

Abstract
A long-standing enigma concerns the geographic and ecological origins of the intensively studied vinegar fly, Drosophila melanogaster. This globally distributed human commensal is thought to originate from sub-Saharan Africa, yet until recently, it had never been reported from undisturbed wilderness environments that could reflect its precommensal niche. Here, we document the collection of 288 D. melanogaster individuals from multiple African wilderness areas in Zambia, Zimbabwe, and Namibia. The presence of D. melanogaster in these remote woodland environments is consistent with an ancestral range in southern-central Africa, as opposed to equatorial regions. After sequencing the genomes of 17 wilderness-collected flies collected from Kafue National Park in Zambia, we found reduced genetic diversity relative to town populations, elevated chromosomal inversion frequencies, and strong differences at specific genes including known insecticide targets. Combining these genomes with existing data, we probed the history of this species’ geographic expansion. Demographic estimates indicated that expansion from southern-central Africa began ∼13,000 years ago, with a Saharan crossing soon after, but expansion from the Middle East into Europe did not begin until roughly 1,800 years ago. This improved model of demographic history will provide an important resource for future evolutionary and genomic studies of this key model organism. Our findings add context to the history of D. melanogaster, while opening the door for future studies on the biological basis of adaptation to human environments.
Keywords: population genomics, Drosophila, Africa, demographic history, wilderness collection, commensal evolution
Introduction
Drosophila melanogaster is a globally widespread species which “has invariably appeared to be a strict human commensal” (Lachaise and Silvain 2004). In spite of the importance of this species for diverse fields of biological inquiry, it remains “among the commonest species whose exact place of origin and even ancestry have never been satisfactorily explained” (Lachaise and Silvain 2004). Its relatives are primarily distributed across sub-Saharan Africa and nearby islands, and a biogeographic analysis proposed an ancestral range in western and central Africa (Lachaise et al. 1988). Despite considerable efforts to sample D. melanogaster from this region, it was never discovered in undisturbed equatorial wilderness, instead occurring only in human settlements and “seminatural habitats” in areas such as the Taï Rainforest of Côte d'Ivoire and Mount Nimba in Guinea (Lachaise et al. 1988).
More recently, population genomic analysis indicated that D. melanogaster may have instead originated in southern-central Africa: populations from Zambia and Zimbabwe have the highest levels of genetic variation, whereas other populations may have lost diversity due to founder event bottlenecks during geographic expansion (Pool et al. 2012). These findings motivated the hypothesis that D. melanogaster had originated (and might still persist) in wild environments of southern-central Africa (Pool et al. 2012), which are primarily characterized by seasonally dry Miombo and Mopane woodlands (Ryan et al. 2016). These woodlands feature important seasonal fluctuations in fruit resources (Walker 1995), temperature, and especially precipitation (fig. 1). An origin from such variable environments (as opposed to humid equatorial forest) might contribute to the species’ more robust thermal, desiccation, and starvation tolerances than related species (Stanley et al. 1980; van Herrewege and David 1997). Drosophila melanogaster has occasionally been sampled from lodges or camps near natural areas in this region, including from the Sengwa Research Station in Zimbabwe (Begun and Aquadro 1993) from Matobo and Victoria Falls National Parks in Zimbabwe, and from Chobe National Park in Botswana (Dubill 1996). However, it has been unclear whether the species consistently occupies wilderness environments in this region.
Fig. 1.

Collection environment and genomic differentiation of the Kafue population of Drosophila melanogaster. (A) Locations of five D. melanogaster wilderness collections. (B–F) Satellite imagery of these sites. (G) Miombo woodland environment. (H) A sample of the diverse array of fruits present in this environment. (I) Climate graph for Livingstone, Zambia, depicting an extended dry season and a wide range of temperatures.
Here, we report the sampling of D. melanogaster from five different wilderness areas in southern-central Africa, including one recently described collection (Mansourian et al. 2018). We compare sequenced genomes from a wilderness collection in Zambia to those from nearby town populations, revealing noteworthy genome-wide and locus-specific patterns of genetic diversity. Based on this expanded set of genomes, we perform a demographic analysis that refines our understanding of the expansion of D. melanogaster within and beyond Africa.
Results and Discussion
Drosophila melanogaster Survives in African Wilderness
We report the collection of D. melanogaster in five distinct natural areas of Zambia, Zimbabwe, and Namibia (fig. 1). These locations represent a gradient of remoteness from human habitation, ranging from 1.4 to >55 km from the nearest camp or village (table 1). At Matobo National Park, the revelation of this species’ strong affinity for the local marula fruit (Mansourian et al. 2018) enabled the collection of 255 wilderness individuals. Especially given that human hunter–gatherers who formerly occupied this region also favored marula (Walker 1995), it may be that some commensal evolution has occurred in the histories of these wilderness populations. Regardless, the existence of remote wilderness populations in the Miombo and Mopane forests, but not elsewhere on the African continent (Lachaise and Silvain 2004), is consistent with the hypothesis of a precommensal origin from these environments (Pool et al. 2012). These findings should motivate additional collection efforts to further clarify the wilderness ecology of D. melanogaster, including consistent sampling across space and time.
Table 1.
Collection Information for Wilderness Samples of D. melanogaster.
| Location | Latitude | Longitude | Females | Males | Month (season) | Collectors | Woodland | Site Information |
|---|---|---|---|---|---|---|---|---|
| Matobo NP, ZW | −20.48 | 28.51 | 74 | 142 | March 2017 (late wet) | Stensmyr/Mansourian | Mixed | Several sites >3 km from lodge or camp site, mostly marula groves |
| Gonarezhou NP, ZW | −21.72 | 31.71 | 2 | 0 | February 2015 (late wet) | Rebecca Harris | Mopane | >35 km from camp and >55 km from town |
| Kafue NP, ZM | −14.69 | 26.39 | 10 | 12 | November 2014 (early wet) | Stensmyr/Mansourian | Miombo | 1 male >4 km from McBride's camp site, others with 4 km of camp |
| S Luangwa NP, ZM | −13.07 | 31.79 | 12 | 15 | July/August 2015 (late dry) | Matute/Cooper | Mopane | 1.4 km from Mfuwe Lodge |
| NE Namibia | −17.52 | 24.53 | 8 | 13 | July/August 2015 (late dry) | Matute/Cooper | Miombo | 4.3 km from Zambezi Mubala Lodge |
Wild D. melanogaster Have Distinct Patterns of Genomic Diversity
In Kafue National Park, Zambia, we sampled a single male in Miombo wilderness over 4 km from any human structure, whereas multiple females and males were caught within ∼4 km of an intermittently used camp site (table 1). To gain insights into the biology of wilderness D. melanogaster and its relationship to town populations, we sequenced individual genomes of 5 females and 12 males from this first wilderness collection, including the more remote male individual, using previously described methods (Lack et al. 2015).
