

Ibrexafungerp (IBX), formerly SCY-078, is a novel, oral and intravenous, semisynthetic triterpenoid glucan synthase inhibitor in clinical development for treating multiple fungal infections, including invasive candidiasis. Intra-abdominal candidiasis (IAC) is one of the most common types of invasive candidiasis associated with high mortality largely due to poor drug exposure in infected lesions.
KEYWORDS: ibrexafungerp, intra-abdominal candidiasis, lesion, drug penetration, matrix-assisted desorption ionization–mass spectrometry imaging (MALDI-MSI), laser capture microdissection (LCM)
ABSTRACT
Ibrexafungerp (IBX), formerly SCY-078, is a novel, oral and intravenous, semisynthetic triterpenoid glucan synthase inhibitor in clinical development for treating multiple fungal infections, including invasive candidiasis. Intra-abdominal candidiasis (IAC) is one of the most common types of invasive candidiasis associated with high mortality largely due to poor drug exposure in infected lesions. To better understand the potential of IBX to treat such infections, we investigated its penetration at the site of infection. Using matrix-assisted laser desorption ionization–mass spectrometry imaging (MALDI-MSI) and laser capture microdissection (LCM)-directed high-pressure liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS), we investigated tissue distribution and lesion-specific drug exposure of IBX in a clinically relevant IAC mouse model. After a single-dose treatment, IBX quickly distributed into tissues and efficiently accumulated within lesions. Drug concentrations of IBX within the liver abscesses were almost 100-fold higher than the serum concentration. In addition, drug penetration after repeated treatment of IBX was compared with micafungin. IBX exhibited robust and long-lasting lesion penetration after repeated treatment. These data indicate that IBX penetrates into intra-abdominal abscesses highly efficiently and holds promise as a potential therapeutic option for IAC patients.
TEXT
Intra-abdominal candidiasis (IAC) is a commonly encountered type of invasive candidiasis yet is poorly understood compared to candidemia (1–5). Often, IAC is associated with high mortality rates and is devastating among the immunocompromised patient population. Abscesses are the major pathological manifestation of IAC and occur over subsequent days as Candida cells invade abdominal organs from the peritoneal cavity (1, 3, 6, 7). Azoles, polyenes, and echinocandins are the three main classes of antifungal agents that are available for the treatment of this invasive infection (8). The typical treatment for IAC requires several weeks of antifungal therapy. Echinocandins are recommended as first-line agents; however, their clinical effectiveness is highly variable, with known potential for breakthrough resistance, and parenteral administration is the only option (9). Drug exposure within lesions is a major factor limiting therapeutic response. Thus, there is an unmet need for the development of new therapeutic agents.
Ibrexafungerp (IBX) is a novel, first-in-class, semisynthetic triterpenoid glucan synthase inhibitor in clinical development for treating invasive fungal infections caused by Candida spp., Aspergillus spp., Pnecumocystis spp., and other rare fungal pathogens (10–15). Despite sharing the same drug target with echinocandins, IBX exerts its antifungal activity in a way both overlapping with and distinct from what has been uncovered for echinocandin drugs (16). This underlying mechanism explains the finding that IBX has potent activities against not only the wild-type (WT) Candida species but strains carrying fks mutations that confer resistance to echinocandins (12, 14, 17, 18). IBX also differs from echinocandins, which are intravenous (i.v.)-only drugs, since it is available for administration in both i.v. and oral formulations (12, 19, 20). Extensive in vitro and in vivo preclinical studies (10–12, 14, 16–18, 20–25) and phase I and II clinical studies (8, 26) have been performed demonstrating an excellent safety profile, favorable pharmacokinetic (PK) properties, and strong overall efficacy. Nevertheless, like for many existing drugs and those in clinical development, we generally lack knowledge of how IBX penetrates into heavily infected tissue lesions and whether it could be a therapeutic option for deep-seated difficult-to-treat intra-abdominal infections.
The objective of this study was to evaluate the penetration of IBX at the site of infection in a clinically relevant intra-abdominal candidiasis (IAC) mouse model due to C. albicans. Spatial distribution of IBX in infected liver was assessed using matrix-assisted laser desorption ionization–mass spectrometry imaging (MALDI-MSI) post-single and -repeated treatment. Drug exposure in distinct compartments of infected liver tissue was quantified and compared with that of therapeutic dosing of micafungin (MCF) using laser capture microdissection (LCM)-directed high-pressure liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). In vivo efficacy against intra-abdominal infections was also evaluated for IBX and compared with MCF in a head-to-head fashion.
RESULTS
Serum pharmacokinetics (PK) of IBX in mice with IAC.
The serum drug levels of IBX were measured in IAC mice after receiving a single oral dose of IBX at 30 mg/kg on day 3 postinfection. The drug concentration-time profile (Fig. 1) was consistent with what was previously observed in an invasive candidiasis mouse model (23). At this dosing level, a mean maximum serum drug concentration (Cmax) of 1.695 μg/ml was observed at 3 h postdose, and an average of 0.380 μg/ml IBX was circulating 24 h after dosing.
FIG 1.
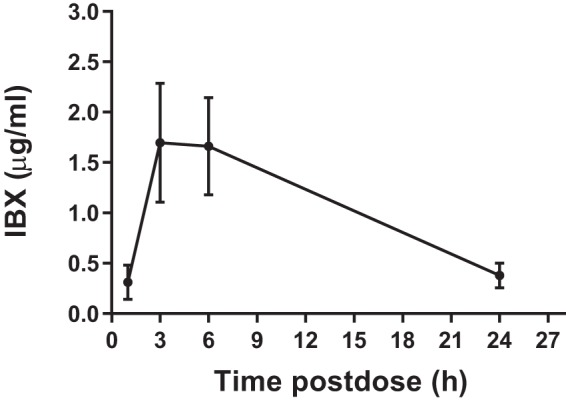
Serum drug concentration-time curve after a single oral administration of IBX at 30 mg/kg.
