G.Y., Kim S. Rational Engineering of Enzyme Allosteric Regulation through Sequence Evolution Analysis. PLoS Comput. Biol. 2012;8:e1002612. doi: 10.1371/journal.pcbi.1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dror R.O., Green H.F., Valant C., Borhani D.W., Valcourt J.R., Pan A.C., Arlow D.H., Canals M., Lane J.R., Rahmani R., et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 118.Nussinov R., Tsai C.J. The Design of Covalent Allosteric Drugs. Annu. Rev. Pharmacol. Toxicol. 2015;55:249–267. doi: 10.1146/annurev-pharmtox-010814-124401. [DOI] [PubMed] [Google Scholar]
- 119.Xu X., Chen Y., Fu Q., Ni D., Zhang J., Li X., Lu S. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J. Enzym. Inhib. Med. Chem. 2019;34:361–374. doi: 10.1080/14756366.2018.1553167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., Sausville E.L., Lukow D.A., Liu L., Chait A.R., et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019;11:eaaw8412. doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith R.D., Lu J., Carlson H.A. Are there physicochemical differences between allosteric and competitive ligands? PLoS Comput. Biol. 2017;13:e1005813. doi: 10.1371/journal.pcbi.1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Raman S. Systems Approaches to Understanding and Designing Allosteric Proteins. Biochemistry. 2018 doi: 10.1021/acs.biochem.7b01094. [DOI] [PubMed] [Google Scholar]
- 123.Baxter J.D., Chasanov W.M. An Update on HIV-1 Protease Inhibitor Resistance. J. AIDS Clin. Res. 2016;7:1–7. doi: 10.4172/2155-6113.1000581. [DOI] [Google Scholar]
- 124.Kagan R.M., Dunn K.J., Snell G.P., Nettles R.E., Kaufman H.W. Trends in HIV-1 Drug Resistance Mutations from a U.S. Reference Laboratory from 2006 to 2017. AIDS Res. Hum. Retroviruses. 2019;35:698–709. doi: 10.1089/aid.2019.0063. [DOI] [PubMed] [Google Scholar]
- 125.Rabahi M.F., da Silva Júnior J.L.R., Ferreira A.C.G., Tannus-Silva D.G.S., Conde M.B. Tuberculosis treatment. J. Bras. Pneumol. 2017;43:472–486. doi: 10.1590/s1806-37562016000000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mega E.R. Alarming surge in drug-resistant HIV uncovered. Nature. 2019 doi: 10.1038/d41586-019-02316-x. [DOI] [PubMed] [Google Scholar]
- 127.Centers for Disease Control and Prevention . Antibiotic Resistance Threats in the United States. Centers for Disease Control and Prevention; Atlanta, GA, USA: 2013. [Google Scholar]
- 128.Zhong W., Cui L., Goh B.C., Cai Q., Ho P., Chionh Y.H., Yuan M., Sahili A.E., Fothergill-Gilmore L.A., Walkinshaw M.D., et al. Allosteric pyruvate kinase-based “logic gate” synergistically senses energy and sugar levels in Mycobacterium tuberculosis. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-02086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mishra A., Mamidi A.S., Rajmani R.S., Ray A., Roy R., Surolia A. An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a novel combinatorial therapy. EMBO Mol. Med. 2018;10:1–21. doi: 10.15252/emmm.201708038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wellington S., Nag P.P., Michalska K., Johnston S.E., Jedrzejczak R.P., Kaushik V.K., Clatworthy A.E., Siddiqi N., McCarren P., Bajrami B., et al. A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 2017;13:943–950. doi: 10.1038/nchembio.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rzomp K.A., Scholtes L.D., Briggs B.J., Whittaker G.R., Scidmore M.A. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 2003;71:5855–5870. doi: 10.1128/IAI.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cortes C., Rzomp K.A., Tvinnereim A., Scidmore M.A., Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple rab GTPases. Infect. Immun. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bruce E.A., Digard P., Stuart A.D. The Rab11 Pathway Is Required for Influenza A Virus Budding and Filament Formation. J. Virol. 2010;84:5848–5859. doi: 10.1128/JVI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kumar A.P., Lukman S. Allosteric binding sites in Rab11 for potential drug candidates. PLoS ONE. 2018;13:e0198632. doi: 10.1371/journal.pone.0198632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hernández Alvarez L., Barreto Gomes D.E., Hernández González J.E., Pascutti P.G. Dissecting a novel allosteric mechanism of cruzain: A computer-aided approach. PLoS ONE. 2019;14:e0211227. doi: 10.1371/journal.pone.0211227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen Q., Wang G., Li S., Liu X., Lu S., Chen Z., Song K., Yan J., Geng L., Huang Z., et al. ASD v3.0: Unraveling Allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Res. 2016;44:D527–D535. doi: 10.1093/nar/gkv902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Abdel-Magid A.F. Allosteric Modulators: An Emerging Concept in Drug Discovery. ACS Med. Chem. Lett. 2015 doi: 10.1021/ml5005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Van Westen G.J.P., Gaulton A., Overington J.P. Chemical, Target, and Bioactive Properties of Allosteric Modulation. PLoS Comput. Biol. 2014;10:e1003559. doi: 10.1371/journal.pcbi.1003559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Häberle J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther. Clin. Risk Manag. 2011:327. doi: 10.2147/TCRM.S12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Van Wagenen B., Moe S., Balandrin M., DelMar E., Nemeth E. Calcium Receptor-Active Compounds. US6211244B1. 2001 Apr 3;
- 142.Adjeroud S., Tonon M.C., Leneveu E., Lamacz M., Danger J.M., Gouteux L., Cazin L., Vaudry H. VI. The benzodiazepine agonist clonazepam potentiates the effects of γ-aminobutyric acid on α-MSH release from neurointermediate lobes in vitro. Life Sci. 1987 doi: 10.1016/0024-3205(87)90046-4. [DOI] [PubMed] [Google Scholar]
