

Ceftolozane-tazobactam is a potent β-lactam/β-lactamase inhibitor combination approved for the treatment of complicated intraabdominal and complicated urinary tract infections and, more recently, the treatment of hospital-acquired and ventilator-associated bacterial pneumonia. Although the activities of ceftolozane are not enhanced by tazobactam against Pseudomonas aeruginosa, it remains the most potent antipseudomonal agent approved to date.
KEYWORDS: antimicrobial resistance, aminoglycoside, ceftolozane, Pseudomonas, synergy, tobramycin
ABSTRACT
Ceftolozane-tazobactam is a potent β-lactam/β-lactamase inhibitor combination approved for the treatment of complicated intraabdominal and complicated urinary tract infections and, more recently, the treatment of hospital-acquired and ventilator-associated bacterial pneumonia. Although the activities of ceftolozane are not enhanced by tazobactam against Pseudomonas aeruginosa, it remains the most potent antipseudomonal agent approved to date. Emerging data worldwide has included reports of microbiological failure in patients with serious bacterial infections caused by multidrug-resistant (MDR) P. aeruginosa as a result of ceftolozane resistance developed within therapy. The objective of this study is to compare the efficacy of a tobramycin homodimer plus ceftolozane versus ceftolozane-tazobactam alone against MDR and extensively drug-resistant (XDR) P. aeruginosa. Tobramycin homodimer, a synthetic dimer of two monomeric units of tobramycin, was developed to abrogate the ribosomal properties of tobramycin with a view to mitigating aminoglycoside-related toxicity and resistance. Herein, we report that tobramycin homodimer, a nonribosomal aminoglycoside derivative, potentiates the activities of ceftolozane versus MDR/XDR P. aeruginosa in vitro and delays the emergence of resistance to ceftolozane-tazobactam in the wild-type PAO1 strain. This combination is also more potent than a standard ceftazidime-avibactam combination against these isolates. Conversely, a tobramycin monomer with intrinsic ribosomal properties does not potentiate ceftolozane under similar conditions. Susceptibility and checkerboard studies were assessed using serial 2-fold dilution assays, following the Clinical and Laboratory Standards Institute (CLSI) guidelines. This strategy provides an avenue to further preserve the clinical utility of ceftolozane and enhances its spectrum of activity against one of the most difficult-to-treat pathogens in hospitals.
INTRODUCTION
β-Lactam antibiotics (penicillins, monobactams, cephalosporins, and carbapenems) are a mainstay in the treatment of serious bacterial infections caused by multidrug resistant (MDR) and extensively drug resistant (XDR) Gram-negative bacilli (1). They act by covalently binding to penicillin-binding proteins (PBPs) involved in cross-linking of peptidoglycan during cell wall synthesis, leading to lysis and cell death. The global phenotypic and genotypic resistance to this class of drugs is, in part, driven by their ubiquitous usage (1, 2). Signature resistance mechanisms to β-lactam antibiotics include hyperproduction of chromosomal AmpC, acquisition of plasmid-borne extended-spectrum β-lactamases (ESBLs), loss of outer membrane (OM) porins (OprD and OprF), upregulation of multidrug efflux pumps, and modifications in the active sites of PBPs (2–5). Clinical strategies to circumvent these resistance mechanisms involve coadministration of β-lactam antibiotics with β-lactamase inhibitors (2) and the use of newer generations of β-lactam antibiotics that are less affected by some or all of these known mechanisms in targeted bacterial populations (6, 7).
Ceftolozane is a new cephalosporin antibiotic that was approved in 2014 (as a ceftolozane-tazobactam combination) for the treatment of complicated intra-abdominal and complicated urinary tract infections (8, 9), and recently gained approval for the treatment of hospital-acquired and ventilator-acquired bacterial pneumonia in 2019 (https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-hospital-acquired-and-ventilator-associated-bacterial-pneumonia). It is an oxyimino-aminothiazolyl cephalosporin with a pyrazole substituent at the 3-position side chain instead of the lighter pyridinium present in ceftazidime (Fig. 1). Ceftolozane is the most potent cephalosporin against Pseudomonas aeruginosa because of its ability to evade the multitude of resistance mechanisms employed by the pathogen, including efflux pumps, reduced porin uptake, and modifications of PBPs (6, 10). P. aeruginosa, a member of the ESKAPE (11) group of pathogens and a common cause of nosocomial infections in debilitated patients and chronic respiratory infections in cystic fibrosis patients, is well known for its extraordinary capacity to develop resistance to almost any available antibiotic upon exposure, using a variety of mechanisms (12–15). The addition of tazobactam, a β-lactam-based β-lactamase inhibitor, does not have a major impact on the activity of ceftolozane against P. aeruginosa, although it is believed to expand its coverage versus ESBL-producing Enterobacterales (6, 16). One comparative study recently demonstrated an overall improved activity of ceftazidime-avibactam compared to ceftolozane-tazobactam (C/T) versus ESBL-producing Escherichia coli and P. aeruginosa (16). Occasional microbiological failure to C/T has also been documented in clinics worldwide (16–18). Because C/T is frequently used in critical clinical situations for resistant, MDR, and XDR P. aeruginosa, it is natural to question whether its utility can be further optimized by adding a second antibiotic or an adjuvant. Indeed, only a few agents have exhibited demonstrable synergy with C/T. For example, amikacin and colistin exhibit different (synergistic or additive/indifferent) interactions with C/T against different P. aeruginosa strains in vitro (19), while a separate study reported a clinical treatment success of a C/T-resistant P. aeruginosa strain due to synergy with tobramycin (18). The mechanism of synergism in this clinical success was hypothesized to be due to extensive outer membrane disruption by tobramycin (18). In both cases, the isolates were either susceptible or intermediately resistant to the potentiating antibiotics (i.e., MIC of amikacin = 2 to 8 μg/ml; colistin = 1 to 2 μg/ml; tobramycin ≤1 to 4 μg/ml) (18, 19), consistent with known synergism between β-lactam antibiotics and ribosomal aminoglycosides (20, 21). However, these combinations may result in different outcomes in isolates that are highly resistant to aminoglycosides and/or colistin. Moreover, clinical use of colistin is often limited by both pharmacokinetic limitations and significant toxicities (22, 23), while the use of aminoglycosides is typically associated with nephrotoxicity as well as a propensity to cause irreversible hearing loss, an effect that has been linked to the lack of precise selectivity for prokaryotic ribosomes (24–26). Synthetic aminoglycoside analogs with lower bacterial and human mitochondrial ribosome specificities have been shown to exhibit reduced ototoxic potentials in cochlear explants, in culture and in guinea pig (27), and we further hypothesize that a nonribosomal aminoglycoside will exhibit lower idiosyncratic toxicities and drug-induced hearing loss. The objective of this study was therefore to evaluate the efficacy of ceftolozane in combination with a tobramycin homodimer adjuvant that does not possess canonical activities of aminoglycosides. Antibiotic adjuvants without overt antibacterial activity as standalone agents are less likely to select for resistance (28, 29). A number of nonribosomal tobramycin molecules that potentiate different antibiotics (including β-lactams) against P. aeruginosa have been documented in literature but none has been reported to synergize with ceftolozane (28, 30–34). Recently, we identified a tobramycin homodimer scaffold that is: (i) more potent than the gold standard OM permeabilizer, polymyxin B nonapeptide (PMBN); (ii) nonhemolytic against red blood cells; (iii) noncytotoxic against human embryonic kidney (HEK293) and liver (HepG2) cells; and (iv) nontoxic in vivo against Galleria mellonella wax moths where colistin was toxic (34). In the current study, we investigated a combination of tobramycin homodimer in combination with ceftolozane and compared its activity with standard β-lactam/β-lactamase inhibitor combinations against Gram-negative bacteria in vitro, using the current CLSI broth microdilution methodology. We also assessed a combination of ceftolozane and tobramycin monomer to investigate the role of intrinsic aminoglycoside activity and resistance mechanisms on the combination therapy.