Distribution and penetration of IBX at infected tissue sites after a single-dose treatment.
By employing MALDI-MSI, we visualized IBX distribution in infected tissues at 1, 3, 6, and 24 h after single dosing. IBX was administered at the empirical peak time of lesion formation in intra-abdominal organs, among which liver has demonstrated the most consistent abscess phenotype (27). Therefore, we chose liver tissues as representative target tissues for all downstream drug exposure analysis. The IBX-specific ion maps (Fig. 2) acquired at early time points (1 ∼ 6 h postdose) displayed an exuberant drug distribution in nonlesion tissues yet a minimal to marginal level of partitioning in lesions. This distribution pattern was visibly reversed at 24 h postdose, when higher drug signals were emitted from lesions, in contrast to largely decreased drug signals captured from surrounding uninvolved tissues. To investigate whether drug was delivered to its target cells, we analyzed the histopathology (Fig. 2) of the hematoxylin and eosin (H&E)-stained and Gomori methenamine-silver (GMS)-stained tissue sections adjacent to those subjected to MALDI-MSI evaluation. We found that liver lesions/abscesses consisted of collections of macrophage and neutrophil infiltrates with a heterogeneous presence of necrosis in the lesion center. GMS staining showed entangled fungal networks being confined within lesions, indicating that only the drug that entered the lesion pocket represents the “true” drug exposure over fungal cells exerting antifungal effect.
FIG 2.

Drug distribution in infected liver tissues after a single dose of IBX. (Upper row) Ion maps of IBX in representative liver tissues collected 1, 3, 6, and 24 h after a single oral dose of IBX at 30 mg/kg. The signal intensity color bar is fixed for IBX, with gradually increased intensity from blue (no signal) to red (max signal). H&E (middle row) and GMS (bottom row) staining of adjacent sections are shown below each ion map. Scale bars, 3 mm. Outlines highlight the lesion area on each tissue section. Dashed lines on 24-h samples denote that the upper left part of the nonlesion tissue seen on H&E- and GMS-stained sections was trimmed off the section subjected to MALDI-MSI.
To confirm the MALDI-MSI observations, we next applied LCM followed by LC-MS/MS to precisely measure drug exposure in lesion and nonlesion areas of infected liver tissues at each postdose time point as mentioned above (Fig. 3). The average drug levels were measured at 0.2, 2.5, and 3.8 μg/g in lesions versus 17.7, 78.1, and 86.6 μg/g in nonlesion tissues at 1, 3, and 6 h postdose, respectively. This drug distribution pattern with most of the drug persisting in surrounding tissues but very little drug delivered into abscesses inverted at 24 h postdose, when the average IBX concentration was measured at 35.9 μg/g within abscesses versus 21.1 μg/g in nonlesion tissues. The almost 10-fold increase of drug concentration within lesions from 6 h to 24 h postdose was accompanied by a 4- and 5-fold drug level declination in uninvolved tissues and serum, respectively, demonstrating a prolonged drug retention and accumulation within abscesses.
FIG 3.

Quantification of drug exposure in infected livers. IBX concentration was measured in abscesses (lesion) and uninvolved (nonlesion) tissues dissected from liver sections collected 1, 3, 6, and 24 h after a single oral dose of IBX at 30 mg/kg. Error bars show the standard deviation from 3 liver samples.
Drug accumulation in liver abscesses after repeated doses of IBX and comparison with that of MCF.
Taking the single dosing results forward, we then asked whether drug accumulation within abscesses holds steady after repeated treatment of IBX with a humanized dosing regimen. In this experiment, mice were treated twice daily (BID) with IBX at 15 mg/kg for 2 days. (This is a humanized dose that matches 750 mg/day in humans based on internal communication with Scynexis, Inc. This dosing regimen results in a systemic exposure comparable to the targeted concentration in humans for efficacy.) Livers were collected at 24 h and 48 h after the last dose and analyzed by MALDI-MSI (Fig. 4). The 24-h liver sample scan showed a high level of drug penetration into the lesion pocket, similar to that observed at 24 h post-single dosing. A more striking lesion drug accumulation was seen at 48 h in the liver image, further supporting the profound drug exposure of IBX at infected tissue sites (drug concentrations were measured at 20.9 μg/g within lesions versus 3.3 μg/g in uninvolved areas using LCM-directed LC-MS/MS).
FIG 4.
