

Abstract
We report three patients with Feingold 2 syndrome with the novel features of growth hormone deficiency associated with adenohypophyseal compression, aortic dilation, phalangeal joint contractures, memory, and sleep problems in addition to the typical features of microcephaly, brachymesophalangy, toe syndactyly, short stature, and cardiac anomalies. Microdeletions of chromosome 13q that include the MIR17HG gene were found in all three. One of the patients was treated successfully with growth hormone. In addition to expanding the phenotype of Feingold 2 syndrome, we suggest management of patients with Feingold 2 syndrome include echocardiography at the time of diagnosis in all patients and consideration of evaluation for growth hormone deficiency in patients with short stature.
Keywords: aortic dilation, Feingold syndrome, growth hormone deficiency, MIR17HG
1 |. INTRODUCTION
Feingold syndrome Types 1 and 2 are autosomal dominant Mendelian disorders characterized by microcephaly, short stature, brachymesophalangy, thumb hypoplasia, and toe syndactyly. Feingold 1 syndrome (FG1) is caused by point mutations and deletions resulting in haploinsufficiency of MYCN, while deletions of 13q resulting in haploinsufficiency of MIR17HG have been implicated as the cause of Feingold 2 syndrome (FG2). The core features common to almost all patients with FG2 are microcephaly, learning disabilities, short stature, and brachymesophalangy. Patients with FG2 have been noted in prior publications to have “mild dysmorphisms” with the most frequently described features being epicanthal folds and shortened palpebral fissures. Phenotypically, the absence of gastrointestinal atresia in FG2 is the major distinguishing feature between FG1 and FG2. Additional features of FG2 may include hearing impairment and cardiac anomalies including bicuspid aortic valve with aortic valve stenosis and Tetralogy of Fallot (Grote, Repnikova, & Amudhavalli, 2015; Low, Buxton, & Newbury-Ecob, 2015; Valdes-Miranda et al., 2014). It has been suggested that the overlapping phenotypes of FG1 and FG2 may be due to miR-17~92 (one of the microRNAs produced by MIR17HG) mediating the action of MYCN (de Pontual et al., 2011). This notion is supported by demonstration of the direct binding of MYC transcription factors to MIR17HG (Li, Choi, Casey, Dill, & Felsher, 2014) and by the rescue of the skeletal abnormalities of Mycn-knockout mice by overexpression of miR-17~92 (Mirzamohammadi et al., 2018). Recent evidence that the skeletal abnormalities in mice with deletion of MYCN or miR-17~92 are mediated by separate pathways could help explain the differences in phenotypes between FG1 and FG2 (Mirzamohammadi et al., 2018).
Heterozygous germline deletion of the polycistronic miR-17~92 miRNA cluster on 13q is the cause of FG2 syndrome. The deletions range in size from 165 kb to 17.2 Mb in an area that includes 141 additional genes (arr[hg19]chr13:81,484,523–99,930,889; Wierenga, Jiang, Yang, Mulvihill, & Tsinoremas, 2013), including 31 genes with an entry in OMIM, and 7 with an associated phenotype. As of April 2017, the total number of cases of FG2 described in the literature was 12 (three in one family, two in another, all others presumed de novo; Grote et al., 2015; Sirchia et al., 2017). Additionally, of 18 cases in DECIPHER (Firth et al.) with a deletion encompassing MIR17HG there were five who have some phenotypic features of FG2 syndrome. The phenotypes and management of FG2 are still being defined and expanded as more cases receive attention.
We report three patients with FG2 with several novel clinical findings including adenohypophyseal compression with concomitant growth hormone insufficiency, aortic dilation, chronic myofascial pain, recurrent soft tissue injury, sleep, and memory problems. These findings expand the clinical spectrum of FG2 syndrome and may help to direct preventive and diagnostic care in patients with deletions of MIR17HG.
2 |. METHODS AND MATERIALS
A single nucleotide polymorphism (SNP)-based microarray was performed on plasma samples from two patients. For the first patient, the Human Infinium CytoSNP-850K v.1.1 Beadchip was used, which contains over 850,000 markers. The data were then analyzed using CNV partition 2.4.4.0 with Human Genome Build 37/hg19. For the second patient, the Affymetrix Cytoscan HD platform was used, which contains over 743,000 SNP probes and 1,953,000 nonpolymorphic copy number probes. The data were analyzed using Chromosome Analysis Suite with Human Genome Build 37/hg19.