FIG 1.

Chemical structures of reference and synthetic compounds. Compound 1 is a synthetic dimer of tobramycin conjugated at the C-5 position.
RESULTS AND DISCUSSION
P. aeruginosa susceptibility.
The isolates used in this study were all multidrug resistant clinical isolates (except wild-type PAO1). All isolates, except PAO1 and PA101885, were carbapenem-resistant (meropenem MIC ≥4 μg/ml). Two isolates were resistant to colistin (MIC ≥4 μg/ml) and only one isolate (PA259-96918) expressing IMP-18 metallo-β-lactamase (MBL) was resistant to ceftolozane-tazobactam (C/T; MIC ≥16/4 μg/ml). All clinical isolates, except PA101243, were either resistant or intermediately resistant to ciprofloxacin (MIC ≥2 μg/ml). Whereas some isolates were susceptible to tobramycin monomer (MIC ≤4 μg/ml), none of the isolates studied was susceptible to tobramycin homodimer 1 (MIC >128 μg/ml), suggesting a loss of ribosomal function in compound 1 (a synthetic dimer of tobramycin conjugated at the C-5 position), as previously shown for related tobramycin analogs (31). These data are presented in Table 1 and susceptibility breakpoints were interpreted according to CLSI standards.
TABLE 1.
Susceptibility profiles of different P. aeruginosa isolates used in the study
| Isolate | MIC (μg/ml)a |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TZP | ATM | CRO | FEP | CAZ | C/T | IPM | MEM | CIP | MXF | TOB | GEN | AMK | TGC | MIN | CST | |
| PAO1b | 8 | 4 | ND | ND | 2 | 0.5 | ND | 1 | 0.125 | 1 | 1 | 1 | ND | 4 | 8 | 1 |
| PA100036 | 8 | 16 | 32 | 4 | 8 | 0.5 | 8 | 4 | >16 | >16 | 128 | >32 | 32 | 32 | 16 | 2 |
| PA101885 | 16 | 16 | 32 | 8 | 8 | 0.5 | 1 | 4 | >16 | >16 | 1 | ≤0.5 | ≤1 | 8 | 8 | 1 |
| PA259-96918c | 64 | 32 | >64 | >64 | 512 | >16 | 32 | 512 | >16 | >16 | 256 | >32 | >64 | 32 | 32 | 1 |
| PA260-97103 | 128 | 64 | >64 | 16 | 32 | 1 | 32 | 16 | 16 | >16 | 32 | >32 | 4 | 16 | 16 | 1 |
| PA262-101856 | 64 | 32 | 64 | 32 | 16 | 1 | 32 | 32 | >16 | >16 | 1024 | >32 | >64 | 32 | 64 | 1 |
| PA264-104354 | 256 | 64 | >64 | 32 | 128 | 2 | 32 | 64 | >16 | >16 | 128 | >32 | 8 | 32 | 32 | 1 |
| PA91433 | 64 | 512 | >64 | 16 | 1024 | 0.5 | 32 | 16 | 2 | 16 | 16 | 32 | >32 | 32 | 16 | 4 |
| PA101243 | 128 | 32 | >64 | 64 | 64 | 1 | 16 | 16 | 1 | 8 | 128 | >32 | >64 | ND | 2 | 1024 |
| PA86052 | 256 | 64 | >64 | 32 | 64 | 2 | 32 | 16 | >16 | >16 | 1 | 4 | 16 | ND | ND | 1 |
| PA88949 | 256 | 32 | >64 | 32 | 64 | 2 | >32 | 16 | 4 | 8 | 4 | 16 | 32 | ND | ND | 2 |
| PA107092 | 512 | 64 | >64 | 64 | 128 | 2 | 32 | 32 | >16 | >16 | >64 | >32 | 8 | ND | ND | 1 |
| PA108590 | 256 | 32 | >64 | 32 | 32 | 2 | 32 | 16 | 4 | 16 | 4 | 16 | 32 | ND | ND | 1 |
| PA109084 | 512 | 64 | >64 | 32 | 64 | 2 | 32 | 16 | 4 | 16 | 4 | 16 | 32 | ND | ND | 1 |
TZP, piperacillin-tazobactam; ATM, aztreonam; CRO, ceftriaxone; FEP, cefepime; CAZ, ceftazidime; C/T, ceftolozane-tazobactam; IPM, imipenem; MEM, meropenem; CIP, ciprofloxacin; MXF, moxifloxacin; TOB, tobramycin; GEN, gentamicin; AMK, amikacin; TGC, tigecycline; MIN, minocycline; CST, colistin; ND, not determined.
Wild type.
MBL-producing.
Comparison of different β-lactam/β-lactamase inhibitor combinations against MDR P. aeruginosa.