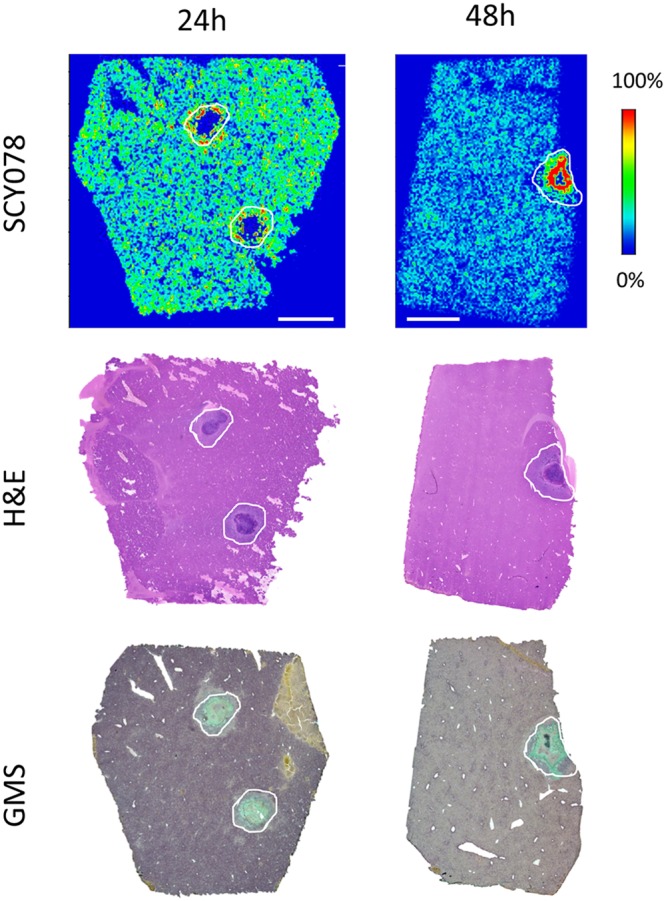
Drug distribution in infected liver tissues after repeated doses of IBX. (Upper row) Ion maps of IBX in representative liver tissues collected 24 and 48 h after the last dose of a 2-day treatment of IBX at 15 mg/kg, BID. The signal intensity color bar is fixed for IBX, with gradually increased intensity from blue (no signal) to red (max signal). The middle and bottom rows show H&E and GMS staining, respectively, of adjacent sections at each corresponding time point. Scale bars, 3 mm.
In light of our previous finding that MCF (representative echinocandin as the standard of care for invasive candidiasis) has limited penetration into liver abscesses (27), we next performed experiments to compare both drug exposure and in vivo antifungal efficacy for IBX and MCF side by side. For this purpose, IAC mice were treated with either IBX (15 mg/kg, BID) or MCF (5 mg/kg, once a day [QD]) at the corresponding humanized therapeutic dose and dosing frequency (28, 29) for 2 or 3 days. As shown in Fig. 5, trough (24 h after 2 days and 3 days of treatment) serum drug levels were not significantly different between the two drugs, although IBX concentrations (224.3 ± 65.0 ng/ml and 379.6 ± 123.2 ng/ml, respectively) were numerically lower than MCF (257.8 ± 167.2 ng/ml and 456.7 ± 225.9 ng/ml, respectively) at both time points. Drug exposure in infected livers (Fig. 6) after 2 days of treatment was quantified as 18.2 ± 9.4 and 13.2 ± 4.0 μg/g of IBX in lesion and nonlesion areas, respectively, versus 2.9 ± 1.1 and 0.3 ± 0.2 μg/g of MCF in the corresponding subcompartments. After a continuous 3-day treatment (Fig. 6), IBX showed boosted drug accumulation in lesions (25.7 ± 8.2 μg/g), while drug levels in uninvolved tissues kept steady (14.2 ± 4.6 μg/g). MCF levels at this time point were measured at 2.1 ± 1.4 μg/g in lesions and 0.2 ± 0.2 μg/g in nonlesion tissues, similar to what was observed after 2 days of treatment.
FIG 5.
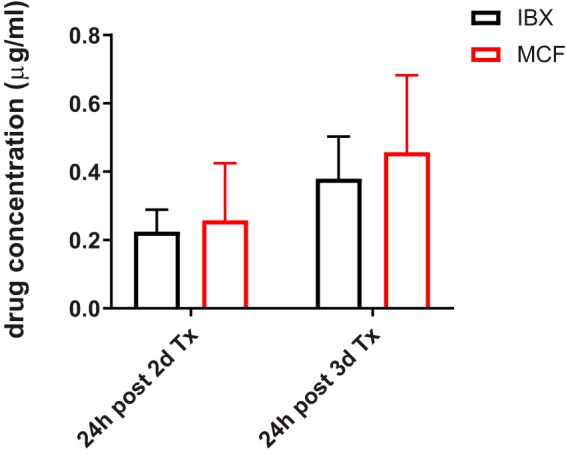
Serum drug concentrations of IBX and MCF 24 h after 2-day and 3-day treatments. IBX was given orally at 15 mg/kg, BID. MCF was given i.p. at 5 mg/kg, QD.
FIG 6.

Drug accumulation comparison in infected liver compartments between repeated doses of MCF and IBX. Drug concentration was measured in lesions and nonlesion areas dissected from liver tissues collected 24 h after 2-day and 3-day treatments with MCF (5 mg/kg, i.p., QD) or IBX (15 mg/kg, orally [p.o.], BID).
We also evaluated liver fungal burdens at each of the drug exposure measurement time points, as well as 24 h after a single dose of each drug and predose (Fig. 7). The singly dosed drug did not significantly decrease fungal burdens in the liver. However, a significant 0.52 log10 CFU/g average burden reduction was achieved in mice that received 2-day treatment of IBX, with complete liver sterilization in one out of eight mice. This antifungal effect was further boosted when IBX therapy extended to 3 days, with an average 0.8 log10 CFU/g liver burden reduction and liver fungal clearance achieved in 3 out of 8 mice in this treatment group. In comparison, 2 days of treatment with MCF resulted in a merely 0.18 log10 CFU/g average liver burden reduction, and mice that received 3-day MCF treatment had liver burdens as high as vehicle control-treated mice.