Based on sequence comparisons (Patterson et al. 2006; Corbett-Detig and Hartl 2012) against town-sampled genomes with previously inferred karyotypes (Lack et al. 2016), the Kafue population displayed some of the highest chromosomal inversion frequencies ever observed in D. melanogaster. The overall proportion of inverted chromosome arms was 2.3 times higher in Kafue than in the Siavonga, Zambia town population (supplementary table S1, Supplementary Material online; two proportion z-test P < 0.0001), and higher than the previous D. melanogaster collections scored by cytological (Aulard et al. 2002) or genomic methods (Kapun et al. 2016; Lack et al. 2016) depicted in figure 2.
Fig. 2.
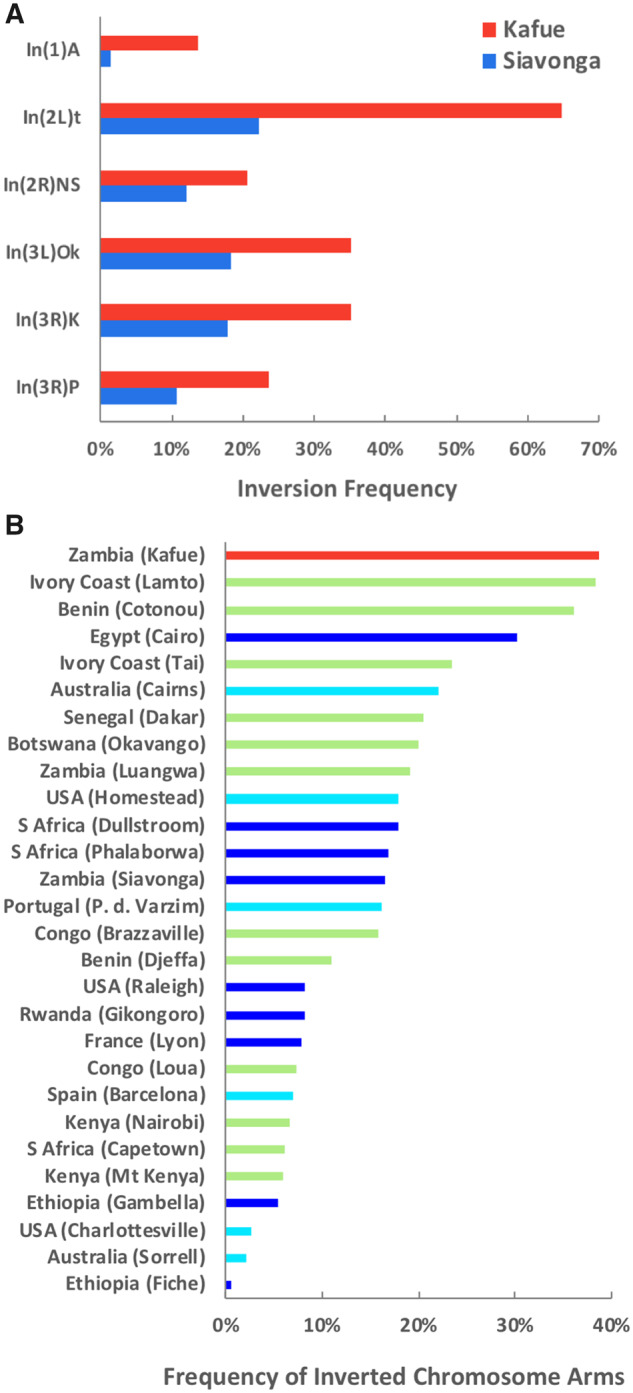
Elevated inversion frequencies in the Kafue population. (A) For all six chromosomal inversions common in this region, the Kafue park population yielded higher rearrangement frequencies than the Siavonga town population. (B) Kafue had a higher proportion of inverted chromosome arms (across X/2L/2R/3L/3R) than previous Drosophila melanogaster collections from Africa or elsewhere. Inversions were computationally inferred from individual genomes (dark blue) or pooled genomic samples (light blue), or scored cytologically (green). Frequencies were compiled for populations with sample sizes of at least 20, and for the six inversions in (A), plus In(1)Be, In(3L)P, and In(3R)Mo, which are not common in Zambia. Among pooled samples, only the populations yielding the highest and lowest inversion frequencies from each continent are depicted.
At least three distinct hypotheses could account for this preponderance of inversions. First, although these inversions all represent derived states, high inversion frequencies could be an ancestral characteristic, with frequencies declining during the adaptation and geographic expansion of commensal populations (analogous to the decline of inversion frequencies as the species has adapted to colder environments; Adrion et al. 2015; Pool et al. 2017). Second, high inversion frequencies could represent a derived characteristic, with local inversion haplotypes creating coadapted gene complexes (Kirkpatrick and Barton 2006) that link wilderness-adapted alleles in this park population to buffer against migration from larger town populations. Third, if the Kafue population represents a relatively small local deme (as suggested below), in which inbreeding may frequently expose recessive lethal/deleterious alleles (Greenberg and Crow 1960), then inversion heterozygotes might reduce such fitness costs in light of their lower levels of genomic homozygosity. Further experiments and analysis will be needed to clarify the evolutionary forces shaping inversion frequencies in park and town populations.
Principle components analysis (PCA) revealed overall genetic similarity between Kafue flies and southern African town populations, in line with a modest FST of 0.028 between Kafue and Siavonga (supplementary fig. S1, Supplementary Material online). The more remote wilderness-collected individual clustered by PCA with other Kafue genomes, consistent with their membership in the same local population. Surprisingly, the Kafue genomes largely held a subset of the variation contained in the Siavonga, Zambia town population. Nucleotide diversity was reduced by 17% in the park sample (0.731% vs. 0.876%; permutation P < 0.00001), which could not be explained by differences in haploid versus diploid genotype calling (see Materials and Methods section). This diversity reduction was present on all five major chromosome arms (supplementary fig. S2, Supplementary Material online), but it was more pronounced on the X chromosome, which can be more affected by population size reductions (Pool and Nielsen 2007), and on the two autosomal arms that showed sharper inversion frequency differences (2L and 3R; supplementary table S1, Supplementary Material online).