We evaluated the susceptibilities of nine different MDR/XDR P. aeruginosa phenotypes, exhibiting multiple resistance patterns, to different combinations of β-lactam/β-lactamase inhibitors such as aztreonam-avibactam, ceftazidime-avibactam, ceftolozane-avibactam and ceftolozane-tazobactam (C/T). Avibactam is a non-β-lactam-based diazabicyclooctane β-lactamase inhibitor that binds covalently and reversibly to β-lactamases, while tazobactam is a β-lactam-based sulfone β-lactamase inhibitor that binds irreversibly to the active site of β-lactamases (35). Both avibactam and tazobactam target the active sites of serine β-lactamases, with avibactam being able to inhibit class A, C, and some D enzymes, while tazobactam only inhibits some class A enzymes (36). Thus, ceftazidime-avibactam couples a well-known cephalosporin with a novel non-β-lactam β-lactamase inhibitor while C/T combines a novel cephalosporin with an established β-lactam β-lactamase inhibitor. A ceftolozane-avibactam combination was tested to investigate the spectrum of serine-based β-lactamases on the potency of ceftolozane. We found that neither ceftolozane nor avibactam was active against MBLs (MICPA259 >16 μg/ml) (Table 2), consistent with the literature (37, 38). Aztreonam, a monobactam, is resistant to inactivation by MBLs (class B β-lactamases) but not serine-β-lactamases (39), hence, a combination of aztreonam-avibactam was investigated. Coadministration of different concentrations (4, 8, or 16 μg/ml) of avibactam with aztreonam or ceftazidime resulted in variable levels of susceptibilities for the MDR P. aeruginosa isolates, but not with ceftolozane (Table 2). Also, tazobactam did not improve the activity of ceftolozane against any of the isolates up to the highest concentration (16 μg/ml) tested (Table 2). For strains presumed to express AmpC enzymes (negative for the carbapenemases KPC, OXA-48, NDM, IMP, VIM, and GES), microbiological response to the inhibitory actions of avibactam seem to be optimal at 4 μg/ml, and higher concentrations of avibactam did not confer commensurate benefit on the combination (Table 2). Overall, the potent activity of C/T observed in this study was comparable to that of ceftazidime-avibactam against P. aeruginosa isolates presumed to express AmpC, an observation that is consistent with a previous study of a larger sample size (36). However, C/T appears to be more potent in other clinical isolates where AmpC expression may not be the predominant mechanism of resistance.
TABLE 2.
Comparison of different β-lactam antibiotics and β-lactamase inhibitors at different concentrations against MDR/XDR P. aeruginosa clinical isolatesi
| Strain | MIC (μg/ml)j |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATM | A/Aa | A/Ab | A/Ac | CAZ | CZAa | CZAb | CZAc | CTZ | C/Td | C/Te | C/Tf | CTZ/Ac | |
| Non-AmpC-expressing strains | |||||||||||||
| PAO1g | 4 | 4 | 4 | 4 | 2 | 2 | 2 | 2 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| PA259-96918h | 32 | 32 | 16 | 16 | 512 | 512 | 512 | 256 | >16 | >16 | >16 | >16 | >16 |
| PA264-104354 | 64 | 16 | 16 | 16 | 128 | 32 | 32 | 16 | 2 | 2 | 2 | 2 | 0.5 |
| AmpC-expressing strains | |||||||||||||
| PA260-97103 | 32 | 8 | 8 | 4 | 32 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 0.5 |
| PA86052 | 32 | 2 | 1 | 0.5 | 64 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| PA88949 | 32 | 8 | 4 | 4 | 64 | 4 | 4 | 2 | 2 | 2 | 2 | 1 | 1 |
| PA107092 | 64 | 32 | 32 | 16 | 128 | 16 | 8 | 8 | 2 | 2 | 2 | 2 | 1 |
| PA108590 | 32 | 8 | 8 | 8 | 32 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| PA109084 | 64 | 8 | 16 | 16 | 64 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 1 |
Avibactam at 4 μg/ml.
Avibactam at 8 μg/ml.
Avibactam at 16 μg/ml.
Tazobactam at 4 μg/ml.
Tazobactam at 8 μg/ml.
Tazobactam at 16 μg/ml.
Wild type.
MBL-producing.
All isolates (except PA259-96918) were negative for the carbapenemases KPC, OXA-48, NDM, IMP, VIM, and GES.
ATM, aztreonam; A/A, aztreonam-avibactam; CAZ, ceftazidime; CZA, ceftazidime-avibactam; CTZ, ceftolozane; C/T, ceftolozane-tazobactam; CTZ/A, ceftolozane-avibactam.
Checkerboard study of ceftolozane and tobramycin homodimer 1.
In a study of archived P. aeruginosa isolates collected >10 years before the release of avibactam, an 18% resistance rate to ceftazidime-avibactam was found to be mediated by decreased cell wall permeability and increased efflux, rather than changes in PBPs or novel β-lactamases (40). This means that development of new antimicrobial agents targeted for the treatment of MDR bacteria may still encounter clinically important resistance, and that permeability and efflux is a threat to all future drug development. The intrinsic potency of ceftolozane against all P. aeruginosa clinical isolates studied (except PA259-96918) and its lack of synergistic interaction with β-lactamase inhibitors (avibactam and tazobactam) suggest that ceftolozane is inert to the actions of class A, C, and D β-lactamases, but not class B MBLs. Low susceptibility of P. aeruginosa to ceftolozane is presumed to be mediated by production of MBLs (41). To investigate whether ceftolozane suffers from either of the intrinsic resistance mechanisms of outer membrane permeability and efflux, we assessed its potency, alone and in combination with compound 1, against efflux-competent MDR and efflux-deficient P. aeruginosa isolates. Compound 1, a molecule derived by dimerization of short-chain amphiphilic tobramycins, has been previously reported as a nontoxic and more potent outer membrane permeabilizer than polymyxin B nonapeptide (PMBN) (34). A checkerboard assay was therefore used to assess the interactions between ceftolozane and compound 1, and the results were interpreted as a function of the fractional inhibitory concentration index (FICI) (42). To exhibit synergism, compound A at ≤¼ MIC must lower the MIC of compound B by at least four fold. Compound 1, with MIC ≥256 μg/ml against Gram-negative bacteria (Tables 3 and 4), has a maximum theoretical synergistic value of ≥64 μg/ml. However, a maximum working concentration of 16 μg/ml (7 μM) was used in this study, as this concentration may be readily achieved in human plasma based on the achievable plasma concentrations of aminoglycosides (20 to 200 μM, i.e., 10 to 113 μg/ml) (43, 44). Surprisingly, compound 1 was found to potentiate the activity of ceftolozane (4- to 32-fold) against wild-type, MDR/XDR, and efflux mutants of P. aeruginosa, but not against an MBL-expressing PA259-96918 isolate (Table 3). Neither tobramycin monomer nor tazobactam could potentiate ceftolozane against any of these isolates (Table 3). It should be noted that most of these isolates are resistant to aminoglycosides (Table 1), indicating that the potentiating effect of compound 1 is unaffected by multitudes of aminoglycoside resistance mechanisms. The MICs of ceftolozane in efflux-deficient mutants PAO200 and PAO750 (lacking different clinically relevant efflux pumps) and wild-type PAO1 were also the same (MIC = 0.5 μg/ml) (Table 3), suggesting that ceftolozane is unlikely to be a substrate of resistance nodulation division (RND) efflux pumps. However, compound 1 potentiates ceftolozane in these strains, suggesting a mechanism that is independent of RND efflux pumps. Compound 1 can also potentiate several other β-lactam antibiotics, such as meropenem, piperacillin, ceftazidime, aztreonam, etc. against these isolates.