FIG 7.

Liver burden comparison at predose, 24 h after a single dose, and 24 h after 2-day and 3-day treatments with IBX, MCF, and vehicle control. Each symbol represents the liver burden of a single animal. Short lines are the mean burdens determined for 4 ∼ 8 mice in each treatment group. Symbols on the x axis represent mice with no liver burden (sterilization). The percentages of mice with liver sterilization were plotted as bars on the right y axis underneath burdens of each corresponding group. The significant burden difference was labeled as *(P < 0.05) and **(P < 0.01).
DISCUSSION
A well established and widely accepted concept for antimicrobial agents is that drug penetration at a target tissue site is one of the key determinants for the fate of infections, ranging from therapeutic cure to breakthrough infections with or without drug resistance development. Herein, we investigated this important perspective for the novel investigational antifungal drug IBX in a clinically relevant IAC mouse model. IAC is a common form of invasive candida infection, but it is difficult to treat, and the clinical outcomes are variable and often dissatisfying with existing regimens, of which echinocandins are the recommended standard of care (3, 4). This variable effectiveness of echinocandins in IAC patients relative to that achieved in candidemia patients, along with the fact that echinocandin resistance is rising, particularly with Candida glabrata infections and more frequently seen with IAC patients (30, 31), has raised concerns about the uncertain penetration of the echinocandin drugs into tissue lesions laden with infecting fungal cells at the current dosing level used in clinical settings. Recently, we took the initiative to unravel drug penetration at the site of infection for two echinocandin drugs (MCF and rezafungin) and found that MCF at a humanized therapeutic dose did not penetrate into intra-abdominal abscesses at a sufficient level to promote efficient killing to clear the infection from the tissue (27). In the present study, we applied the same strategy to evaluate the penetration of IBX and assess its lesion exposure properties.
Even though blocking of β-1,3-glucan synthesis is the common mechanism for echinocandins and IBX to achieve antifungal activity, IBX is a molecule distinct from the echinocandin drug class, with a unique structure and different physiological and PK properties. With the understanding that IBX can be readily delivered to a variety of body sites, including vaginal tissues and secretions, at pharmacologically meaningful levels (25), we sought to evaluate lesion penetration of this drug in IAC. After a single oral administration, the distribution of IBX in nonlesion areas of infected liver tissue was visualized using MALDI-MSI. The nonlesion liver tissue-to-serum drug concentration ratios were stable at all postdose time points (56.9:1, 46.1:1, 52.1:1, and 55.5:1 at 1, 3, 6, and 24 h, respectively), indicating that IBX kinetics in nonlesion liver tissues follows the same changing pattern as that circulating in blood. The approximately 50-fold-higher drug exposure in livers relative to blood was also consistent with the previous report using whole-body autoradiography (24). In comparison, we found that lesion PK of the IBX, however, was substantially different from that of nonlesion tissues and blood. The drug was able to penetrate into and accumulate efficiently within liver abscesses, although the initial penetration was slow, with lesion drug concentrations far below that in uninvolved tissue during the first 6 h postdose but rapidly elevating at 24 h and exceeding the nonlesion tissue level by 1.7-fold. Thereafter, we confirmed that repeated dosing of humanized therapeutic IBX resulted in sustained high levels of drug being retained within abscesses (18.2 and 25.7 μg/ml at 24 h after 2 and 3 days of treatment, respectively), regardless of the serum drug concentration dropping to trough levels. Using the mean 24-h lesion drug concentration after repeated dosing (21.9 μg/ml), we expect a steady-state lesion 24-h area under the concentration curve (AUC0–24h) of ∼526.7 μg · h/ml, far exceeding (47-fold) target therapeutic exposure in plasma (11.2 μg · h/ml) (23). Thus, it is not surprising to see that this level of drug exposure was associated with an average of 0.8 log10 CFU/g fungal burden reduction in liver and complete liver clearance in 37.5% of the mice that received the treatment. Of note, as a standard of care control in our study, MCF at the humanized therapeutic dose did not achieve any significant in vivo efficacy against IAC at any of three evaluation time points. Lesion MCF exposure was measured at ∼2.5 μg/ml after repeated daily dosing. Assuming that this is the drug level of MCF being maintained within the lesion compartment, the steady-state lesion AUC0–24h would then be 60 μg · h/ml, below the pharmacodynamic target exposure (75 to 139 μg · h/ml) set for both pediatric and adult human patients (32–34).
It has been proposed that IBX and echinocandins are Cmax/MIC- and AUC/MIC-driven drugs to achieve efficacy, and the AUC/MIC is considered the most optimal pharmacodynamic (PD) index for these drugs (21, 23, 28, 35). In previous preclinical studies, the static PD targets against C. albicans were identified as total drug AUC/MIC (tAUC/MIC) of 500 for IBX and 5,299 for MCF, using serum/plasma-based measurement (21, 35). In our study, the lesion 24-h tAUC/MIC (MICs of C. albicans strain SC3514 were 0.03 μg/ml for IBX and 0.016 μg/ml for MCF) was estimated as 17,556 for IBX and 3,750 for MCF. It is apparent that lesion IBX exposure was well above (35-fold) the PD target required to inhibit cell growth in the disseminated candidiasis mouse model, yet MCF failed to reach the static tAUC/MIC target in abscesses, which likely explained why we did not see an obvious antifungal effect with MCF in the IAC model. We recognize that lesion tAUC/MIC obtained in our study was not quite comparable with blood PD targets set for the disseminated infection. However, given that lesion PD targets are yet to be established and that fungal cells circulating in blood in the disseminated model are largely confined within intra-abdominal abscesses in the IAC model, such an analogy is reasonable and makes pharmacological sense. It is noteworthy that free drug AUC/MIC was not calculated here because protein binding in lesions is unknown for both drugs, and current plasma protein binding factors determined by in vitro analytics were very similar for IBX (99.8% in mice) (23) and MCF (99.8% in mice and 99.76 ∼ 99.8% in human) (36, 37). Nevertheless, future studies to comprehensively assess the lesion PD target and better understand lesion protein binding are warranted.
