

Abstract
Chemical conjugation of anti-epidermal growth factor receptor monoclonal antibodies (anti-EGFR mAbs) to organic-inorganic hybrid liposomal immunocerasomes via maleimide-thiol coupling chemistry is explored as a mechanism for selectively targeting cancer cells. The cellular uptake and internalization of immunocerasomes are investigated in A431 cells that express an abnormally high level of EGFR, DU145 cells that overexpress EGFR, and HL-60 cells that are used as a negative control. The internalization study reveals a strong correlation between the receptor-mediated endocytosis of immunocerasomes and the membrane expression of EGFR. Further, free anti-EGFR mAbs and immunocerasomes conjugated with anti-EGFR mAbs at nanomolar doses display similar anti-proliferative effects on A431 cells. Additionally, serum proteins greatly reduce the cellular uptake of cerasomes that is mediated by non-specific receptors, but have no adverse effects on the specific EGFR-mediated delivery of immunocerasomes to A431 cells.
Keywords: Organic-inorganic hybrid lipid nanovesicles, Anti-EGFR antibody, Selective targeting, Cancer cells
1. Introduction
Selective targeting is an important parameter in the design and synthesis of therapeutic and/or imaging nanocarriers. Conventionally, the enhanced permeability and retention (EPR) effect in tumor vasculature has been exploited for the selective targeting of macromolecular drugs or drug-loaded nanocarriers to cancer cells [1]. A long circulation time is necessary for drugs or drug-loaded carriers to accumulate in tumor tissues, much more than in normal tissues. This, however, does not completely prevent drugs or drug-loaded carriers from accumulating in healthy tissues and causing tissue damage [2,3]. Compared to the passive EPR-targeting selective delivery, ligands that are specific to cell-surface receptors can be conjugated to nanoparticles, creating therapeutic and/or imaging nanocarriers with an improved ability to selectively target cancer cells [4,5]. Various ligands, including short peptides [4], aptamers [6] and antibodies [5], have been exploited for selective targeting. Of note, antibodies that specifically bind to cell-surface receptors enable the selective delivery of drug-loaded carries to target cells and may possess therapeutic effects as well [7]. As an example, anti-CD20 antibodies selectively bind to and kill CD20-expressing B-lymphoma cells [8].
In this study, epidermal growth factor receptors (EGFR), which are overexpressed by several human cancers [9], are exploited as a promising target for the selective delivery of nanocarriers. Specifically, organic-inorganic liposomal cerasomes are conjugated with anti-EGFR monoclonal antibodies (mAbs) that target the EGFR glycoproteins on cell surfaces [10], creating immunocerasomes for selective delivery. Previously, Leung et al. reported the engineering of cerasomes with greatly enhanced morphological stability over conventional liposomes [11]. As membrane-mimetic nanocarriers, liposomes are among the first drug carriers that have been used in clinical applications [12]. Notably, membrane-mimetic materials display an impressive ability to inhibit platelet adhesion/activation, suppress protein adsorption, and retain the bioactivities of immobilized biomolecules [13,14]. Consequently, biomedical devices such as stents and nanoparticle-based carriers can be coated with phospholipid polymers, acquiring excellent blood compatibility and creating membrane-mimetic structures that can be readily functionalized with different bioactive molecules [14,15]. However, membrane-mimetic materials often lack sufficient morphological stability. A variety of strategies have been proposed to improve their biochemical and physical stabilities, so that their applications may be extended to areas in which durability is highly relevant [16]. An organic-inorganic hybridization approach has thus been utilized to engineer highly stable liposomal cerasomes in which the phosphate heads of phospholipids are replaced with triethoxysilyl moieties [17,18]. The hydrolysis and polymerization of triethoxysilyl moieties and the subsequent formation of polysiloxane networks on the surfaces of cerasomes render the lipid nanovesicles morphologically stable. The use of cerasomes as nanocarriers in gene [17] and drug delivery [11,19] has been examined. Herein, the ability of anti-EGFR-conjugated immunocerasomes to selectively target cancer cells is investigated.