Although further genomic analysis of wilderness populations will be needed to assess the generality of these diversity relationships, the lower diversity of the Kafue genomes does not align with the predictions of a domestication bottleneck in the history of our town populations. Speculatively, the reduced diversity of Kafue could hint that this local park population is part of a metapopulation in which individual demes have lower and potentially fluctuating population sizes that reduce local genomic diversity, whereas the broader wilderness metapopulation might conceivably hold as much or more diversity than a comparable set of town samples. Alternatively, this pattern does not exclude the possibility of the Kafue population being reintroduced from a Siavonga-like commensal source population, and experiencing a founder event or subsequent bottleneck. Thus, we pursued demographic inference to clarify the history of divergence and migration among park and town populations.
The History of D. melanogaster
The availability of population genomic data from park and town populations offers the potential to illuminate the history of this species within and beyond its ancestral range. We therefore used fastsimcoal (Excoffier et al. 2013), a frequency-based demographic inference method, to estimate historical parameters relating the Kafue sample and previously sequenced town samples (Lack et al. 2016). We deployed an iterative model-building approach to estimate the species’ expansion history, which eliminated the need to estimate a prohibitive number of demographic parameters simultaneously. We began with a core set of town populations (Cameroon, Ethiopia, Rwanda, Zambia), since many of them have larger sample sizes than Kafue and their somewhat greater differentiation may yield clearer estimates of divergence times, before extending this model to include other town populations and Kafue. Since we could not investigate every possible population topology, we focused on the Dxy distance tree obtained from genomic data (Pool et al. 2012), except for ambiguous nodes for which we compared likelihoods from multiple topologies. We separately obtained results for chromosome arms X, 2R, and 3L, because these arms gave adequate samples of inversion-free genomes for most populations of interest. Sample sizes ranged between 9 and 172 depending on the population and chromosome arm. For brevity, we mainly focus below on median arm point estimates and their associated 95% confidence intervals (CIs) for each parameter discussed below (Materials and Methods), whereas full results are presented in supplementary table S2, Supplementary Material online. Note that these CIs reflect uncertainty in demographic parameters only, and not uncertainty in the assumed mutation rates (5.21 × 10−9 for autosomes and 5.07 × 10−9 for the X chromosome; Huang et al. 2016) and generation time (15 per year; Turelli and Hoffmann 1995; Pool 2015), at least the latter of which may vary across space and time (Cooper et al. 2014).
Among the town populations, we estimated that Zambia and Rwanda populations of D. melanogaster split ∼12,887 years ago (CI 11,091–17,113; fig. 3). This split may reflect the timing of D. melanogaster expanding beyond its southern-central African ancestral range, and it aligns with the archaeologically inferred peak of marula usage by hunter–gatherers in Zimbabwe (Walker 1995). We estimated that the Cameroon–Oku sample split from the Rwanda lineage ∼12,607 years ago (CI 10,975–16,243), which overlaps with the Zambia–Rwanda split, suggesting a relatively rapid onward expansion into west-central Africa. Our estimates indicate that our Ethiopian populations split off an estimated 9,571 years ago (CI 8,916–10,547). The above divergence times suggest sub-Saharan expansion within a broad interval (7–16 kya) in which African environments were becoming warmer and wetter following the Last Glacial Maximum (Chevalier and Chase 2015), potentially impacting both the distribution of wild habitat for D. melanogaster and human activity in the region. These results contrast with one study’s 72 kya divergence estimate between Zambia and a lumped panel of four western African populations (Kapopoulou et al. 2018). However, they are similar to the ten pairwise sub-Saharan divergence times from another recent study (Kern and Hey 2017). Extending our core model, we estimated a relatively recent divergence time between our lowland and highland Ethiopian samples (based on only the X chromosome, which met sample size thresholds) at 2,693 years (CI 2,339–3,061), suggesting that occupation of the Ethiopian plateau was a more recent event.
Fig. 3.

An estimated history of sampled Drosophila melanogaster populations. (A) For the focal population tree identified from distance relationships and model likelihood comparisons, median arm point estimates are depicted for each estimated divergence time and population size. (B) The locations of the included populations are indicated. Colors of population branches (A) and circles (B) indicate populations and lineages within the apparent ancestral range (red), and those in regions occupied by roughly 12 kya (yellow) and by ∼2 kya (blue).
Significant interest has centered on the timing of the species’ expansion out of sub-Saharan Africa, entailing a population bottleneck that created a major axis of genetic differentiation. Previous demographic studies (Li and Stephan 2006; Thornton and Andolfatto 2006; Duchen et al. 2013; Arguello et al. 2019) were limited to single African (Zimbabwe) and European (the Netherlands) populations, and often assumed that the split between these populations occurred at the same time as the trans-Saharan founder event. In contrast, we investigated more geographically detailed models in which an Egypt/France branch split off from either the Cameroon, Ethiopia, or Rwanda lineages, or from internal branches between them. Of these, a split from the internal branch between the Rwanda and Cameroon lineages offered the highest likelihood for all three arms (supplementary table S3, Supplementary Material online), with an estimated split time of 12,406 years ago (CI 12,366–12,593). Hence, the species’ trans-Saharan expansion may have extended from its expansion across equatorial Africa, at a time when the Sahara was beginning to grow less arid (Chevalier and Chase 2015). There is evidence that fig cultivation in the Middle East began more than 11,000 years ago (Kislev et al. 2006), which may have aided the persistence of commensal D. melanogaster outside of sub-Saharan Africa. Our estimated Saharan crossing ∼12 kya is more recent than some previous population genetic estimates of ∼16 to 23 kya (Li and Stephan 2006; Thornton and Andolfatto 2006; Duchen et al. 2013; Arguello et al. 2019). This difference partly stems from our use of an estimate of 15 generations per year (Turelli and Hoffmann 1995; Pool 2015), versus the order of magnitude conjecture of 10 used in other studies. Our estimates may also be aided by the inclusion of equatorial African populations, whereas most previous studies fixed the out-of-Africa bottleneck time to equal the divergence time between European and southern African populations.
We also report the first estimate of the expansion of D. melanogaster from the Middle East into Europe, based on an Egypt–France split just 1,840 years ago based on arms X/3L (joint CI 1,528–1,942). This estimate offers context for the expansion of D. melanogaster into cooler environments, although it may not have reached northern Europe until much later (Keller 2007). The European expansion may have been facilitated by historical factors such as Roman and Byzantine rule of the eastern Mediterranean region, and by the concurrent expansion of citrus and grape cultivation in southern Europe (Watson 1983; Unwin 2005).
