

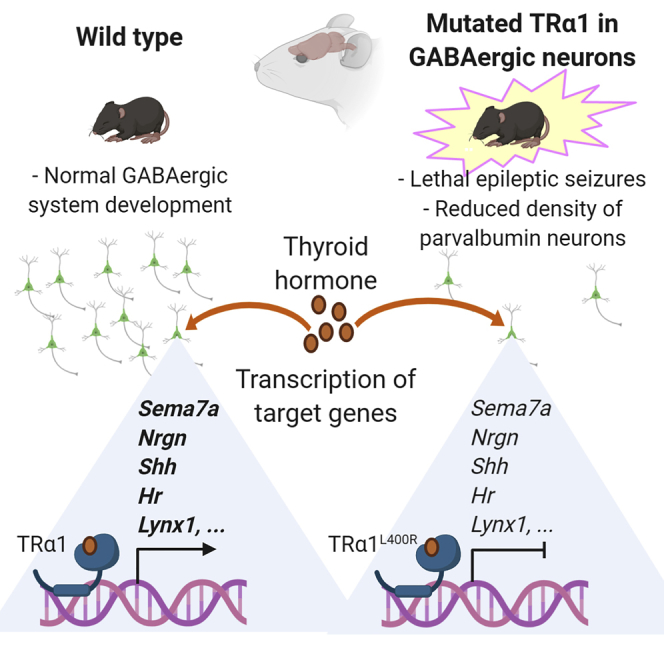
Summary
Mammalian brain development critically depends on proper thyroid hormone signaling, via the TRα1 nuclear receptor. The downstream mechanisms by which TRα1 impacts brain development are currently unknown. In order to investigate these mechanisms, we used mouse genetics to induce the expression of a dominant-negative mutation of TRα1 specifically in GABAergic neurons, the main inhibitory neurons in the brain. This triggered post-natal epileptic seizures and a profound impairment of GABAergic neuron maturation in several brain regions. Analysis of the transcriptome and TRα1 cistrome in the striatum allowed us to identify a small set of genes, the transcription of which is upregulated by TRα1 in GABAergic neurons and which probably plays an important role during post-natal maturation of the brain. Thus, our results point to GABAergic neurons as direct targets of thyroid hormone during brain development and suggest that many defects seen in hypothyroid brains may be secondary to GABAergic neuron malfunction.
Subject Areas: Neuroscience, Molecular Neuroscience, Developmental Neuroscience
Graphical Abstract
Highlights
-
•
GABAergic neurons are a direct target of thyroid hormone in the developing brain
-
•
Impaired TH/TRα1 signaling severely impairs the development of GABAergic neurons
-
•
GABAergic-specific genes directly targeted by TH/TRα1 signaling have been listed
-
•
A defect in the GABAergic system is expected in patients with a THRA mutation
Neuroscience; Molecular Neuroscience; Developmental Neuroscience
Introduction
Thyroid hormones (TH, including thyroxine, or T4, and 3,3′,5-triiodo-L-thyronine, or T3, its active metabolite) exert a broad influence on neurodevelopment. If untreated soon after birth, congenital hypothyroidism, i.e. early TH deficiency, affects brain development and a number of cognitive functions (Rovet, 2014). Severe cases display mental retardation, autism spectrum disorders (ASD), and epilepsy (Fetene et al., 2017). TH mainly acts by binding to nuclear receptors called TRα1, TRβ1, and TRβ2, which are encoded by the THRA and THRB genes (Thra and Thrb in mice, formerly TRα and TRβ). These receptors form heterodimers with other nuclear receptors, the retinoid X receptors (RXRs), and bind chromatin at specific locations (thyroid hormone response elements), acting as TH-dependent transcription activators of neighboring genes. In the developing brain, the predominant type of receptor is TRα1 (Bradley et al., 1989). Accordingly, THRA germline mutations, which have currently been reported in only 45 patients, cause a syndrome named RTHα (resistance to thyroid hormone due to THRA mutations), resembling congenital hypothyroidism, with mental retardation and a high occurrence of ASD and epilepsy (van Gucht et al., 2017).
Cellular alterations caused by early TH deficiency or germline mutations have been extensively studied in rodents. Many neurodevelopmental processes depend on proper TH signaling, and virtually all glial and neuronal cell populations are affected by TH deficiency (Berbel et al., 2014, Bernal et al., 2003). However, in the mouse cerebellum, we have previously found that, although Thra expression is ubiquitous in this brain region, only a subset of cell types displayed a direct, cell-autonomous, response to TH (Fauquier et al., 2014). More specifically, Cre/loxP technology, used to express a dominant-negative variant of TRα1 (TRα1L400R), has provided genetic evidence that the cell-autonomous influence of TRα1 is limited to astrocytes and GABAergic neurons (Fauquier et al., 2014). By altering the differentiation of GABAergic neurons, TRα1L400R prevented the secretion of several growth factors and neurotrophins. This indirectly altered the proliferation and differentiation of granule cells and oligodendrocytes (Picou et al., 2012, Picou et al., 2014). Therefore, GABAergic neurons occupy a pivotal position during cerebellum development, amplifying the initial TH signal. This allows TH to synchronize cellular interactions and the maturation of neuronal networks during the first post-natal weeks (Flamant et al., 2017). As defects in TH signaling are known to alter GABAergic neurons outside the cerebellum (Berbel et al., 1996, Harder et al., 2018, Wallis et al., 2008), we asked whether the direct role of TH in GABAergic neurons, initially observed in the cerebellum, could be generalized to other brain regions.
In the present study, we used the same genetic strategy to block TH response in the entire GABAergic lineage by expressing TRα1L400R from early developmental stages specifically in GABAergic neurons in all brain areas. This had dramatic neurodevelopmental consequences on the development of the GABAergic system and caused lethal epileptic seizures. Genome-wide analyses allowed us to pinpoint the genetic defects induced by the mutation and to identify a small set of genes activated by TH in GABAergic neurons. These genes are likely to play a key role in the neurodevelopmental function of TH.