In the present study, organic-inorganic liposomal cerasomes are prepared using a combined sol-gel and self-assembly process and anti-EGFR mAbs are conjugated to cerasomes via maleimide-thiol coupling chemistry, creating immunocerasomes. Fluorescent lipid NBD-DPPE is also included in the preparation of immunocerasomes, enabling the imaging analysis of cellular uptake, internalization, and intracellular trafficking of the lipid nanovesicles. Next, EGFR expression is examined in A431 epidermoid carcinoma cells, DU145 prostate carcinoma cells, and HL-60 acute promyelocytic leukemia cells, and the selective targeting of immunocerasomes to the three cell lines is analyzed. This is to establish a correlation between the selective delivery of immunecerasomes and the differing EGFR expression by the target cells. Last, the inhibitory effects of immunocerasomes on cell proliferation are investigated. It has been reported that EGFR overexpression by cancer cells can intensify downstream signals and promote the aggressive growth of tumors [20], and that anti-EGFR mAbs can block the binding of growth factors to EGFR, suppressing tumor growth [21,22]. However, chemical modification of the monoclonal antibodies may compromise their therapeutic effects. This study tests the ability of the lipid nanocarriers to retain the therapeutic effects of the chemically conjugated anti-EGFR mAbs.
2. Materials and methods
2.1. Materials
N-[N-(3-Triethoxysilyl)propylsuccinamoly]dihexadecylamine (i.e., CFL) was synthesized according to a previously reported method [18]. An N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamide (NBD-DPPE), an Alexa Flour® 488 goat anti-mouse IgG, Measure-IT™ thiol assay kit, and ProLong® gold antifade reagent with DAPI and LIVE/DEAD® viability/cytotoxicity kit for mammalian cells were purchased from Invitrogen (Carlsbad, CA), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG (2000) Maleimide) was from Avanti Polar Lipids (Alabaster, AL). Anti-EGFR (Ab-3) mouse mAb (225) was from EMD Millipore (Billerica, MA). Cholesterol and thiazolyl blue tetrazolium bromide (MTT) were from Alfa Aesar (Ward Hill, MA). Traut’s reagent (2-Iminothiolane•HCl) was from Thermo Scientific (Waltham, MA). A Bio-Rad protein assay kit was from Bio-Rad (Hercules, CA). Zeba spin desalting columns and Sepharose® CL-6B were from Sigma-Aldrich (St. Louis, MO). The A43 and HL-60 cell lines were purchased from ATCC (Manassas, VA). The DU-145 cell line was a generous gift from Dr. Daruka Mahadevan (Arizona Cancer Center, Univ. of Arizona).
2.2. Preparation of cerasomes
Cerasomes were prepared using a combined ethanol injection and extrusion method. First, 140 mM of CFL and 52 mM of cholesterol in acidic ethanol were incubated overnight at pH 3.0 and room temperature, permitting the hydrolysis of CFL. Stock solutions of DSPE-PEG (2000) Maleimide (MAL-DSPE-PEG) at a concentration of 6.8 mM were prepared in chloroform, and stock solutions of NBD-DPPE at a concentration of 0.5 mM were prepared in a 1:1 (v/v) acidic ethanol and chloroform mixture. The solutions were then mixed, leading to a lipid mixture containing CFL, cholesterol, NBD-DPPE, and MAL-DSPE-PEG at a 14.2:9.4:0.2:1 molar ratio. The lipid mixture was injected to degassed PBS at 70 °C and sonicated for 15 min in a bath-type sonicator, forming multilamellar cerasomes. The use of degassed PBS at 70 °C helped remove organic solvents, including chloroform with a boiling point 61.2 °C. To obtain unilamellar cerasomes, the multilamellar cerasomes were extruded through a 100 nm polycarbonate membrane mounted on the LiposoFast extruder (Avestin, Canada). The obtained cerasomes were allowed to further polymerize at room temperature for 1 hr, and then filtered using a 0.2 μm syringe filter and sterilized with ultraviolet (UV) exposure for 30 min.
2.3. Preparation of immunocerasomes
Anti-EGFR mAbs were conjugated to cerasomes via maleimide-thiol coupling chemistry. In brief, 0.5 mg/ml anti-EGFR mAbs were thiolated by reacting with a 20-fold molar excess of 2 mg/ml Traut’s reagent in degassed PBS at pH 7.0 and room temperature in darkness under agitation for 1 hr. Excessive Traut’s reagents were separated from thiolated anti-EGFR mAbs using a Zeba Spin desalting column. The number of sulfhydryl groups on thiolated anti-EGFR mAbs was determined by a measure-IT™ thiol assay.