Having addressed the history of range expansion, we returned to the Kafue sample. We explored four models in which Kafue branched from 1) the lineage ancestral to all town populations, 2) the shared Zambia–Siavonga and South Africa branch, 3) the Zambia–Siavonga terminal branch, or 4) the internal branch ancestral to Rwanda and the other nonsouthern populations. Of these, model 1, with Kafue as the sister to all other populations, had the best likelihood for two of three arms (supplementary table S3, Supplementary Material online), and so we focused on this topology. Model 4, which was supported by arm 2R, would still imply that the Kafue lineage had split from the others by roughly 13 kya. For model 1, we obtained an estimate of 36,872 years for the divergence between Kafue and the town populations, with a fairly broad CI (27,087–58,786). Although this topology tentatively places Kafue outside the group of town populations, we emphasize that the history of this species’ adaptation to human settlements may have been spatially and temporally complex, and that this topology is not a necessary condition for important evolutionary differences between wilderness and town populations. Our estimated split time between geographically proximate Zambia populations is consistent with D. melanogaster having a deep history in this region, and suggests that the Kafue population was not recently reintroduced from a Siavonga-like town population.
Concurrently, we added a South African population to our model, resulting in a divergence time estimate of 1,324 years from the Siavonga lineage (CI 991–1,643). A surprisingly recent expansion southward from the ancestral range could relate to Bantu groups’ arrival in this region beginning ∼1,500 years ago and the accompanying onset of agriculture (Newman 1995).
Because we focused on the chronology of range expansion and analyzed many populations, we implemented a simple model of population size, with two epochs in the ancestral population and one for each internal/terminal branch (fig. 3). Our estimates implied growth in the ancestral range, in line with the excess of rare alleles observed in sub-Saharan populations (Lack et al. 2015, 2016). Natural selection may also contribute to such long-term growth signals, while downwardly biasing estimates of Ne (Ewing and Jensen 2016; Schrider et al. 2016). However, the relatively recent parameters that differentiate our populations should be less affected by selective sweeps (Lange and Pool 2018). The population sizes of other branches align with the progressive loss of genetic diversity during the species’ geographic expansion (Pool et al. 2012). Whereas ancestral Ne begins at ∼2.1 million and reaches ∼4.9 million in the Zambia lineage, the lowest value is roughly 150,000 for the Egypt/France lineage encompassing the trans-Saharan expansion (although the true initial founder event size within that lineage was surely lower than our constant size estimate). Estimated migration rates ranged from roughly 10−9 to 10−5, with the highest values occurring between geographically proximate populations (supplementary table S2, Supplementary Material online).
We conducted neutral simulations based on the demographic parameters estimated from each chromosomal arm, and found that resulting patterns of genetic diversity and differentiation corresponded reasonably well to empirical estimates (supplementary table S4, Supplementary Material online). We also performed a validation exercise to confirm the reliability of our demographic estimates. We conducted coalescent simulations using all parameter point estimates for the X chromosome (for which all populations could be included). We then precisely replicated our inference pipeline, including its addition of new populations to a core model as described above, independently for 100 simulated data sets. In general, we found that parameter estimates aligned well with the simulated values (supplementary table S5, Supplementary Material online), suggesting that our demographic inference functioned as intended. Among the few exceptions, the divergence time between Kafue and the town populations was not well-estimated by this procedure. Here, the empirical estimate (the true simulated value) was slightly below the 90% CI of the simulation estimates, whereas the median simulated estimate was over 3-fold higher. This result is in line with the heterogeneity observed among estimates from the three analyzed chromosome arms, which had already indicated uncertainty in the timing of the split between wilderness and town populations. Only one other parameter out of 41 examined, the timing of ancestral population growth, had a validation CI that did not quite overlap with the empirical estimate CI (supplementary table S5, Supplementary Material online). Additional wilderness sampling and analysis may further illuminate the historical and contemporary relationships between wilderness and town populations of D. melanogaster.
Potential Adaptive Differences between Park and Town Populations
The overall genetic similarity of park and town populations should improve our ability to detect allele frequency differences at specific genes that may result from local adaptation, including genetic changes related to adaptation to modern settlements. We therefore applied Population Branch Excess (PBE, a three population FST-based statistic; Yassin et al. 2016) to identify genomic windows with unusually strong frequency differences between the Kafue population and both the Zambia–Siavonga and South Africa–Dullstroom town populations.
Notably, the six strongest outlier regions included significantly higher PBE values than expected under neutrality from any window in the genome based on simulations using our demographic estimates (fig. 4A;supplementary table S6, Supplementary Material online; see Materials and Methods section). The top window was centered on the transcription factor lilliputian, in which a 5′ UTR variant absent from 156 analyzed town genomes was present in 13 of 20 Kafue alleles. In light of this gene’s diverse functions, which impact cell size, developmental patterning, and behavior, its specific role in local adaptation between park and town populations remains an open question. Aside from a low recombination region very close to the third chromosome centromere (which includes 36 genes), the strongest outlier regions also included two regions centered on poorly understood genes that both reach their highest expression level in the testes: the predicted phosphatase inhibitor CG12620 and the predicted ubiquitination gene CG31807.
Fig. 4.

Potential genomic targets of adaptive differences between wild and town populations. (A) A plot of PBE indicates genomic windows (averaging 4 kb in length) that have elevated genetic differentiation between the Kafue population and town populations, with labeled examples described in the text. The dotted line indicates genome-wide significance based on neutral demographic simulations. (B) The GO categories most enriched for PBE outliers are given along with raw P values. These GO categories include biological process (green) and molecular activity (blue) functions, after excluding redundant categories. Only the top-listed category has a P value low enough to attain genome-wide significance after correction for multiple tests (adjusted P = 0.003).
The other two top outliers both centered on neurotransmission-related genes that encode known insecticide targets: Acetylcholine esterase (Ace) and nicotinic Acetylcholine Receptor α5 (nAChRα5). nAChRα5 is part of the receptor targeted by neonicotinoids and some other insecticides (Perry et al. 2008; Watson et al. 2010). Ace is a major determinant of resistance to organophosphate and carbamate insecticides (Mutero et al. 1994). Known resistance mutations at Ace have been primarily reported among non-African populations (Karasov et al. 2010). Three such variants (at residues 161, 265, and 330) segregate among our Zambian genomes, with all showing lower frequencies in the park population (3% each in Kafue, vs. 13%, 5%, and 5% in Siavonga). These modest differences are overshadowed by intronic single nucleotide polymorphisms (SNPs) that display frequency differences of ∼50% between these park and town populations. It will therefore be of interest to test for additional resistance variants in African populations, and for differences in insecticide resistance between wilderness and town populations.