Results
Mouse Models Designed to Target TRα1 in GABAergic Neurons
We generated new mouse models by combining existing and novel “floxed” Thra alleles with the Gad2Cre transgene (Figure 1A). This transgene drives the expression of Cre recombinase in all GABAergic neurons and their progenitors from an early prenatal stage (around E12.5) (Taniguchi et al., 2011). In the context of the modified Thra alleles used in the present study, Cre recombinase eliminates a transcriptional stop cassette and triggers the expression of TRα1 variants. The ThraAMI allele (formerly TRαAMI) (Quignodon et al., 2007) encodes TRα1L400R (Figure 1B), which exerts a permanent transcriptional repression on target genes, even in the presence of TH. This is due to its inability to recruit transcription coactivators, which normally interact with the C-terminal helix (AA 398-407) of TRα1, and permanent interaction with transcription corepressors (Figure S1). This allelic design, which eliminates alternate splicing, increases the expression of the mutant receptor over that of the wild-type receptor, as shown by comparing peak surfaces after Sanger sequencing: in the striatum of ThraAMI/gn mice, the ThraAMI allele represents 65 ± 4% of all Thra transcripts (mean ± standard deviation, n = 4; see also (Markossian et al., 2018)). This ensures a complete inhibition of TH response in heterozygous cells (Markossian et al., 2018). As complete deprivation of TH (Mansouri et al., 1998), ubiquitous expression of TRα1L400R results in the death of heterozygous mice 2–3 weeks after birth (Quignodon et al., 2007). The second modified Thra allele, named ThraSlox, differs from ThraAMI only by an additional frameshift mutation, which eliminates the C-terminal helix of TRα1 (Markossian et al., 2018). As for ThraAMI, expression of the ThraSlox allele exceeds that of the wild-type Thra allele: in the striatum of ThraSlox/gn mice, the ThraSlox allele represents 59 ± 1% of all Thra transcripts (mean ± standard deviation, n = 6). The ThraSlox allele encodes TRα1E395fs401X (Figure 1B), which is nearly identical to a pathological variant found in a patient (van Mullem et al., 2012, van Mullem et al., 2014). TRα1E395fs401X is expected to be functionally equivalent to TRα1L400R and behaves similarly in in vitro assays (Markossian et al., 2018). The third modified Thra allele used in the present study is ThraTAG, a novel construct that encodes a functional receptor, TRα1TAG, with a fragment of protein G at its N-terminus (Burckstummer et al., 2006). This tag has a high affinity for IgGs, which makes it suitable to address chromatin occupancy (Chatonnet et al., 2013). Transient expression assays show that the N-terminal GS tag does not impair the transactivation capacity of TRα1 (Figure S2). Double heterozygous mice, combining the presence of GADad2Cre and of a modified Thra allele, express TRα1L400R, TRα1E395fs401X, or TRα1TAG in the GABAergic cell lineage only. They will be respectively designated as ThraAMI/gn, ThraSlox/gn, and ThraTAG/gn in the following,/gn being used to indicate that the modified Thra alleles were expressed specifically in GABAergic neurons. In all phenotyping experiments, littermates carrying only ThraAMI, ThraSlox, or GAD2Cre were used as controls.
Figure 1.

Thra Alleles and Survival Curves
(A) Schematic representation of Thra alleles in ThraAMI, ThraSlox and ThraTAG mice. In all 3 alleles, the coding sequence is preceded with a floxed stop cassette (PGKNeo Poly(A). The intronless structure eliminates alternate splicing and internal promoter and thus prevents the production of TRα2, TRΔα1, and TRΔα2 non-receptor protein. The dispensable IRES Tau-lacZ reporter part was not included in the ThraTAG construct.
(B) C-terminal amino acid sequence of Thra gene products used in the present study, starting from AA393. Shaded amino acids differ from wild-type TRα1. ThraAMI mutation results in a single amino acid substitution within TRα1 helix 12. ThraSlox mutation is a deletion resulting in a +1 frameshift, leading to elimination of helix 12, as in several RTHα patients.
(C) Survival curves of mice expressing a mutated TRα1 in GABAergic neurons (green and red lines) and of control littermates (black line).
See also Figures S1–S3 and Videos S1 and S2.
Post-natal Lethality Caused by Thra Mutations in GABAergic Neurons
Born at the expected frequency, ThraAMI/gn mice did not usually survive beyond the third post-natal week (Figure 1C). Video recording of litters in their home cage indicated that most mice started to display epileptic seizures a few days before death. Occasionally, sudden death was observed at the end of a seizure. In most cases, seizures impeded maternal care and this likely precipitated the death of the pups (Videos S1 and S2). Although lethality was also observed in ThraSlox/gn mice (Figure 1C), about one-third of these mice survived into adulthood. Adult ThraSlox/gn mice did not display any obvious epileptic seizure anymore. However, their locomotor behavior was significantly altered, as evidenced in an open-field test (Figure S3). These observations show that, although the two mutations are expected to be equivalent, TRα1E395fs401X is less detrimental than TRα1L400R.
The seizure occurred during the dark phase of the photoperiod and was video recorded using infrared lighting.
Note the unsuccessful attempts of the mother to drag the pup back to the nest.
A Global Impairment in the Differentiation of GABAergic Neurons
In order to label neurons of the GABAergic lineage in a generic way, we combined the ThraAMI and Gad2Cre alleles with the Rosa-tdTomato transgene, which enabled to trace the cells in which Cre/loxP recombination had taken place. The density of tdTomato + cells was not reduced in ThraAMI/gn Rosa-tdTomato mice, arguing against a possible alteration in the proliferation, migration, or survival of GABAergic neuron progenitors. In the hippocampus, notably in the dentate gyrus, the number of tdTomato + cells was even increased (Figure 2).
Figure 2.

Expression of the tdTomato Fluorescent Protein in TRαAMI/gnRosa-tdTomato and Control Littermates at PND14
(A) Low-magnification images illustrating the relative fluorescence intensity in the cortex (Cx), striatum (Str), and hippocampus (Hp) in GAD2Cre Rosa-tdTomato mice.
(B) Representative images allowing to compare the density of tdTomato positive cells in selected brain regions in ThraAMI/gnRosa-tdTomato and control littermates (GAD2Cre Rosa-tdTomato).
(C) Relative density of tdTomato + cells in ThraAMI/gnRosa-tdTomato (“M” in the graph stands for mutants, filled triangles; red lines for the mean and standard deviation) and control littermates (“C”, empty circles; blue lines for the mean and standard deviation) at PND14. *p < 0.05.