3 |. CLINICAL REPORT
3.1 |. Patient 1
This 11-year-old male presented to genetics clinic at 30 months of age for developmental delay, hypotonia, and an abnormal karyotype for a 13q32.1q33 deletion (Table 1). Pregnancy was uncomplicated. He was born at 38 weeks via emergent C-section for decreased fetal heart rate to a then 35-year-old G2P1 mother and 37-year-old father. No consanguinity was reported. Birth weight was 3,147 g (49th centile) and length was 47.6 cm (24th centile). Spinal MRI prompted by a sacral dimple was normal. As an infant he had failure to thrive, gastroesophageal reflux disease, global developmental delay, hypotonia, and developmental dysplasia of the hips managed by a harness through orthopedics. A renal ultrasound and echocardiogram were normal. Parental studies were not completed. The parents did not have any features of FG2 on exam and had no history of learning or growth problems.
TABLE 1.
Summary of clinical features of the patients reported herein in addition to the patients FG2 reviewed previously (Grote et al., 2015; Marcelis et al., 2008)
| Features | Patient 1 | Patient 2 | Patient 3 | FG2 |
|---|---|---|---|---|
| Deletion size | 8 Mb | 1.84 Mb | 1.84 Mb | 165 kb–17.2 Mb |
| Sex | Male | Male | Male | 6 M:11F reported |
| Microcephaly | Y | N | Y | 88% (14/16) |
| Short stature | Y | N | Y | 86% (13/14) |
| DD/ID/LD | Y | Y | Y | 100% (16/16) |
| Brachymesophalangy | Y | Y | Y | 100% (17/17) |
| 5th finger clinodactyly | Y | Y | Y | 100% (9/9) |
| Thumb hypoplasia | N | N | N | 33% (4/12) |
| Toe Syndactyly | N | Y | Y | 64% (9/14) |
| Gastrointestinal atresia | N | N | N | 0% (0/18) |
| Cardiac defect | Ya | Yb | N | 40%c (4/10) |
| Deafness/hard-of-hearing | N | N | N | 66% (2/3) |
| Keratokonus | N | N | N | 1 patient |
| Strabismic amblyopia | N | Y | N | NR |
| Compressed adenohypophysis | Y | N | N | NR |
| Memory impairment | N | Y | Y | NR |
| Insomnia | N | Y | Y | NR |
When no report was made of the presence or absence of a feature, that patient was not included in the denominator. Current study’s patients included in FG2 summary column. NA = not applicable; NR = frequency not reported.
Dilated aortic annulus, aortic root, and sinotubular junction.
Bicuspid aortic valve.
Stenotic bicuspid aortic valve with trivial regurgitation, tetralogy of Fallot, unspecified.
For developmental delays, he received physical, occupational, and speech and language therapies. He sat independently by 9 months, pulled to stand by 17 months, and walked by 19 months. He had a pincer grasp after 1 year of age and at 30 months he had 10–15 words though was difficult to understand. Later, he was diagnosed with attention deficit hyperactivity disorder requiring pharmacotherapy. At age 10, he required special education support in school. He required multiple dental procedures due to erosion and carries, and ear tube placement for recurrent otitis media. Family history was remarkable for history of hip dysplasia in his mother and a healthy older full sister.
On initial evaluation in our clinic at 30 months, he had short stature (75.5 cm, <3rd centile and equivalent to 50th centile for a 12-month-old male), microcephaly (45.5 cm, <3rd centile and equivalent to 50th centile for a 10-month old), bilateral epicanthal folds, small nose with a smooth philtrum, microstomia with micrognathia, posteriorly rotated ears, bilateral 5th finger clinodactyly, proximally placed thumbs, and proportionately small hands (<3rd%tile total hand length) for height (Figure 1). He had mild joint laxity at the elbows and knees and a mild pectus excavatum deformity. Fifth finger brachydactyly was noted at a follow-up visit. A BAC array showed 13q31.3q32.2 (RP11–319L6->RP11–383H17) x1. There were no genes in this deletion at that time that were associated with the patient’s phenotype.
FIGURE 1.