- 143.Rice K.D., Aay N., Anand N.K., Blazey C.M., Bowles O.J., Bussenius J., Costanzo S., Curtis J.K., Defina S.C., Dubenko L., et al. Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973) ACS Med. Chem. Lett. 2012 doi: 10.1021/ml300049d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Desai M.A., Burnett J.P., Ornstein P.L., Schoepp D.D. Cyclothiazide acts at a site on the alpha-amino-3-hydroxy-5-methyl-4- isoxazole propionic acid receptor complex that does not recognize competitive or noncompetitive AMPA receptor antagonists. J. Pharmacol. Exp. Ther. 1995;272:38–43. [PubMed] [Google Scholar]
- 145.Tömösközi Z., Finance O., Arányi P. Drotaverine interacts with the L-type Ca2+ channel in pregnant rat uterine membranes. Eur. J. Pharmacol. 2002;449:55–60. doi: 10.1016/S0014-2999(02)01993-3. [DOI] [PubMed] [Google Scholar]
- 146.Chen J., Yang J., Sun X., Wang Z., Cheng X., Lu W., Cai X., Hu C., Shen X., Cao P. Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/R140Q. Sci. Rep. 2017;7:16458. doi: 10.1038/s41598-017-16427-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Morlock E.V., Czajkowski C. Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Mol. Pharmacol. 2011 doi: 10.1124/mol.110.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Krause R.M., Buisson B., Bertrand S., Corringer P.J., Galzi J.L., Changeux J.P., Bertrand D. Ivermectin: A positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1998 doi: 10.1124/mol.53.2.283. [DOI] [PubMed] [Google Scholar]
- 149.Haefely W., Kulcsar A., Mohler H. Possible involvement of GABA in the central actions of benzodiazepines. Psychopharmacol. Bull. 1975;14:131–151. [PubMed] [Google Scholar]
- 150.Waugh D.J.J., Gaivin R.J., Damron D.S., Murray P.A., Perez D.M. Binding, Partial Agonism, and Potentiation of α1-Adrenergic Receptor Function by Benzodiazepines: A Potential Site of Allosteric Modulation. J. Pharmacol. Exp. Ther. 1999;291:1164–1171. [PubMed] [Google Scholar]
- 151.Garcia-Perez J., Rueda P., Staropoli I., Kellenberger E., Alcami J., Arenzana-Seisdedos F., Lagane B. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem. 2011 doi: 10.1074/jbc.M110.168955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ai N., Wood R.D., Yang E., Welsh W.J. Niclosamide is a Negative Allosteric Modulator of Group I Metabotropic Glutamate Receptors: Implications for Neuropathic Pain. Pharm. Res. 2016;33:3044–3056. doi: 10.1007/s11095-016-2027-9. [DOI] [PubMed] [Google Scholar]
- 153.Ahmed A.H., Oswald R.E. Piracetam defines a new binding site for allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors. J. Med. Chem. 2010 doi: 10.1021/jm901905j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Artsimovitch I., Vassylyeva M.N., Svetlov D., Svetlov V., Perederina A., Igarashi N., Matsugaki N., Wakatsuki S., Tahirov T.H., Vassylyev D.G. Allosteric Modulation of the RNA Polymerase Catalytic Reaction Is an Essential Component of Transcription Control by Rifamycins. Cell. 2005;122:351–363. doi: 10.1016/j.cell.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 155.Janssen P.A., Lewi P.J., Arnold E., Daeyaert F., De Jonge M., Heeres J., Koymans L., Vinkers M., Guillemont J., Pasquier E., et al. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J. Med. Chem. 2005 doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 156.Vilella-Bach M., Nuzzi P., Fang Y., Chen J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin- associated protein kinase activity and G1 progression. J. Biol. Chem. 1999 doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- 157.Sinha N. Ticagrelor: Molecular discovery to clinical evidence ticagrelor: A novel antiplatelet agent. Indian Heart J. 2012 doi: 10.1016/j.ihj.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gilmartin A.G., Bleam M.R., Groy A., Moss K.G., Minthorn E.A., Kulkarni S.G., Rominger C.M., Erskine S., Fisher K.E., Yang J., et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 159.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nyamai D.W., Tastan Bishop Ö. Aminoacyl tRNA synthetases as malarial drug targets: A comparative bioinformatics study. Malar. J. 2019;18:34. doi: 10.1186/s12936-019-2665-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Musyoka T.M., Kanzi A.M., Lobb K.A., Tastan Bishop Ö. Analysis of non-peptidic compounds as potential malarial inhibitors against Plasmodial cysteine proteases via integrated virtual screening workflow. J. Biomol. Struct. Dyn. 2016;34:2084–2101. doi: 10.1080/07391102.2015.1108231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Faya N., Penkler D.L., Tastan Bishop Ö. Human, vector and parasite Hsp90 proteins: A comparative bioinformatics analysis. FEBS Open Bio. 2015;5:916–927. doi: 10.1016/j.fob.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu D., Zhang J., Roy A., Zhang Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins: Struct. Funct. Bioinform. 2011;79:147–160. doi: 10.1002/prot.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pieper U., Webb B.M., Dong G.Q., Schneidman-Duhovny D., Fan H., Kim S.J., Khuri N., Spill Y.G., Weinkam P., Hammel M., et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014;42:D336–D346. doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hatherley R., Brown D.K., Glenister M., Tastan Bishop Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS ONE. 2016;11:e0166698. doi: 10.1371/journal.pone.0166698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Song Y., DiMaio F., Wang R.Y.R., Kim D., Miles C., Brunette T., Thompson J., Baker D. High-Resolution Comparative Modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zimmermann L., Stephens A., Nam S.Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A.N., Alva V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018;430:2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 170.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T.G., Bertoni M., Bordoli L., et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Šali A., Blundell T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 172.Kim D.E., Chivian D., Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Roy A., Kucukural A., Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bradley P., Misura K.M., Baker D. Biochemistry: Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 175.Lee J., Freddolino P.L., Zhang Y. From Protein Structure to Function with Bioinformatics. Springer Netherlands; Dordrecht, The Netherlands: 2017. Ab Initio Protein Structure Prediction; pp. 3–35. Chapter 1. [DOI] [Google Scholar]