We used immunohistochemistry to detect alterations of GABAergic neuron differentiation at postnatal day 14 (PND14) in various brain areas (quantitative data in Tables 1 and S1). Parvalbumin (PV) immunostaining, which labels major populations of GABAergic neurons in several brain areas (DeFelipe et al., 2013), revealed a defect in Purkinje cell arborization and a deficit in basket and stellate GABAergic interneurons in ThraAMI/gn cerebellum as expected from previous data (Fauquier et al., 2011, Markossian et al., 2018) (Figure S4). A drastic reduction in the density of PV + neurons (90%–95% reduction relative to controls) was also visible in the hippocampus, cortex, and striatum of ThraAMI/gn mice (Figure 3A). As a complement to PV labeling, we used Wisteria floribunda lectin (WFA) to label perineuronal nets, as it has been previously reported that a reduction in PV + neuron numbers may be accompanied by a persistence of these extracellular matrix structures (Filice et al., 2016). In ThraAMI/gn mice, WFA labeling was drastically reduced compared with that of control mice, but perineuronal nets were observed around PV-negative cell bodies (Figure S5), suggesting that absence of PV labeling was not necessarily a sign of PV neuron loss. We also used antibodies directed against calretinin (CR), somatostatin (SST), and neuropeptide Y (NPY) to label other key populations of GABAergic neurons (Kepecs and Fishell, 2014). All GABAergic neuron subtypes investigated were affected but in a rather complex pattern. The density of CR + neurons was increased in the cortex and CA2-CA3 area of the hippocampus. In the hippocampal dentate gyrus, where CR + neurons are normally concentrated in the granular cell layer, the limits of this layer were ill-defined and CR + neurons spread into the molecular and polymorph layers (Figure 3B). The density of SST + neurons was also augmented in hippocampal dentate gyrus but not significantly altered in the cortex or striatum (Figure 4A). The density of NPY + neurons was significantly reduced in the cortex, but not in the striatum and hippocampus, where, by contrast, NPY immunoreactivity of the fibers increased (Figures 4B and Table 1).
Table 1.
Relative Abundance of Several GABAergic Neuron Subtypes in ThraAMI/gn, Compared with Control, Mouse Brains at PND14, as Evidenced by Immunohistochemistry
| Cortex |
Hippocampus (DG) |
Hippocampus (CA) |
Striatum |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | ThraAMI/gn | Control | ThraAMI/gn | Control | ThraAMI/gn | Control | ThraAMI/gn | ||
| Parvalbumin neuronal density | Mean | 1.00 | 0.05a | 1.00 | 0.05a | 1.00 | 0.33a | 1.00 | 0.10a |
| SD | 0.14 | 0.01 | 0.50 | 0.13 | 0.34 | 0.21 | 0.48 | 0.15 | |
| n | 11 | 10 | 11 | 12 | 11 | 10 | 10 | 8 | |
| Neuropeptide Y neuronal density | Mean | 1.00 | 0.58a | 1.00 | 0.92 | ND | ND | 1.00 | 1.07 |
| SD | 0.00 | 0.11 | 0.00 | 0.06 | ND | ND | 0.11 | 0.20 | |
| n | 5 | 6 | 3 | 3 | ND | ND | 5 | 6 | |
| Neuropeptide Y fluorescence intensity | Mean | ND | ND | 1.00 | 1.23a | ND | ND | 1.00 | 1.31a |
| SD | ND | ND | 0.00 | 0.08 | ND | ND | 0.06 | 0.25 | |
| n | ND | ND | 3 | 3 | ND | ND | 6 | 6 | |
| Calretinin neuronal density | Mean | 1.00 | 0.75 | ND | ND | 1.00 | 4.90a | ND | ND |
| SD | 0.12 | 0.54 | ND | ND | 0.41 | 4.10 | ND | ND | |
| n | 5 | 4 | ND | ND | 5 | 4 | ND | ND | |
| Somatostatin neuronal density | Mean | 1.00 | 1.12 | 1.00 | 2.55a | 1.00 | 1.08 | 1.00 | 1.16 |
| SD | 0.02 | 0.31 | 0.14 | 0.66 | 0.13 | 0.29 | 0.03 | 0.19 | |
| n | 6 | 5 | 6 | 5 | 6 | 5 | 6 | 5 | |
See also Table S1.
Significantly different from control (p < 0.05).
Figure 3.

Immunohistochemistry for Parvalbumin and Calretinin
Immunohistochemistry for parvalbumin (A) and calretinin (B) in PND14 ThraAMI/gn and control mouse pups in selected brain regions. Right panels: scatterplots illustrating the relative density of immunoreactive cells in control (C, empty circles; blue lines for the mean and standard deviation) and mutant (M, filled triangles; red lines for the mean and standard deviation) mice. Calretinin-immunoreactive neurons could not be quantified in the dentate gyrus, due to the difficulty in delineating individual cells in this area. *p < 0.05. See also Figures S4–S7.
Figure 4.

Immunohistochemistry for Somatostatin and Neuropeptide Y
Immunohistochemistry for somatostatin (A) and neuropeptide Y (B) in PND14 ThraAMI/gn and control mouse pups in the striatum, hippocampus, and cortex. Right panels: scatterplots illustrating the relative density of immunoreactive cells in control (C, empty circles; blue lines for the mean and standard deviation) and mutant (M, filled triangles; red lines for the mean and standard deviation) mice. *p < 0.05. See also Figures S6 and S7.
Most of the above-mentioned immunohistochemistry experiments were also carried out in ThraSlox/gn mice at PND14. In all instances, the defects observed in GABAergic neuron populations were the same as in ThraAMI/gn mice (details in Table S1 and Figure S6). In the surviving ThraSlox/gn adult mice, PV and NPY immunohistochemistry indicated that the differentiation of neurons expressing these markers was not only delayed, but permanently impaired (Figure S7 and Table S1). Taken together, these data indicate that expressing a mutant TRα1 alters the late steps of development of GABAergic neurons, reducing the numbers of PV+ and NPY + cells in some brain areas, while favoring an expansion of the SST+ and CR + cell populations. In the hippocampus, this is accompanied by subtle morphological alterations.
Changes in Gene Expression Caused by TRα1L400R in GABAergic Neurons
Because the impairment in GABAergic neuron differentiation does not appear to be restricted to a specific brain area or a specific neuronal GABAergic subpopulation, we hypothesized that a general mechanism might underlie the involvement of TH signaling in the terminal maturation of GABAergic neurons. In order to decipher this mechanism and identify the TRα1 target genes in GABAergic neurons, we first compared the transcriptome of the cortex in ThraAMI/gn and control littermates at two different post-natal stages, PND7 and PND14, where we previously found clear histological alterations. Differential gene expression analysis pointed out a set of 58 genes whose expression was deregulated in ThraAMI/gn mouse cortex, compared with age-matched control mice. Clustering analysis outlined contrasting patterns of regulations (Figure S8). Exploring available single-cell RNAseq data (https://singlecell.broadinstitute.org) obtained from adult visual cortex (Tasic et al., 2016) suggests that a large part of the observed variations reflect the alterations in neuronal maturation already revealed by immunohistochemistry. For example, many downregulated genes are preferentially expressed in PV + neurons (Akr1c18, Dusp10, Eya4, Flywch2, Me3, Ntn4, Pcp4L1, Ppargc1a, Pvalb, Stac2, Syt2) whereas most overexpressed genes are preferentially expressed in SST+ and/or VIP + GABAergic interneurons (Calb1, Cxcl14, Hap1, Klhl14, Pcdh8, Prox1, Rbp4, Rxfp3, Sema3c, Sst, Tac2, Zcchc12). Interestingly, the analysis also revealed an upregulation of Thra at PND7 in control mice, which was absent in mutant mice. However, we expected a larger set of differentially expressed genes on the basis of previous in vitro analysis (Gil-Ibanez et al., 2014). This probably results from the relatively low representation of GABAergic neurons among cortical cells. We thus decided to focus our investigation on the striatum, where the high abundance of GABAergic neurons, mainly spiny projection neurons, facilitates analysis. As expected, the same differential gene expression analysis identified a larger set of differentially expressed genes in the striatum (191 genes). A single factor Deseq2 analysis pointed out 126 genes whose expression was deregulated at PND7 in the striatum of ThraAMI/gn mice, compared with age-matched control mice. At PND14, this number raised to 215 genes. Overall, 260 genes were found to be deregulated in the striatum of ThraAMI/gn mice at either stage. Eight of these genes (Cxcl14, Flywch2, Glra3, Ppargc1a, Prox1, Pvalb, Sst, Syt2) were also deregulated in the cortex. Hierarchical clustering analysis of the striatum data mainly revealed a sharp transition between PND7 and PND14 in the striatum of control mice, which did not take place in ThraAMI/gn mice (Figure 5).