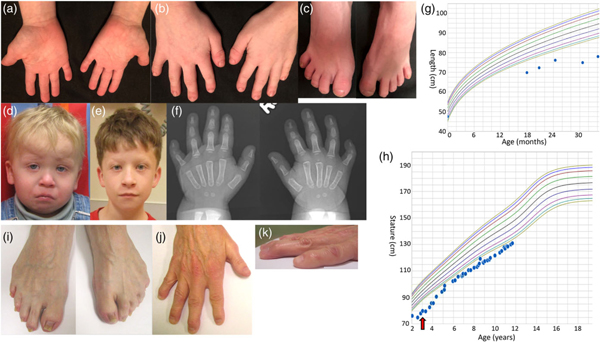
Patient 1’s hands (a,b), feet (c), profile age 2 years 5 months (d), age 10 years 11 months (e), hand radiographs (f), and growth charts (g—infant 0–24 months, h—child 2–12 years). Red arrow indicates initiation of GH therapy. Feet (i) and hand (j,k) of patient 3 [Color figure can be viewed at wileyonlinelibrary.com]
He was evaluated for short stature at age 2, and a brain MRI revealed a compressed adenohypophysis due to a suspected Rathke cleft cyst, a partially empty sella, and a normal neurohypophysis (Figure 2). At 3 years of age, growth hormone deficiency was diagnosed with peak growth hormone level of 2.2 ng/ml, and growth hormone therapy was initiated and maintained at weight-based dosing to the time of this report. He had a delayed bone age of 2 years to 2 years 8 months at a chronological age of 4 years 5 months. The growth velocity was below the 3rd centile prior to initiation of therapy, after the initiation of therapy (age 3.5–5.5 years) was above the 75th centile, and then dropped to the 50th centile or below after age 6. The IGF-1 values were in the high-normal range after initiation of therapy (Supporting Information Table S1). At age 8 years his growth plateaued: a work up to rule out a secondary growth disorder was normal including thyroid studies (Supporting Information Table S1). A follow-up bone age at chronological age 10 years 8 months revealed a bone age between the standard deviation of 11 year 6 months and 12 year 6 months (normal). He continues on growth hormone treatment (0.3 mg/kg/dose).
FIGURE 2.
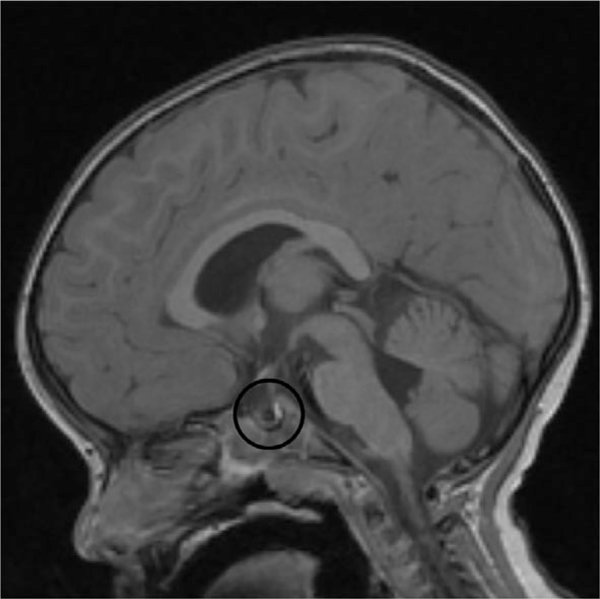
Patient 1’s pituitary MRI demonstrating compressed anterior pituitary, normal posterior pituitary, and possible Rathke cleft cyst
Currently at age 11 years, he plots at the 1st centile for age, which is shorter than his mid-parental height expectation at the 75th percentile. The echocardiogram at age 32 months revealed aortic annulus and aortic root dilation to 1.32 cm (Z-score + 3.02) and 1.88 cm (Z-score + 3.49), respectively. Neither the sinotubular junction nor the ascending aorta were dilated. Repeat echocardiogram at age 10 years showed aortic root, aortic annulus, and sinotubular junction all mildly dilated with measurements of 2.65 cm (Z-score + 3.11), 1.83 cm (Z-score + 2.60), and 2.11 cm (Z-score + 2.86), respectively. Ophthalmology and audiology evaluations were normal. A SNP array was performed to better define his known chromosome 13q deletion and detect other CNVs to consider in the context of his phenotype. The ~8 Mb deletion at 13q31.3q32.3 was defined to include nucleotides 91,491,721–99,522,261 (arr[hg19]) which includes the MIR17HG gene (Figure 3), recently implicated to cause Feingold 2. No other clinically significant CNVs were detected.
FIGURE 3.