We then performed gene ontology (GO) enrichment analysis on PBE outliers to identify functional categories that may hold adaptive differences between the park and town populations. Although this analysis was intended as an exploratory hypothesis generation exercise, one GO category (“morphogenesis of an epithelial fold”) yielded a lower raw P value than would be expected for any GO category by chance (with a comparable P value occurring in only 0.3% of randomized data sets). This and other enriched Kafue PBE GO categories involved developmental and gene regulatory functions that could have a wide range of potential organismal impacts (fig. 4B;supplementary table S7, Supplementary Material online). Among the more specific processes identified were insulin signaling and immune regulation categories (Toll signaling, hemocyte proliferation). Insulin signaling genes have shown strong clinal differentiation in North America (Fabian et al. 2012). Most immunity processes did not show elevated differentiation among five worldwide populations (Early et al. 2017), but some immunity genes displayed parallel SNP frequency changes associated with the parallel adaptation to colder climates (Pool et al. 2017). Although we cannot yet confirm which aspects of fly biology may have been targeted by local adaptation between park and town populations, results of this exploratory analysis provide hypotheses for phenotypes, genes, and processes that may underlie adaptation to wild versus anthropogenic environments. The top GO categories obtained here were distinct from those previously found to differentiate African town populations, which included categories related to olfaction, pigmentation, and splicing (Pool et al. 2012). Hence, the biological nature of adaptive differences between wilderness and town populations may differ from the biology of local adaptation among town populations.
Conclusions
In this study, we have shed light on the origins and history of one of the most widely studied organisms, D. melanogaster. Multiple field expeditions demonstrate that D. melanogaster continues to occupy wild environments in southern-central Africa. Alongside the concurrent discovery that D. melanogaster favors the indigenous marula over any other tested fruit (Mansourian et al. 2018), these results complement population genomic findings (Pool et al. 2012) suggesting that this region represents the precommensal ancestral range of D. melanogaster. Demographic analysis using genomes from wilderness and town areas clarifies important aspects of the expansion history of D. melanogaster within and beyond Africa, including the time-scale of its spread within sub-Saharan Africa, its Saharan crossing, and its spread into Europe, with intriguing parallels to human and climate history.
A wilderness D. melanogaster population from Zambia displays unique genetic features, including 1) reduced genetic diversity, 2) a surprisingly high frequency of polymorphic chromosomal inversions, and 3) elevated genetic differentiation in certain genes related to insecticide resistance, development, immune regulation, and insulin signaling. These patterns merit further study in other wild D. melanogaster populations. More broadly, the opportunity to study wilderness populations of D. melanogaster provides the opportunity to assess behavioral, morphological, and physiological differences between wilderness and commensal populations. Combining these phenotypic analyses with molecular studies, quantitative trait mapping, and population genetics may provide unique opportunities to uncover mechanisms that underlie adaptation to wilderness versus commensal environments.
Materials and Methods
Fly Collection
We collected flies in Zambia, Zimbabwe, and Namibia using either traps constructed from PET bottles (0.5 l) or buckets that were baited with fruit (mango, banana, or local varieties) mixed with baker’s yeast and swept with insect nets. Traps were placed in the field in the morning and hung from trees at ∼1.5 m above ground level and emptied after 12–24 h. Buckets were swept in the morning (8:00–10:00 AM) or evening (5:00–7:00 PM). Drosophila melanogaster was distinguished from other Drosophila species based on morphology (Lachaise et al. 1988; McEvey 2019) of either wild-caught individuals or the F1 male offspring of females. Some collected flies were preserved in 70% ethanol to facilitate later genomic analysis. Collections were performed in coordination with the Zimbabwe Parks and Wildlife Management Authority and the Zambia Wildlife Agency (from which a collection permit was obtained).
Genomic Sequence Data Collection
Genomic sequence data was collected from individual wild-caught, ethanol-preserved flies from the Zambia Kafue sample. These included 5 females (KF4, KF5, KF10, KF11, and KF20.) and 12 males (KF1, KF2, KF3, KF6, KF7, KF8, KF9, KF18, KF21, KF22, KF23, and KF24). Single fly DNA was isolated using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs E7370). Genomic sequencing was conducted using the Illumina HiSeq 2000 platform with 100 bp paired end reads, with ∼300 bp inserts. Sequencing depth per genome ranged from 14× to 51×, with a median depth of 23×. Reference alignment and consensus sequence generation was as previously described in Lack et al. (2015) for inbred strains. To facilitate the analysis of fully heterozygous chromosomes (i.e. all except male X chromosomes), we produced diploid consensus sequences with ambiguity codes representing heterozygous genotype calls. Assessing the outcomes of diploid versus haploid genotype calling, we found that female Kafue X chromosomes had lower nucleotide diversity than male Kafue X chromosomes (0.71% vs. 0.76%). However, X-linked diversity for the (haploid-called) Zambia–Siavonga sample was higher still (0.84%), confirming the lower genetic diversity of the Kafue population regardless of the ploidy of genotype calling. The generated Kafue files, along with published genomes (Lack et al. 2016), were used for the calculation of genome-wide statistics based on genetic distances, and for the analyses described below. Sequence read data is available from NIH Short Read Archive accession PRJNA329555.
PCA and Inversion Detection
To examine potential genetic differentiation between the Kafue sample and previously studied town-collected populations from Southern Africa, we used the PCA implementation of Eigensoft (Patterson et al. 2006) version 6.0.1. Whenever sequences of mixed ploidy were compared (heterozygous Kafue chromosomes versus haploid, homozygous, or hemizygous chromosomes from other populations), sites were categorized as X-linked with individuals labeled as female or male based on ploidy. Since inversions can alter genetic variation across whole chromosome arms (Corbett-Detig and Hartl 2012; Pool et al. 2012), we focused PCA on standard chromosome arms. For published individual strain genomes, inversion calls had previously been documented (Lack et al. 2015). As previously shown (Corbett-Detig and Hartl 2012), inverted chromosome arms consistently have reduced genetic distance in breakpoint regions with respect to each other. For the Kafue genomes, suspected inversions were readily identified via examination of genetic distances around inversion breakpoints, in comparison with published standard and inverted chromosomes from southern Africa genomes (Lack et al. 2015). The inversions present in this region and tested for Kafue genomes were In(1)A, In(2L)t, In(2R)NS, In(3L)OK, In(3R)K, and In(3R)P. These results were consistent with PCA-based groupings when inverted arms were included.