TABLE 3.
Ceftolozane plus compound 1 is more potent than the ceftolozane-tazobactam combination against MDR/XDR P. aeruginosa clinical isolatesg
| Strain | MIC (μg/ml)h |
FICIc | Fold potentiationc | |||||
|---|---|---|---|---|---|---|---|---|
| CTZ | C/Ta | TOB | CTZ/TOBb | Compound 1 | CTZ/1c | |||
| PAO1d | 0.5 | 0.5 | 1 | 0.5 | >128 | 0.125 | <0.31 | 4 |
| PA259-96918e | >16 | >16 | 256 | >16 | >128 | >16 | ND | ND |
| PA260-97103 | 1 | 1 | 128 | 1 | >128 | 0.25 | <0.31 | 4 |
| PA262-101856 | 1 | 1 | 1024 | 1 | >128 | 0.25 | <0.56 | 4 |
| PA264-104354 | 2 | 2 | 128 | 2 | >128 | 0.25 | <0.19 | 8 |
| PA91433 | 0.5 | 0.5 | 16 | 0.5 | >128 | 0.125 | <0.31 | 4 |
| PA100036 | 0.5 | 0.5 | 128 | 0.5 | >128 | 0.125 | <0.31 | 4 |
| PA101243 | 1 | 1 | 128 | 1 | >128 | 0.125 | <0.19 | 8 |
| PA101885 | 0.5 | 0.5 | 1 | 0.5 | >128 | 0.125 | <0.31 | 4 |
| PA108590 | 2 | 1 | 4 | 2 | >128 | 0.25 | <0.19 | 8 |
| PA114228 | 0.5 | 0.5 | 8 | 0.5 | >128 | 0.03125 | <0.13 | 16 |
| PAO200f | 0.5 | 0.5 | 0.5 | 0.5 | >128 | 0.0625 | <0.19 | 8 |
| PAO750f | 0.5 | 0.5 | 0.5 | 0.5 | >128 | 0.0156 | <0.09 | 32 |
Tazobactam at 7 μM (tested up to 50 μM, i.e., 16 μg/ml against all isolates).
Tobramycin at 7 μM (tested up to 120 μM, i.e., >64 μg/ml against tobramycin-resistant isolates).
Tobramycin homodimer at 7 μM (concentration-dependent potentiation).
Wild type.
MBL-producing.
Efflux-deficient mutants: PAO200 (ΔmexA-mexB-oprM); PAO750 (ΔmexAB–oprM, ΔmexCD–oprJ, ΔmexEF–oprN, ΔmexJK, ΔmexXY, and ΔopmH).
Checkerboard assay of ceftolozane with tazobactam, tobramycin, and tobramycin homodimer 1 at equimolar concentrations.
CTZ, ceftolozane; C/T, ceftolozane-tazobactam; TOB, tobramycin monomer; Compound 1, tobramycin homodimer; FICI, fractional inhibitory concentration index (FICI ≤0.5 is synergistic); ND, not determined.
TABLE 4.
Efficacy of ceftolozane-tobramycin homodimer is not different from ceftolozane-tazobactam combination against other wild-type and MDR/XDR Gram-negative bacterial clinical isolates
| Isolated | MIC (μg/ml)c |
FICIb | Fold potentiationb | |||
|---|---|---|---|---|---|---|
| CTZ | C/Ta | Compound 1 | CTZ/1b | |||
| AB ATCC 17978 | 2 | 0.5 | >128 | 1 | <0.56 | 2 |
| AB027 | >16 | >16 | >128 | >16 | ND | ND |
| AB031 | >16 | >16 | >128 | 4 | <0.31 | >4 |
| AB92247 | 2 | 2 | >128 | 1 | <0.56 | 2 |
| AB110193 | >16 | >16 | >128 | 4 | <0.31 | >4 |
| AB LAC-4 | 8 | 8 | >128 | 8 | <1.06 | 1 |
| E. coli 94393 | 0.25 | 0.25 | >128 | 0.25 | <1.06 | 1 |
| E. coli 94474 | 0.25 | 0.25 | >128 | 0.5 | <1.06 | 1 |
| E. coli 107115 | >16 | >16 | >128 | >16 | ND | ND |
| E. cloacae 117029 | 0.25 | 0.25 | >128 | 0.25 | <0.56 | 2 |
| E. cloacae 121187 | 0.125 | 0.125 | >128 | 0.0625 | <0.56 | 2 |
| KP 113250 | 1 | 1 | >128 | 1 | <0.56 | 2 |
| KP 113254 | 0.5 | 0.5 | >128 | 0.5 | <1.06 | 1 |
| KP 116381 | >16 | >16 | >128 | >16 | ND | ND |
Tazobactam at 7 μM (tested up to 50 μM, i.e., 16 μg/ml against all isolates).
Tobramycin homodimer at 7 μM (potentiation observed at higher concentrations).
CTZ, ceftolozane; C/T, ceftolozane-tazobactam; Compound 1, tobramycin homodimer.
AB, Acinetobacter baumannii; KP, Klebsiella pneumoniae; LAC, Los Angeles County isolate.