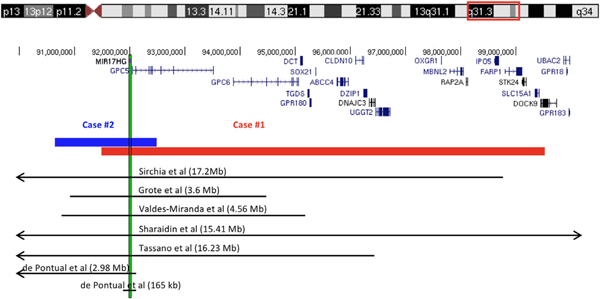
Schematic representation of the deletions identified in Patients 1 and 2 and the genes in the region (only those with OMIM phenotype are included), above the deletions reported in the literature. The vertical bar shows the location of MIR17HG [Color figure can be viewed at wileyonlinelibrary.com]
3.2. |. Patient 2
This 28-year-old male presented to genetics clinic for suspected myopathy, recurrent musculoskeletal pain and soft tissue injuries, and dysmorphic features as listed below (Table 1). He was born full term via vaginal delivery after an unremarkable pregnancy. His birthweight was 2.26 kg (0.6th centile). He met all of his developmental milestones on time. The patient reported comprehension and memory problems and required extra time for tests in college but graduated. Additional medical history included depression, unilateral strabismus that required surgery, and chronic diffuse musculoskeletal pain. He had sleep problems including daytime somnolence and insomnia, and for a time he had a diagnosis of narcolepsy that was subsequently retracted. His mother is of Chinese heritage, and his father is of European heritage; consanguinity is not reported. His father (Patient 3), also has FG2 syndrome.
On evaluation in our clinic at age 28 years, his head circumference was 54 cm (~10th centile), weight 48.9 kg (<5th centile), height 1.67 m (~15th centile), and BMI 17.5. He had epicanthal folds and hypotelorism; inner canthal distance of 2.7 cm (13th centile), interpupillary distance 5.0 cm (1st centile), outer canthal distance 7.7 cm (1st centile), and palpebral fissure length 2.5 cm (17th centile). He had low set nipples, low scapulae with winging, and short 5th digits of all four extremities. He had contractures of both the proximal and distal interphalangeal joints of hands and feet. There was syndactyly of toes 2–4 and scooped toenails. Examination was otherwise normal including skin and hair. Neurology evaluation for suspected muscle disease showed normal muscle strength and deep tendon reflexes as well as normal electromyography and nerve conduction study. In addition, he had Raynaud’s phenomenon, sclerodactyly of the distal interphalangeal skin, and paresthesias in the distal shin and ankles Audiology evaluation was normal.
Other evaluations which were normal included extensive serologic testing for autoimmune and inflammatory disorders, abdominal ultrasound, and brain MRI. Echocardiogram showed a bicuspid aortic valve with trace aortic regurgitation and a normal aorta. A skeletal survey series of X-rays showed mild levoconvex curvature of the thoracic spine and mildly foreshortened distal phalanges of the hands and feet. SNP microarray revealed a 1.8 Mb deletion of 13q31.3 (90,653,738–92,490,593) encompassing MIR17HG and a 650 kb duplication of uncertain significance in 17p13.3 (83,035–735,768; Figure 3). The 17p13.3 duplication contains seven genes annotated in OMIM: RPH3AL, RFLNB, VPS53, FAM57A, GEMIN4, MRM3, and NXN.
3.3 |. Patient 3
This 70-year-old male is the father of patient 2. He was born at term after an uncomplicated pregnancy; a “curved foot” (possible equinovarus) that required casting was noted at birth with no problems noted in the neonatal period. His medical history includes ulcerative colitis, coronary artery disease, inguinal hernia, obstructive sleep apnea, bilateral vitreous detachment, cataracts, erectile dysfunction due to hypogonadotropic hypogonadism which was late-onset (at age 53 years), was mild, and other pituitary hormones were normal. He had mild noncentral hypothyroidism. He has a reported learning disability and lifelong memory problems. He works as an accountant. He did not report a hearing deficit. At age 70, his head circumference was 53.5 cm (3rd centile), height 1.68 m (11th centile), weight 57.9 kg (8th centile). He had a large nose and short palpebral fissures (2.5 cm, <3rd centile). His inner canthal distance of 3.5 cm (80th centile), interpupillary distance 5.9 cm (40th centile), and outer canthal distance 8.0 cm (10th centile). He had short 5th fingers, short toes with 2–3 and 3–4 toe syndactyly bilaterally (Figure 1). Total hand length was 18.4 cm (75th centile), middle finger length 7.7 cm (75th centile), 5th finger length 4.5 cm bilaterally.