Figure 5.

Differentially Expressed Genes in the Striatum
Hierarchical clustering analysis of differentially expressed transcripts in the striatum of ThraAMI/gn mice and control littermates at PND7 and PND14. The analysis is restricted to 260 genes for which the fold-change is > 2 or <0.5 (adjusted p value < 0.05) for at least one developmental stage. High expression is in yellow, low expression is in blue, average in black. Note that the changes in gene expression between PND7 and PND14 are more conspicuous in control than in mutant mice, suggesting that a maturation process is blunted by the mutation. See also Figure S8 and Tables S2 and S4.
These differences in gene expression, as measured by Ampliseq, may have two origins. They can reflect deregulations of the expression of TRα1 target genes in GABAergic neurons, but they can also reflect various indirect consequences of these deregulations, such as a change in the composition of the cell population. In order to pinpoint the TRα1 target genes within the set of differentially expressed genes in striatum, we crossed the above results with a dataset obtained in wild-type mice, comparing different hormonal statuses. We assumed that the expression of TRα1 target genes should be quickly modified by changes in TH levels, whereas indirect consequences should be much slower. In the dataset, 181 genes were found to be deregulated in the striatum of hypothyroid mice, whereas 86 genes responded to a 2-day TH treatment of hypothyroid mice (Dataset S1). RT-qPCR was used to confirm some of these observations (Tables S2–S4). In this dataset, however, the response of GABAergic neurons to TH cannot be distinguished from the response of other cell types present in the striatum, which also express TRα1. The overlap between the two datasets pointed out a set of 38 TH-activated genes whose expression pattern suggested a direct regulation by TRα1 in GABAergic neurons, whereas only 1 gene displayed an expression pattern that suggests a negative regulation by T3-bound TRα1 (Figure 6A).
Figure 6.

Identifying a Core Set of TRα1 Target Genes in GABAergic Neurons of the Striatum by Combining RNAseq and Chip-Seq Analyses
(A) RNAseq identifies a set of 38 genes, the expression pattern of which is fully consistent with a positive regulation by TRα1, and only 1 gene that has the opposite expression pattern.
(B) Extract of the Mus musculus genome browser, around the Hr gene, a well-characterized TRα1 target gene. The 3 upper boxes indicate TRBSs identified as significant by the MACS2 algorithm. Note that a DR4-like element (lower track, red asterisks), as defined below, is found in only one of the 3 peaks.
(C) Consensus sequence found in TRBSs identified by de novo motif search is close to the DR4 consensus (5′AGGTCANNNNAGGTCA-3′).
(D) Combinations of RNAseq and Chip-Seq data. In the Venn diagrams, each fraction gives the number of genes with a proximal TRBS (<30 kb for transcription start site, large lettering) and, among these genes, those in which a DR4 element was identified (small lettering). A set of 35 genes fulfill the criteria for being considered as genuine TRα1 target genes: they are downregulated in hypothyroid and mutant mice and upregulated after TH treatment of hypothyroid mice. For 17 of these genes, the TRBS contains a recognizable DR4-like element.
See also Data S1 and Tables S2–S5.
Identification of Direct TRα1 Target Genes in GABAergic Neurons of the Striatum
To complete the identification of TRα1 target genes in striatal GABAergic neurons, we analyzed chromatin occupancy by TRα1 at a genome-wide scale by ChIPseq. Using ThraTAG/gn mice, we precipitated the DNA/protein complexes, which contain the tagged TRα1 from the whole striatum, to address chromatin occupancy in GABAergic neurons only. This experiment revealed the existence of 7,484 chromatin sites occupied by TRα1TAG (thyroid hormone receptor binding sites = TRBSs) in the genome (Figure 6B). In agreement with our previous study (Chatonnet et al., 2013) de novo motif discovery (http://meme-suite.org/tools/meme-chip) and enrichment analysis revealed a single consensus sequence for the binding of TRα1/RXR heterodimers. The sequence is the so-called DR4 element (Figure 6C). Assuming that proximity (<30kb) between the TRα1TAG binding site and the transcription start site is sufficient for direct transcriptional regulation by TRα1 would lead to consider a large fraction of genes as putative target genes (3,979/23,931 annotated genes in the mouse genome mm10 version; 16.6%). Among the 38 genes that are sensitive to TRα1L400R expression and hypothyroidism and responsive to TH in hypothyroid mice, genes with a proximal TRBS were overrepresented (35/38: 92%; enrichment of 5.5 compared with the whole set of annotated genes). Interestingly, this enrichment was more striking if we considered only the TRBSs where a DR4 element was identified (3,813/7,484; 51%): DR4 elements were present within 30 kb of 7.5% of annotated genes (1,786/23,931) and of 45% of the putative TRα1 target genes identified in the present study (17/38) (Figure 6D). The same reasoning leads to the conclusion that the transcription of genes that are downregulated after T3 treatment is not regulated by chromatin-bound TRα1 (Figure 6D). Overall, the ChipSeq dataset suggests that a large fraction of the TRBS does not reflect the binding of TRα1/RXR heterodimers to DR4 elements but corresponds to other modes of chromatin association, which do not necessarily promote TH-mediated transactivation. However, as we cannot rule out that other types of response elements are also used, this analysis leaves us with 35 genes, which meet all the criteria for being considered as TRα1 direct target genes. Although some of these genes are known to have a neurodevelopmental function, they do not fall into a specific ontological category (Table S5). This implies that TH promotes GABAergic neuron maturation by simultaneously acting on different cell compartments and cellular pathways.