To summarize, our study shows for the first time that IBX is able to penetrate effectively into intra-abdominal abscesses containing C. albicans. In comparison with MCF, the representative drug of the current standard of care for IAC patients, IBX demonstrated superior lesion penetration properties with lesion drug exposure upon completion of 3-day treatment, far exceeding the target therapeutic exposure by 47-fold. More importantly, this prominent lesion drug exposure was associated with a significant PD endpoint in the IAC mouse model, which was not achieved with MCF using a humanized dosing regimen in parallel. These data suggest that IBX may be a promising oral option to treat patients with IAC.
MATERIALS AND METHODS
Antifungal agents.
IBX, IBX-D9 (Scynexis, Inc., Jersey City, NJ, USA), and micafungin (Astellas Pharma, Inc., Tokyo, Japan) were obtained as standard powders from their manufacturer. For dosing, IBX was formulated in 0.5% methylcellulose and adjusted to pH 6.5 prior to oral administration.
Strain.
Candida albicans strain SC5314 was grown in yeast-extract-peptone-dextrose (YPD) broth at 37°C with shaking overnight. Cells were washed, counted, and prepared in a sterile stool matrix containing a final concentration of 1 × 108 CFU/ml for infection as previously described (27).
Animals.
Female 18- to 20-g CD1 mice (Charles River, Wilmington, MA, USA) were housed in the animal biosafety level-2 research animal facility at the Center for Discovery and Innovation at Hackensack Meridian Health (HMH). All experimental procedures were performed in accordance with National Research Council guidelines and approved by the HMH Institutional Animal Care and Use Committee (IACUC).
Mouse model of intra-abdominal candidiasis, PK, and efficacy evaluation.
A well-established, immunocompetent IAC mouse model was used for this study (38). Mice were infected intraperitoneally (i.p.) with a 100-μl cell suspension prepared in sterile stool matrix containing 1 × 107 CFU of C. albicans SC5314 as previously described (27). All drug treatment was initiated on day 3 postinfection. For the single-dose PK study, mice received a single oral treatment of IBX at 30 mg/kg and were sacrificed 1, 3, 6, and 24 h postdose (3 mice per time point). Livers were explored for abscesses (Fig. 2), dissected, placed on a cryohistology tray, snap-frozen in liquid nitrogen, and stored at –80°C for MALDI-MSI and LCM-directed drug quantification. Blood was also collected at each time point for serum drug concentration measurement. For repeated-dose experiments, mice received oral BID IBX at 15 mg/kg, once-daily i.p. treatment of MCF at 5 mg/kg, or vehicle control for 2 or 3 days. Livers and blood were collected 24 h after each regimen for both drug exposure (5 mice per time point per drug) and efficacy evaluation (8 mice per group per time point). Liver fungal burden was also assessed predose and 24 h post-single-dose treatment (4 mice per group).
Tissue sectioning.
Using a Leica CM 1860 UV cryostat (Buffalo Grove, IL), 12-μm thick tissue sections were cut and mounted onto stainless steel slides for MALDI-MSI analysis and onto frosted glass microscope slides for GMS and H&E staining. Adjacent 25-μm-thick tissue sections were mounted onto a thin polymer membrane slide (PET) for laser capture microdissection. All slides were air-dried for 10 min, sealed in a small airtight sealable bag and transferred to –80°C storage until analysis.
Matrix application.
Using an HTX TM-sprayer (Chapel Hill, NC) operating with a 50 μl/min flow rate, 60°C nozzle temperature, and 5 lb/in2 of pressure, 2,5-dihydroxybenzoic acid (DHB) (25 mg/ml in 50% methanol) containing 50 μl of internal standard (1 mg of IBX-D9 dissolved in 1 ml of 100% methanol) was applied to the surface. Thirty passes over the tissue were performed.
MALDI-MSI analysis.
Imaging analysis was performed using a MALDI LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) with a resolution of 60,000× at 400 m/z, with full width at half maximum (39). The resolution was sufficient to resolve IBX peaks from the background without the requirement of MS/MS and subsequent loss of signal. However, drug peak identities were confirmed by acquiring several MS/MS spectra directly from the dosed tissues. Standards of IBX were analyzed directly from the stainless-steel target plate and spiked into drug-naive liver tissue to optimize instrument parameters. The limit of detection (LOD) for MALDI-MSI analysis of IBX was 2.5 μg/g of liver tissue. Spectra were acquired in the 700 to 780 m/z range, using positive mode ionization. Data visualization was performed using Thermo ImageQuest software. Normalized ion images of IBX were generated by dividing IBX [M+H]+ signal (730.528 ± 0.005 m/z) by IBX-D9 [M+H]+ signal (739.585 ± 0.005 m/z).
Laser-capture microdissection.