To conjugate anti-EGFR mAbs to cerasomes, the thiolated antibodies were incubated with cerasomes at a 1385:1 lipid/antibody molar ratio at 4°C in darkness under agitation for 24 hr. Unconjugated anti-EGFR mAbs was separated from immunocerasomes by size-exclusion chromatography using a Sepharose CL-6B column. The amount of conjugated anti-EGFR mAbs was determined by a Bio-Rad protein assay. The molecular weight of anti-EGFR mAbs is 150 kDa.
2.4. Cell culture
DU 145 prostate carcinoma cells were cultured in an RPMI 1640 medium (Cellgro, VA) supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (Gemini Bio-Product, CA). A431 epidermoid carcinoma cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Cellgro, VA) supplemented with 10% FBS. HL-60 acute promyelocytic leukemia cells were cultured in Iscove’s Modified Dulbecco’s Medium (ATCC, VA) supplemented with 20% FBS. All the cells were cultured in a humidified incubator at 37 °C and 5% CO2.
2.5. Cellular uptake of cerasomes
In a microscopic study of cellular uptake, DU145 and A431 cells were cultured overnight on a 1.5-inch coverslip at a density of 2.5 × 105 cells and HL-60 cells were cultured overnight in 24 well plates at a density of 2.5 × 105 cells per well. The cells were incubated with 185 μM NBD-DPPE labeled cerasomes or immunocerasomes for 3 hr, rinsed three times using PBS, and fixed with a picric acid fixing solution containing 4% paraformaldehyde and 0.4% picric acid in PBS for 10 min. After fixation, the HL-60 cells were spun at 3500 rpm and 4 °C for 5 min so that the cells were settled onto a round coverslip of 12 mm in diameter. The samples were then rinsed three times with PBS, air dried, and incubated with the ProLong ® Gold antifade reagent with DAPI overnight. Finally, the samples were mounted on a glass slide and imaged using a Leica DMI 4000B inverted microscope workstation, which consists of a 63X air objective lens, a SensiCam QE cooled digital 12 bit CCD camera (Cooke, MI), a HXP short-arc mercury vapor lamp light sources (OSRAM, Germany), a UV excitation Leica A4 filter cube, a green excitation Leica N2.1 filter cube, and a blue excitation Leica L5 filter cube.
The cellular uptake of cerasomes and immunocerasomes were also quantified by the relative NBD-DPPE fluorescence intensity of the internalized nanovesicles. DU145, A431, and HL-60 cells were seeded in 6-well plates at a density of 2.5 × 105 cells per well and incubated for 24 hr, allowing the cells to establish adhesion to the substrate. The cells were then incubated with 185 μM NBD-DPPE labeled cerasomes or immunocerasomes for 3 hr. Finally, the cells were rinsed three times using PBS and lysed with 200 μl of 0.1% Triton X-100 in PBS for 30 min. The fluorescence intensity of the lysates were measured using a Synergy 2 SL Luminescence Microplate Reader (BioTek, VT) at an excitation wavelength of 460/40 nm and an emission wavelength of 528/20 nm [23].
2.6. Immunostaining of EGFR
DU145, A431, and HL-60 cells were seeded and fixed onto coverslips following the aforementioned protocol. The samples were rinsed with PBS and blocked by heat-inactivated normal goal serum (NGS) for 1 hr at room temperature. Then, 1–2 μg of anti-EGFR mAbs were added to the blocking solution, and the samples were incubated for 2 hr. Finally, the samples were rinsed three times with NGS, incubated with 2 ug of Alexa Flour® 488 goat anti-mouse IgG secondary antibody in 1 ml of PBS for 30 min, rinsed again three times with PBS, and imaged using the fluorescence microscope.