- 176.AlQuraishi M. End-to-End Differentiable Learning of Protein Structure. Cell Syst. 2019;8:292–301.e3. doi: 10.1016/j.cels.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Melo F., Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 178.Benkert P., Tosatto S.C.E., Schomburg D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Struct. Funct. Bioinform. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 179.Wiederstein M., Sippl M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lüthy R., Bowie J.U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 181.McGuffin L.J. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24:586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- 182.Shen M.Y., Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sali A. MODELLER A Program for Protein Structure Modeling Release 9v4, r6262. [(accessed on 21 January 2020)];2008 Available online: https://salilab.org/modeller/9v4/manual/
- 184.Taştan Bishop Ö., De Beer T.A., Joubert F. Protein homology modelling and its use in South Africa. S. Afr. J. Sci. 2008;104:2–6. [Google Scholar]
- 185.Mobley D.L., Bannan C.C., Rizzi A., Bayly C.I., Chodera J.D., Lim V.T., Lim N.M., Beauchamp K.A., Slochower D.R., Shirts M.R., et al. Escaping Atom Types in Force Fields Using Direct Chemical Perception. J. Chem. Theory Comput. 2018;14:6076–6092. doi: 10.1021/acs.jctc.8b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Huang Z., Zhu L., Cao Y., Wu G., Liu X., Chen Y., Wang Q., Shi T., Zhao Y., Wang Y., et al. ASD: A comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011;39:663–669. doi: 10.1093/nar/gkq1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang Z., Mou L., Shen Q., Lu S., Li C., Liu X., Wang G., Li S., Geng L., Liu Y., et al. ASD v2.0: Updated content and novel features focusing on allosteric regulation. Nucleic Acids Res. 2014;42:D510–D516. doi: 10.1093/nar/gkt1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Astl L., Verkhivker G.M. Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim. Biophys. Acta Gen. Subj. 2019 doi: 10.1016/j.bbagen.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 189.Pu L., Govindaraj R.G., Lemoine J.M., Wu H.C., Brylinski M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019;15:e1006718. doi: 10.1371/journal.pcbi.1006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Xu Y., Wang S., Hu Q., Gao S., Ma X., Zhang W., Shen Y., Chen F., Lai L., Pei J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018;46:W374–W379. doi: 10.1093/nar/gky380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hetényi C., Van Der Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006 doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 192.Hetényi C., van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2009;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hassan N.M., Alhossary A.A., Mu Y., Kwoh C.K. Protein-Ligand Blind Docking Using QuickVina-W with Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017 doi: 10.1038/s41598-017-15571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Iorga B., Herlem D., Barré E., Guillou C. Acetylcholine nicotinic receptors: Finding the putative binding site of allosteric modulators using the “blind docking” approach. J. Mol. Model. 2006;12:366–372. doi: 10.1007/s00894-005-0057-z. [DOI] [PubMed] [Google Scholar]
- 195.Grant B.J., Lukman S., Hocker H.J., Sayyah J., Brown J.H., McCammon J.A., Gorfe A.A. Novel Allosteric Sites on Ras for Lead Generation. PLoS ONE. 2011;6:e25711. doi: 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pavlovicz R.E., Henderson B.J., Bonnell A.B., Boyd R.T., McKay D.B., Li C. Identification of a Negative Allosteric Site on Human α4β2 and α3β4 Neuronal Nicotinic Acetylcholine Receptors. PLoS ONE. 2011;6:e24949. doi: 10.1371/journal.pone.0024949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jin T., Yu H., Huang X.F. Selective binding modes and allosteric inhibitory effects of lupane triterpenes on protein tyrosine phosphatase 1B. Sci. Rep. 2016;6:20766. doi: 10.1038/srep20766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chen J., Jiang H., Li F., Hu B., Wang Y., Wang M., Wang J., Cheng M. Computational insight into dengue virus NS2B-NS3 protease inhibition: A combined ligand- and structure-based approach. Comput. Biol. Chem. 2018;77:261–271. doi: 10.1016/j.compbiolchem.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 199.Hetényi C., Van Der Spoel D. Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Sci. 2011 doi: 10.1002/pro.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ghersi D., Sanchez R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins: Struct. Funct. Bioinform. 2009;74:417–424. doi: 10.1002/prot.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 202.Jain A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 203.Venkatachalam C., Jiang X., Oldfield T., Waldman M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003;21:289–307. doi: 10.1016/S1093-3263(02)00164-X. [DOI] [PubMed] [Google Scholar]