Discussion
Using two mouse models expressing mutant forms of TRα1 specifically in GABAergic neurons, we present evidence showing that TH, bound to TRα1, is required for the late steps of development of GABAergic neurons. As in the present study we have used GABAergic-specific somatic mutations, we can ascertain that the observed defects are cell-autonomous consequences of impaired TH signaling. We found that the defect in GABAergic differentiation is not restricted to a specific brain area, nor to a specific subtype of GABAergic neurons, as several subtypes of both projecting neurons and interneurons were affected by TRα1 mutations. This suggests that, although GABAergic neurons of different brain areas have different embryonal origins (Leto et al., 2006, Marin and Muller, 2014), they share a common pathway of maturation that depends on TH/TRα1 signaling. Transcriptome analysis revealed a significant overlap between the regulated genes of mutant mice in the cortex, where most GABAergic neurons are interneurons, and in the striatum, which is mainly populated by projecting GABAergic neurons, i.e., medium spiny neurons.
These results extend previous findings, obtained either in hypothyroid rodents or in mice with Thra mutations, demonstrating the role of TH in GABAergic neurons in the cerebellum (Fauquier et al., 2011, Manzano et al., 2007), striatum (Diez et al., 2008), cortex (Wallis et al., 2008), hippocampus (Navarro et al., 2015), and hypothalamus (Harder et al., 2018). The major contribution of the present work is to demonstrate that the effect of TH/TRα1 on GABAergic neuron development is cell-autonomous. In many respects, neurodevelopmental damage caused by TRα1L400R and TRα1E395fs401X appears to be similar to, but more dramatic than, that reported for the ubiquitous TRα1R384C mutation (Wallis et al., 2008), which is impaired in its affinity for TH, but possesses a residual capacity to transactivate gene expression (Tinnikov et al., 2002). The effects reported here with Thra knock-in mutations are also congruent with, although much stronger than, those previously reported in the brains of Thra KO mice (Guadano-Ferraz et al., 2003). This difference is not a surprise and has been previously shown to derive from the permanent transcriptional repression exerted by TRα1L400R and TRα1E395fs401X in presence or absence of TH (Flamant et al., 2017). The transcriptional repression effect exerted by unliganded TRα is completely lost in Thra KO mice, resulting in a much milder phenotype.
The present data indicate that TRα1 plays a major role in the late steps of development of several categories of GABAergic neurons. This is most obvious for PV + interneurons, which almost disappear from several brain areas in ThraAMI/gn mice. However, their progenitors appear to be present, as evidenced by the use of tdTomato as a reporter for cells of the GABAergic lineage. Thus, we can exclude that TRα1 plays a major role in the first steps of GABAergic neuron development, i.e., progenitor proliferation and migration.
In the hippocampus, the density of tdTomato+ and CR + cells was higher in ThraAMI/gn mice than in controls, and the limits of the granular layer were blurred. These results are congruent with previous studies. Indeed, blurring of the borders of the granular layers has been observed in hypothyroid rats (Navarro et al., 2015). An increase in CR + cells has also been reported in the hippocampus of mice expressing TRαR384C (Hadjab-Lallemend et al., 2010). Thus, we can speculate that TH signaling in GABAergic neurons plays a role in the process of lamination in the hippocampus. In particular, the increase in tdTomato + cells in the hippocampus of ThraAMI/gn mice may result from impaired cell apoptosis, possibly affecting CR neurons.
The fate of the progenitors that fail to express PV in ThraAMI/gn mice is unclear. One hypothesis is that they commit to a different GABAergic lineage. This could notably explain the excess of SST + cells in the hippocampus, because PV+ and SST + cortical interneurons share the same precursors (Hu et al., 2017, Mukhopadhyay et al., 2009). However, the results obtained in the cortex do not support such hypothesis, because the density of SST + cells in the cortex did not differ between ThraAMI/gn mice and their control littermates. An alternative hypothesis would be that the effects observed in different categories of GABAergic neurons are secondary to the near disappearance of PV + interneurons. Indeed, many defects caused by hypothyroidism in the brain are indirect, some of them being secondary to a defect in neurotrophin secretion in the microenvironment (Bernal, 2002, Giordano et al., 1992, Neveu and Arenas, 1996, Yu et al., 2015).
Our genome-wide search pinpointed a small set of genes fulfilling the criteria, which lead us to consider them as genuine TRα1 target genes in GABAergic neurons: (1) the mRNA level of these genes is TH responsive, (2) it is decreased in the striatum of ThraAMI/gn mice, and (3) TRα1 occupies a chromatin binding site located at a limited distance of their transcription start site. The last criterion is important, as it helps to differentiate between direct and indirect influences of TRα1 on gene regulation. Thus, we have addressed chromatin occupancy by TRα1 in vivo, in the striatum. Importantly, we have used a genetic strategy enabling to selectively identify GABAergic neuron-specific TRα1 binding to DNA within a heterogeneous tissue. The large number of chromatin binding sites that we have identified (7,484 TRBS) contrasts with the small set of 35 genes that we have identified as being directly regulated by TRα1 in GABAergic neurons. As we have used stringent statistical thresholds, we have probably overlooked some genuine TRα1 genes. For example, Klf9 is a known target gene (Chatonnet et al., 2015), which is not present in our list, due to a below-threshold downregulation in the striatum of hypothyroid mice. However, even with liberal statistical thresholds, the number of presumptive target genes would not exceed 100, a number which is still small compared with the number of genes with a proximal TRBS. Such a contrast has previously been observed in other systems (Ayers et al., 2014, Chatonnet et al., 2013, Grontved et al., 2015, Ramadoss et al., 2014) and suggests that only a small fraction of the TRBSs are involved in TH-mediated transactivation. Finally, the low level of correspondence between TRBSs and genes that are downregulated after T3 treatment (Figure 6D) suggests that the negative regulation of gene expression exerted by TH is not directly mediated by chromatin-bound TRα1, but perhaps an indirect consequence of the upregulation of transcription inhibitors. Further investigations will be required to better define these active TRBSs and better establish the correspondence between chromatin occupancy and transcriptional regulation by TRα1.
Most of the TRα1 target genes identified in the striatum have already been identified as being sensitive to the local TH level in various brain areas and at different developmental stages (see Table S1 in Chatonnet et al., 2015). This reinforces the hypothesis that they belong to a common genetic program that is regulated by TH, via TRα1, and that promotes the proper maturation of several categories of GABAergic neurons. Although their function in neurons is for a large part unknown, these genes can be grouped according to the putative function of their protein products: Shh and Fgf16 encode secreted proteins, which play major roles in cellular interactions. Sema7a and Nrtn are involved in axon growth and pathfinding. Others are likely to define the electrophysiological properties of neurons by encoding ion channels (Kctd17), transporters of small metabolites (Slc22a3, Slc26a10), or modulators of synaptic activity (Nrgn, Lynx1).