To investigate the breadth of the observed synergistic interactions, we assessed the combination against other clinically relevant Gram-negative bacteria, including Acinetobacter baumannii, Escherichia coli, Enterobacter cloacae, and Klebsiella pneumoniae. We found that potentiation and/or synergistic interaction between ceftolozane and compound 1 was more pronounced in P. aeruginosa (Table 3) than in other Gram-negative bacteria (Table 4). Ceftolozane, alone or in combination with tazobactam, was found to be generally less potent against A. baumannii (Table 4), consistent with prior studies (45, 46), however, compound 1 dose-dependently restored susceptibility in 2 of 4 resistant isolates tested (Table 4). Our data revealed that a low concentration (<1 μM, i.e., 2 μg/ml) of compound 1 is enough to cause a measurable effect in P. aeruginosa, while a higher concentration (≥4 μM) might be necessary for a similar effect in other Gram-negative bacteria.
Time-kill assays.
To further investigate the kinetics of bacterial growth and cidality of the adjunctive therapy, time-kill assays were performed using two multidrug-resistant P. aeruginosa strains, PA264-104354 and PA114228. Bactericidal activity is defined as a ≥3 log10 reduction in the total CFU/ml from the original inoculum over 24 h, while bacteriostatic activity is defined as maintenance of a <3 log10 reduction in the total CFU/ml from the original inoculum (47). Synergy is defined as a ≥2 log10 decrease in the number of CFU/ml between the combination and the most active component of the combination after 24 h, where at least one of the drugs must be present at a concentration that does not affect the growth curve of the test organism (48). The growth curves of both strains when treated with 16 μg/ml (7 μM) of compound 1 were identical to their respective controls (without drug), suggesting the lack of intrinsic antibacterial potency for compound 1. For PA264-104354 and PA114228, ceftolozane alone (at 2 × MIC) was bactericidal after 9 h and 6 h, respectively, and regrowth occurred after 24 h (Fig. 2). However, a combination of ceftolozane (at 2 × MIC) and compound 1 (at 7 μM) exhibited bactericidal and synergistic interactions against both strains after 6 h and prevented regrowth of bacteria after 24 h (Fig. 2). Indeed, cultures treated with the combination therapy were completely sterilized after 9 h of incubation, representing a >5 log10 reduction from the starting inoculum (Fig. 2). Overall, the bacterial burden reduction in the time-kill assays is consistent with synergistic interactions between ceftolozane and compound 1 in the checkerboard assay.
FIG 2.

Time-kill synergy graphs. The activities of ceftolozane at 2 × MIC alone (orange line) and in combination with compound 1 (green line) against multidrug-resistant Pseudomonas aeruginosa. (A) PA264-104354, MIC ceftolozane = 2 μg/ml. (B) PA114228, MIC ceftolozane = 0.5 μg/ml. Each data point represents an average of three independent determinations.
Tobramycin homodimer 1 delays development of resistance to ceftolozane.
To investigate the dynamics and possible mechanism(s) of development of resistance to the combinations, we exposed wild-type PAO1 to sub-MICs of ceftazidime, C/T, and C/T/compound 1 combinations by serial passaging. The basal MICs of these compounds in PAO1 strain were 2, 0.5, and 0.125 μg/ml, respectively. The development (rate and extent) of resistance was fastest for ceftazidime, with a 32-fold change in MIC (i.e., from 2 to 64 μg/ml) after 7 days, followed by a much slower rate of development of resistance to C/T, with modal concentrations reaching only 4 × MIC after 7 days (Fig. 3). In contrast, we were unable to obtain mutants resistant to a combination of C/T/compound 1 after the completion of the 7-day experiment (Fig. 3).
FIG 3.
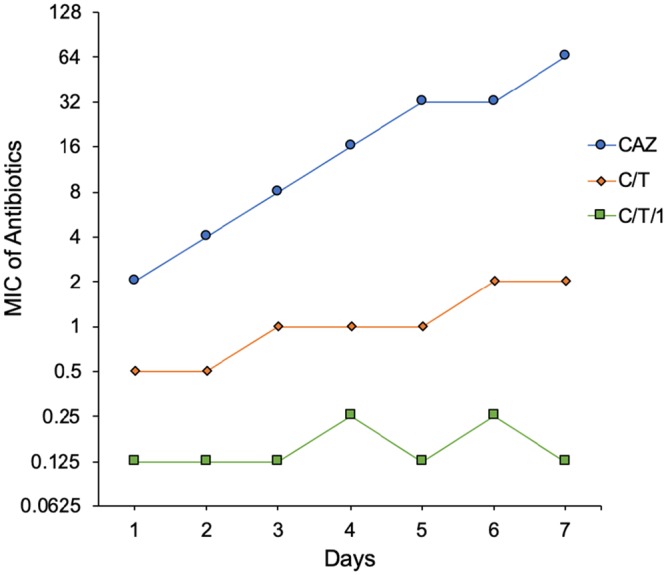
Emergence of resistance study. Serial passaging of P. aeruginosa wild-type PAO1 exposed to sub-MICs of CAZ, C/T, and C/T/1 for 7 days. CAZ, ceftazidime; C/T, ceftolozane-tazobactam; C/T/1, ceftolozane-tazobactam/compound 1.
To gain insights into the emerging mechanism(s) of resistance and investigate whether there is an induction of cross-resistance to other antibiotics, susceptibility profiles of day 7 mutants were assessed against a panel of antibiotics and compared to the wild-type PAO1 strain (Table 5). Mutants selected upon exposure to sub-MIC levels of C/T developed increased (4-fold) resistance to all β-lactam antibiotics tested, but not against ciprofloxacin, rifampin, and minocycline (Table 5). Global pleiotropic effects associated with a severe fitness cost as been hypothesized as a reason for the slow and low-level resistance development to C/T by PAO1 (10). Modification of intrinsic (AmpC) and horizontally acquired β-lactamases appears to be a major mechanism that may lead to C/T resistance in MDR P. aeruginosa (49). The significant increase in susceptibility of the mutant to colistin (16-fold decrease in MIC) is indicative of an associated fitness cost for moderate C/T resistance in PAO1. For mutants obtained upon exposure to sub-MICs of C/T/compound 1, cross-resistance to β-lactam antibiotics and rifampin was not detected, however increased susceptibility to ciprofloxacin, minocycline, and colistin was observed (Table 5). The increased susceptibility to ciprofloxacin and minocycline may be a consequence of reduced expression of genes belonging to the MDR efflux pumps MexXY-OprM and MexCD-OprJ, as revealed in a previous whole-genome characterization of C/T resistance mechanisms in PAO1 (10). Tetracyclines (32) and fluoroquinolones (50, 51) are known substrates for RND efflux pumps. To assess whether compound 1 induces resistance to tobramycin, we compared the susceptibility of the generated mutants and wild-type PAO1 to tobramycin. We found that the MICs of tobramycin across these isolates were identical (Table 5), suggesting that compound 1 does not trigger resistance development to aminoglycosides. This observation is consistent with a previous report on studies with A. baumannii (34).