Demographic Estimation
To better understand the expansion history of D. melanogaster, demographic parameters were estimated using fastsimcoal 2.6 (Excoffier et al. 2013). In addition to the Kafue genomes, we added data from other published D. melanogaster genomes from the Drosophila Genome Nexus (Lack et al. 2015, 2016), including town-collected samples from Zambia (ZI), South Africa (SD), Rwanda (RG), Cameroon (CO), Ethiopia (lowland EA and highland EF), Egypt (EG), and France (FR). For each model investigated, we ran fastsimcoal for 100 independent runs, each with 100,000 coalescent simulations and 50 expectation-maximization cycles, and then selected the model with the smallest difference between observed and expected likelihoods to obtain point estimates. Estimated parameters included population split times, symmetric migration rates between geographically adjacent populations, and population sizes (two epochs in the ancestral population, one size for each internal and terminal branch). Ranges of uniform priors are given in supplementary table S2, Supplementary Material online. Population size calculations were based on mutation rates of 5.21 × 10−9 for autosomes and 5.07 × 10−9 for the X chromosome (Huang et al. 2016). Conversion of generations (provided directly by fastsimcoal) to years was based on an estimate of 15 generations per year (Pool 2015).
Parameter estimates were separately obtained for chromosome arms X, 2R, and 3L. The unscaled output of fastsimcoal (raw generations, allelic migration probabilities) helps to make estimates from the X chromosome and autosomes more comparable. However, we might naturally expect lower population size estimates for the X, and we therefore rescale our reported X-linked Ne estimates by a factor of 4/3, corresponding to the assumption of Nf = Nm. In general, we obtained similar results from X-linked and autosomal data (supplementary table S2, Supplementary Material online).
Because inversions may have distinct histories reflecting the action of natural selection and can affect genetic variation across whole arms (Corbett-Detig and Hartl 2012; Pool et al. 2012), we included only genomes that were inversion-free for a given chromosome arm. Higher inversion frequencies on arms 2L and 3R led us to omit them from our demographic analysis. To minimize the impact of direct selection on our analyzed sites, we included only short intron (bp 8 to 30 of introns ≤65 bp in length; Halligan and Keightley 2006) and 4-fold synonymous sites. To reduce the impact of linked selection, we only analyzed sites with regional sex-averaged recombination rates of at least 1 cM/Mb (Comeron et al. 2012).
To obtain input data from each population for each analyzed chromosome arm, we chose sample sizes that jointly maximized this quantity along with the number of retained sites. We then excluded any site that failed to meet the chosen sample size threshold in any analyzed population, and we randomly down-sampled when more alleles than the target sample size were available. Populations were excluded for a given arm if the average available sample size was trivially small; this was the case for EA for 2R and 3L, and EG for 2R only. Sample sizes used for X/2R/3L were: CO 9/9/10, EA 14/0/0, EF 44/25/11, EG 21/0/11, FR 60/53/40, KF 12/20/12, RG 22/21/25, SD 43/11/10, ZI 172/150/141. In interpreting data across chromosome arms, we consider (e.g.), that for arm 2R, the split time of the FR lineage (without EG) from its sub-Saharan ancestral branch refers to the same historical event as the split time of the shared EG/FR lineage for the X chromosome.
Rather than investigate the over 2 million possible rooted topologies of the above nine populations, we focused on the topology supported by genetic distances (fig. 3). We implemented an iterative model-building approach to limit the number of simultaneously estimated parameters. We began with four populations representing the species’ expansion within sub-Saharan Africa: ZI, RG, CO, and EA (or alternately EF). Next, we independently expanded this core model in two directions: 1) by adding the KF and SD samples to complete our southern African sampling, and 2) by adding the EG and FR populations to encompass the species expansion beyond sub-Saharan Africa. For the X chromosome only, we also extended the core model a third time to add a second Ethiopian sample, EF, to a model that already contained EA, to investigate the timing of the species’ expansion into the Ethiopian highlands. When adding additional populations to our core model, we fixed previous point estimates for parameters already estimated that were not obviously connected to the newly added populations.
Only two branch placements were found to vary among neighbor joining trees (Kumar et al. 2016) from the three analyzed chromosome arms, and we therefore conducted model choice simulations to evaluate their placement. We evaluated whether the Kafue population should be placed as the basal branch (most likely topology; fig. 3; supplementary table S3, Supplementary Material online), or else split from the ZI/SD internal branch, the ZI terminal branch, or the internal branch leading to the nonsouthern African populations. We also evaluated whether the branch leading to Egypt/France should split from the terminal branches leading to Rwanda, Cameroon, or Ethiopia, or else from the two internal branches within that population group; of these, a split from the internal branch preceding the Cameroon/Ethiopia split was most likely (fig. 3; supplementary table S3, Supplementary Material online). Model choice was also used in cases where the temporal sequence of unrelated population splitting events needed to be defined, and again the most likely model was the focus of analysis. For any of these cases, fastsimcoal does not offer a formal test of model choice, and we therefore make no claims about the statistical significance of any given topology; we simply focus on the most likely tree and the demographic estimates associated with it.
Simulations via ms (Hudson 2002) implementing point estimate models were conducted to confirm agreement with basic summaries of the observed data (supplementary table S4, Supplementary Material online). CIs for each parameter estimate were obtained by nonparametric bootstrapping—generating 100 bootstrap data sets by resampling with replacement from the empirical data, and performing 100 independent fastsimcoal runs on each of them (mirroring our empirical data analysis). To leverage our estimates from these three chromosome arms, we report the median arm point estimate for each demographic parameter, in addition to arm-specific values. We also report 95% CIs for this median arm estimate in addition to CIs for each separate chromosome arm. To generate CIs for the arm median point estimate, one bootstrap parameter estimate was randomly drawn for each chromosome arm, and the middle value was returned.
We also performed a validation analysis to assess the reliability of our demographic inference pipeline. We conducted coalescent simulations via ms (Hudson 2002) to generate 100 data sets that mirrored empirical data from the X chromosome, simulating a history defined by our X chromosome point estimates for each demographic parameter. We chose the X chromosome because it featured data from all analyzed populations, and 100 data sets was the maximum feasible computational scale of this analysis (entailing millions of cpu hours). Mirroring our empirical data analysis, fastsimcoal was applied to produce parameter estimates and CIs for our core model populations (ZI, RG, CO, EA), and then to add inferences for the remaining populations in three separate runs (KF + SD, EG + FR, and EF alone). The resulting 100 independent estimates for each parameter were examined for bias, and CIs were evaluated to check for any evidence that uncertainty in our estimates had been understated.