Overall, the broad influence of TRα1 mutations on GABAergic neuron differentiation and maturation is expected to greatly and permanently impair brain function, notably in the cortex, where a subtle equilibrium between different GABAergic neuron subtypes is necessary for normal development and plasticity (Butt et al., 2017). In the mouse models presented here, epileptic seizures appear to be a main cause of mortality, which sheds light on the cause of the lethality that had been previously observed in mice with different Thra mutations (Fraichard et al., 1997, Kaneshige et al., 2001, Tinnikov et al., 2002) as well as in mice suffering from complete TH deprivation (Flamant et al., 2002, Mansouri et al., 1998). Of note, the increase in NPY intensity that we have reported in the striatum and hippocampus of mutant mice may be secondary to the occurrence of epileptic seizures in these mice. Indeed, epileptic seizures are known to increase the expression of NPY in various brain regions, including the hippocampus (Husum et al., 2002, Vezzani and Sperk, 2004). The epileptic phenotype induced in mice by expressing a Thra mutation in GABAergic neurons is highly relevant to human pathology, as a history of epilepsy has been reported for several of the rare patients with a THRA mutation (Moran and Chatterjee, 2016). Autism spectrum disorders (ASD), whose comorbidity with epilepsy is well documented, have also been reported in these patients (Kalikiri et al., 2017, Yuen et al., 2015). It is likely that these pathological traits are also due to a defect in GABAergic neuron maturation, and our data suggest that these patients might benefit from a treatment with GABA receptor agonists.
Limitations of the Study
The modified Thra alleles used in the present study (ThraAMI, ThraSlox and ThraTAG) result from an extensive remodeling of the Thra gene, which eliminates all the Thra-encoded proteins except for the mutated receptor. We have shown previously that such elimination of alternate splicing from the Thra locus resulted in a moderate overexpression of the mutant allele, compared with the wild-type allele. This overexpression is likely to lead to exaggerated phenotypic manifestations (Markossian et al., 2018). Such exaggerated phenotype has proved efficient in bringing to light key mechanisms by which TH/TRα1 signaling impacts brain development, but the clinical relevance of these mouse models is questionable. Importantly, a similar, albeit milder phenotype has been evidenced in GABAergic neurons of CRISPR/Cas9-generated mice with Thra mutations, which are more relevant models of the human RTHα disease (Markossian et al., 2018). Moreover, the epileptic phenotype observed in ThraAMI/gn and ThraSlox/gn mice appears to be congruent with the high occurrence of epilepsy, which has been reported in patients with RTHα (Moran and Chatterjee, 2015, van Gucht et al., 2017). Thus, the mouse models used in the present study appear as relevant for the study of the mechanisms of TH/TRα1 during brain development, even though they do not faithfully mimic the human disease.
A second limitation lies in the use of TRBSs as indicators of direct TRα1 target genes. The conventional strategy, which only takes into account the distance between a TRBS and the nearest transcription start site, has limited value, as nuclear receptors sometimes act at very long distances due to chromosomal looping (Bagamasbad et al., 2008, Buisine et al., 2015, Buisine et al., 2018) Further investigations will be required to better define the correspondence between chromatin occupancy by TRα1 and transcriptional regulation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Catherine Etter, Lucas Jacquin, FlorentDelannoy, and Victor Valcárcel, who contributed to histological studies, TiphanyLaurens, who contributed to behavioral analyses, and Karine Gauthier, who performed in vivo experiments with propylthiouracil/TH treatments. We thank Nadine Aguilera, Marie Teixeira, and the ANIRA-PBES facility for help in transgenesis and mouse breeding. We also thank Benjamin Gillet, Sandrine Hughes, and the PSI platform of IGFL for deep DNA sequencing. Finally, we thank Emmanuel Quemener from Center Blaise Pascal/ENSL for the development and maintenance of the ENS Galaxy portal with the help of SIDUS (Single Instance Distributing Universal System). This work was supported by grants from the French Agence Nationale de la Recherche (Thyromut2 program; ANR-15-CE14-0011-01) and from the European Union's Horizon 2020 research and innovation program under grant agreement no. 825753 (ERGO).
Author Contributions
FF, SR, and RG conceived the study. SM and DA created the genetically modified mouse lines. SR characterized the histological and behavioral phenotype of the mice. RG, MRM, MP, and FF carried out the transcriptome and ChipSeq experiments. FF and SR wrote the manuscript. All authors reviewed and commented on the final manuscript.
Declaration of Interests
The authors declare no competing interest.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100899.
Data and Code Availability
RNASeq and ChipSeq data are accessible through GEO Series accession number GSE143933 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE143933).
Supplemental Information
References
- Ayers S., Switnicki M.P., Angajala A., Lammel J., Arumanayagam A.S., Webb P. Genome-wide binding patterns of thyroid hormone receptor Beta. PLoS One. 2014;9:e81186. doi: 10.1371/journal.pone.0081186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagamasbad P., Howdeshell K.L., Sachs L.M., Demeneix B.A., Denver R.J. A role for basic transcription element-binding protein 1 (BTEB1) in the autoinduction of thyroid hormone receptor beta. J. Biol. Chem. 2008;283:2275–2285. doi: 10.1074/jbc.M709306200. [DOI] [PubMed] [Google Scholar]
- Berbel P., Marco P., Cerezo J.R., DeFelipe J. Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci. Lett. 1996;204:65–68. doi: 10.1016/0304-3940(96)12318-1. [DOI] [PubMed] [Google Scholar]
- Berbel P., Navarro D., Roman G.C. An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Front. Endocrinol. 2014;5:146. doi: 10.3389/fendo.2014.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal J. Action of thyroid hormone in brain. J. Endocrinol. Invest. 2002;25:268–288. doi: 10.1007/BF03344003. [DOI] [PubMed] [Google Scholar]
- Bernal J., Guadano-Ferraz A., Morte B. Perspectives in the study of thyroid hormone action on brain development and function. Thyroid. 2003;13:1005–1012. doi: 10.1089/105072503770867174. [DOI] [PubMed] [Google Scholar]
