

Abstract
Compound 1 is a curcumin di-O-2,2-bis(hydroxymethyl)propionate that shows significant in vitro and in vivo inhibitory activity against MDA-MB-231 cells with eight to ten-fold higher potency than curcumin. Here, we modified the α-position (C-4 position) of the central 1,3-diketone moiety of 1 with polar or nonpolar functional groups to afford a series of 4,4-disubstituted curcuminoid 2,2-bis(hydroxymethyl)propionate derivatives and evaluated their anticancer activities. A clear structure–activity relationship of compound 1 derivatives focusing on the functional groups at the C-4 position was established based on their anti-proliferative effects against the MDA-MB-231 and HCT-116 cell lines. Compounds 2–6 are 4,4-dimethylated, 4,4-diethylated, 4,4-dibenzylated, 4,4-dipropargylated and 4,4-diallylated compound 1, respectively. Compounds 2m–6m, the ester hydrolysis products of compounds 2–6, respectively, were synthesized and assessed for anticancer activity. Among all compound 1 derivatives, compound 2 emerged as a potential chemotherapeutic agent for colon cancer due to the promising in vivo anti-proliferative activities of 2 (IC50 = 3.10 ± 0.29 μM) and its ester hydrolysis product 2m (IC50 = 2.17 ± 0.16 μM) against HCT-116. The preliminary pharmacokinetic evaluation of 2 implied that 2 and 2m are main contributors to the in vivo efficacy. Compound 2 was further evaluated in an animal study using HCT-116 colon tumor xenograft bearing nude mice. The results revealed a dose-dependent efficacy that led to tumor volume reductions of 27%, 45%, and 60% at 50, 100, and 150 mg/kg doses, respectively. The established structure–activity relationship and pharmacokinetic outcomes of 2 is the guidance for future development of 4,4-disubstituted curcuminoid 2,2-bis(hydroxymethyl)- propionate derivatives as anticancer drug candidates.
Keywords: curcuminoid derivatives, prodrug, colon cancer, breast cancer, active metabolites
1. Introduction
Curcumin [(1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione, the structure of which is shown in Scheme 1], the naturally occurring phytochemical from the rhizome of Curcuma longa L., is a polyphenol with a symmetrical structure composed of two ortho-methoxyphenol rings connected to each other through a flexible conjugated hydrocarbon chain. It is a versatile therapeutic agent against cancer [1] and exhibits diverse pharmacological effects, including antidiabetic [2], antiviral [3], analgesic [4], nephroprotective [5], and cardioprotective effects [6], via regulating the gene expression [7] and protein binding [8]. Similar to some phenolic natural products, curcumin is a multi-target anticancer agent [9] with the ability to interfere with several signaling pathways associated with tumor progression, metastasis, apoptosis, and angiogenesis [10].
Scheme 1.

PLE mediated hydrolysis of 1.
While curcumin shows promising therapeutic properties, its low solubility and poor chemical and metabolic stability hinder further drug development [11]. The low chemical stability of curcumin is demonstrated by its rapid decomposition upon exposure to light or high temperatures (>70 °C) [12]. The compound decomposes into several degradation products, including a bicyclopentadione derivative, vanillin, feruloylmethane, and ferulic acid. The degradation of curcumin presumably results from its autoxidation [13], wherein the starting radical species is formed through hydrogen atom removal from the phenolic moiety. The phenolic group with an easily abstractable proton and the keto-enol system display significant hydrogen-donating abilities [14] that could initiate the autoxidative degradation of curcumin. The poor metabolic stability of curcumin can be ascribed to the presence of two phenolic hydroxyl groups that are prone to the formation of glucuronide and sulfate conjugates through the phase II metabolic process [15]. Moreover, the enone moiety of curcumin is accessible to nicotinamide adenine dinucleotide phosphate (NADHP) cytochrome P450 reductase or alcohol dehydrogenases, allowing curcumin to be readily reduced to the hydrogenated metabolites dihydrocurcumin, tetrahydrocurcumin, hexahydrocurcumin, and hexahydrocurcuminol [16].
Medicinal chemists have performed structural optimizations of curcumin to improve its stability and activity. Most of these attempts were focused on modifying the phenolic hydroxyl groups, keto-enol system, and enone moiety of curcumin [17,18,19,20]. However, the first two abovementioned structural features are required for radical scavenging activity, which is regarded as vital for curcumin chemopreventive and anti-inflammatory activities [21]. In addition, the enone moiety of curcumin, serving as the electrophilic Michael acceptor, can interact with cysteine or selenocysteine residues in proteins to mediate biological activities [22]. Thus, finding a balance between structural stability and biological potency can become challenging. According to the FDA [23], about 30% of the curcumin clinical trials stalled at stage II, mostly due to discrepancies between in vitro activity and in vivo response. Accordingly, to achieve success in clinical trials, obtaining a more definite picture of signaling pathways and ADME profiling for curcumin is recommended.
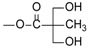
We previously developed a series of curcuminoid prodrugs by modifying the phenolic hydroxyl groups of curcumin into a 2,2-bis(hydroxymethyl)propionate group [24]. Among the derivatives, the curcumin 2,2-bis(hydroxymethyl)propionate 1 exhibited stability and solubility superior to those of curcumin. Particularly, 1 showed comparable anticancer activity against MDA-MB-231 cells in vitro and in vivo and had 8 to 10 times more potency than curcumin. Subsequently, the α-position in the central 1,3-diketone moiety (C-4 position) of 1 were alkylated with methyl, ethyl, benzyl, and propargyl groups to give compounds 2, 3, 4, and 5, respectively. Among them, 2, 3, and 5 showed more potent in vitro anti-proliferative activity than 1 against HCT-116 cells; therefore, were recommended them as potential therapeutic agents for colon cancer [24]. In our recent in vitro studies on the enzymatic hydrolysis of 1, we demonstrated that treating 1 with porcine liver esterase (PLE) cleaves its two ester groups gradually to form curcumin (see supporting information). Compounds 2-5 are ester-based derivatives and structurally correlated to 1. Therefore, we predicted that a high concentration of esterase, as particularly found in the intestinal tract and mucosa [25], would hydrolyze 2, 3, 4, and 5 into their metabolites 2m, 3m, 4m, and 5m, respectively (Scheme 2). However, the anticancer activity of 2m–5m should be evaluated prior to the further in vivo investigations. Compounds 2–5 were substituted with two nonpolar alkyl chains at the C-4 position with the loss of keto-enol tautomerism. The anticancer activity of compound 1 derivatives, which were substituted with two hydrophilic units or two polar side chains at the C-4 position, still remains unclear.
Scheme 2.

In vivo enzymatic hydrolysis of 2–5.
Herein, we report the successful replacement of the acidic α-hydrogens at the C-4 position of compound 1 with polar or nonpolar functional groups to produce compounds 6, 9, 10, 11, 12, and 14. Compounds 2m–6m were prepared through the chemical hydrolysis of corresponding parent compounds 2–6. The anticancer activities of 2–6, 9–12, 14 and 2m–6m were evaluated in vitro. A selected compound was submitted for preliminary pharmacokinetic study and antitumor study. The detailed descriptions of the synthetic design, in vitro screening, and in vivo study of the abovementioned compounds are presented as follows.
2. Results and Discussion
2.1. Chemistry
The detailed synthesis of compound 1 derivatives (substituted at the C-4 position with allyl (6), hydroxymethyl (10), acetoxymethyl (11), methoxymethyl (12)), novel 2,6-dimethyl curcumin (DMC) derivatives (13 and 15), and the ester hydrolysis products of derivatives showing potential anticancer activity (2m, 3m, 4m, 5m, 6m, 16, and 17) are depicted in Scheme 3 and Scheme 4. As shown in Scheme 3, compound 6 was synthesized according to a procedure established for the synthesis of 2–5. Compound 7, prepared by the esterification of curcumin with 2,2,5-trimethyl-1,3-dioxane-5-carboxylic acid, was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and formaldehyde in THF to produce intermediary compound 8. The latter was used without column chromatographic purification in acid-promoted hydrolysis at room temperature to produce 9 with an overall yield of 6% for two synthetic steps.