TABLE 5.
Susceptibility profiles of wild-type P. aeruginosa PAO1 compared with resistant mutants generated from seven serial passagesa
| Antibiotic | Wild-type | C/T mutant | C/T/1 mutant |
|---|---|---|---|
| Ceftazidime | 2 | 8 | 4 |
| Meropenem | 1 | 4 | 2 |
| Aztreonam | 4 | 16 | 8 |
| Ceftolozane | 0.5 | 2 | 1 |
| Ciprofloxacin | 0.125 | 0.125 | 0.0156 |
| Rifampicin | 16 | 8 | 16 |
| Minocycline | 8 | 4 | 2 |
| Colistin | 1 | 0.0625 | 0.25 |
| Tobramycin | 1 | 2 | 1 |
| CTZ/1 | 0.125 | 0.125 | 0.125 |
MIC (in μg/ml) of day 7 mutants are reported. Concentration of compound 1 = 7 μM. CTZ, ceftolozane; C/T, ceftolozane-tazobactam; 1, compound 1.
In conclusion, we show that reduced permeability across the outer membrane is an important mechanism by which P. aeruginosa restricts the influx of ceftolozane into the cell. With the exception of MBL-producing PA259-96918, tobramycin homodimer (1), but not the monomeric unit of tobramycin or tazobactam, potentiated the activity of ceftolozane against all MDR/XDR P. aeruginosa clinical isolates tested (Table 3), resulting in susceptibility levels far below the CLSI breakpoint of 4 μg/ml. Our data also suggest that moderate C/T resistance in PAO1 may be associated with an important fitness cost, and that compound 1 delays the evolution of resistance to this combination. We also demonstrated that a combination of ceftolozane and compound 1 offers better microbiological potency than a standard ceftazidime-avibactam combination against MDR/XDR P. aeruginosa isolates. Thus, a ceftolozane-compound 1 combination is envisaged as a potential treatment option for MDR P. aeruginosa infections, minimizing the development of self- and cross-resistance to other potential antipseudomonal agents.
MATERIALS AND METHODS
Chemical synthesis.
The design, synthesis, and full characterization of tobramycin homodimer (1) has been previously reported (34).
Bacterial strains.
Bacteria isolates were obtained from either the American Type Culture Collection (ATCC), the Canadian National Intensive Care Unit (CAN-ICU), or the Canadian Ward (CANWARD) surveillance studies (41). Clinical isolates were cultured from body fluids and tissues of patients suffering from presumed “clinically significant” infectious diseases. Antimicrobial susceptibilities of clinical isolates were evaluated (using ATCC strains as quality control strains) and categorized, where appropriate, as either multidrug resistant (MDR), extensively drug-resistant (XDR), or pan drug-resistant (PDR) (52).
Susceptibility assay.
Susceptibility of bacteria to antimicrobial agents was assessed by the broth microdilution method, in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines (47). At least three replicates of the MIC assays were performed for each chemical entity.
Checkerboard assay.
Combination studies with different antibiotics were performed in 96-well plates as previously described (32). Briefly, the antibiotic of interest was serially diluted in Mueller-Hinton broth (MHB) along the abscissa while the adjuvant was serially diluted along the ordinate. Overnight bacterial cultures were diluted in 0.85% wt/vol saline to achieve a 0.5 McFarland turbidity, followed by 1:50 dilution in MHB. Equal volume of this bacterial culture was then added to each well and incubated at 37°C for 18 h. The fractional inhibitory concentration (FIC) for each antibiotic was calculated by dividing the MIC of the antibiotic in the presence of adjuvant by the MIC of the antibiotic alone. Similarly, the FIC of adjuvant was calculated by dividing the MIC of the adjuvant in the presence of antibiotic by the MIC of the adjuvant alone. FIC index is the sum of both FICs. FIC indices were interpreted as follows: ≤0.5, synergistic; >0.5 to 4, no interaction; and >4, antagonistic (42).
Time-kill assays.
Time-kill curve analyses were performed by culturing P. aeruginosa in LB medium in the presence of antibiotics, alone and in combination with compound 1. A 30-μl aliquot of a 0.5-McFarland inoculum of each strain was diluted to 3 ml of LB broth (containing different combinations of antimicrobial agents and adjuvants) and incubated at 37°C with shaking at 250 rpm. At specific time intervals (0, 1, 3, 6, 9, and 24 h), 100 μl was taken from each sample, serially diluted in sterile phosphate-buffered saline (PBS), plated on LB agar plates, and incubated at 37°C in a humid 5% CO2-enriched atmosphere. Bacterial colonies were counted after 20 h of incubation.
Development of resistance study.
The emergence of resistance to ceftolozane-tazobactam plus compound 1 was determined by serial passaging, as previously described (30, 34). Briefly, wild-type P. aeruginosa PAO1 cells were grown in 1 ml MHB media containing ceftolozane-tazobactam (at ¼ × MIC, ½ × MIC, 1 × MIC, 2 × MIC, and 4 × MIC), alone and in combination with compound 1. Tazobactam was used at a fixed concentration of 4 μg/ml throughout the experiment, while ceftazidime alone was included as a control. At 24-h intervals, the cultures were assessed for growth. Cultures from the second highest concentrations that allowed visible growth were harvested and diluted to 0.5 McFarland in sterile PBS, followed by 1:50 dilution into fresh MHB media containing ¼ × MIC, ½ × MIC, 1 × MIC, 2 × MIC, and 4 × MIC of each antibiotic. This serial passaging was repeated for 7 days. For ceftolozane-tazobactam plus compound 1 combinations, the concentration of compound 1 was kept constant at 7 μM throughout the experiment. For cultures that grew at higher than the MIC levels, cultures in the highest concentrations that permitted growth were passaged on drug-free LB plates and MICs were determined by broth microdilution in MHB.