- 204.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 205.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Trott O., Olson A. NIH Public Access. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334.AutoDock. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Corbeil C.R., Williams C.I., Labute P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012;26:775–786. doi: 10.1007/s10822-012-9570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zhao H., Caflisch A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorgan. Med. Chem. Lett. 2013;23:5721–5726. doi: 10.1016/j.bmcl.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 209.Ruiz-Carmona S., Alvarez-Garcia D., Foloppe N., Garmendia-Doval A.B., Juhos S., Schmidtke P., Barril X., Hubbard R.E., Morley S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014;10:e1003571. doi: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Allen W.J., Balius T.E., Mukherjee S., Brozell S.R., Moustakas D.T., Lang P.T., Case D.A., Kuntz I.D., Rizzo R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015;36:1132–1156. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang Z., Sun H., Yao X., Li D., Xu L., Li Y., Tian S., Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016;18:12964–12975. doi: 10.1039/C6CP01555G. [DOI] [PubMed] [Google Scholar]
- 212.Lepšík M., Řezáč J., Kolář M., Pecina A., Hobza P., Fanfrlík J. The Semiempirical Quantum Mechanical Scoring Function for In Silico Drug Design. ChemPlusChem. 2013;78:921–931. doi: 10.1002/cplu.201300199. [DOI] [PubMed] [Google Scholar]
- 213.Pecina A., Haldar S., Fanfrlík J., Meier R., Řezáč J., Lepšík M., Hobza P. SQM/COSMO Scoring Function at the DFTB3-D3H4 Level: Unique Identification of Native Protein–Ligand Poses. J. Chem. Inf. Model. 2017;57:127–132. doi: 10.1021/acs.jcim.6b00513. [DOI] [PubMed] [Google Scholar]
- 214.Fanfrlik J., Bronowska A.K., Rezac J., Prenosil O., Konvalinka J., Hobza P. A Reliable Docking/Scoring Scheme Based on the Semiempirical Quantum Mechanical PM6-DH2 Method Accurately Covering Dispersion and H-Bonding: HIV-1 Protease with 22 Ligands. J. Phys. Chem. B. 2010;114:12666–12678. doi: 10.1021/jp1032965. [DOI] [PubMed] [Google Scholar]
- 215.Brahmkshatriya P.S., Dobes P., Fanfrlik J., Rezac J., Paruch K., Bronowska A., Lepsík M., Hobza P. Quantum Mechanical Scoring: Structural and Energetic Insights into Cyclin-Dependent Kinase 2 Inhibition by Pyrazolo[1,5-a]pyrimidines. Curr. Comput. Aided-Drug Des. 2013;9:118–129. doi: 10.2174/1573409911309010011. [DOI] [PubMed] [Google Scholar]
- 216.Dobes P., Rezac J., Fanfrlik J., Otyepka M., Hobza P. Semiempirical Quantum Mechanical Method PM6-DH2X Describes the Geometry and Energetics of CK2-Inhibitor Complexes Involving Halogen Bonds Well, While the Empirical Potential Fails. J. Phys. Chem. B. 2011;115:8581–8589. doi: 10.1021/jp202149z. [DOI] [PubMed] [Google Scholar]
- 217.Abdizadeh H., Guven G., Atilgan A.R., Atilgan C. Perturbation response scanning specifies key regions in subtilisin serine protease for both function and stability. J. Enzym. Inhib. Med. Chem. 2015;30:867–873. doi: 10.3109/14756366.2014.979345. [DOI] [PubMed] [Google Scholar]
- 218.Penkler D.L., Sensoy Ö., Atilgan C., Tastan Bishop Ö. Perturbation-Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017;57:1359–1374. doi: 10.1021/acs.jcim.6b00775. [DOI] [PubMed] [Google Scholar]
- 219.Hatherley R., Brown D.K., Musyoka T.M., Penkler D.L., Faya N., Lobb K.A., Tastan Bishop Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015;7:29. doi: 10.1186/s13321-015-0080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Song K., Li Q., Gao W., Lu S., Shen Q., Liu X., Wu Y., Wang B., Lin H., Chen G., et al. AlloDriver: A method for the identification and analysis of cancer driver targets. Nucleic Acids Res. 2019;47:W315–W321. doi: 10.1093/nar/gkz350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Huang M., Song K., Liu X., Lu S., Shen Q., Wang R., Gao J., Hong Y., Li Q., Ni D., et al. AlloFinder: A strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 2018;46:W451–W458. doi: 10.1093/nar/gky374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li S., Shen Q., Su M., Liu X., Lu S., Chen Z., Wang R., Zhang J. Alloscore: A method for predicting allosteric ligand-protein interactions. Bioinformatics. 2016;32:1574–1576. doi: 10.1093/bioinformatics/btw036. [DOI] [PubMed] [Google Scholar]
- 223.Guarnera E., Tan Z.W., Zheng Z., Berezovsky I.N. AlloSigMA: Allosteric signaling and mutation analysis server. Bioinformatics. 2017;33:3996–3998. doi: 10.1093/bioinformatics/btx430. [DOI] [PubMed] [Google Scholar]
- 224.Kaya C., Armutlulu A., Ekesan S., Haliloglu T. MCPath: Monte Carlo path generation approach to predict likely allosteric pathways and functional residues. Nucleic Acids Res. 2013;41:249–255. doi: 10.1093/nar/gkt284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Clarke D., Sethi A., Li S., Kumar S., Chang R.W., Chen J., Gerstein M. Identifying Allosteric Hotspots with Dynamics: Application to Inter- and Intra-species Conservation. Structure. 2016;24:826–837. doi: 10.1016/j.str.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Atilgan C., Atilgan A.R. Perturbation-Response Scanning Reveals Ligand Entry-Exit Mechanisms of Ferric Binding Protein. PLoS Comput. Biol. 2009;5 doi: 10.1371/journal.pcbi.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bahar I., Lezon T.R., Bakan A., Shrivastava I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mech Membrane Proteins. Chem. Rev. 2010;110:1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gerek Z.N., Ozkan S.B. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS Comput. Biol. 2011;7:18–25. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Vijayabaskar M., Vishveshwara S. Interaction Energy Based Protein Structure Networks. Biophys. J. 2010;99:3704–3715. doi: 10.1016/j.bpj.2010.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Di Paola L., Giuliani A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015;31:43–48. doi: 10.1016/j.sbi.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 231.Dokholyan N.V. Controlling Allosteric Networks in Proteins. Chem. Rev. 2016 doi: 10.1021/acs.chemrev.5b00544. [DOI] [PubMed] [Google Scholar]