His SNP microarray revealed a 1.8 Mb deletion of 13q31.3 (90,653,738–92,490,593) encompassing MIR17HG but not the duplication.
4. |. DISCUSSION
We present three patients with features of Feingold 2 syndrome (FG2) associated with interstitial deletions encompassing MIR17HG on chromosome 13 who have previously unreported phenotypic features that could impact clinical management including aortic dilation, adenohypophyseal compression with growth hormone deficiency, chronic myofascial pain, and sleep and memory problems. In addition to other common associated issues such as hearing loss, problems with vision, intellectual, or learning disabilities (Table 1), the features presented herein highlight several treatable medical issues that may occur in patients with FG2.
The pathogenesis and genetic determinants of the different phenotypic manifestations of FG2 syndrome have not been clearly delineated. MIR17HG encodes a polycistronic miRNA transcript and has been implicated as the minimum common deleted region in all patients with FG2 syndrome (de Pontual et al., 2011; Grote et al., 2015). While the majority of the phenotypes can be presumed to be related to deletion of the miR72~19 cluster, uncommon phenotypes such as keratoconus have been suggested to be caused by deletion of nearby genes (Sirchia et al., 2017). Haploinsufficiency of GPC5, (one of the genes closest to MIR17HG), has also been hypothesized to be related biologically to the limb phenotypes of FG2 (de Pontual et al., 2011, Quélin et al., 2009). This is supported by GPC5 expression studies in mice showing restricted expression in the developing CNS, limb, and kidney (Saunders, Paine-Saunders, & Lander, 1997). However, patients in DECIPHER with deletion of only GPC5 did not have skeletal phenotypes (Sharaidin et al., 2013) leading to the view that deletion of MIR17HG is responsible for the core clinical findings of FG2.
Cardiac phenotypes appear to be relatively common in FG2 as, with the addition of our patients (Table 1), 50% of those patients with FG2 in whom a cardiac examination was reported had an anomaly. Patient 1 is the first patient with aortic dilation and patient 2 is the second patient with bicuspid aortic valve (BAV). BAV is the most common congenital heart defect with an estimated prevalence of 1.3% of the population worldwide and it is associated with aortic dilation/aneurysm 20–84% of the time as well as varying degrees of valvular dysfunction (Verma & Siu, 2014). Although the molecular basis of the BAV-aortic aneurysm spectrum is not known at this time, it is known to be inherited in an autosomal dominant manner with reduced penetrance and variable expressivity (Kent et al., 2013). Established guidelines exist for the monitoring and management of BAV (Verma & Siu, 2014) including echocardiography for evaluation of the aortic valve and ascending aorta, which should be considered at the time of diagnosis for any patient with FG2.
Patient 1 was treated for short stature with growth hormone with some success. This is the first report of successful growth hormone therapy in FG2. The two previously reported patients with FG2 have been given growth hormone; one without apparent impact on height (Grote et al., 2015; 25–50th centile for height at birth, 31st centile at age 14) and one without improvement (Sirchia et al., 2017). No radiographic evaluation of the pituitary gland of these patients was reported. It could be that all patients with FG2 who have GH deficiency also have pituitary abnormalities on imaging, or in some cases the dysregulation of genes affected by the MIR17HG locus could result in GH deficiency. SOX21 has been suggested as a potential candidate gene responsible for GH deficiency and short stature in 13q deletions (Amor, Voullaire, Bentley, Savarirayan, & Choo, 2005). This gene is included in patient 1’s deletion and in the deletion of one other patient with FG2 (Sharaidin, Knipe, Bain, & Goel, 2013). However, many deletions in patients with FG2 do not include SOX21 including one patient with FG2 and GH deficiency (Grote et al., 2015) and many other patients with short stature and FG2 who have not had their HPA axis evaluated. In a review of 14 patients with 13q deletions of various sizes, the minimal overlapping region of patients who had short stature (all below −2.5 SD) was in band 13q31.3, approximately 2 Mb between 89.5 and 91.6 Mb in band 13q31.3 (Kirchhoff et al., 2009). A report of patient with mosaicism for a ring chromosome and a deletion of 13q31.1-q32.3 had hand malformations and growth hormone deficiency whose episodic hypo-glycemia and stature improved with GH therapy (Amor et al., 2005); precise breakpoints of the deletion were not reported. Given patient 1’s success and the success of GH replacement in patients with larger deletions of 13q, referral to endocrinology and testing for growth hormone deficiency in patients with FG2 syndrome should be considered when short stature is present.