Scheme 3.

Reagents and Conditions: (a) Formaldehyde in THF, DBU, 0 °C to rt, 2 h; (b) HCl, MeOH, rt, 1 h, 6% for two steps; (c) Et3N, acetyl chloride, DCM, rt, 12 h; then HCl, MeOH, rt, 1 h, 3% for three steps; (d) K2CO3, CH3I, DMF, rt, 6 h; then HCl, MeOH, rt, 1 h, 4% for three steps; (e) Formaldehyde in THF, DBU, 0 °C to rt, 2 h, 42%; (f) Compound 13, Et3N, DCM, rt, 12 h; and (g) HCl, MeOH, 1 h, 7% for two steps.
Scheme 4.

Reagents and Conditions: (a) NaOMe, MeOH, rt, 2 h, 82–87%; (b) K2CO3, alkyl iodide, DMF, rt, 20 h; and (c) NaOMe, MeOH, rt, 3 h, 28–33% for two steps.
Compound 9, however, proved unstable, slowly decomposing even when kept at 4 °C and presenting an NMR spectrum comprising a messy jungle of peaks after being dissolved in DMSO-d6 for 1 week at room temperature. The low stability of 9 may have been due to self- or cross-hydrolysis, such that the two hydroxyl groups at the C-4 alkyl chains cleaved the ester linkage of the 2,2-bis(hydroxymethyl)propionate group. Previously, we had found that upon exposure to alcohol for several hours, the ester linkages of 1 were broken under neutral or basic conditions, leading to the conversion of 1 to curcumin and several unknown products. Therefore, we modified the hydroxymethyl group of 9 and were able to produce compounds 10 and 11. To prepare 10, compound 9 was acetylated with acetyl chloride in the presence of Et3N, and the resulting di-acetylation intermediate was hydrolyzed by hydrochloric acid. Compound 11 was obtained with an overall yield of 5% following a similar three-step reaction modified for methylation. The stability of these two products at room temperature may have been due to the conversion of the labile hydroxyl groups of the C-4 alkyl chains of 10 and 11 into non-nucleophilic methyl and acetyl groups, respectively, which prevented degradation.
Because DMC is a dimethylated derivative of curcumin [26] that exhibits better anticancer activity and stability than curcumin, we synthesized a novel DMC derivative (compound 12) in which C-4 was substituted with two bulky 2,2-bis(hydroxymethyl)propionate groups. To produce this derivative, commercially available DMC was reacted with DBU and formaldehyde in THF. The reaction of 12 with 2,2,5-trimethyl-1,3-dioxane-5-carbonyl chloride 13 [27] in the presence of Et3N provided a diol intermediate, which was then hydrolyzed by hydrochloric acid in MeOH to yield compound 14. In addition, as illustrated in Scheme 4, compounds 2m–6m were prepared through the NaOMe-mediated hydrolysis of their corresponding parent compounds 2–6, with high yield.
For SAR analysis, compounds 15 and 16, which were substituted with two straight alkyl chains with three or four carbons at the C4 position, were prepared from curcumin bisacetate 17 [28] via a two-step procedure comprising K2CO3-induced dialkylation and NaOMe-mediated hydrolysis. For the synthesis of four possible phase I metabolites of compound 2, compound 2m was subjected to hydrogenation reactions. As shown in Scheme 5, compound 18 is a partially hydrogenated product that can be prepared in 19% yield by treating compound 2m with H2(g) (1.0 atm, balloon) and a catalytic amount of 10% w/w Pd/C in ethyl acetate. When the solvent was changed from ethyl acetate to methanol, the hydrogenation reaction yielded the fully hydrogenated diketone 19 in 52% yield and β-hydroxy ketone 20 in 23% yield. Finally, the reduction of 20 with NaBH4 in MeOH at room temperature (rt) for 2 h provided the desired diol compound 21 in 31% yield.
Scheme 5.

Reagents and Conditions: (a) 10% w/w Pd/C, ethyl acetate, rt, 1 h, 19%; (b) 10% w/w Pd/C, MeOH, rt, 2 h; and (c) NaBH4, MeOH, 0 °C to rt, 2 h, 31%.
2.2. Structure–Activity Relationship
As mentioned above, we synthesized 2–5 and evaluated their anti-proliferative activity against the MDA-MB-231 and HCT-116 cell lines (72-h treatment). These compounds, along with all newly synthesized compounds, were screened for anti-proliferative activity against the same TNBC and colon cancer cell lines. The results of the in vitro assay are compiled in Table 1.
Table 1.
The anti-proliferative effects of curcumin derivatives against MDA-MB-231 and HCT-116 cell lines.
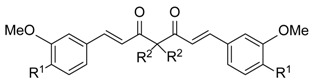
| Compound | R1 | R2 | IC50 a (M)/ 48 h | |
|---|---|---|---|---|
| MDA-MB-231 | HCT-116 | |||
| Curcumin | OH | H | 22.05 ± 2.97 | 26.33 ± 1.38 |
| 1 |
|
H | 3.06 ± 0.18 | 6.25 ± 0.60 |
| 2 |
|
methyl | 2.25 ± 0.10 | 3.10 ± 0.29 |
| 3 |
|
ethyl | 3.02 ± 0.15 | 1.93 ± 0.01 |
| 4 |
|
benzyl | 2.45 ± 0.24 | 7.66 ± 0.61 |
| 5 |
|
propargyl | 5.87 ± 0.10 | 5.81 ± 0.23 |
| 6 |
|
allyl | 3.13 ± 0.54 | 0.92 ± 0.06 |
| 9 |
|
CH2OH | >100 | >100 |
| 10 |
|
CH2OAc | 6.55 ± 0.04 | 6.53 ± 0.01 |
| 11 |
|
CH2OMe | 84.92 ± 1.67 | 69.08 ± 3.11 |
| 12 | OCH3 | CH2OH | 53.50 ± 3.45 | 58.00 ± 0.60 |
| 14 | OCH3 |
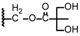
|
57.43 ± 2.06 | 40.10 ± 1.61 |
| 2m | OH | methyl | 4.17 ± 0.15 | 2.17 ± 0.16 |
| 3m | OH | ethyl | 9.12 ± 0.40 | 8.18 ± 0.57 |
| 4m | OH | benzyl | 36.52 ± 1.66 | 9.40 ± 0.02 |
| 5m | OH | propargyl | 3.10 ± 0.15 | 1.38 ± 0.06 |
| 6m | OH | allyl | 6.88 ± 0.36 | 4.40 ± 0.53 |
| 15 | OH | propyl | 18.17 ± 0.53 | 10.73 ± 1.63 |
| 16 | OH | butyl | >100 | 67.00 ± 3.18 |
a All data presented are means from at least three experiments with standard deviations of the value quoted.
The anticancer effects of 2–5 against both cell lines were still better or at least similar to that of 1, although the drug treatment duration was reduced to 48 h. The newly synthesized compound 6 had a higher anti-proliferative activity against HCT-116. Compound 9 was less potent against the cell lines than curcumin and 1, which was used as a reference compound in the current study. We hypothesize that the two polar and hydrophilic side chains at the C-4 position in 9 are the cause for the weak anticancer effects observed for this compound. Another possibility is that 9 decomposed during the screening due to its low stability. Compound 10, having two acetoxymethyl groups rather than hydroxymethyl groups at the C-4 side chains, showed significant improvement in in vitro anticancer activities compared to 9; however, it was still less potent than 1. Compound 11, substituted with two methoxymethyl groups at the C-4 position, displayed poor anticancer activity with high IC50 values for both cell lines used.
After listing the compounds in descending order based on their potency against HCT-116 (6 > 3 > 2 > 5 > 1 > 10 > 4 >> 11 >> 9), we concluded that higher polarity and hydrophilicity of the C-4 side chain are detrimental to the anticancer activity of compound 1 derivatives. Alternatively, the DMC derivative 4,4-dihydroxymethyl dimethoxycurcumin (compound 12) displayed low anti-proliferative activity. When the two hydroxyl moieties of 12 both esterified with 2,2-bis(hydroxymethyl)propionic acid, the resulting compound 14 exhibited activities comparable to those of 12, which correlates with our previous findings regarding the unfavorable effect of having a hydrophilic moiety at the C-4 position.