- 232.Feher V.A., Durrant J.D., Van Wart A.T., Amaro R.E. Computational approaches to mapping allosteric pathways. Curr. Opin. Struct. Biol. 2014;25:98–103. doi: 10.1016/j.sbi.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Stolzenberg S., Michino M., LeVine M.V., Weinstein H., Shi L. Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim. Biophys. Acta Biomembr. 2016;1858:1652–1662. doi: 10.1016/j.bbamem.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ricci C.G., Silveira R.L., Rivalta I., Batista V.S., Skaf M.S. Allosteric Pathways in the PPARγ-RXRα nuclear receptor complex. Sci. Rep. 2016;6:19940. doi: 10.1038/srep19940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bhattacharya S., Vaidehi N. Differences in Allosteric Communication Pipelines in the Inactive and Active States of a GPCR. Biophys. J. 2014;107:422–434. doi: 10.1016/j.bpj.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Guo J., Pang X., Zhou H.X. Two Pathways Mediate Interdomain Allosteric Regulation in Pin1. Structure. 2015;23:237–247. doi: 10.1016/j.str.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Blacklock K., Verkhivker G.M. Allosteric Regulation of the Hsp90 Dynamics and Stability by Client Recruiter Cochaperones: Protein Structure Network Modeling. PLoS ONE. 2014;9:e86547. doi: 10.1371/journal.pone.0086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ghosh A., Sakaguchi R., Liu C., Vishveshwara S., Hou Y.M. Allosteric Communication in Cysteinyl tRNA Synthetase. J. Biol. Chem. 2011;286:37721–37731. doi: 10.1074/jbc.M111.246702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sethi A., Eargle J., Black A.A., Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rivalta I., Sultan M.M., Lee N.S., Manley G.A., Loria J.P., Batista V.S. Allosteric pathways in imidazole glycerol phosphate synthase. Proc. Natl. Acad. Sci. USA. 2012;109:E1428–E1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ming D., Wall M.E. Quantifying allosteric effects in proteins. Proteins: Struct. Funct. Bioinform. 2005;59:697–707. doi: 10.1002/prot.20440. [DOI] [PubMed] [Google Scholar]
- 242.Ming D., Wall M.E. Interactions in Native Binding Sites Cause a Large Change in Protein Dynamics. J. Mol. Biol. 2006;358:213–223. doi: 10.1016/j.jmb.2006.01.097. [DOI] [PubMed] [Google Scholar]
- 243.Mitternacht S., Berezovsky I.N. Binding Leverage as a Molecular Basis for Allosteric Regulation. PLoS Comput. Biol. 2011;7:e1002148. doi: 10.1371/journal.pcbi.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bowman G.R., Geissler P.L. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. USA. 2012;109:11681–11686. doi: 10.1073/pnas.1209309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.McClendon C.L., Friedland G., Mobley D.L., Amirkhani H., Jacobson M.P. Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles. J. Chem. Theory Comput. 2009;5:2486–2502. doi: 10.1021/ct9001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ausiello G., Firmani D., Laura L. The (betweenness) centrality of critical nodes and network cores; Proceedings of the 2013 9th International Wireless Communications and Mobile Computing Conference (IWCMC); Sardinia, Italy. 1–5 July 2013; pp. 90–95. [DOI] [Google Scholar]
- 247.Kimuda M.P., Laming D., Hoppe H.C., Bishop O.T. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules. 2019;24:142. doi: 10.3390/molecules24010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Süel G.M., Lockless S.W., Wall M.A., Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 249.Lockless S.W., Ranganathan R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 250.Halabi N., Rivoire O., Leibler S., Ranganathan R. Protein Sectors: Evolutionary Units of Three-Dimensional Structure. Cell. 2009;138:774–786. doi: 10.1016/j.cell.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.McLaughlin R.N., Poelwijk F.J., Raman A., Gosal W.S., Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012 doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Marino Buslje C., Teppa E., Di Doménico T., Delfino J.M., Nielsen M. Networks of High Mutual Information Define the Structural Proximity of Catalytic Sites: Implications for Catalytic Residue Identification. PLoS Comput. Biol. 2010;6:e1000978. doi: 10.1371/journal.pcbi.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Simonetti F.L., Teppa E., Chernomoretz A., Nielsen M., Marino Buslje C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013;41:W8–W14. doi: 10.1093/nar/gkt427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Aguilar D., Oliva B., Marino Buslje C. Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS ONE. 2012 doi: 10.1371/journal.pone.0041430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.de Juan D., Pazos F., Valencia A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 256.Socolich M., Lockless S.W., Russ W.P., Lee H., Gardner K.H., Ranganathan R. Evolutionary information for specifying a protein fold. Nature. 2005;437:512–518. doi: 10.1038/nature03991. [DOI] [PubMed] [Google Scholar]
- 257.Morcos F., Pagnani A., Lunt B., Bertolino A., Marks D.S., Sander C., Zecchina R., Onuchic J.N., Hwa T., Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA. 2011;108 doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wang N., Lodge J.M., Fierke C.A., Mapp A.K. Dissecting allosteric effects of activator-coactivator complexes using a covalent small molecule ligand. Proc. Natl. Acad. Sci. USA. 2014;111:12061–12066. doi: 10.1073/pnas.1406033111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Nishi H., Hashimoto K., Panchenko A.R. Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure. 2011;19:1807–1815. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chakrabarti S., Panchenko A.R. Coevolution in defining the functional specificity. Proteins: Struct. Funct. Bioinform. 2009;75:231–240. doi: 10.1002/prot.22239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Chakrabarti S., Panchenko A.R. Structural and Functional Roles of Coevolved Sites in Proteins. PLoS ONE. 2010;5:e8591. doi: 10.1371/journal.pone.0008591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Martin L.C., Gloor G.B., Dunn S.D., Wahl L.M. Using information theory to search for co-evolving residues in proteins. Bioinformatics. 2005;21:4116–4124. doi: 10.1093/bioinformatics/bti671. [DOI] [PubMed] [Google Scholar]