ACKNOWLEDGMENTS
We thank A. Kumar (University of Manitoba) for the gift of efflux-deficient P. aeruginosa strains (PAO200 and PAO750).
This work was supported by a discovery grant (DG 2018-06047) from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
REFERENCES
- 1.Bush K, Bradford PA. 2016. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med 6:a025247. doi: 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paterson DL, Bonomo RA. 2005. Extended-spectrum beta-lactamases: a clinical update. Clin Microbiol Rev 18:657–686. doi: 10.1128/CMR.18.4.657-686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X-Z, Plésiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacoby GA. 2009. AmpC beta-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhanel GG, Chung P, Adam H, Zelenitsky S, Denisuik A, Schweizer F, Lagacé-Wiens PRS, Rubinstein E, Gin AS, Walkty A, Hoban DJ, Lynch JP, Karlowsky JA. 2014. Ceftolozane/tazobactam: a novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant Gram-negative bacilli. Drugs 74:31–51. doi: 10.1007/s40265-013-0168-2. [DOI] [PubMed] [Google Scholar]
- 7.Zhanel GG, Golden AR, Zelenitsky S, Wiebe K, Lawrence CK, Adam HJ, Idowu T, Domalaon R, Schweizer F, Zhanel MA, Lagacé-Wiens PRS, Walkty AJ, Noreddin A, Lynch Iii JP, Karlowsky JA. 2019. Cefiderocol: a siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 79:271–289. doi: 10.1007/s40265-019-1055-2. [DOI] [PubMed] [Google Scholar]
- 8.Solomkin J, Hershberger E, Miller B, Popejoy M, Friedland I, Steenbergen J, Yoon M, Collins S, Yuan G, Barie PS, Eckmann C. 2015. Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin Infect Dis 60:1462–1471. doi: 10.1093/cid/civ097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagenlehner FM, Umeh O, Steenbergen J, Yuan G, Darouiche RO. 2015. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: a randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet 385:1949–1956. doi: 10.1016/S0140-6736(14)62220-0. [DOI] [PubMed] [Google Scholar]
- 10.Cabot G, Bruchmann S, Mulet X, Zamorano L, Moyà B, Juan C, Haussler S, Oliver A. 2014. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob Agents Chemother 58:3091–3099. doi: 10.1128/AAC.02462-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 12.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 14.Breidenstein EBM, de la Fuente-Núñez C, Hancock R. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Bonomo RA, Szabo D. 2006. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin Infect Dis 43:S49–S56. doi: 10.1086/504477. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz de la Rosa J-M, Nordmann P, Poirel L. 2019. ESBLs and resistance to ceftazidime/avibactam and ceftolozane/tazobactam combinations in Escherichia coli and Pseudomonas aeruginosa. J Antimicrob Chemother 74:1934–1939. doi: 10.1093/jac/dkz149. [DOI] [PubMed] [Google Scholar]
- 17.Haidar G, Philips NJ, Shields RK, Snyder D, Cheng S, Potoski BA, Doi Y, Hao B, Press EG, Cooper VS, Clancy CJ, Nguyen MH. 2017. Ceftolozane-tazobactam for the treatment of multidrug-resistant Pseudomonas aeruginosa infections: clinical effectiveness and evolution of resistance. Clin Infect Dis 65:110–120. doi: 10.1093/cid/cix182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.So W, Shurko J, Galega R, Quilitz R, Greene JN, Lee GC. 2019. Mechanisms of high-level ceftolozane/tazobactam resistance in Pseudomonas aeruginosa from a severely neutropenic patient and treatment success from synergy with tobramycin. J Antimicrob Chemother 74:269–271. doi: 10.1093/jac/dky393. [DOI] [PubMed] [Google Scholar]
- 19.Caballero R, Abuhussain A, Kuti JL, Nicolau DP. 2018. Efficacy of human-simulated exposures of ceftolozane-tazobactam alone and in combination with amikacin or colistin against multidrug-resistant Pseudomonas aeruginosa in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 62:e02384-17. doi: 10.1128/AAC.02384-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poole K, Gilmour C, Farha MA, Parkins MD, Klinoski R, Brown ED. 2018. Meropenem potentiation of aminoglycoside activity against Pseudomonas aeruginosa: involvement of the MexXY-OprM multidrug efflux system. J Antimicrob Chemother 73:1247–1255. doi: 10.1093/jac/dkx539. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura A, Hosoda M, Kato T, Yamada Y, Itoh M, Kanazawa K, Nouda H. 2000. Combined effects of meropenem and aminoglycosides on Pseudomonas aeruginosa in vitro. J Antimicrob Chemother 46:901–904. doi: 10.1093/jac/46.6.901. [DOI] [PubMed] [Google Scholar]
- 22.Yapa WS, Li J, Patel K, Wilson JW, Dooley MJ, George J, Clark D, Poole S, Williams E, Porter CJH, Nation RL, McIntosh MP. 2014. Pulmonary and systemic pharmacokinetics of inhaled and intravenous colistin methanesulfonate in cystic fibrosis patients: targeting advantage of inhalational administration. Antimicrob Agents Chemother 58:2570–2579. doi: 10.1128/AAC.01705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheah S-E, Wang J, Nguyen VTT, Turnidge JD, Li J, Nation RL. 2015. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70:3291–3297. doi: 10.1093/jac/dkv267. [DOI] [PubMed] [Google Scholar]
- 24.Böttger EC, Springer B, Prammananan T, Kidan Y, Sander P. 2001. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep 2:318–323. doi: 10.1093/embo-reports/kve062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. 2008. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A 105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matt T, Ng CL, Lang K, Sha S-H, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Böttger EC. 2012. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci U S A 109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulman E, Belakhov V, Wei G, Kendall A, Meyron-Holtz EG, Ben-Shachar D, Schacht J, Baasov T. 2014. Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity: a strategy for the treatment of genetic diseases. J Biol Chem 289:2318–2330. doi: 10.1074/jbc.M113.533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domalaon R, Idowu T, Zhanel GG, Schweizer F. 2018. Antibiotic hybrids: the next generation of agents and adjuvants against Gram-negative pathogens? Clin Microbiol Rev 31:e00077-17. doi: 10.1128/CMR.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyers M, Wright GD. 2019. Drug combinations: a strategy to extend the life of antibiotics in the 21st century. Nat Rev Microbiol 17:141–155. doi: 10.1038/s41579-018-0141-x. [DOI] [PubMed] [Google Scholar]