Distinct subcompartments of infected liver tissue (e.g., abscesses and surrounding uninvolved tissue) totaling 1 million to 3 million μm2 were carefully dissected from 4 to 6 serial liver tissue sections using a Leica LMD 6 (CC7000) system (Buffalo Grove, IL) (40). Lesion areas were identified optically from the bright-field image scan and by comparison to the adjacent sectioned GMS- and H&E-stained tissue. The dissected tissues were collected into 0.25-ml standard PCR tubes and immediately stored in a –80°C freezer unless analyzed immediately.
Drug quantitation by LC-MS/MS.
Prior to analysis, the PCR tubes containing LCM samples were thawed at room temperature. To this, 50-μl of extraction solution (1:1 acetonitrile/methanol [ACN/MeOH] containing 10 ng/ml verapamil internal standard), 10 μl of 1:1 ACN-H2O solution, and 2 μl of phosphate-buffered saline (PBS) buffer were added, and then tubes were sonicated for 10 min and centrifuged at 4,000 rpm for 5 min at room temperature. Standard curve and quality-control tubes were created by combining 50 μl of extraction solution, 10 μl of serial diluted IBX or MCF in ACN-H2O, and 2 μl of liver homogenate from untreated animals (40), followed by sonication and centrifugation as described above. Then, 50 μl of supernatant was transferred from both study samples and standards for LC-MS/MS analysis and diluted with an additional 50 μl of deionized water into a 96-well deep-well plate.
LC-MS analysis was performed on an AB Sciex Qtrap 6500+ instrument (Ontario, Canada) coupled to a Shimadzu high-pressure liquid chromatography (HPLC) system. Chromatography was performed with an Agilent SB-C8 2.1 × 30 mm, particle size 3.5 μm, using a reverse-phase gradient. The mobile phase A was 0.1% formic acid in 100% water, and mobile phase B was 0.1% formic acid in 100% ACN. Drug quantitation in tissue was conducted using the transition m/z 739.39/584.40 for IBX, 1270.40/1190.40 for MCF, and 455.40/165.20 for verapamil, which was used as the internal standard.
Statistical analysis.
Absolute drug concentrations were graphed and statistically analyzed in GraphPad software (Prism 8; GraphPad Software, Inc., San Diego, CA). Drug levels in different tissue compartments and liver burden counts at various time points were compared using one-way analysis of variance (ANOVA), and Dunn’s multiple comparison was used for the post hoc analyses. Statistical significance was defined as a P value of <0.05.
ACKNOWLEDGMENTS
This work was supported by a Scynexis, Inc., research contract to Y.Z. and D.S.P., by NIH grant AI109025 to D.S.P., and by NIH grants S10 OD023524-01 and S10 OD023524-01 to V.D.
We thank Enriko Dolgov, Padmaja Paderu, Min Hee Lee, Nathaly Cabrera, Rosa Hernandez, and Mila Krel for their great help with the animal experiments.
D.S.P. serves on advisory boards for Cidara, Amplyx, Scynexis, Astellas, Matinas, and N8 Medical. D.S.P. has an issued U.S. patent concerning echinocandin resistance. S.B. and D.A. are employees of Scynexis.
REFERENCES
- 1.Blot SI, Vandewoude KH, De Waele JJ. 2007. Candida peritonitis. Curr Opin Crit Care 13:195–199. doi: 10.1097/MCC.0b013e328028fd92. [DOI] [PubMed] [Google Scholar]
- 2.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis 62:e1-50. doi: 10.1093/cid/civ933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vergidis P, Clancy CJ, Shields RK, Park SY, Wildfeuer BN, Simmons RL, Nguyen MH. 2016. Intra-abdominal candidiasis: the importance of early source control and antifungal treatment. PLoS One 11:e0153247. doi: 10.1371/journal.pone.0153247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bassetti M, Marchetti M, Chakrabarti A, Colizza S, Garnacho-Montero J, Kett DH, Munoz P, Cristini F, Andoniadou A, Viale P, Rocca GD, Roilides E, Sganga G, Walsh TJ, Tascini C, Tumbarello M, Menichetti F, Righi E, Eckmann C, Viscoli C, Shorr AF, Leroy O, Petrikos G, De Rosa FG. 2013. A research agenda on the management of intra-abdominal candidiasis: results from a consensus of multinational experts. Intensive Care Med 39:2092–2106. doi: 10.1007/s00134-013-3109-3. [DOI] [PubMed] [Google Scholar]
- 5.Montravers P, Leroy O, Eckmann C. 2015. Intra-abdominal candidiasis: it’s still a long way to get unquestionable data. Intensive Care Med 41:1682–1684. doi: 10.1007/s00134-015-3894-y. [DOI] [PubMed] [Google Scholar]
- 6.Pfaller M, Neofytos D, Diekema D, Azie N, Meier-Kriesche HU, Quan SP, Horn D. 2012. Epidemiology and outcomes of candidemia in 3648 patients: data from the Prospective Antifungal Therapy (PATH Alliance(R)) registry, 2004–2008. Diagn Microbiol Infect Dis 74:323–331. doi: 10.1016/j.diagmicrobio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Messer SA, Jones RN, Fritsche TR. 2006. International surveillance of Candida spp. and Aspergillus spp.: report from the SENTRY Antimicrobial Surveillance Program (2003). J Clin Microbiol 44:1782–1787. doi: 10.1128/JCM.44.5.1782-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D. 2018. Lack of impact by SCY-078, a first-in-class oral fungicidal glucan synthase inhibitor, on the pharmacokinetics of rosiglitazone, a substrate for CYP450 2C8, supports the low risk for clinically relevant metabolic drug-drug interactions. J Clin Pharmacol 58:1305–1313. doi: 10.1002/jcph.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiederhold NP, Lewis JS II. 2007. The echinocandin micafungin: a review of the pharmacology, spectrum of activity, clinical efficacy and safety. Expert Opin Pharmacother 8:1155–1166. doi: 10.1517/14656566.8.8.1155. [DOI] [PubMed] [Google Scholar]