- 263.Gloor G.B., Martin L.C., Wahl L.M., Dunn S.D. Mutual Information in Protein Multiple Sequence Alignments Reveals Two Classes of Coevolving Positions †. Biochemistry. 2005;44:7156–7165. doi: 10.1021/bi050293e. [DOI] [PubMed] [Google Scholar]
- 264.Tillier E.R., Lui T.W. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics. 2003;19:750–755. doi: 10.1093/bioinformatics/btg072. [DOI] [PubMed] [Google Scholar]
- 265.Zhao Y., Wang Y., Gao Y., Li G., Huang J. Integrated Analysis of Residue Coevolution and Protein Structures Capture Key Protein Sectors in HIV-1 Proteins. PLoS ONE. 2015;10:e0117506. doi: 10.1371/journal.pone.0117506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Xu P., Duong D.M., Seyfried N.T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hsu Y.H., Traugh J.A. Reciprocally Coupled Residues Crucial for Protein Kinase Pak2 Activity Calculated by Statistical Coupling Analysis. PLoS ONE. 2010;5:e9455. doi: 10.1371/journal.pone.0009455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Jeon J., Nam H.J., Choi Y.S., Yang J.S., Hwang J., Kim S. Molecular Evolution of Protein Conformational Changes Revealed by a Network of Evolutionarily Coupled Residues. Mol. Biol. Evol. 2011;28:2675–2685. doi: 10.1093/molbev/msr094. [DOI] [PubMed] [Google Scholar]
- 269.Tse A., Verkhivker G.M. Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PLoS ONE. 2016;11:e0166583. doi: 10.1371/journal.pone.0166583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Verkhivker G.M. Integrating genetic and structural data on human protein kinome in network-based modeling of kinase sensitivities and resistance to targeted and personalized anticancer drugs. Biocomputing. 2016 doi: 10.1142/9789814749411_0005. [DOI] [PubMed] [Google Scholar]
- 271.Albano J.M., de Paula E., Pickholz M. Molecular Dynamics. InTechOpen; Melbourne, Australia: 2018. Molecular Dynamics Simulations to Study Drug Delivery Systems. Chapter 5. [DOI] [Google Scholar]
- 272.Proctor E.A., Kota P., Aleksandrov A.A., He L., Riordan J.R., Dokholyan N.V. Rational coupled dynamics network manipulation rescues disease-relevant mutant cystic fibrosis transmembrane conductance regulator. Chem. Sci. 2015 doi: 10.1039/C4SC01320D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kolinski A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004;51:349–371. doi: 10.18388/abp.2004_3575. [DOI] [PubMed] [Google Scholar]
- 274.Kmiecik S., Gront D., Kolinski M., Wieteska L., Dawid A.E., Kolinski A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016;116:7898–7936. doi: 10.1021/acs.chemrev.6b00163. [DOI] [PubMed] [Google Scholar]
- 275.Kmiecik S., Kouza M., Badaczewska-Dawid A., Kloczkowski A., Kolinski A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int. J. Mol. Sci. 2018;19:3496. doi: 10.3390/ijms19113496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Jamroz M., Kolinski A., Kmiecik S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013;41:427–431. doi: 10.1093/nar/gkt332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Krüger D.M., Ahmed A., Gohlke H. NMSim web server: Integrated approach for normal mode-based geometric simulations of biologically relevant conformational transitions in proteins. Nucleic Acids Res. 2012;40:310–316. doi: 10.1093/nar/gks478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Camps J., Carrillo O., Emperador A., Orellana L., Hospital A., Rueda M., Cicin-Sain D., D’Abramo M., Gelpí J.L., Orozco M. FlexServ: An integrated tool for the analysis of protein flexibility. Bioinformatics. 2009;25:1709–1710. doi: 10.1093/bioinformatics/btp304. [DOI] [PubMed] [Google Scholar]
- 279.Ciemny M.P., Badaczewska-Dawid A.E., Pikuzinska M., Kolinski A., Kmiecik S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019;20:606. doi: 10.3390/ijms20030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Tirion M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 281.Bahar I., Atilgan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 282.Haliloglu T., Bahar I., Erman B. Gaussian Dynamics of Folded Proteins. Phys. Rev. Lett. 1997;79:3090–3093. doi: 10.1103/PhysRevLett.79.3090. [DOI] [Google Scholar]
- 283.Doruker P., Atilgan A.R., Bahar I. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: Application to α-amylase inhibitor. Proteins: Struct. Funct. Bioinf. 2000;40:512–524. doi: 10.1002/1097-0134(20000815)40:3<512::AID-PROT180>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 284.Romo T.D., Grossfield A. Validating and improving elastic network models with molecular dynamics simulations. Proteins: Struct. Funct. Bioinform. 2011;79:23–34. doi: 10.1002/prot.22855. [DOI] [PubMed] [Google Scholar]
- 285.Isin B., Doruker P., Bahar I. Functional Motions of Influenza Virus Hemagglutinin: A Structure-Based Analytical Approach. Biophys. J. 2002;82:569–581. doi: 10.1016/S0006-3495(02)75422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Temiz N.A., Bahar I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins: Struct. Funct. Genet. 2002;49:61–70. doi: 10.1002/prot.10183. [DOI] [PubMed] [Google Scholar]