Identification of Candidate Regions for Local Adaptation
The PBE statistic (Yassin et al. 2016) was used to quantify genetic differentiation specific to the Kafue genomes, when compared against two town-collected populations samples from Zambia (ZI) and South Africa (SP) (Lack et al. 2015). Due to limited samples of homozygous standard Kafue chromosomes for arms 2L and 3R, inverted chromosome arms were not excluded from the PBE analysis, but similar results were obtained from standard arms only.
PBE measures the degree to which Kafue-specific allele frequency change exceeds that expected based on locus and genomic differentiation (Yassin et al. 2016). Some loci with strongly positive PBE could reflect adaptation to commensal environments shared by the SP and ZI samples, but absent from the Kafue population. PBE was applied in diversity-scaled genomic windows containing 250 nonsingleton SNPs in the ZI sample. Each window’s PBE quantile was determined based on the proportion of windows on the same chromosome arm with an equal or greater PBE value.
To assess the genome-wide significance of window PBE values, we performed neutral simulations based on an estimated demographic model for these same three populations. For simplicity, we focused on the 2R model which featured an intermediate divergence time between Kafue and the other populations. We typical average window lengths and sample sizes for each population, with a recombination rate of 1 cM/Mb that should be conservative outside centromeric and telomeric regions (Comeron et al. 2012). We generated 1 million ms (Hudson 2002) coalescent simulations with the following command line:
./ms 176 1000000 -t 111 -r 169 4600 -I 3 20 134 22 94.2 -en 0 1 0.450 -en 0 2 2.33 -en 0 3 2.18 -em 0 1 3 71.9 -em 0 3 1 71.9 -em 0 2 3 89.4 -em 0 3 2 89.4 -ej 0.00444 3 2 -en 0.00444000001 2 0.542 -em 0.0044000002 1 2 93.8 -em 0.0044000002 2 1 93.8 -en 0.0479 2 0.283 -em 0.0479000001 1 2 4.16 -em 0.0479000001 2 1 4.16 -ej 0.0660 2 1 -en 0.066000001 1 1.01 -em 0.066000002 1 2 0 -em 0.066000002 2 1 0 -en 0.0894 1 1>KFZISD_test2b.txt.
We then obtained a critical value of PBE representing a 5% probability that any of 25,548 windows would reach this threshold under the neutral model, under the conservative assumption that each window’s PBE value is independent.
The top 1% of PBE quantiles were considered outliers for GO enrichment analysis, under the hypothesis that these outliers will be enriched for genuine targets of local adaptation. GO enrichment was assessed as previously described in Pool et al. (2012). Two or more outlier windows were merged into the same outlier window region if they were separated by no more than four nonoutlier windows (to conservatively avoid counting the same selective sweep more than once). Locations of outlier regions were then randomly permuted, while maintaining their lengths, to properly account for the arrangement and lengths of genes in each functional category. Each outlier region was only allowed to vote for a given GO category one time (from both the empirical and permuted outlier regions), to avoid spurious results from clusters of functionally linked paralogs. For each GO term, a raw P value was defined by the proportion of 1,000,000 randomized data sets in which a greater or equal number of outliers from that category was obtained. Then, by comparing across these randomized data sets, the lowest raw P value for each of them was obtained, and a threshold for analysis-wide significance was defined based on a minimum raw P value observed in 5% or fewer randomized data sets.
Supplementary Material
Acknowledgments
We are grateful for the kind hospitality and assistance we received from Chris and Charlotte McBride of McBride’s Safari Camp. We also thank Russ Corbett-Detig for advice on inversion calling and Nandita Garud for providing the locations of Ace SNPs. This work was funded by grants from the Crafoord Foundation and Vetenskapsrådet to MCS, by the National Institutes of Health (R01 GM111797 to J.E.P.), (R01 GM121750 to D.R.M.), and (R35 GM124701 to B.S.C.); and by the National Science Foundation (1737752 to D.R.M.).
References
- Adrion JR, Hahn MW, Cooper BS.. 2015. Revisiting classic clines in Drosophila melanogaster in the age of genomics. Trends Genet. 31(8):434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello JR, Laurent S, Clark AG.. 2019. Demographic history of the human commensal Drosophila melanogaster. Genome Biol Evol. 11(3):844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulard S, David JR, Lemeunier F.. 2002. Chromosomal inversion polymorphism in Afrotropical populations of Drosophila melanogaster. Genet Res. 79(1):49–63. [DOI] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF.. 1993. African and North American populations of Drosophila melanogaster are very different at the DNA level. Nature 365(6446):548–550. [DOI] [PubMed] [Google Scholar]
- Chevalier M, Chase BM.. 2015. Southeast African records reveal a coherent shift from high- to low-latitude forcing mechanisms along the east African margin across last glacial–interglacial transition. Quat Sci Rev. 125:117–130. [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S.. 2012. The many landscapes of recombination in Drosophila melanogaster. PLoS Genet. 8(10):e1002905.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BS, Hammad LA, Montooth KL.. 2014. Thermal adaptation of cellular membranes in natural populations of Drosophila melanogaster. Funct Ecol. 28(4):886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Hartl DL.. 2012. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 8(12):e1003056.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubill AJ. 1996. The evolution and biogeography of sexual isolation in southern African Drosophila [bachelor's thesis]. [Princeton (NJ)]: Princeton Univ.