- 10.Berkow EL, Angulo D, Lockhart SR. 2017. In vitro activity of a novel glucan synthase inhibitor, SCY-078, against clinical isolates of Candida auris. Antimicrob Agents Chemother 61:e00435-17. doi: 10.1128/AAC.00435-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larkin E, Hager C, Chandra J, Mukherjee PK, Retuerto M, Salem I, Long L, Isham N, Kovanda L, Borroto-Esoda K, Wring S, Angulo D, Ghannoum M. 2017. The emerging pathogen Candida auris: growth phenotype, virulence factors, activity of antifungals, and effect of SCY-078, a novel glucan synthesis inhibitor, on growth morphology and biofilm formation. Antimicrob Agents Chemother 61:e02396-16. doi: 10.1128/AAC.02396-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfaller MA, Messer SA, Rhomberg PR, Borroto-Esoda K, Castanheira M. 2017. Differential activity of the oral glucan synthase inhibitor SCY-078 against wild-type and echinocandin-resistant strains of Candida species. Antimicrob Agents Chemother 61:e00161-17. doi: 10.1128/AAC.00161-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghannoum M, Long L, Larkin EL, Isham N, Sherif R, Borroto-Esoda K, Barat S, Angulo D. 2018. Evaluation of the antifungal activity of the novel oral glucan synthase inhibitor SCY-078, singly and in combination, for the treatment of invasive aspergillosis. Antimicrob Agents Chemother 62:e00244-18. doi: 10.1128/AAC.00244-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez-Ortigosa C, Paderu P, Motyl MR, Perlin DS. 2014. Enfumafungin derivative MK-3118 shows increased in vitro potency against clinical echinocandin-resistant Candida species and Aspergillus species isolates. Antimicrob Agents Chemother 58:1248–1251. doi: 10.1128/AAC.02145-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamoth F, Alexander BD. 2015. Antifungal activities of SCY-078 (MK-3118) and standard antifungal agents against clinical non-Aspergillus mold isolates. Antimicrob Agents Chemother 59:4308–4311. doi: 10.1128/AAC.00234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jimenez-Ortigosa C, Perez WB, Angulo D, Borroto-Esoda K, Perlin DS. 2017. De novo acquisition of resistance to SCY-078 in Candida glabrata involves FKS mutations that both overlap and are distinct from those conferring echinocandin resistance. Antimicrob Agents Chemother 61:e00833-17. doi: 10.1128/AAC.00833-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiederhold NP, Najvar LK, Jaramillo R, Olivo M, Pizzini J, Catano G, Patterson TF. 2018. Oral glucan synthase inhibitor SCY-078 is effective in an experimental murine model of invasive candidiasis caused by WT and echinocandin-resistant Candida glabrata. J Antimicrob Chemother 73:448–451. doi: 10.1093/jac/dkx422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghannoum M, Long L, Isham N, Hager C, Wilson R, Borroto-Esoda K, Barat S, Angulo D. 2019. Activity of a novel 1,3-beta-d-glucan synthase inhibitor, ibrexafungerp (formerly SCY-078), against Candida glabrata. Antimicrob Agents Chemother 63:e01510-19. doi: 10.1128/AAC.01510-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D. 2019. Clinical pharmacokinetics and drug-drug interaction potential for coadministered SCY-078, an oral fungicidal glucan synthase inhibitor, and tacrolimus. Clin Pharmacol Drug Dev 8:60–69. doi: 10.1002/cpdd.588. [DOI] [PubMed] [Google Scholar]
- 20.Scorneaux B, Angulo D, Borroto-Esoda K, Ghannoum M, Peel M, Wring S. 2017. SCY-078 is fungicidal against candida species in time-kill studies. Antimicrob Agents Chemother 61:e01961-16. doi: 10.1128/AAC.01961-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lepak AJ, Marchillo K, Andes DR. 2015. Pharmacodynamic target evaluation of a novel oral glucan synthase inhibitor, SCY-078 (MK-3118), using an in vivo murine invasive candidiasis model. Antimicrob Agents Chemother 59:1265–1272. doi: 10.1128/AAC.04445-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schell WA, Jones AM, Borroto-Esoda K, Alexander BD. 2017. Antifungal activity of SCY-078 and standard antifungal agents against 178 clinical isolates of resistant and susceptible Candida species. Antimicrob Agents Chemother 61:e01102-17. doi: 10.1128/AAC.01102-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wring SA, Randolph R, Park S, Abruzzo G, Chen Q, Flattery A, Garrett G, Peel M, Outcalt R, Powell K, Trucksis M, Angulo D, Borroto-Esoda K. 2017. Preclinical pharmacokinetics and pharmacodynamic target of SCY-078, a first-in-class orally active antifungal glucan synthesis inhibitor, in murine models of disseminated candidiasis. Antimicrob Agents Chemother 61:e02068-16. doi: 10.1128/AAC.02068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wring S, Borroto-Esoda K, Solon E, Angulo D. 2018. SCY-078, a novel fungicidal agent, demonstrates distribution to tissues associated with fungal infections