- Bradley D.J., Young W.S., 3rd, Weinberger C. Differential expression of alpha and beta thyroid hormone receptor genes in rat brain and pituitary. Proc. Natl. Acad. Sci. U S A. 1989;86:7250–7254. doi: 10.1073/pnas.86.18.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisine N., Ruan X., Bilesimo P., Grimaldi A., Alfama G., Ariyaratne P., Mulawadi F., Chen J., Sung W.K., Liu E.T. Xenopus tropicalis genome Re-scaffolding and Re-annotation reach the resolution required for in vivo ChIA-PET analysis. PLoS One. 2015;10:e0137526. doi: 10.1371/journal.pone.0137526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisine N., Ruan X., Ruan Y., Sachs L.M. Chromatin interaction analysis using paired-end-tag (ChIA-PET) sequencing in tadpole tissues. Cold Spring Harb. Protoc. 2018 doi: 10.1101/pdb.prot104620. [DOI] [PubMed] [Google Scholar]
- Burckstummer T., Bennett K.L., Preradovic A., Schutze G., Hantschel O., Superti-Furga G., Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- Butt S.J., Stacey J.A., Teramoto Y., Vagnoni C. A role for GABAergic interneuron diversity in circuit development and plasticity of the neonatal cerebral cortex. Curr. Opin. Neurobiol. 2017;43:149–155. doi: 10.1016/j.conb.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Chatonnet F., Flamant F., Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim. Biophys. Acta. 2015;1849:122–129. doi: 10.1016/j.bbagrm.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Chatonnet F., Guyot R., Benoit G., Flamant F. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc. Natl. Acad. Sci. U S A. 2013;110:E766–E775. doi: 10.1073/pnas.1210626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J., Lopez-Cruz P.L., Benavides-Piccione R., Bielza C., Larranaga P., Anderson S., Burkhalter A., Cauli B., Fairen A., Feldmeyer D. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 2013;14:202–216. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez D., Grijota-Martinez C., Agretti P., De Marco G., Tonacchera M., Pinchera A., de Escobar G.M., Bernal J., Morte B. Thyroid hormone action in the adult brain: gene expression profiling of the effects of single and multiple doses of triiodo-L-thyronine in the rat striatum. Endocrinology. 2008;149:3989–4000. doi: 10.1210/en.2008-0350. [DOI] [PubMed] [Google Scholar]
- Fauquier T., Chatonnet F., Picou F., Richard S., Fossat N., Aguilera N., Lamonerie T., Flamant F. Purkinje cells and Bergmann glia are primary targets of the TRalpha1 thyroid hormone receptor during mouse cerebellum postnatal development. Development. 2014;141:166–175. doi: 10.1242/dev.103226. [DOI] [PubMed] [Google Scholar]
- Fauquier T., Romero E., Picou F., Chatonnet F., Nguyen X.N., Quignodon L., Flamant F. Severe impairment of cerebellum development in mice expressing a dominant-negative mutation inactivating thyroid hormone receptor alpha1 isoform. Dev. Biol. 2011;356:350–358. doi: 10.1016/j.ydbio.2011.05.657. [DOI] [PubMed] [Google Scholar]
- Fetene D.M., Betts K.S., Alati R. Mechanisms IN endocrinology: maternal thyroid dysfunction during pregnancy and behavioural and psychiatric disorders of children: a systematic review. Eur. J. Endocrinol. 2017;177:R261–R273. doi: 10.1530/EJE-16-0860. [DOI] [PubMed] [Google Scholar]
- Filice F., Vorckel K.J., Sungur A.O., Wohr M., Schwaller B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain. 2016;9:10. doi: 10.1186/s13041-016-0192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamant F., Gauthier K., Richard S. Genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr. Top. Dev. Biol. 2017;125:303–335. doi: 10.1016/bs.ctdb.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Flamant F., Poguet A.L., Plateroti M., Chassande O., Gauthier K., Streichenberger N., Mansouri A., Samarut J. Congenital hypothyroid Pax8(-/-) mutant mice can be rescued by inactivating the TRalpha gene. Mol. Endocrinol. 2002;16:24–32. doi: 10.1210/mend.16.1.0766. [DOI] [PubMed] [Google Scholar]
- Fraichard A., Chassande O., Plateroti M., Roux J.P., Trouillas J., Dehay C., Legrand C., Gauthier K., Kedinger M., Malaval L. The T3R alpha gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J. 1997;16:4412–4420. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Ibanez P., Bernal J., Morte B. Thyroid hormone regulation of gene expression in primary cerebrocortical cells: role of thyroid hormone receptor subtypes and interactions with retinoic acid and glucocorticoids. PLoS One. 2014;9:e91692. doi: 10.1371/journal.pone.0091692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano T., Pan J.B., Casuto D., Watanabe S., Arneric S.P. Thyroid hormone regulation of NGF, NT-3 and BDNF RNA in the adult rat brain. Brain Res. Mol. Brain Res. 1992;16:239–245. doi: 10.1016/0169-328x(92)90231-y. [DOI] [PubMed] [Google Scholar]
- Grontved L., Waterfall J.J., Kim D.W., Baek S., Sung M.H., Zhao L., Park J.W., Nielsen R., Walker R.L., Zhu Y.J. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat. Commun. 2015;6:7048. doi: 10.1038/ncomms8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadano-Ferraz A., Benavides-Piccione R., Venero C., Lancha C., Vennstrom B., Sandi C., DeFelipe J., Bernal J. Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol. Psychiatry. 2003;8:30–38. doi: 10.1038/sj.mp.4001196. [DOI] [PubMed] [Google Scholar]
- Hadjab-Lallemend S., Wallis K., van Hogerlinden M., Dudazy S., Nordstrom K., Vennstrom B., Fisahn A. A mutant thyroid hormone receptor alpha1 alters hippocampal circuitry and reduces seizure susceptibility in mice. Neuropharmacology. 2010;58:1130–1139. doi: 10.1016/j.neuropharm.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Harder L., Dudazy-Gralla S., Muller-Fielitz H., Hjerling Leffler J., Vennstrom B., Heuer H., Mittag J. Maternal thyroid hormone is required for parvalbumin neurone development in the anterior hypothalamic area. J. Neuroendocrinol. 2018;30:e12573. doi: 10.1111/jne.12573. [DOI] [PubMed] [Google Scholar]
- Hu J.S., Vogt D., Sandberg M., Rubenstein J.L. Cortical interneuron development: a tale of time and space. Development. 2017;144:3867–3878. doi: 10.1242/dev.132852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husum H., Gruber S.H., Bolwig T.G., Mathe A.A. Extracellular levels of NPY in the dorsal hippocampus of freely moving rats are markedly elevated following a single electroconvulsive stimulation, irrespective of anticonvulsive Y1 receptor blockade. Neuropeptides. 2002;36:363–369. doi: 10.1016/s0143-4179(02)00086-0. [DOI] [PubMed] [Google Scholar]
- Kalikiri M.K., Mamidala M.P., Rao A.N., Rajesh V. Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients. Autism Res. 2017;10:1919–1928. doi: 10.1002/aur.1838. [DOI] [PubMed] [Google Scholar]