Subsequently, the activities of the ester hydrolysis products 2m–6m were explored. Unexpectedly, 5m displayed substantial improvement in anticancer activity by two- to four-fold relative to that of parent 5. Compound 2m exhibited activity similar to that of 2, whereas 3m, 4m, and 6m showed inferior activity than their corresponding parent compounds 3, 4, and 6. Further screening of 15 and 16 revealed an obvious SAR, such that the anticancer activity of these hydrolysis compounds decreased with the increasing length of the C-4 alkyl side chains. In addition, lower structural volume was seemingly preferred to sterically congested side chains, as demonstrated by the analysis of 2m, 3m, 15, 4m, and 16.
Among all examined compounds, 2 and its ester hydrolysis product 2m possessed significant anticancer activity against MDA-MB-231 and HCT-116; therefore, 2 was chosen for further studies. Notably, the solubility of 2 was more significant than that of curcumin as was evident from the vehicle of 2 for the following in vivo study. The maximum solubility of 2 in the aqueous vehicle comprising 5% ethanol, 10% Tween 80, and 85% saline was approximately 200 mg/mL, while curcumin was almost insoluble in the vehicle. Based on the logic for the metabolism of curcumin [29] and an ester-type prodrug [30], we speculated that the in vivo metabolism of 2 would readily produce 2m. Subsequently, the latter would be converted into glucuronide 22 and sulfate 23 through the phase II metabolism conjugation of hydrogenated products 18, 19, 20, and 21 via the phase I metabolic pathway (Scheme 6). To verify our assumptions, the preliminary pharmacokinetic evaluation of 2 was executed.
Scheme 6.

In vivo metabolism of 2.
2.3. The Preliminary Pharmacokinetic Evaluation of 2
The preliminary in vivo pharmacokinetic evaluation of 2 was designed to identify the anticipated metabolites and evaluate the plasma stability of 2 and 2m. The four possible phase I metabolites 18, 19, 20, and 21 were synthesized and subjected to anti-proliferative screening against MDA-MB-231 and HCT-116. Compound 18 exhibited moderate to weak anti-proliferative activity (MDA-MB-231, IC50 = 36.48 ± 0.34 μM; HCT-116, IC50 = 9.64 ± 0.21 μM) and the others are inactive against both cell lines (IC50 ≥ 100 μM). The assessment of 2 and relevant metabolites was performed in male Sprague-Dawley rats after the oral administration of 2 at a dose of 100 mg/kg. The LC signals of 2, 2m, and 18–21 was recognized unequivocally by comparison to the signals of standard compounds. The direct analysis of 22 and 23 was not easily attainable due to the low extraction efficiency and high probability of sample loss in LC column. It has been well-documented that curcumin glucuronide and curcumin sulfate can be transformed back into curcumin by the enzyme-mediated hydrolysis reaction [31]. Accordingly, the treatment of 22 and 23 with enzyme that contains sulfatase and glucuronidase is supposed to produce 2m. In the current study, the amounts of 22 and 23 were not calculated individually but counted as a summary value based on the disparities between the LC signal of 2m in enzyme-treated and -untreated serum samples.
In practice, LC-MS analysis of one serum sample which was collected at 20 min post-dosing demonstrated the existence of 2, 2m, 18, 19, 20, 21, 22 and 23 (Figure S1 in the Supporting Information). Shown in Figure 1 are the LC signal count-time profiles for 2, 2m, 18, 19, and the comparison of 2m, 22, and 23, following oral administration. The analysis of 20 and 21 is not illustrated due to their low LC signal response and non-significant anticancer activity. The maximum blood content of 2 was achieved at 20 min, while the second and third highest appeared at 120 min and 360 min, respectively, following administration. The multiple peaks of 2 observed in the profile could be attributed to an enterohepatic circulation cycle.
Figure 1.

The amount of 2, 2m, 18, 19, 22, and 23 in serum at different time points following a single oral administration of 2 at a dose of 100 mg/kg. The blood samples were collected at 5, 10, 20, 30, 60, 120, 240, 360, and 480 min after the administration of 2. The samples were deproteinated and analyzed by HPLC.
Such behavior, extensively studied in the phenolic drugs, was beneficial in maintaining the circulating concentration of free-form curcumin [32] and may extend the pharmacological effect of drug [33]. The hydrolysis metabolite 2m emerged from 10 min and reached its maximum blood content at 120 min, indicating that a medium-to-high esterase-mediated biotransformation proceeded in vivo to yield the efficient absorption of 2m. Subsequently, the phase II metabolic process of 2m progressed quickly to produce 22 and 23. The ratio of 2m to its phase II metabolites (22 and 23) is about 1:4 based on the area under the curves in part (e) of Figure 1.
The LC signal count-time profiles of 18 and 19 are analogous to that of 2m, implying that phase I reduction reaction also occurred rapidly. Compared to non-formulated curcumin with a short elimination period of 28 min in the rat model [34], 2 exhibited superior plasma stability. Otherwise, the active metabolite 2m is equally potent as 2 with regard to anticancer activity, and the maximum blood content of 2m appeared after that of 2 could be considered as an extension of drug efficacy of 2. Thus, the pharmacokinetic outcome was to guide the following in vivo efficacy study of 2.
2.4. In Vivo Antitumor Efficacy of 2
The therapeutic effect of 2 was evaluated on HCT-116 colon tumor xenograft bearing nude mice. The oral administration of 2 at doses of 50, 100, or 150 mg/kg bw per day were performed after tumors reached approximately 100 mm3 and continued for 30 consecutive days. 5-Fluorouracil treatment (30 mg/kg bw, i.p., QOD) was used as a positive control. The group treated with compound 2 (100 mg/kg) had a statistically significant reduction in tumor volumes, with a 45% tumor inhibition ratio observed when compared with the cancerous control mice. Positive control group exhibited a capability to cause 40% tumor-growth inhibition. There was a clear dose- and time-dependent inhibitory effect on tumor size in the other two groups, in which at 50 and 150 mg/kg doses the volume of the HCT-116 colon tumor was reduced to 73% and 40% of the control, respectively (Figure 2). The body weight of the mice was recorded prior to dosing every 3 d, and the average body weight of each group is depicted in Figure 3. Otherwise, they were monitored for visible signs of toxicity and behavioral changes 1 h after each administration. No obvious adverse effects were observed between the 2-administrated and control groups.
Figure 2.

The efficacy study of 2. In vivo antitumor activity of compound 2 was evaluated in HCT-116 tumor xenograft bearing nude mice. Compound 2 (50, 100, or 150 mg/kg, p.o.) was administered once daily and 5-Fluorouracil was administrated (30 mg/kg, i.p.) every other day. The results are the mean ± SEM of six mice in each group. *** P < 0.001 compared to untreated control.
Figure 3.

Mean body weight-time profile of 2- administrated and control groups.
3. Materials and Methods
3.1. General Information
The reactions were performed under an air atmosphere unless otherwise stated. All solvents and reagents were employed as received. Analytical thin layer chromatography (TLC) was performed on SiO2 60 F254 plates and flash column chromatography was carried out using SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh), both of which are available from E. Merck (Darmstadt, Germany). Visualization was performed under UV irradiation at 254 nm followed by staining with aqueous potassium permanganate [KMnO4 (3 g) and K2CO3 (20 g) in 300 mL of H2O containing 5 mL of an aqueous solution of NaOH (5%, w/v)] and charring by heat gun. 1H- and 13C-NMR spectra were recorded on a 500 FT NMR instrument (Bruker, Billerica, MA, USA). Chloroform-d and methanol-d were used as solvents and TMS (δ = 0.00 ppm) as an internal standard. Chemical shifts are reported as δ values in ppm as referenced to TMS. Multiplicities are recorded as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextet), sept (septet), dd (doublet of doublets), dt (doublet of triplets), br (broad), m (multiplet). Coupling constants (J) are expressed in Hz. LRMS and HRMS were measured by a JMS-HX110 spectrometer (JEOL, Tokyo, Japan) and spectroscopic data were recorded as m/z values. Melting points were measured using an Electrothermal instrument (Anatec Yanaco Inc., Kyoto, Japan).