- 30.Lyu Y, Yang X, Goswami S, Gorityala BK, Idowu T, Domalaon R, Zhanel GG, Shan A, Schweizer F. 2017. Amphiphilic tobramycin−lysine conjugates sensitize multidrug resistant gram-negative bacteria to rifampicin and minocycline. J Med Chem 60:3684–3702. doi: 10.1021/acs.jmedchem.6b01742. [DOI] [PubMed] [Google Scholar]
- 31.Gorityala BK, Guchhait G, Fernando DM, Deo S, McKenna SA, Zhanel GG, Kumar A, Schweizer F. 2016. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew Chem Int Ed Engl 55:555–559. doi: 10.1002/anie.201508330. [DOI] [PubMed] [Google Scholar]
- 32.Idowu T, Arthur G, Zhanel GG, Schweizer F. 2019. Heterodimeric rifampicin–tobramycin conjugates break intrinsic resistance of Pseudomonas aeruginosa to doxycycline and chloramphenicol in vitro and in a Galleria mellonella in vivo model. Eur J Med Chem 174:16–32. doi: 10.1016/j.ejmech.2019.04.034. [DOI] [PubMed] [Google Scholar]
- 33.Idowu T, Ammeter D, Arthur G, Zhanel GG, Schweizer F. 2019. Potentiation of β-lactam antibiotics and β-lactam/β-lactamase inhibitor combinations against multidrug and extensively drug-resistant Pseudomonas aeruginosa using non-ribosomal tobramycin–cyclam conjugates. J Antimicrob Chemother 74:2640–2648. doi: 10.1093/jac/dkz228. [DOI] [PubMed] [Google Scholar]
- 34.Idowu T, Ammeter D, Rossong H, Zhanel GG, Schweizer F. 2019. Homodimeric tobramycin adjuvant repurposes novobiocin as an effective antibacterial agent against Gram-negative bacteria. J Med Chem 62:9103–9115. doi: 10.1021/acs.jmedchem.9b00876. [DOI] [PubMed] [Google Scholar]
- 35.Ehmann DE, Jahic H, Ross PL, Gu R-F, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Duin D, Bonomo RA. 2016. Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation β-lactam/β-lactamase inhibitor combinations. Clin Infect Dis 63:234–241. doi: 10.1093/cid/ciw243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda S, Nakai T, Wakai Y, Ikeda F, Hatano K. 2007. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob Agents Chemother 51:826–830. doi: 10.1128/AAC.00860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castanheira M, Mills JC, Costello SE, Jones RN, Sader HS. 2015. Ceftazidime-avibactam activity tested against Enterobacteriaceae isolates from U.S. hospitals (2011 to 2013) and characterization of β-lactamase-producing strains. Antimicrob Agents Chemother 59:3509–3517. doi: 10.1128/AAC.00163-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Queenan AM, Bush K. 2007. Carbapenemases: the versatile β-lactamases. Clin Microbiol Rev 20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winkler ML, Papp-Wallace KM, Hujer AM, Domitrovic TN, Hujer KM, Hurless KN, Tuohy M, Hall G, Bonomo RA. 2015. Unexpected challenges in treating multidrug-resistant Gram-negative bacteria: resistance to ceftazidime-avibactam in archived isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother 59:1020–1029. doi: 10.1128/AAC.04238-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walkty A, Adam H, Baxter M, Lagacé-Wiens P, Karlowsky JA, Hoban DJ, Zhanel GG. 2018. In vitro activity of ceftolozane/tazobactam versus antimicrobial non-susceptible Pseudomonas aeruginosa clinical isolates including MDR and XDR isolates obtained from across Canada as part of the CANWARD study, 2008–16. J Antimicrob Chemother 73:703–708. doi: 10.1093/jac/dkx468. [DOI] [PubMed] [Google Scholar]
- 42.Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 43.Pai MP, Nafziger AN, Bertino JS. 2011. Simplified estimation of aminoglycoside pharmacokinetics in underweight and obese adult patients. Antimicrob Agents Chemother 55:4006–4011. doi: 10.1128/AAC.00174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, Walkty A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP, Karlowsky JA. 2012. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther 10:459–473. doi: 10.1586/eri.12.25. [DOI] [PubMed] [Google Scholar]
- 45.Sader HS, Farrell DJ, Flamm RK, Jones RN. 2014. Ceftolozane/tazobactam activity tested against aerobic Gram-negative organisms isolated from intra-abdominal and urinary tract infections in European and United States hospitals (2012). J Infect 69:266–277. doi: 10.1016/j.jinf.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Farrell DJ, Sader HS, Flamm RK, Jones RN. 2014. Ceftolozane/tazobactam activity tested against Gram-negative bacterial isolates from hospitalised patients with pneumonia in US and European medical centres (2012). Int J Antimicrob Agents 43:533–539. doi: 10.1016/j.ijantimicag.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 47.Clinical and Laboratory Standards Institute. 2019. Performance standards for antimicrobial susceptibility testing; 29th informational supplement. CLSI M100 Wayne, PA: Clinical and Laboratory Standards Institute. [Google Scholar]
- 48.Pournaras S, Vrioni G, Neou E, Dendrinos J, Dimitroulia E, Poulou A, Tsakris A. 2011. Activity of tigecycline alone and in combination with colistin and meropenem against Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae strains by time-kill assay. Int J Antimicrob Agents 37:244–247. doi: 10.1016/j.ijantimicag.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 49.Fraile-Ribot PA, Cabot G, Mulet X, Periañez L, Martín-Pena ML, Juan C, Pérez JL, Oliver A. 2018. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J Antimicrob Chemother 73:658–663. doi: 10.1093/jac/dkx424. [DOI] [PubMed] [Google Scholar]
- 50.Idowu T, Schweizer F. 2017. Ubiquitous nature of fluoroquinolones: the oscillation between antibacterial and anticancer activities. Antibiotics 6:26. doi: 10.3390/antibiotics6040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poole K. 2000. Efflux-mediated resistance to fluoroquinolones in Gram-negative bacteria. Antimicrob Agents Chemother 44:2233–2241. doi: 10.1128/aac.44.9.2233-2241.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]