- 287.Xu C., Tobi D., Bahar I. Allosteric Changes in Protein Structure Computed by a Simple Mechanical Model: Hemoglobin T-R2 Transition. J. Mol. Biol. 2003;333:153–168. doi: 10.1016/j.jmb.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 288.Taly A., Delarue M., Grutter T., Nilges M., Le Novère N., Corringer P.J., Changeux J.P. Normal Mode Analysis Suggests a Quaternary Twist Model for the Nicotinic Receptor Gating Mechanism. Biophys. J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Shrivastava I.H., Bahar I. Common Mechanism of Pore Opening Shared by Five Different Potassium Channels. Biophys. J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Liu X., Xu Y., Li H., Wang X., Jiang H., Barrantes F.J. Mechanics of Channel Gating of the Nicotinic Acetylcholine Receptor. PLoS Comput. Biol. 2008;4:e19. doi: 10.1371/journal.pcbi.0040019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Isin B., Tirupula K.C., Oltvai Z.N., Klein-Seetharaman J., Bahar I. Identification of motions in membrane proteins by elastic network models and their experimental validation. Methods Mol. Biol. 2012 doi: 10.1007/978-1-62703-23-6_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ross C.J., Atilgan A.R., Tastan Bishop Ö., Atilgan C. Unraveling the Motions behind Enterovirus 71 Uncoating. Biophys. J. 2018;114:822–838. doi: 10.1016/j.bpj.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zheng W., Brooks B.R., Thirumalai D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc. Natl. Acad. Sci. USA. 2006;103:7664–7669. doi: 10.1073/pnas.0510426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Hyeon C., Lorimer G.H., Thirumalai D. Dynamics of allosteric transitions in GroEL. Proc. Natl. Acad. Sci. USA. 2006;103:18939–18944. doi: 10.1073/pnas.0608759103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Stan G., Lorimer G.H., Thirumalai D., Brooks B.R. Coupling between allosteric transitions in GroEL and assisted folding of a substrate protein. Proc. Natl. Acad. Sci. USA. 2007;104:8803–8808. doi: 10.1073/pnas.0700607104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Chennubhotla C., Bahar I. Markov propagation of allosteric effects in biomolecular systems: application to GroEL–GroES. Mol. Syst. Biol. 2006;2:36. doi: 10.1038/msb4100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chennubhotla C., Bahar I. Signal Propagation in Proteins and Relation to Equilibrium Fluctuations. PLoS Comput. Biol. 2007;3:e172. doi: 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Bahar I., Chennubhotla C., Tobi D. Intrinsic dynamics of enzymes in the unbound state and relation to allosteric regulation. Curr. Opin. Struct. Biol. 2007;17:633–640. doi: 10.1016/j.sbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chennubhotla C., Yang Z., Bahar I. Coupling between global dynamics and signal transduction pathways: A mechanism of allostery for chaperonin GroEL. Mol. BioSyst. 2008;4:287–292. doi: 10.1039/b717819k. [DOI] [PubMed] [Google Scholar]
- 300.Yang Z., Májek P., Bahar I. Allosteric Transitions of Supramolecular Systems Explored by Network Models: Application to Chaperonin GroEL. PLoS Comput. Biol. 2009;5 doi: 10.1371/journal.pcbi.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Zheng W., Tekpinar M. Large-scale evaluation of dynamically important residues in proteins predicted by the perturbation analysis of a coarse-grained elastic model. BMC Struct. Biol. 2009;9:1–17. doi: 10.1186/1472-6807-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Zheng W., Brooks B., Thirumalai D. Allosteric Transitions in Biological Nanomachines are Described by Robust Normal Modes of Elastic Networks. Curr. Protein Pept. Sci. 2009;10:128–132. doi: 10.2174/138920309787847608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Yang L., Song G., Jernigan R.L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. USA. 2009;106:12347–12352. doi: 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Pande V.S. Understanding protein folding using Markov state models. Adv. Exp. Med. Biol. 2014;797:101–106. doi: 10.1007/978-94-007-7606-7_8. [DOI] [PubMed] [Google Scholar]
- 305.Shukla D., Hernández C.X., Weber J.K., Pande V.S. Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 2015;48:414–422. doi: 10.1021/ar5002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Shukla S., Shamsi Z., Moffett A.S., Selvam B., Shukla D. Application of Hidden Markov Models in Biomolecular Simulations. In: Westhead D.R., Vijayabaskar M.S., editors. Hidden Markov Models. Volume 1552. Springer New York; New York, NY, USA: 2017. pp. 29–41. Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 307.Husic B.E., Pande V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018;140:2386–2396. doi: 10.1021/jacs.7b12191. [DOI] [PubMed] [Google Scholar]
- 308.McGibbon R.T., Schwantes C.R., Pande V.S. Statistical Model Selection for Markov Models of Biomolecular Dynamics. J. Phys. Chem. B. 2014;118:6475–6481. doi: 10.1021/jp411822r. [DOI] [PubMed] [Google Scholar]
- 309.Wu H., Paul F., Wehmeyer C., Noé F. Multiensemble Markov models of molecular thermodynamics and kinetics. Proc. Natl. Acad. Sci. USA. 2016;113:E3221–E3230. doi: 10.1073/pnas.1525092113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Prinz J.H., Wu H., Sarich M., Keller B., Senne M., Held M., Chodera J.D., Schtte C., Noé F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011;134 doi: 10.1063/1.3565032. [DOI] [PubMed] [Google Scholar]
- 311.Harrigan M.P., Sultan M.M., Hernández C.X., Husic B.E., Eastman P., Schwantes C.R., Beauchamp K.A., McGibbon R.T., Pande V.S. MSMBuilder: Statistical Models for Biomolecular Dynamics. Biophys. J. 2017;112:10–15. doi: 10.1016/j.bpj.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Cronkite-Ratcliff B., Pande V. MSMExplorer: Visualizing Markov state models for biomolecule folding simulations. Bioinformatics. 2013;29:950–952. doi: 10.1093/bioinformatics/btt051. [DOI] [PubMed] [Google Scholar]
- 313.Bowman G.R., Noé F. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. In: Bowman G.R., Pande V.S., Noé F., editors. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. Volume 797. Springer; Berlin/Heidelberg, Germany: 2014. p. 148. Chapter 11. [DOI] [Google Scholar]