3.1.1. [(1E,6E)-4,4-Diallyl-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (6)
Mp = 195–197 °C; 1H-NMR (DMSO-d6) δ 7.59 (d, J = 15.5 Hz, 2H), 7.48 (s, 2H), 7.35 (dd, J = 8.2, 1.5 Hz, 2H), 7.12–7.04 (m, 4H), 5.59–5.52 (m, 2H), 5.12–5.04 (m, 4H), 4.82–4.80 (m, 4H), 3.78 (s, 6H), 3.60 (s, 8H), 2.84 (d, J = 7.2 Hz, 4H), 1.17 (s, 6H); 13C NMR (DMSO-d6) δ 196.8, 173.3, 151.7, 143.1, 142.1, 133.3, 133.2, 123,9, 122.9, 122.8, 119.5, 113.4, 67.4, 64.0, 56.7, 51.0, 35.0, 17.3; HRMS [ESI]+ calculated for C37H45O12: 681.2911 [M + H]+; found: 681.2903.
3.1.2. [(1E,6E)-4,4-bis(Hydroxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis(2,2,5-trimethyl-1,3-dioxane-5-carboxylate) (8)
To a stirred solution of 7 (1.00 g, 1.47 mmol) in THF (7.3 mL, containing 0.6 M formaldehyde) was added DBU (38.9 mg, 0.04 mmol) at 0 °C. The reaction mixture was stirred at the same temperature for 30 min and then warm to room temperature. After 2 h, the solution was concentrated under vacuum to provide crude compound, which was identified by LRMS [ESI]+ calculated for C39H49O14: 741.31 [M + H]+; found: 741.33 and used for next step without purification.
3.1.3. [(1E,6E)-4,4-bis(Hydroxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (9)
A solution of crude 8 (0.34 g) and 6 N HCl (0.15 mL) in MeOH (1.83 mL) was stirred at room temperature for 1 h. The reaction mixture was concentrated under vacuum to give the crude product, which was purified by flash chromatography on silica gel with MeOH/CH2Cl2 (1:20) to afford compound 9 (40 mg) as an orange oil. Yield 6% for two steps. 1H-NMR (DMSO-d6) δ 7.52 (d, J = 15.6 Hz, 2H), 7.44 (s, 2H), 7.30 (d, J = 8.2 Hz, 2H), 7.08–7.04 (m, 4H), 5.16 (br s, 6H), 4.19 (s, 4H), 3.78 (s, 6H), 3.61 (s, 8H), 1.17 (s, 6H); LRMS [ESI]+ calculated for C33H40O14: 661.25 [M + H]+; found: 661.4.
3.1.4. [(1E,6E)-4,4-bis(Acetoxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (10)
To a stirred solution of crude 8 (0.450 g) in DCM (10 mL) were added NEt3 (1.0 mL) and acetyl chloride (0.140 g, 1.81 mmol) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 12 h. Then H2O (10 mL) and diluted HCl(aq) (1 N, 3 mL) were added to quench the reaction. The aqueous layer was separated and extracted with CH2Cl2 (3 × 8 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated to give the crude product, which was then treated with HCl(aq) (6 N, 0.2 mL) in MeOH (2 mL) at room temperature. The reaction mixture was stirred at room temperature for 1h and then concentrated under vacuum to give the crude product, which was purified on silica gel with MeOH/CH2Cl2 (1:20) to afford 10 (33 mg) as a yellow solid (mp = 194–196 °C). Yield 3% for three steps; 1H-NMR (DMSO-d6) δ 7.68 (d, J = 15.5 Hz, 2H), 7.51 (s, 2 H), 7.37 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 15.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 4.85 (t, J = 5.0 Hz, 8H), 3.80 (s, 6 H), 3.65-3.59 (m, 8H), 1.97 (s, 6H), 1.19 (s, 6H); 13C-NMR (DMSO-d6) δ 193.1, 173.3, 170.5, 151.7, 144.6, 142.5, 132.9, 123.9, 122,7, 121.7, 113.6, 64.0, 61.9, 56.7, 51.0, 20.9, 17.3; HRMS [ESI]+ calculated for C37H44NaO16: 767.2527 [M + Na]+; found: 767.2521.
3.1.5. [(1E,6E)-4,4-bis(Methoxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (11)
To a stirred solution of crude 8 (0.453 g) in DMF (1.0 mL) were added K2CO3 (0.240 g, 1.73 mmol) and MeI (0.247 g, 1.73 mmol) at 0 oC. The reaction mixture was warmed to room temperature and stirred for 6 h. Then H2O (10 mL) and diluted HCl(aq) (1 N, 3 mL) were added to quench the reaction. The aqueous layer was separated and extracted with EtOAc (3 × 8 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated to give the crude product, which was then treated with HCl(aq) (6 N, 0.25 mL) in MeOH (2 mL) at room temperature. The reaction mixture was stirred at room temperature for 1h and then concentrated under vacuum to give the crude product, which was purified on silica gel with MeOH/CH2Cl2 (1:20) to afford 11 (73 mg) as a yellow solid (mp = 187–188 °C). Yield 5% for three steps; 1H-NMR (DMSO-d6) δ 7.65 (d, J = 16.0 Hz, 2H), 7.49 (s, 2 H), 7.31 (d, J = 10.0 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 6.69 (d, J = 16.0 Hz, 2H), 4.85 (t, J = 5.5 Hz, 4 H), 3.80 (s, 6H), 3.74 (s, 6 H), 3.66-3.60 (m, 8H), 1.20 (s, 6H); 13C-NMR (DMSO-d6) δ 193.1, 173.4, 167.1, 151.6, 144.4, 141.8, 133.2, 123.8, 121,9, 118.4, 112.6, 64.0, 56.3, 51.9, 51.0, 17.3; HRMS [ESI]+ calculated for C35H44NaO14: 711.2629 [M + Na]+; found: 711.2633.
3.1.6. (1E,6E)-1,7-bis(3,4-Dimethoxyphenyl)-4,4-bis(hydroxymethyl)hepta-1,6-diene-3,5-dione (12)
To a stirred solution of DMC (1.270 g, 3.20 mmol) in THF (20 mL, containing 0.6 M formaldehyde) was added DBU (15 mg, 0.10 mmol). The reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was concentrated under vacuum and then purified by flash chromatography on silica gel with MeOH/CH2Cl2 (1:32) to afford 12 (558 mg, 42% yield) as a yellow solid; mp = 174–176 °C; 1H-NMR (DMSO-d6) δ 7.49 (d, J = 15.5 Hz, 2H), 7.48 (d, J = 2.0 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 6.97–6.91 (m, 4H), 4.78 (t, J = 4.5 Hz, 2H), 4.19 (d, J = 4.5 Hz, 4H), 3.79 (s, 12H); 13C-NMR (DMSO-d6) δ 196.2, 151.7, 149.1, 142.8, 127.4, 123.8, 120,9, 112.1, 111.3, 71.6, 60.1, 56.1, 56.0; HRMS [ESI]+ calculated for C25H28NaO8: 479.1682 [M + Na]+; found: 479.1677.
3.1.7. 2,2-bis[(E)-3-(3,4-Dimethoxyphenyl)acryloyl]propane-1,3-diylbis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (14)
To a solution of 12 (0.170 g, 0.37 mmol) in CH2Cl2 (4 mL) was added NEt3 (0.15 mL, 1.10 mmol), DMAP (0.110 g, 0.92 mmol) and 2,2,5-trimethyl-1,3-dioxane-5-carbonyl chloride 13 (0.210 g, 1.10 mmol) sequentially at room temperature. The reaction mixture was stirred at the same temperature for 12h and then concentrated under vacuum to give the crude product, which without purification was treated with HCl(aq) (6 N, 0.1 mL) in MeOH (0.5 mL). The resulting mixture was stirred at room temperature for 1h and then concentrated under vacuum to give the crude product, which was purified by flash chromatography on silica gel with MeOH/CH2Cl2 (1:19) to afford compound 14 (16 mg, 7% for two steps) as a yellow solid; mp > 300 °C; 1H-NMR (DMSO-d6) δ 7.64 (d, J = 15.5 Hz, 2H), 7.34–7.30 (m, 4H), 7.03 (d, J = 15.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 4.74 (s, 4H), 4.63 (t, J = 5.5 Hz, 4H), 3.79 (s, 12H), 3.40-3.37 (m, 8H), 0.95(s, 6H); 13C-NMR (DMSO-d6) δ 193.3, 174.6, 152.1, 149.4, 145.0, 127.1, 124.7, 119,5, 112.0, 111.4, 66.9, 63.9, 62.3, 56.1, 56.1, 50.7, 17.0; HRMS [ESI]+ calculated for C35H44NaO14: 711.2629 [M + Na]+; found: 711.2620.