during mass balance studies with intravenous and oral [(14)C]SCY-078 in albino and pigmented rats. Antimicrob Agents Chemother 63:e02119–18. doi: 10.1128/AAC.02119-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larkin EL, Long L, Isham N, Borroto-Esoda K, Barat S, Angulo D, Wring S, Ghannoum M. 2019. A novel 1,3-beta-d-glucan inhibitor, ibrexafungerp (formerly SCY-078), shows potent activity in the lower pH environment of vulvovaginitis. Antimicrob Agents Chemother 63:e02611-18. doi: 10.1128/AAC.02611-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spec A, Mycoses Study Group, Pullman J, Thompson GR, Powderly WG, Tobin EH, Vazquez J, Wring SA, Angulo D, Helou S, Pappas PG. 2019. MSG-10: a phase 2 study of oral ibrexafungerp (SCY-078) following initial echinocandin therapy in non-neutropenic patients with invasive candidiasis. J Antimicrob Chemother 74:3056–3062. doi: 10.1093/jac/dkz277. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Prideaux B, Nagasaki Y, Lee MH, Chen PY, Blanc L, Ho H, Clancy CJ, Nguyen MH, Dartois V, Perlin DS. 2017. Unraveling drug penetration of echinocandin antifungals at the site of infection in an intra-abdominal abscess model. Antimicrob Agents Chemother 61:e01009-17. doi: 10.1128/AAC.01009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andes DR, Diekema DJ, Pfaller MA, Marchillo K, Bohrmueller J. 2008. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. glabrata in a neutropenic murine candidiasis model. Antimicrob Agents Chemother 52:3497–3503. doi: 10.1128/AAC.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard SJ, Livermore J, Sharp A, Goodwin J, Gregson L, Alastruey-Izquierdo A, Perlin DS, Warn PA, Hope WW. 2011. Pharmacodynamics of echinocandins against Candida glabrata: requirement for dosage escalation to achieve maximal antifungal activity in neutropenic hosts. Antimicrob Agents Chemother 55:4880–4887. doi: 10.1128/AAC.00621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farmakiotis D, Tarrand JJ, Kontoyiannis DP. 2014. Drug-resistant Candida glabrata infection in cancer patients. Emerg Infect Dis 20:1833–1840. doi: 10.3201/eid2011.140685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shields RK, Nguyen MH, Press EG, Clancy CJ. 2014. Abdominal candidiasis is a hidden reservoir of echinocandin resistance. Antimicrob Agents Chemother 58:7601–7605. doi: 10.1128/AAC.04134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seibel NL, Schwartz C, Arrieta A, Flynn P, Shad A, Albano E, Keirns J, Lau WM, Facklam DP, Buell DN, Walsh TJ. 2005. Safety, tolerability, and pharmacokinetics of micafungin (FK463) in febrile neutropenic pediatric patients. Antimicrob Agents Chemother 49:3317–3324. doi: 10.1128/AAC.49.8.3317-3324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuse ER, Chetchotisakd P, da Cunha CA, Ruhnke M, Barrios C, Raghunadharao D, Sekhon JS, Freire A, Ramasubramanian V, Demeyer I, Nucci M, Leelarasamee A, Jacobs F, Decruyenaere J, Pittet D, Ullmann AJ, Ostrosky-Zeichner L, Lortholary O, Koblinger S, Diekmann-Berndt H, Cornely OA, Micafungin Invasive Candidiasis Working Group. 2007. Micafungin versus liposomal amphotericin B for candidaemia and invasive candidosis: a phase III randomised double-blind trial. Lancet 369:1519–1527. doi: 10.1016/S0140-6736(07)60605-9. [DOI] [PubMed] [Google Scholar]
- 34.Hope WW, Kaibara A, Roy M, Arrieta A, Azie N, Kovanda LL, Benjamin DK Jr. 2015. Population pharmacokinetics of micafungin and its metabolites M1 and M5 in children and adolescents. Antimicrob Agents Chemother 59:905–913. doi: 10.1128/AAC.03736-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, Lepak A. 2010. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother 54:2497–2506. doi: 10.1128/AAC.01584-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamato Y, Kaneko H, Hashimoto T, Katashima M, Ishibashi K, Kawamura A, Terakawa M, Kagayama A. 2002. Pharmacokinetics of the antifungal drug micafungin in mice, rats and dogs, and its in vitro protein binding and distribution to blood cells. Jpn J Chemother 50:74–79. [Google Scholar]
- 37.Hebert MF, Smith HE, Marbury TC, Swan SK, Smith WB, Townsend RW, Buell D, Keirns J, Bekersky I. 2005. Pharmacokinetics of micafungin in healthy volunteers, volunteers with moderate liver disease, and volunteers with renal dysfunction. J Clin Pharmacol 45:1145–1152. doi: 10.1177/0091270005279580. [DOI] [PubMed] [Google Scholar]
- 38.Cheng S, Clancy CJ, Xu W, Schneider F, Hao B, Mitchell AP, Nguyen MH. 2013. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J Infect Dis 208:1529–1537. doi: 10.1093/infdis/jit335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, Chen C, Kaya F, Weiner DM, Chen P-Y, Song T, Lee M, Shim TS, Cho JS, Kim W, Cho SN, Olivier KN, Barry CE, Dartois V. 2015. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med 21:1223–1227. doi: 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman M, Blanc L, Chen PY, Dartois V, Prideaux B. 2018. Spatial quantification of drugs in pulmonary tuberculosis lesions by laser capture microdissection liquid chromatography mass spectrometry (LCM-LC/MS). J Vis Exp doi: 10.3791/57402. [DOI] [PMC free article] [PubMed] [Google Scholar]