2.7. Cell proliferation
The anti-proliferative effects of cerasomes, anti-EGFR mAbs, and anti-EGFR-conjugated immunocerasomes on DU145 cells were examined by an MTT assay, and their effects on A431 cells were analyzed by a Live/Dead assay. The cells were seeded at a density of 5,000 cells per well for the MTT assay and 125,000 cell per well for the Live/Dead assay in 96-well plates and cultured overnight. Prior to experiments, the cells were incubated in medium without FBS for 1.5 hr. Cerasomes, immunocerasomes or anti-EGFR mAbs at given concentrations were added into the medium and the cells were incubated for 48 hr at 37 °C and 5% CO2. In the MTT assay, 20 μL of MTT solution was added into each well and the samples were incubated for 3.5 hr before the medium in each well was replaced by 200 μL of DMSO. The UV absorbance of each sample was examined by a microplate reader (ELx800UV, Bio-Tek) at a wavelength of 570 nm. In the Live/Dead assay, the medium in each well was replaced by 100 μL of PBS containing 2.6 mM of Ethidium homodimer-1 and 5.1 mM of Calcein AM followed by a 45 min incubation. The fluorescence intensities of each sample were measured using a luminescence microplate reader at 485 ± 10 (ex)/ 530 ± 12.5 nm (em) for Calcein AM and 530 ± 12.5 (ex)/ 645 ± 20 nm (em) for Ethidium homodimer-1. All results were represented as the percentage of viable cells as referenced to the untreated control.
2.8. Data analysis
A Student’s t-test was performed using Microsoft Excel to assess the statistical significance of the experimental results. Statistical significance at p < 0.05 was considered a difference.
3. Results and discussion
3.1. Formation of immunocerasomes
As illustrated in Fig. 1, ethanol injection was employed as a simple method for the engineering of cerasomes. In acidic conditions, the organoalkoxysilane heads of CFL were hydrolyzed and the hydrolyzed CFL are polymerized into dimers, trimers, and larger n-mers (n > 3), as previously shown in a mass spectroscopy study [18]. While all the cerasome components, including CFL, cholesterol, NBD-DPPE, and MAL-DSPE-PEG are well dissolved in ethanol, chloroform, or an ethanol and chloroform mixture, the hydrophobic lipids are highly non-soluble in water. When the lipid mixture of CFL, cholesterol, NBD-DPPE, and MAL-DSPE-PEG at a 14.2:9.4:0.2:1 molar ratio is injected into PBS, multilamellar cerasomes are formed. Relatively uniform, unilamellar cerasomes with the most probable diameter of 140 nm were obtained by extruding the multilamellar cerasomes through a 100 nm polycarbonate membrane [11]. In the organic-inorganic hybrid cerasomes, the hydrolyzed and partially polymerized organoalkoxysilane heads of CFL form polysiloxane networks, rendering the nanovesicles remarkably stable in PBS and lipid-dissolving surfactants [11]. Further, the cerasomes contain maleimide terminal groups, which can readily conjugate with thiolated anti-EGFR mAbs.