3.1.8. General Procedure for the Synthesis of Compounds 2m–6m
The general procedure is illustrated immediately below with compound 2m as a specific example.
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylhepta-1,6-diene-3,5-dione (2m)
To a stirred solution of compound 2 (0.450 g, 0.72 mmol) in CH3OH (6 mL) were added NaOMe (0.116 g, 2.16 mmol) at room temperature. The reaction mixture was stirred at the same temperature for 2 h and then H2O (5 mL) and diluted HCl(aq) (1 N, 1 mL) were added to quench the reaction. The aqueous layer was separated and extracted with CH2Cl2 (3 × 6 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated to give the crude product, which was then purified by flash chromatography on silica gel with EtOAc/n-hexane/CH2Cl2 (1:2:1) to afford 2m (0.245 g, 86% yield) as an orange solid; mp = 104−106 °C; 1H-NMR (CDCl3) δ 7.69 (d, J = 15.5 Hz, 2H), 7.12 (dd, J = 8.0, 1.5 Hz, 2H), 7.01 (s, 2H), 6.91 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 15.5 Hz, 2H), 5.97 (s, 2H), 3.94 (s, 6H), 1.48 (s, 6H); 13C-NMR (CDCl3) δ 198.2, 148.6, 146.8, 144.5, 126.8, 124.1, 119.1, 114.8, 109.8, 60.8, 56.0, 14.1; HRMS [ESI]+ calculated for C23H24NaO6: 419.1471 [M + Na]+; found: 419.1480
(1E,6E)-4,4-Diethyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (3m)
Yield: 87%; mp = 114–115 °C; 1H-NMR (CDCl3) δ 7.64 (d, J = 15.5 Hz, 2H), 7.05 (dd, J = 8.3, 1.5 Hz, 2H), 6.95 (d, J = 2.0 Hz, 2H), 6.85 (d, J = 8.0 Hz, 2H), 6.59 (d, J = 15.5 Hz, 2H), 5.88 (s, 2H), 3.88 (s, 6H), 2.06 (q, J = 7.5 Hz, 4H), 0.71 (t, J = 7.5 Hz, 6H); 13C-NMR (CDCl3) δ 197.9, 148.5, 146.7, 144.0, 126.8, 124.2, 119.6, 114.7, 109.7, 69.0, 56.1, 21.6, 7.7; HRMS [ESI]+ calculated for C25H28NaO6: 447.1784 [M + Na]+; found: 447.1788.
(1E,6E)-4,4-Dibenzyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (4m)
Yield: 82%; mp = 134–135 °C; 1H-NMR (CDCl3) δ 7.72 (d, J = 15.0 Hz, 2H), 7.26–7.21 (m, 6H), 7.15–7.13 (m, 4H), 7.07–7.05 (m, 2H), 6.90–6.89 (m, 4H), 6.58 (dd, J = 15.5, 1.0 Hz, 2H), 5.31 (s, 2H), 3.89 (s, 6H), 3.42 (s, 4H); 13C-NMR (CDCl3) δ 196.9, 148.7, 146.8, 144.0, 136.6, 130.4, 128.1, 126.8, 126.6, 124.3, 120.8, 114.8, 109.8, 70.1, 56.1, 37.7; HRMS [ESI]+ calculated for C25H32NaO6: 571.2097 [M + Na]+; found: 571.2090.
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-di(prop-2-yn-1-yl)hepta-1,6-diene-3,5-dione (5m)
Yield: 83%; mp = 201–203 °C; 1H-NMR (CDCl3) δ 7.75 (d, J = 15.0 Hz, 2H), 7.13 (dd, J = 8.0, 1.5 Hz, 2H), 7.03 (d, J = 2.0 Hz, 2H), 6.92 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 15.0 Hz, 2H), 5.97 (s, 2H), 3.95 (s, 6H), 3.19 (s, 4H), 2.04 (s, 4H); 13C-NMR (CDCl3) δ 193.5, 148.9, 145.9, 126.5, 124.7, 117.8, 114.8, 109.8, 79.1, 71.9, 66.9, 56.1, 21.2; HRMS [ESI]+ calculated for C27H28O6: 471.1784 [M + Na]+; found: 471.1783.
(1E,6E)-4,4-Diallyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (6m)
Yield: 85%; mp = 160–162 °C; 1H-NMR (CDCl3) δ 7.65 (d, J = 15.5 Hz, 2H), 7.06 (dd, J = 8.3, 1.5 Hz, 2H), 6.95 (d, J = 2.0 Hz, 2H), 6.85 (d, J = 8.0 Hz, 2H), 6.61 (d, J = 15.5 Hz, 2H), 5.87 (s, 2H), 5.54–5.49 (m, 2H), 5.07–5.04 (m 4H), 3.88 (s, 6H), 2.78 (d, J = 7.5 Hz, 4H); 13C-NMR (CDCl3) δ 194.6, 148.7, 146.8, 144.6, 132.3, 126.7, 119.3, 119.1, 114.7, 109.7, 67.8, 56.1, 34.4; HRMS [ESI]+ calculated for C27H24NaO6: 467.1471 [M + Na]+; found: 467.1477.
3.1.9. General Procedure for the Synthesis of Compounds 15 and 16
The general procedure is illustrated below with compound 15 as a specific example. To a solution of curcumin bisacetate 17 (0.76 g, 1.68 mmol) in DMF (8 mL) was added K2CO3 (0.58 g, 4.20 mmol) and propyl bromide (0.435 g, 3.53 mmol) sequentially at 0 °C. The reaction mixture was stirred at room temeperatre for 20 h and then H2O (10 mL) and diluted HCl(aq) (1 N, 5 mL) were added to quench the reaction. The aqueous layer was separated and extracted with EtOAc (3 × 10 mL). The combined organic layer was dried over MgSO4, filtered and concentrated to give the crude product, which was subjected without any purification to the base-promoted hydrolysis reaction [NaOMe (0.190 g, 3.53 mmol), MeOH (5 mL)]. The reaction was followed by TLC until no starting material was present. The resulting mixture was then concentrated to give the crude product, which was then purified by flash chromatography on silica gel with EtOAc/n-hexane/CH2Cl2 (1:2:1) to afford compound 15 (0.213 g, 28% yield for two steps) as a yellow solid.
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dipropylhepta-1,6-diene-3,5-dione (15)
mp = 137–139 °C; 1H-NMR (CDCl3) δ 7.69 (d, J = 15.0 Hz, 2H), 7.11 (dd, J = 8.3, 1.5 Hz, 2H), 7.01 (d, J = 1.5 Hz, 2H), 6.91 (d, J = 8.0 Hz, 2H), 6.65 (d, J = 15.5 Hz, 2H), 5.90 (s, 2H), 3.94 (s, 6H), 2.05–2.01 (m, 4H), 1.15–1.08 (m, 4H), 0.95 (t, J = 7.5 Hz, 6H); 13C-NMR (CDCl3) δ 197.9, 148.5, 146.7, 144.0, 126.8, 124.1, 119.5, 114.7, 109.7, 68.4, 56.1, 31.8, 16.8, 14.7; HRMS [ESI]+ calculated for C27H32NaO6: 475.2097 [M + Na]+; found: 475.2100.