- Kaneshige M., Suzuki H., Kaneshige K., Cheng J., Wimbrow H., Barlow C., Willingham M.C., Cheng S. A targeted dominant negative mutation of the thyroid hormone alpha 1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc. Natl. Acad. Sci. U S A. 2001;98:15095–15100. doi: 10.1073/pnas.261565798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A., Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto K., Carletti B., Williams I.M., Magrassi L., Rossi F. Different types of cerebellar GABAergic interneurons originate from a common pool of multipotent progenitor cells. J. Neurosci. 2006;26:11682–11694. doi: 10.1523/JNEUROSCI.3656-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A., Chowdhury K., Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat. Genet. 1998;19:87–90. doi: 10.1038/ng0598-87. [DOI] [PubMed] [Google Scholar]
- Manzano J., Cuadrado M., Morte B., Bernal J. Influence of thyroid hormone and thyroid hormone receptors in the generation of cerebellar gamma-aminobutyric acid-ergic interneurons from precursor cells. Endocrinology. 2007;148:5746–5751. doi: 10.1210/en.2007-0567. [DOI] [PubMed] [Google Scholar]
- Marin O., Muller U. Lineage origins of GABAergic versus glutamatergic neurons in the neocortex. Curr. Opin. Neurobiol. 2014;26:132–141. doi: 10.1016/j.conb.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markossian S., Guyot R., Richard S., Teixeira M., Aguilera N., Bouchet M., Plateroti M., Guan W., Gauthier K., Aubert D. CRISPR/Cas9 editing of the mouse Thra gene produces models with variable resistance to thyroid hormone. Thyroid. 2018;28:139–150. doi: 10.1089/thy.2017.0389. [DOI] [PubMed] [Google Scholar]
- Moran C., Chatterjee K. Resistance to thyroid hormone due to defective thyroid receptor alpha. Best Pract. Res. Clin. Endocrinol. Metab. 2015;29:647–657. doi: 10.1016/j.beem.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran C., Chatterjee K. Resistance to thyroid hormone alpha-emerging definition of a disorder of thyroid hormone action. J. Clin. Endocrinol. Metab. 2016;101:2636–2639. doi: 10.1210/jc.2016-2317. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A., McGuire T., Peng C.Y., Kessler J.A. Differential effects of BMP signaling on parvalbumin and somatostatin interneuron differentiation. Development. 2009;136:2633–2642. doi: 10.1242/dev.034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro D., Alvarado M., Navarrete F., Giner M., Obregon M.J., Manzanares J., Berbel P. Gestational and early postnatal hypothyroidism alters VGluT1 and VGAT bouton distribution in the neocortex and hippocampus, and behavior in rats. Front. Neuroanat. 2015;9:9. doi: 10.3389/fnana.2015.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu I., Arenas E. Neurotrophins promote the survival and development of neurons in the cerebellum of hypothyroid rats in vivo. J.Cell Biol. 1996;133:631–646. doi: 10.1083/jcb.133.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picou F., Fauquier T., Chatonnet F., Flamant F. A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol. Endocrinol. 2012;26:608–618. doi: 10.1210/me.2011-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picou F., Fauquier T., Chatonnet F., Richard S., Flamant F. Deciphering direct and indirect influence of thyroid hormone with mouse genetics. Mol. Endocrinol. 2014;28:429–441. doi: 10.1210/me.2013-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignodon L., Vincent S., Winter H., Samarut J., Flamant F. A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol. Endocrinol. 2007;21:2350–2360. doi: 10.1210/me.2007-0176. [DOI] [PubMed] [Google Scholar]
- Ramadoss P., Abraham B.J., Tsai L., Zhou Y., Costa-e-Sousa R.H., Ye F., Bilban M., Zhao K., Hollenberg A.N. Novel mechanism of positive versus negative regulation by thyroid hormone receptor beta1 (TRbeta1) identified by genome-wide profiling of binding sites in mouse liver. J. Biol. Chem. 2014;289:1313–1328. doi: 10.1074/jbc.M113.521450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovet J.F. The role of thyroid hormones for brain development and cognitive function. Endocr. Dev. 2014;26:26–43. doi: 10.1159/000363153. [DOI] [PubMed] [Google Scholar]
- Taniguchi H., He M., Wu P., Kim S., Paik R., Sugino K., Kvitsiani D., Fu Y., Lu J., Lin Y. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B., Menon V., Nguyen T.N., Kim T.K., Jarsky T., Yao Z., Levi B., Gray L.T., Sorensen S.A., Dolbeare T. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinnikov A., Nordstrom K., Thoren P., Kindblom J.M., Malin S., Rozell B., Adams M., Rajanayagam O., Pettersson S., Ohlsson C. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor alpha1. EMBO J. 2002;21:5079–5087. doi: 10.1093/emboj/cdf523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gucht A.L.M., Moran C., Meima M.E., Visser W.E., Chatterjee K., Visser T.J., Peeters R.P. Resistance to thyroid hormone due to heterozygous mutations in thyroid hormone receptor alpha. Curr. Top. Dev. Biol. 2017;125:337–355. doi: 10.1016/bs.ctdb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- van Mullem A., van Heerebeek R., Chrysis D., Visser E., Medici M., Andrikoula M., Tsatsoulis A., Peeters R., Visser T.J. Clinical phenotype and mutant TRα1. N. Engl. J. Med. 2012;366:1451–1453. doi: 10.1056/NEJMc1113940. [DOI] [PubMed] [Google Scholar]
- van Mullem A.A., Visser T.J., Peeters R.P. Clinical consequences of mutations in thyroid hormone receptor-alpha1. Eur. Thyroid J. 2014;3:17–24. doi: 10.1159/000360637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A., Sperk G. Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides. 2004;38:245–252. doi: 10.1016/j.npep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Wallis K., Sjogren M., van Hogerlinden M., Silberberg G., Fisahn A., Nordstrom K., Larsson L., Westerblad H., Morreale de Escobar G., Shupliakov O. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J. Neurosci. 2008;28:1904–1915. doi: 10.1523/JNEUROSCI.5163-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Iwasaki T., Xu M., Lesmana R., Xiong Y., Shimokawa N., Chin W.W., Koibuchi N. Aberrant cerebellar development of transgenic mice expressing dominant-negative thyroid hormone receptor in cerebellar Purkinje cells. Endocrinology. 2015;156:1565–1576. doi: 10.1210/en.2014-1079. [DOI] [PubMed] [Google Scholar]
- Yuen R.K., Thiruvahindrapuram B., Merico D., Walker S., Tammimies K., Hoang N., Chrysler C., Nalpathamkalam T., Pellecchia G., Liu Y. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015;21:185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The seizure occurred during the dark phase of the photoperiod and was video recorded using infrared lighting.
Note the unsuccessful attempts of the mother to drag the pup back to the nest.
Data Availability Statement
RNASeq and ChipSeq data are accessible through GEO Series accession number GSE143933 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE143933).