Musculoskeletal symptoms have not been previously reported in association with FG. While brachymesophalangy is considered a core feature of FG2, and other digital anomalies such as 5th finger clinodactyly, hypoplastic thumbs, and toe syndactyly are common (Table 1). However, chronic pain, sclerodactyly, and joint contractures have not been previously described. One previously reported patient did have joint stiffness of the distal joints; this patient also had a “congenital heart defect” (Valdes-Miranda et al., 2014). Interestingly, the proximal breakpoint of that patient’s deletion (90789654–95,350,099) was very close to that of patient 2. While the 17q13.3 duplication is of uncertain significance and has not been associated with disease, it is possible that this or another currently unidentified variant could explain parts of this patient’s phenotype not commonly seen in FG2.
In murine models, direct evidence has been found implicating the miR17~19 polycistron in bone, lung, and heart development, as well as developmental and cognitive deficits. A role for miR17~19 have been found in growth and skeletal maturation by regulation of osteoblast proliferation and differentiation (Zhou, Ma, Chen, Chen, & Yu, 2014) in a mouse model with hemizygous deletion of miR17~92 (de Pontual et al., 2011). The functional cooperation and specialization of the miR17~92 polycistron was shown in mice by allelic series of miR-17~92 deletions (Han et al., 2015). Deletion of miR-17 specifically modulated axial patterning, and deletions of various combinations of the cluster led perinatal lethality, lung hypoplasia, and defective cardiac development. The latter was present only in homozygous deletions of the entire cluster.
While intellectual disability and developmental delay are common in FG2, psychiatric problems including obsessions, anxiety, mood dysregulation, and disruptive behavior have only been described in one patient (Ganjavi, Siu, Speevak, & MacDonald, 2014). Sleep and memory problems have not been previously reported in FG2 syndrome. There is evidence that deletion of the miR17~19 cluster leads to developmental and cognitive deficits in mice. Neurobehavioral abnormalities have been found in mice with heterozygous deletions of miR17~92; in addition to decreased body growth, these mice had impaired vocalization, deficits in spatial ability, social novelty recognition, and memory span, as well as altered dopamine and serotonin levels in the prefrontal cortex and hippocampus (Fiori et al., 2015). Hence, sleep and memory problems may be part of FG2.
5. |. CONCLUSIONS
In conclusion, we present three patients with microdeletions of 13q13.3 that expand the clinical features of Feingold 2 syndrome. We believe consideration of testing for GH deficiency and echocardiography should be standard for patients with FG2 syndrome. In addition, physicians caring for patients with FG2 should be aware of the potential for problems with mood (Valdes-Miranda et al., 2014), memory, and sleep.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank the patients and their families for participation in this study.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
CONFLICT OF INTEREST
The authors declare no conflicts of interest related to the publication of this article.