(1E,6E)-4,4-Dibutyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (16)
mp = 144–146 °C; 1H-NMR (CDCl3) δ 7.68 (d, J = 15.5 Hz, 2H), 7.11 (dd, J = 8.3, 1.5 Hz, 2H), 7.01 (s, 2H), 6.91 (d, J = 8.2 Hz, 2H), 6.65 (d, J = 15.5 Hz, 2H), 6.01 (s, 2H), 3.93 (s, 6H), 2.07–2.03 (m, 4H), 1.37–1.33 (m, 4H), 1.10–1.03 (m, 4H), 0.90 (t, J = 7.3 Hz, 6H); 13C-NMR (CDCl3) δ 198.0, 148.5, 146.8, 144.0, 126.9, 124.1, 119.5, 114.7, 109.7, 68.3, 56.1, 29.1, 25.5, 23.2, 13.9; HRMS [ESI]+ calculated for C29H36NaO6: 503.2410 [M + Na]+; found: 503.2400.
3.1.10. (E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylhept-1-ene-3,5-dione (18)
To a stirred solution of compound 2m (0.740 g, 1.87 mmol) in EtOAc (10 mL) was added Pd/C (37 mg, 5% w/w) in one portion. The resulting mixture was hydrogenated under 1 atmosphere of H2 at room temperature. After 1 h, the mixture was filtered by celite and concentrated to give the crude product, which was purified by flash chromatography on silica gel with EtOAc/n-hexane/CH2Cl2 (1:2:1) to afford compound 18 (140 mg, 19% yield) as a light-yellow oil and recover compound 2m (500 mg, 67% yield). 1H-NMR (CDCl3) δ 7.61 (d, J = 15.5 Hz, 1H), 7.07 (dd, J = 8.5, 1.5 Hz, 1H), 6.95–6.93 (m, 2H), 6.75 (d, J = 8.0 Hz, 1H), 6.65–6.62 (m, 2H), 6.49 (d, J = 15.5 Hz, 1H), 6.07 (s, 1H), 5.45 (s, 1H), 3.94 (s, 3H), 3.79 (s, 3H), 2.83 (t, J = 7.5 Hz, 2H), 2.74 (t, J = 7.0 Hz, 2H), 1.38 (s, 6H); 13C-NMR (CDCl3) δ 209.4, 197.7, 148.7, 146.8, 146.3, 144.7, 143.8, 132.6, 126.9, 123.9, 121.0, 118.2, 114.9, 114.2, 111.0, 110.0, 61.7, 56.0, 55.7, 40.7, 29.6, 21.0; HRMS [ESI]+ calculated for C23H26NaO6: 421.1627 [M + Na]+; found: 421.1233.
3.1.11. 1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylheptane-3,5-dione (19) and 5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-4,4-dimethylheptan-3-one (20)
To a stirred solution of compound 2m (0.885 g, 2.23 mmol) in MeOH (10 mL) was added Pd/C (45 mg, 10% w/w) in one portion. The resulting mixture was hydrogenated under 1 atmosphere of H2 at room temperature. After 2 h, the mixture was filtered by celite and concentrated to give the crude product, which was purified by flash chromatography on silica gel with EtOAc/n-hexane/ (1:2) to afford compound 19 (465 mg, 52% yield) as a colorless oil and compound 20 (206 mg, 23% yield) as a colorless oil.
Compound 19: 1H-NMR (CDCl3) δ 6.83 (d, J = 8.0 Hz, 2H), 6.65 (d, J = 2.0 Hz, 2H), 6.63–6.61 (m, 2H), 5.51 (s, 2H), 3.87 (s, 6H), 2.76 (t, J = 7.0 Hz, 4H), 2.58 (t, J = 7.0 Hz, 4H), 1.29 (s, 6H); 13C-NMR (CDCl3) δ 208.9, 146.4, 143.9, 132.7, 120.9, 114.3, 114.9, 111.7, 62.4, 55.9, 40.5, 29.4, 21.0; HRMS [ESI]+ calculated for C23H28NaO6: 423.1784 [M + Na]+; found: 423.1780.
Compound 20: 1H-NMR (CDCl3) δ 6.86–6.83 (m, 2H), 6.72–6.66 (m, 4H), 5.62–5.61 (br s, 2H), 3.89 (s, 3H), 3.87 (s, 3H), 3.71–3.69 (m, 1H), 2.90–2.71 (m, 5H), 2.61–2.51 (m, 2H), 1.71–1.55 (m, 2H), 1.13 (s, 3H), 1.08 (s, 3H); 13C-NMR (CDCl3) δ 216.5, 146.4, 143.9, 143.7, 133.9, 133.1, 120.9, 120.8, 114.4, 114.3, 111.2, 111.1, 55.9, 51.7, 40.3, 33.7, 32.6, 29.4, 21.7, 19.3; HRMS [ESI]+ calculated for C23H30NaO6: 425.4768 [M + Na]+; found: 425.4760.
3.1.12. 1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylheptane-3,5-diol (21)
To a solution of compound 20 (110 mg, 0.27 mmol) in MeOH (3 mL) was added NaBH4 (30 mg, 0.81 mmol) at 0 °C. The resulting mixture was allowed to stir at room temperature for 2 h, and then H2O (3 mL) was added. The aqueous layer was separated and extracted with CH2Cl2 (3 × 6 mL). The combined organic extracts were washed brine, dried over MgSO4, filtered and concentrated to give the crude residue, which was purified by flash chromatography on silica gel with EtOAc/n-hexane/ (1:1) to afford compound 21 (34 mg, 31% yield) as a colorless oil. 1H-NMR (CDCl3) δ 6.86 (d, J = 8.0 Hz, 2H), 6.74–6.71 (m, 4H), 5.63 (s, 2H), 3.89 (s, 6H), 3.59–3.56 (m, 2H), 3.11 (s, 2H), 2.85–2.71 (m, 2H), 2.61–2.55 (m, 2H), 1.81–1.72 (m, 4H), 0.90 (s, 6H); 13C-NMR (CDCl3) δ 146.5, 143.7, 134.1, 134.0, 120.9, 114.3, 111.1, 78.7, 55.9, 40.3, 33.9, 32.8, 21.0; HRMS [ESI]+ calculated for C23H32NaO6: 427.4928 [M + Na]+; found: 427.4933.
3.2. Biological Assays
3.2.1. In Vitro MTT [3-(4,5-Dimethylthiazaol-2-yl)-2,4-diphenyltetrazolium bromide] Assay
Cell viability was evaluated by measuring the reduction in MTT to yield blue formazan. Cells were cultured in 96-well plates, allowed to attach overnight, and then treated with compounds. After treatment, MTT solution (1 mg/mL) was added to each well, and plates were incubated for another 2 h. Medium was removed, blue formazan was dissolved in DMSO, and the absorbance was read at 570 nm by Multiskan Go spectrophotometer (Thermo Scientific, Madison, WI, USA).
3.2.2. In Vivo Antitumor Activity Assay
Male nu/nu mice (5 weeks old) from National Laboratory Animal Center (Taipei, Taiwan) were maintained under the procedures and guidelines provided by the Institutional Animal Care and Use Committee of the National Health Research Institutes (Taipei, Taiwan). All experiments were supervised under the Institutional Animal Care and Use Committee, China Medical University, Taichung, Taiwan with a protocol number (CMUIACUC-2018-278). HCT-116 colon cancer cells (5 × 106 cells per mouse) were suspended in 0.1 mL of Matrigel solution (50% v/v Matrigel in PBS) and inoculated into the mammary fat pads of the mice. When the tumor size reached 100 mm3, the tumor-bearing mice were randomly divided into four groups for treating with vehicle (5% ethanol and 10% tween 80 in 85% saline, 0.1 mL) and 2 (p.o. 50, 100 and 150 mg/kg/day body weight) respectively. Compounds was administered via oral route daily for 30 days. 5-Fluorouracil-treated group (i.p. 30 mg/kg) is the positive control. Tumor size and mouse body weight were measured once every 3 days, and tumor volume (mm3) was calculated using the equation: length × (width)2 × 0.5. At the end of experiments, mice were sacrificed.