Fig. 1.
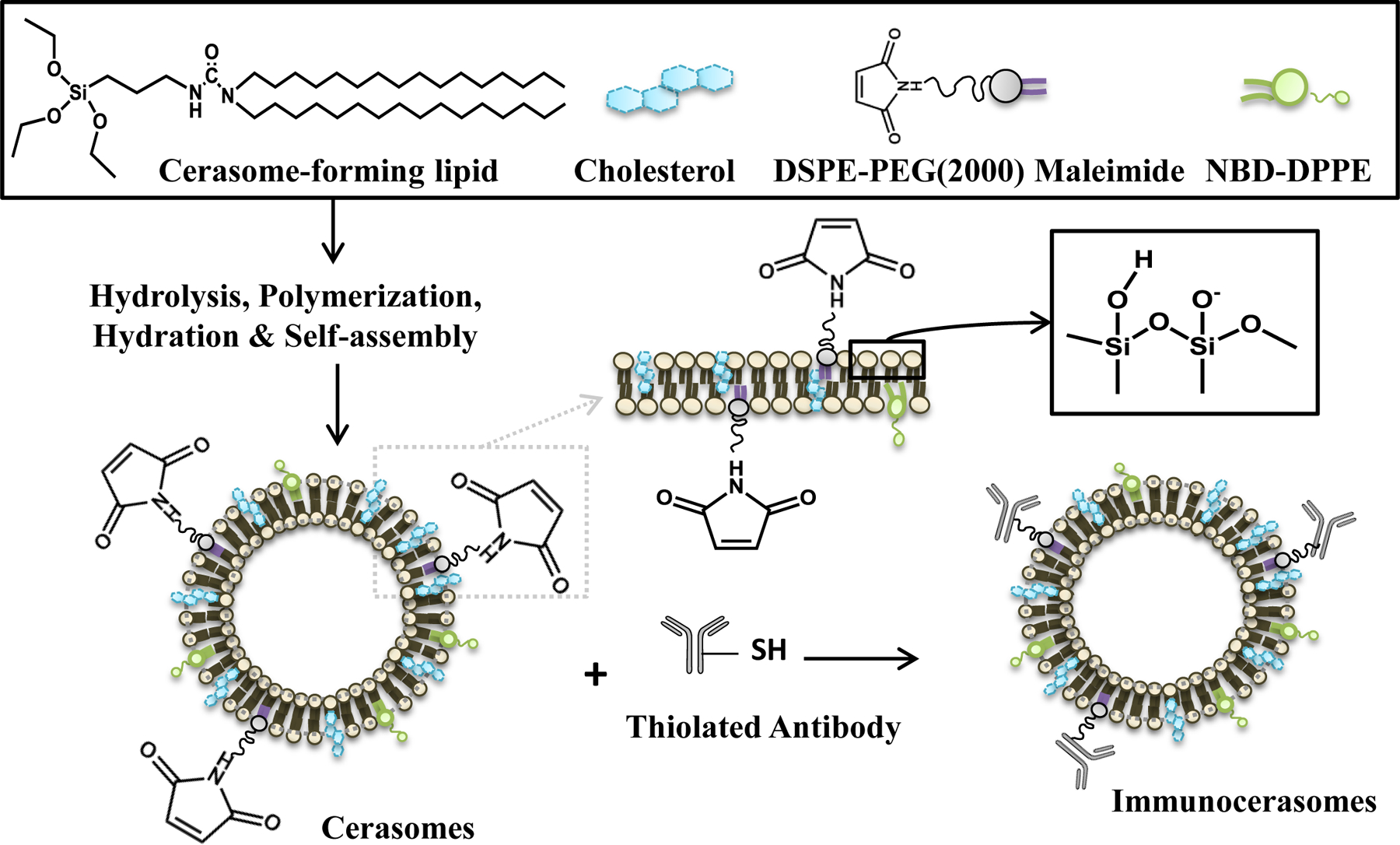
A schematic illustration of immunocerasome formation. Hydrolyzed and partially polymerized cerasome-forming lipids (CFL) in acidic ethanol, cholesterol in acidic ethanol, NBD-DPPE in the mixture of ethanol and chloroform, and MAL-DSPE-PEG in chloroform were mixed and injected into PBS, creating organic-inorganic liposomal cerasomes. The polysiloxane networks formed on the surfaces of nanovesicles provide morphological stability. The maleimide-containing cerasomes were readily conjugated with thiolated anti-EGFR mAbs via maleimide-thiol coupling, forming immunocerasomes.
Anti-EGFR mAbs were thiolated using Traut’s reagents. A measure-IT™ thiol assay suggests that the thiolated anti-EGFR mAbs possess 23 ± 5 sulfhydryl groups per antibody. The cerasomes that contain maleimide terminal groups were incubated with the thiolated anti-EGFR mAbs at a 1,385:1 lipid/antibody molar ratio for 24 hr to prepare immunocerasomes. Using a Bio-Rad protein assay, the efficiency of maleimide-thiol coupling was determined to be 36.52 ±1.54%. An early (1994) study shows that a liposome of 100 nm in size is comprised of 80,000 phospholipid molecules [24]. Assuming that immunocerasomes are structurally similar to liposomes and considering that the most probable diameter of immunocerasomes is 140 nm, we may convert the amount of the conjugated thiolated anti-EGFR mAb into the number of mAbs per nanovesicle. The average number of anti-EGFR mAbs on an immunocerasome was estimated to be 60 ± 13. To create fluorescently labeled immunocerasomes, 0.8% of NBD-DPPE was incorporated into the lipid bilayers during the ethanol injection process.
3.2. Selective targeting of immunocerasomes
The selective targeting of immunocerasomes to cancer cells are mediated by high-affinity antibody-EGFR interactions, and therefore greatly influenced by the membrane expression of EGFR in cancer cells. To understand how antibody-EGFR interactions may affect the cellular uptake and internalization of immunocerasomes, we first examined the differing expression of EGFR in three different cancer cells using direct fluorescent imaging analysis. The three cell lines include A431 epidermoid carcinoma cells, DU145 prostate carcinoma cells, and HL-60 acute promyelocytic leukemia cells. As shown in Fig. 2, A431 cells express an abnormally high level of EGFR; DU145 cells display a high level of EGFR expression; and no detectable expression of EGFR is observed in the non-epidermal HL-60 cells, which are used as a negative control in this study. It is worthwhile mentioning that the overexpression of EGFR in prostate cancer cells was also revealed in comparison to normal prostate cells [25] and NIH/3T3 fibroblasts (Fig. S1).