REFERENCES
- Amor DJ, Voullaire L, Bentley K, Savarirayan R, & Choo KHA (2005). Mosaic monosomy of a neocentric ring chromosome maps brachyphalangy and growth hormone deficiency to 13q31.1–13q32.3. American Journal of Medical Genetics, 133A, 151–157. 10.1002/ajmg.a.30527 [DOI] [PubMed] [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, … Amiel J (2011). Germline deletion of the miR-17–92 cluster causes growth and skeletal defects in humans. Nature Genetics, 43, 1026–1030. 10.1038/ng.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori E, Babicola L, Andolina D, Coassin A, Pascucci T, Patella L, … Ventura R (2015). Neurobehavioral alterations in a genetic murine model of Feingold syndrome 2. Behavior Genetics, 45, 547–559. 10.1007/s10519-015-9724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganjavi H, et al. (2014). BMJ Case Rep. 10.1136/bcr2014-207501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote LE, Repnikova EA, & Amudhavalli SM (2015). Expanding the phenotype of feingold syndrome-2. American Journal of Medical Genetics, 167, 3219–3225. 10.1002/ajmg.a.37368 [DOI] [PubMed] [Google Scholar]
- Han Y-C, Vidigal JA, Mu P, Yao E, Singh I, González AJ, … Ventura A (2015). An allelic series of miR-17~92 mutant mice uncovers functional specialization and cooperation among members of a miRNA polycistron. Nature Genetics, 47, 766–775. 10.1038/ng.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent KC, Crenshaw ML, Goh DLM, Dietz HC (2013). Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J Thorac Cardiovasc Surg, 146, 158–165.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff M, Bisgaard A-M, Stoeva R, Dimitrov B, Gillessen-Kaesbach G, Fryns J-P, … Stefanova M (2009). Phenotype and 244k array-CGH characterization of chromosome 13q deletions: An update of the phenotypic map of 13q21.1-qter. American Journal of Medical Genetics, 149A, 894–905. 10.1002/ajmg.a.32814 [DOI] [PubMed] [Google Scholar]
- Li Y, Choi PS, Casey SC, Dill DL, & Felsher DW (2014). MYC through miR-17–92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell, 26, 262–272. 10.1016/j.ccr.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low KJ, Buxton CC, & Newbury-Ecob RA (2015). Tetralogy of Fallot, microcephaly, short stature and brachymesophalangy is associated with hemizygous loss of noncoding MIR17HG and coding GPC5. Clinical Dysmorphology, 24, 113–114. 10.1097/MCD.0000000000000069 [DOI] [PubMed] [Google Scholar]
- Marcelis CLM, Hol FA, Graham GE, Rieu PNMA, Kellermayer R, Meijer RPP, … de Brouwer APM (2008). Genotype–phenotype correlations in MYCN-related Feingold syndrome. Human Mutation, 29, 1125–1132. 10.1002/humu.20750 [DOI] [PubMed] [Google Scholar]
- Mirzamohammadi F, Kozlova A, Papaioannou G, Paltrinieri E, Ayturk UM, & Kobayashi T (2018). Distinct molecular pathways mediate Mycn and Myc-regulated miR-17–92 microRNA action in Feingold syndrome mouse models. Nature Communications, 9 10.1038/s41467-018-03788-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quélin C, Bendavid C, Dubourg C, de la Rochebrochard C, Lucas J, Henry C, … Pasquier L (2009). Twelve new patients with 13q deletion syndrome: Genotype–phenotype analyses in progress. European Journal of Medical Genetics, 52, 41–46. 10.1016/j.ejmg.2008.10.002 [DOI] [PubMed] [Google Scholar]
- Saunders S, Paine-Saunders S, & Lander AD (1997). Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb, and brain. Developmental Biology, 190, 78–93. 10.1006/dbio.1997.8690 [DOI] [PubMed] [Google Scholar]
- Sharaidin HS, Knipe S, Bain N, & Goel H (2013). Clinical features associated with a 15.41 Mb deletion of chromosome 13q encompassing the MIR17HG locus. Clinical Dysmorphology, 22, 68–70. 10.1097/MCD.0b013e32835f56b3 [DOI] [PubMed] [Google Scholar]
- Sirchia F, Di Gregorio E, Restagno G, Grosso E, Pappi P, Talarico F, … Brusco A (2017). A case of Feingold type 2 syndrome associated with keratoconus refines keratoconus type 7 locus on chromosome 13q. European Journal of Medical Genetics, 60, 224–227. 10.1016/j.ejmg.2017.01.010 [DOI] [PubMed] [Google Scholar]
- Valdes-Miranda JM, Soto-Alvarez JR, Toral-Lopez J, González-Huerta L, Perez-Cabrera A, Gonzalez-Monfil G, … Cuevas-Covarrubias S (2014). A novel microdeletion involving the 13q31.3–q32.1 region in a patient with normal intelligence. European Journal of Medical Genetics, 57, 60–64. 10.1016/j.ejmg.2014.01.006 [DOI] [PubMed] [Google Scholar]
- Verma S, Siu SC (2014). Aortic Dilatation in Patients with Bicuspid Aortic Valve. New England Journal of Medicine, 370, 1920–1929. [DOI] [PubMed] [Google Scholar]
- Wierenga KJ, Jiang Z, Yang AC, Mulvihill JJ, & Tsinoremas NF (2013). A clinical evaluation tool for SNP arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genetics in Medicine, 15, 354–360. 10.1038/gim.2012.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Ma J, Chen S, Chen X, & Yu X (2014). MicroRNA-17–92 cluster regulates osteoblast proliferation and differentiation. Endocrine, 45, 302–310. 10.1007/s12020-013-9986-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.