3.2.3. The Preliminary Pharmacokinetic Evaluation of 2 in Rat
Drug administration and blood collection
Male Sparague-Dawley rat (420 g) from National Laboratory Animal Center, Taipei, Taiwan were maintained under the regulations of the Institutional Animal Care and Use Committee of the National Health Research Institutes, Taiwan. The animal studies were supervised under the Institutional Animal Care and Use Committee, China Medical University, Taichung, Taiwan with a protocol number (CMUIACUC-2018-278). Rat was fasted for 16 h before dosing. Compound 2 was dissolved in aqueous solution (5% ethanol and 10% Tween80 in 85% saline) and administrated via oral gavage at a dosage of 100 mg/kg. Blood samples were collected at selected time points (5, 10, 20, 30, 60, 120, 240, 360, 480 min) post dose and centrifuged at 10,000× g for 15 min to provide serum samples, which were stored at −80 °C until subsequent sample extraction.
3.2.4. Analysis of 2, 2m, 18, 19, 20, 21, 22 and 23 in Serum
Methods for the preparation of 2, 2m, 18, 19, 20 and 21 serum sample are delineated as follows. 50 μL of acetate buffer (pH 5.0), 50 μL of ascorbic acid (200 mg/mL), and 50 μL of 0.1 N HCl solution was added into 100 μL of serum which was collected at different time points). The resulting mixture was partitioned with 250 μL of ethyl acetate (containing 5.0 μg/mL of butyl paraben as the internal standard). After centrifuging at 10,000× g for 15 min, the upper organic layer was separated and dried under nitrogen gas to provide the crude sample, which was diluted with an appropriate volume of acetonitrile for LC-MS analysis.
The amount of 22 and 23 were analyzed by the serum samples before and after treatment with enzyme solution. The procedure for enzyme treatment is shown as follows. 100 μL of serum was treated with enzyme solution (50 μL; containing 1000 units/mL of sulfatase and 39861 units/mL of β-glucuronidase in pH 5.0 acetate buffer), 50 μL of 0.1 N HCl solution and 50 μL of ascorbic acid (200 mg/mL) in the light protected tubes at 37 °C. For optimal hydrolysis efficiency, the incubation time was decided as 2 h based on a previous study [35]. The mixture was then partitioned with 250 μL of ethyl acetate (containing 5.0 μg/mL of butyl paraben as the internal standard). After centrifuging at 10,000× g for 15 min, the upper organic layer was separated and dried under nitrogen gas to provide the crude sample, which was diluted with an appropriate volume of acetonitrile for LC analysis.
3.2.5. LC-MS Analysis Method
All samples obtained from in vivo pharmacokinetic studies were assayed using LC-MS for the analysis of 2 and the corresponding metabolites. The Waters ACQUITY UPLC I-Class system (Waters Corp., Milford, MA, USA) was utilized. System control and all of the mass spectrometry data were acquired and analyzed using UNIFI software (Waters Corp.). For sample separation, the LC was equipped with an Inertsil ph-3 (50 × 2.1 mm, 2 μm particle size) RPLC column (GL Sciences Inc. Tokyo, Japan) in which the column temperature was held at 35 °C, injection volume was 7.5 μL, and the flow rate of the mobile phase was 200 μL/min. The elution started from 50% mobile phase A (ultrapure water + 0.1% formic acid) and 50% mobile phase B (100% methanol + 0.1% formic acid), held at 50% B for 0.5 min, raised to 95% B in 5.5 min, held at 95% B for 1 min, and then lowered to 50% B in 1 min. Before sample injection, the column was equilibrated by pumping 50% B for 4 min. The analysis of target compounds for all samples was performed by using a Waters Vion IMS QTof mass spectrometer. Data were acquired in the electrospray ionization (ESI) positive ion MSE mode with range of m/z 100–1000 and 0.5 s scan time. Parameters were set as capillary voltage of 2.5 kV, source temperature of 100 °C, desolvation temperature at 250 °C, cone gas maintained at 10 L/h, desolvation gas maintained at 600 L/h. The acquired m/z and isotope pattern were processed by UNIFI software to illustrate chromatogram, and the amounts of compound was calculated with the integrated peak area of signals.
4. Conclusions
Here, structural modifications at the C-4 position of compound 1, a 2,2-bis(hydroxymethyl)-propionate curcumin prodrug, with polar and nonpolar functional groups were performed to form a series of 4,4-disubstituted analogs of 1 and DMC. The in vitro anti-proliferative assay of these derivatives elucidated an obvious SAR in which the enhancement of polarity on C-4 resulted in the erosion of anticancer activity. The ester prodrug characteristics of 2–6 motivated us to synthesize their hydrolysis compounds 2m–6m, 15, and 16, whose anticancer activities were highly dependent on the volume and length of the C-4 alkyl chains. The preliminary pharmacokinetic evaluations indicated that the esterase-mediated hydrolysis of 2 proceeded smoothly to produce 2m, which in turn was reduced to 18 and 19 as major phase I metabolites. The phase II metabolic pathway was confirmed by the detection of glucuronide 22 and sulfate 23. However, it is still possible that reduction precedes ester hydrolysis to provide the hydrogenated counterparts of 2, which possess novel chemical skeletons with unknown biological activity.
Our preliminary pharmacokinetic results also shed light on future directions for the structural modification of the original compounds. Additional modifications have been scheduled to reduce the high propensity towards ester hydrolysis by converting the methyl of 2,2-bis(hydroxymethyl)-propionate group into more bulky alkyl groups. Pharmacokinetic studies have been designed to identify, biologically evaluate, and quantify the possible metabolites. Apart from this, compound 2 and 2m may behave like 1 which acts on multiple targets and signaling pathways. The differences between the mechanism of action of 2, 2m and 1 are not definitively understood. Currently, we are actively investigating the mechanism of action and pharmacokinetic parameters of 2 and 2m, including T1/2, AUC, and Cmax, and the results will be reported in due course. The animal study showed that compound 2 inhibits tumor growth by 45% at a dose of 100 mg/kg in HCT-116 xenograft nude mice. Thus, twice-a-day (BID) dosing formulations for achieving higher therapeutic efficacy at lower doses should be considered in further development of title curcuminoid derivatives as anticancer drug candidates.
Supplementary Materials
The following are available online. Scheme S1: PLE mediated hydrolysis of curcumin 2,2-bis(hydroxymethyl)propionate 1. Figure S1: The HPLC analysis of compound 18, 19, 20, 21, 22 and 23. Copies of 1H and 13C NMR spectra of the new products.