- 314.Bowman G.R. A Tutorial on Building Markov State Models with MSMBuilder and Coarse-Graining Them with BACE. In: Livesay D.R., editor. Protein Dynamics: Methods and Protocols. Volume 1084. Springer; Totowa, NJ, USA: 2014. pp. 141–158. Methods in Molecular Biology. Chapter 8. [DOI] [PubMed] [Google Scholar]
- 315.Hart K.M., Ho C.M.W., Dutta S., Gross M.L., Bowman G.R. Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat. Commun. 2016;7:12965. doi: 10.1038/ncomms12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Sengupta U., Strodel B. Markov models for the elucidation of allosteric regulation. Philos. Trans. R. Soc. B Biol. Sci. 2018;373 doi: 10.1098/rstb.2017.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Rosvall M., Bergstrom C.T. An information-theoretic framework for resolving community structure in complex networks. Proc. Natl. Acad. Sci. USA. 2007;104:7327–7331. doi: 10.1073/pnas.0611034104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rosvall M., Bergstrom C.T. Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. USA. 2008;105:1118–1123. doi: 10.1073/pnas.0706851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Rosvall M., Bergstrom C.T. Mapping Change in Large Networks. PLoS ONE. 2010;5:e8694. doi: 10.1371/journal.pone.0008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Rosvall M., Bergstrom C.T. Multilevel Compression of Random Walks on Networks Reveals Hierarchical Organization in Large Integrated Systems. PLoS ONE. 2011;6:e18209. doi: 10.1371/journal.pone.0018209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Rosvall M., Esquivel A.V., Lancichinetti A., West J.D., Lambiotte R. Memory in network flows and its effects on spreading dynamics and community detection. Nat. Commun. 2014;5:4630. doi: 10.1038/ncomms5630. [DOI] [PubMed] [Google Scholar]
- 322.Delvenne J.C., Lambiotte R., Rocha L.E.C. Diffusion on networked systems is a question of time or structure. Nat. Commun. 2015;6:7366. doi: 10.1038/ncomms8366. [DOI] [PubMed] [Google Scholar]
- 323.Kawamoto T., Rosvall M. Estimating the resolution limit of the map equation in community detection. Phys. Rev. E. 2015;91:012809. doi: 10.1103/PhysRevE.91.012809. [DOI] [PubMed] [Google Scholar]
- 324.Aslak U., Rosvall M., Lehmann S. Constrained information flows in temporal networks reveal intermittent communities. Phys. Rev. E. 2018;97:062312. doi: 10.1103/PhysRevE.97.062312. [DOI] [PubMed] [Google Scholar]
- 325.Lambiotte R., Rosvall M., Scholtes I. From networks to optimal higher-order models of complex systems. Nat. Phys. 2019;15:313–320. doi: 10.1038/s41567-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Naithani A., Taylor P., Erman B., Walkinshaw M.D. A Molecular Dynamics Study of Allosteric Transitions in Leishmania mexicana Pyruvate Kinase. Biophys. J. 2015;109:1149–1156. doi: 10.1016/j.bpj.2015.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Roca C., Requena C., Sebastián-Pérez V., Malhotra S., Radoux C., Pérez C., Martinez A., Antonio Páez J., Blundell T.L., Campillo N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzym. Inhib. Med. Chem. 2018;33:1034–1047. doi: 10.1080/14756366.2018.1476502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Bowerman S., Wereszczynski J. Methods in Enzymology. Volume 578. Academic Press; Cambridge, MA, USA: 2016. Detecting Allosteric Networks Using Molecular Dynamics Simulation; pp. 429–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Singh B., Bulusu G., Mitra A. Understanding the thermostability and activity of bacillus subtilis lipase mutants: Insights from molecular dynamics simulations. J. Phys. Chem. B. 2015;119:392–409. doi: 10.1021/jp5079554. [DOI] [PubMed] [Google Scholar]
- 330.Khan S., Farooq U., Kurnikova M. Exploring protein stability by comparative molecular dynamics simulations of homologous hyperthermophilic, mesophilic, and psychrophilic proteins. J. Chem. Inf. Model. 2016;56:2129–2139. doi: 10.1021/acs.jcim.6b00305. [DOI] [PubMed] [Google Scholar]
- 331.Karamzadeh R., Karimi-Jafari M.H., Sharifi-Zarchi A., Chitsaz H., Salekdeh G.H., Moosavi-Movahedi A.A. Machine Learning and Network Analysis of Molecular Dynamics Trajectories Reveal Two Chains of Red/Ox-specific Residue Interactions in Human Protein Disulfide Isomerase. Sci. Rep. 2017;7:3666. doi: 10.1038/s41598-017-03966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.David C.C., Jacobs D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wolf A., Kirschner K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J. Mol. Model. 2013;19:539–549. doi: 10.1007/s00894-012-1563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Kumar V., Pandey P., Idrees D., Prakash A., Lynn A. Delineating the effect of mutations on the conformational dynamics of N-terminal domain of TDP-43. Biophys. Chem. 2019;250:106174. doi: 10.1016/j.bpc.2019.106174. [DOI] [PubMed] [Google Scholar]
- 335.Mehmood A., Khan M.T., Kaushik A.C., Khan A.S., Irfan M., Wei D.Q. Structural Dynamics Behind Clinical Mutants of PncA-Asp12Ala, Pro54Leu, and His57Pro of Mycobacterium tuberculosis Associated With Pyrazinamide Resistance. Front. Bioeng. Biotechnol. 2019;7:1–16. doi: 10.3389/fbioe.2019.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Ricci-López J., Vidal-Limon A., Zunñiga M., Jimènez V.A., Alderete J.B., Brizuela C.A., Aguila S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PLoS ONE. 2019;14:e0213028. doi: 10.1371/journal.pone.0213028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Tzul F.O., Vasilchuk D., Makhatadze G.I. Evidence for the principle of minimal frustration in the evolution of protein folding landscapes. Proc. Natl. Acad. Sci. USA. 2017;114:E1627–E1632. doi: 10.1073/pnas.1613892114. [DOI] [PMC free article] [PubMed] [Google Scholar]