- Duchen P, Živković D, Hutter S, Stephan W, Laurent S.. 2013. Demographic inference reveals African and European admixture in the North American Drosophila melanogaster population. Genetics 193(1):291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early AM, Arguello JR, Cardoso-Moreira M, Gottipati S, Grenier JK, Clark AG.. 2017. Survey of global genetic diversity within the Drosophila immune system. Genetics 205(1):353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing GB, Jensen JD.. 2016. The consequences of not accounting for background selection in demographic inference. Mol Ecol. 25(1):135–141. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M.. 2013. Robust demographic inference from genomic and SNP data. PLoS Genet. 9(10):e1003905.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian DK, Kapun M, Nolte V, Kofler R, Schmidt PS, Schlötterer C, Flatt T.. 2012. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol Ecol. 21(19):4748–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R, Crow JF.. 1960. A comparison of the effect of lethal and detrimental chromosomes from Drosophila populations. Genetics 45(8):1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD.. 2006. Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 16(7):875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Lyman RF, Lyman RA, Carbone MA, Harbison ST, Magwire MM, Mackay TF.. 2016. Spontaneous mutations and the origin and maintenance of quantitative genetic variation. eLife 5:e14625.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18(2):337–338. [DOI] [PubMed] [Google Scholar]
- Kapopoulou A, Pfeifer SP, Jensen JD, Laurent S.. 2018. The demographic history of African Drosophila melanogaster. Genome Biol Evol. 10(9):2338–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapun M, Fabian DK, Goudet J, Flatt T.. 2016. Genomic evidence for adaptive inversion clines in Drosophila melanogaster. Mol Biol Evol. 33(5):1317–1336. [DOI] [PubMed] [Google Scholar]
- Karasov T, Messer PW, Petrov DA.. 2010. Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet. 6(6):e1000924.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A. 2007. Drosophila melanogaster’s history as a human commensal. Curr Biol. 17(3):R77–81. [DOI] [PubMed] [Google Scholar]
- Kern AD, Hey J.. 2017. Exact calculation of the joint allele frequency spectrum for isolation with migration models. Genetics 207(1):241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick M, Barton N.. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173(1):419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislev ME, Hartmann A, Bar-Yosef O.. 2006. Early domesticated fig in the Jordan Valley. Science 312(5778):1372–1374. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise D, Cariou ML, David JR, Lemeunier F, Tsacas L, Ashburner M.. 1988. Historical biogeography of the Drosophila melanogaster species subgroup In: Hecht MK, Wallace B, Prance GT, editors. Evolutionary biology. New York: Plenum Press; p. 159–225. [Google Scholar]
- Lachaise D, Silvain J-F.. 2004. How two Afrotropical endemics made two cosmopolitan commensals: the Drosophila melanogaster–D. simulans paleogeographic riddle. Genetica 120(1–3):17–39. [DOI] [PubMed] [Google Scholar]
- Lack JB, Cardeno CM, Crepeau MW, Taylor W, Corbett-Detig RB, Stevens KA, Langley CH, Pool JE.. 2015. The Drosophila Genome Nexus: a population genomic resource of 623 Drosophila melanogaster genomes, including 197 from a single ancestral range population. Genetics 199(4):1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack JB, Lange JD, Tang AD, Corbett-Detig RB, Pool JE.. 2016. A thousand fly genomes: an expanded Drosophila Genome Nexus. Mol Biol Evol. 33(12):3308–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange JD, Pool JE.. 2018. Impacts of recurrent hitchhiking on divergence and demographic inference in Drosophila. Genome Biol Evol. 10(8):1882–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Stephan W.. 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet. 2(10):e166.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansourian S, Enjin A, Jirle EV, Ramesh V, Rehermann G, Becher PG, Pool JE, Stensmyr MC.. 2018. Wild African populations of Drosophila melanogaster are seasonal specialists on marula fruit. Curr Biol. 28(24):3960–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvey S. 2019. Drosophila melanogaster and D. simulans (Diptera: Drosophilidae)—morphological differences [Internet]. Available from: https://figshare.com/articles/Drosophila_melanogaster_and_D_simulans_Diptera_Drosophilidae_morphological_differences/7981505/1, last accessed November 26, 2019.
- Mutero A, Pralavorio M, Bride JM, Fournier D.. 1994. Resistance-associated point mutations in insecticide-insensitive acetylcholinesterase. Proc Natl Acad Sci U S A. 91(13):5922–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JL. 1995. The peopling of Africa: a geographic interpretation. New Haven (CT: ): Yale University Press. [Google Scholar]
- Patterson N, Price AL, Reich D.. 2006. Population structure and eigenanalysis. PLoS Genet. 2(12):e190.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T, Heckel DG, McKenzie JA, Batterham P.. 2008. Mutations in Dα1 or Dβ2 nicotinic acetylcholine receptor subunits can confer resistance to neonicotinoids in Drosophila melanogaster. Insect Biochem Mol Biol. 38(5):520–528. [DOI] [PubMed] [Google Scholar]
- Pool JE. 2015. The mosaic ancestry of the Drosophila genetic reference panel and the D. melanogaster reference genome reveals a network of epistatic fitness interactions. Mol Biol Evol. 32(12):3236–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Braun DT, Lack JB.. 2017. Parallel evolution of cold tolerance within Drosophila melanogaster. Mol Biol Evol. 34(2):349.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Corbett-Detig RB, Sugino RP, Stevens KA, Cardeno CM, Crepeau MW, Duchen P, Emerson JJ, Saelao P, Begun DJ, et al. 2012. Population genomics of Sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet. 8(12):e1003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R.. 2007. Population size changes reshape genomic patterns of diversity. Evolution 61(12):3001–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CM, Pritchard R, McNicol I, Owen M, Fisher JA, Lehmann C.. 2016. Ecosystem services from southern African woodlands and their future under global change. Philos Trans R Soc B 371(1703):20150312.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Shanku AG, Kern AD.. 2016. Effects of linked selective sweeps on demographic inference and model selection. Genetics 204(3):1207–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SM, Parsons PA, Spence GE, Weber L.. 1980. Resistance of species of the Drosophila melanogaster subgroup to environmental extremes. Aust J Zool. 28(3):413–421. [Google Scholar]
- Thornton K, Andolfatto P.. 2006. Approximate Bayesian inference reveals evidence for a recent, severe bottleneck in a Netherlands population of Drosophila melanogaster. Genetics 172(3):1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA.. 1995. Cytoplasmic incompatibility in Drosophila simulans: dynamics and parameter estimates from natural populations. Genetics 140(4):1319–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin T. 2005. Wine and the vine: an historical geography of viticulture and the wine trade. London: Routledge. [Google Scholar]
- van Herrewege J, David JR.. 1997. Starvation and desiccation tolerances in Drosophila: comparison of species from different climatic origins. Ecoscience 4(2):151–157. [Google Scholar]
- Walker NJ. 1995. Late Pleistocene and Holocene hunter–gatherers of the Matopos: an archaeological study of change and continuity in Zimbabwe. Studies in African archaeology. Uppsala (Sweden: ): Societas Archaeologica Upsaliensis. [Google Scholar]
- Watson AM. 1983. Agricultural innovation in the early Islamic world; the diffusion of crops and farming techniques, 700–1100. Cambridge: Cambridge University Press. [Google Scholar]
- Watson GB, Chouinard SW, Cook KR, Geng C, Gifford JM, Gustafson GD, Hasler JM, Larrinua IM, Letherer TJ, Mitchell JC, et al. 2010. A spinosyn-sensitive Drosophila melanogaster nicotinic acetylcholine receptor identified through chemically induced target site resistance, resistance gene identification, and heterologous expression. Insect Biochem Mol Biol. 40(5):376–384. [DOI] [PubMed] [Google Scholar]
- Yassin A, Debat V, Bastide H, Gidaszewski N, David JR, Pool JE.. 2016. Recurrent specialization on a toxic fruit in an island Drosophila population. Proc Natl Acad Sci U S A. 113(17):4771–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.