Author Contributions
D.-Y.L. performed the preliminary pharmacokinetic analysis and wrote experimental section and supplementary material file. Y.-C.H. designed the preliminary pharmacokinetic study and integrated the experimental data. J.-S.Y. and H.-Y.L. performed the in vivo assay and in vivo animal study. K.-H.L. and T.-Y.C. reviewed and revised the manuscript. S.-C.K. designed the target compounds and interpreted the experimental data. M.-T.H. designed and synthesized targeted compounds. S.-C.K. and M.-T.H. are the responsible researchers, who wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to Ministry of Science and Technology, Taiwan (MOST 107-2320-B-039-028) for financial support. Partial support was also provided by the “Chinese Medicine Research Center, China Medical University” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (CMRC-CHM-5) awarded to S. C. Kuo.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Strimpakos A.S., Sharma R.A. Curcumin: Preventive and therapeutic properties in laboratory studies and clinical trials. Antioxid. Redox Signal. 2008;10:511–546. doi: 10.1089/ars.2007.1769. [DOI] [PubMed] [Google Scholar]
- 2.Nabavi S.F., Thiagarajan R., Rastrelli L., Daglia M., Sobarzo-Sánchez E., Alinezhad H., Nabavi S.M. Curcumin: A natural product for diabetes and its complications. Curr. Top Med. Chem. 2015;15:2445–2455. doi: 10.2174/1568026615666150619142519. [DOI] [PubMed] [Google Scholar]
- 3.Mathew D., Hsu W.L. Antiviral potential of curcumin. J. Funct. Foods. 2018;40:692–699. doi: 10.1016/j.jff.2017.12.017. [DOI] [Google Scholar]
- 4.Sahebkar A., Henrotin Y. Analgesic efficacy and safety of curcuminoids in clinical practice: A systematic review and meta-analysis of randomized controlled trials. Pain Med. 2016;17:1192–1202. doi: 10.1093/pm/pnv024. [DOI] [PubMed] [Google Scholar]
- 5.Osawa T. Nephroprotective and hepatoprotective effects of curcuminoids. Adv. Exp. Med. Biol. 2007;595:407–423. doi: 10.1007/978-0-387-46401-5_18. [DOI] [PubMed] [Google Scholar]
- 6.Miriyala S., Panchatcharam M., Rengarajulu P. Cardioprotective effects of curcumin. Adv. Exp. Med. Biol. 2007;595:359–377. doi: 10.1007/978-0-387-46401-5_16. [DOI] [PubMed] [Google Scholar]
- 7.Shishodia S. Molecular mechanisms of curcumin action: Gene expression. Biofactors. 2013;39:37–55. doi: 10.1002/biof.1041. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S.C., Prasad S., Kim J.H., Patchva S., Webb L.J., Priyadarsini I.K., Aggarwal B.B. Multitargeting by curcumin as revealed by molecular interaction studies. Nat. Prod. Rep. 2011;28:1937–1955. doi: 10.1039/c1np00051a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bordoloi D., Roy N.K., Monisha J., Padmavathi G., Kunnumakkara A.B. Multi-targeted agents in cancer cell chemosensitization: What we learn from curcumin thus far. Recent Pat. Anti-Canc. 2016;11:67–97. doi: 10.2174/1574892810666151020101706. [DOI] [PubMed] [Google Scholar]
- 10.Kunnumakkara A.B., Anand P., Aggarwal B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Nelson K.M., Dahlin J.L., Bisson J., Graham J., Pauli G.F., Walters M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017;60:1620–1637. doi: 10.1021/acs.jmedchem.6b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schieffer G.W. Pressurized liquid extraction of curcuminoids and curcuminoid degradation products from turmeric (curcuma longa) with subsequent HPLC assays. J. Liq. Chromatogr. Relat. Technol. 2002;25:3033–3044. doi: 10.1081/JLC-120015889. [DOI] [Google Scholar]
- 13.Schneider C., Gordon O.N., Edwards R.L., Luis P.B. Degradation of curcumin: From mechanism to biological implications. J. Agric. Food Chem. 2015;63:7606–7614. doi: 10.1021/acs.jafc.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Priyadarsini K.I., Maity D.K., Naik G.H., Kumar M.S., Unnikrishnan M.K., Satav J.G., Mohan H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003;35:475–484. doi: 10.1016/S0891-5849(03)00325-3. [DOI] [PubMed] [Google Scholar]
- 15.Sharma R.A., Steward W.P., Gescher A.J. Pharmacokinetics and pharmacodynamics of curcumin. Adv. Exp. Med. Biol. 2007;595:453–470. doi: 10.1007/978-0-387-46401-5_20. [DOI] [PubMed] [Google Scholar]
- 16.Ireson C., Orr S., Jones D.J., Verschoyle R., Lim C.K., Luo J.L., Howells L., Plummer S., Jukes R., Williams M., et al. Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res. 2001;61:1058–1064. [PubMed] [Google Scholar]
- 17.Rodrigues F.C., Anil Kumar N.A., Thakur G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J Med. Chem. 2019;177:76–104. doi: 10.1016/j.ejmech.2019.04.058. [DOI] [PubMed] [Google Scholar]
- 18.Tomeh M.A., Hadianamrei R., Zhao X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019;20:1033. doi: 10.3390/ijms20051033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Martino R.M.C., De Simone A., Andrisano V., Bisignano P., Bisi A., Gobbi S., Rampa A., Fato R., Bergamini C., Perez D.I., et al. Versatility of the curcumin scaffold: Discovery of potent and balanced dual BACE-1 and GSK-3b inhibitors. J. Med. Chem. 2016;59:531–544. doi: 10.1021/acs.jmedchem.5b00894. [DOI] [PubMed] [Google Scholar]
- 20.Noureddin S.A., El-Shishtawy R.M., Al-Footy K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019;82:11631–11671. doi: 10.1016/j.ejmech.2019.111631. [DOI] [PubMed] [Google Scholar]
- 21.Barzegar A., Moosavi-Movahedi A.A. Intracellular ROS protection efficiency and free radical-scavenging activity of curcumin. PLoS ONE. 2011;6:e26012. doi: 10.1371/journal.pone.0026012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epstein J., Sanderson I.R., Macdonald T.T. Curcumin as a therapeutic agent: The evidence of in vitro, animal and human studies. Br. J. Nutr. 2010;103:1545–1557. doi: 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- 23.For the clinical progress of curcumin, refer to https://clinicaltrials.gov/
- 24.Hsieh M.T., Chang L.C., Hung H.Y., Lin H.Y., Shih M.H., Tsai C.H., Kuo S.C., Lee K.H. New bis(hydroxymethyl) alkanoate curcuminoid derivatives exhibit activity against triple-negative breast cancer in vitro and in vivo. Eur. J Med. Chem. 2017;131:141–151. doi: 10.1016/j.ejmech.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Berry L.M., Wollenberg L., Zhao Z. Esterase activities in the blood, liver and intestine of several preclinical species and humans. Drug Metab. Lett. 2009;3:70–77. doi: 10.2174/187231209788654081. [DOI] [PubMed] [Google Scholar]
- 26.Shi Q., Shih C.C., Lee K.H. Novel anti-prostate cancer curcumin analogues that enhance androgen receptor degradation activity. Anticancer Agents Med. Chem. 2009;9:904–912. doi: 10.2174/187152009789124655. [DOI] [PubMed] [Google Scholar]
- 27.2,2,5-trimethyl-1,3-dioxane-5-carbonyl chloride 13 was formed by treating the acid counterpart with thionyl chloride. For detailed procedure, see: R. Giri, X. Chen, J.Q. Yu, Palladium-catalyzed asymmetric iodination of unactivated C-H bonds under mild conditions. Angew Chem. Int. Ed. Engl. 2005;29:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 28.Basile V., Ferrari E., Lazzari S., Belluti S., Pignedoli F., Imbriano C. Curcumin derivatives: Molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009;78:1305–1315. doi: 10.1016/j.bcp.2009.06.105. [DOI] [PubMed] [Google Scholar]
- 29.Prasad S., Tyagi A.K., Aggarwal B.B. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: The golden pigment from golden spice. Cancer Res. Treat. 2014;46:2–18. doi: 10.4143/crt.2014.46.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liederer B.M., Borchardt R. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006;95:1177–1195. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]
- 31.Kunihiro A.G., Luis P.B., Brickey J.A., Julia A., Jen B.F., Chow H.H.S., Schmeider C., Funk J.L. Beta-glucuronidase catalyzes deconjugation and activation of curcumin-glucuronide in bone. J. Nat. Prod. 2019;82:500–509. doi: 10.1021/acs.jnatprod.8b00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozawa H., Imaizumi A., Sumi Y., Hashimoto T., Kanai M., Makino Y., Tsuda T., Takahashi N., Kakeya H. Curcumin β-D-glucuronide plays an important role to keep high levels of free-form curcumin in the blood. Biol. Pharm. Bull. 2017;40:1515–1524. doi: 10.1248/bpb.b17-00339. [DOI] [PubMed] [Google Scholar]
- 33.Ge S., Tu Y., Hu M. Challenges and opportunities with predicting in vivo phase II metabolism via glucuronidation from in vitro data. Curr. Pharmacol. Rep. 2016;2:326–338. doi: 10.1007/s40495-016-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang K.Y., Lin L.C., Tseng T.Y., Wang S.C., Tsai T.H. Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC−MS/MS. J. Chromatogr. B Anal. Technol.Biomed. Life Sci. 2007;853:183–189. doi: 10.1016/j.jchromb.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh Y.W., Huang C.Y., Yang S.Y., Peng Y.H., Yu C.P., Lee Chao P.D., Chi H.Y. Oral intake of curcumin markedly activated CYP 3A4: In vivo and ex-vivo studies. Sci. Rep. 2014;4:6587–6594. doi: 10.1038/srep06587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.