Fig. 2.
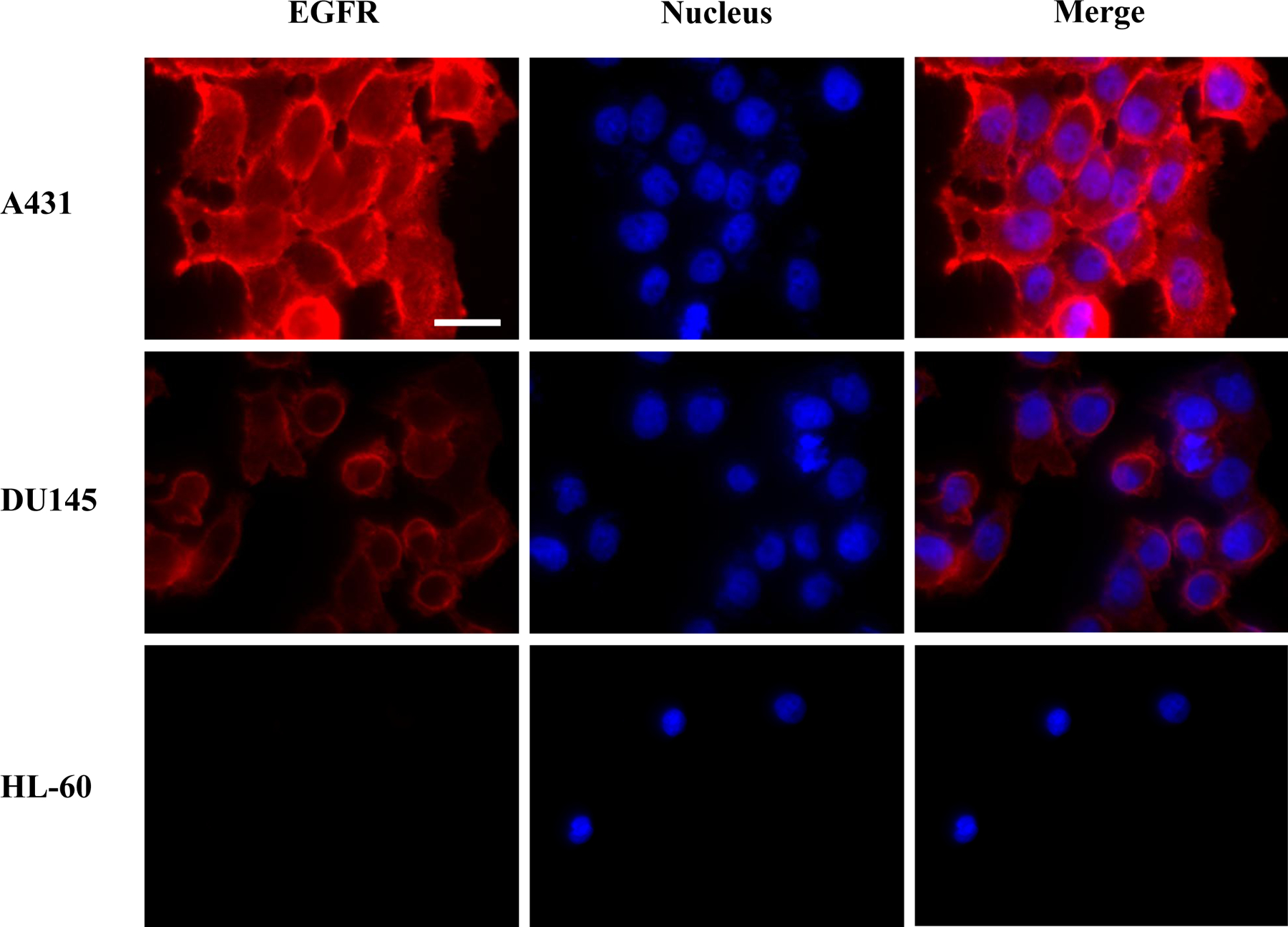
EGFR expression in A431, DU145, and HL-60 cells. EGFR was stained by anti-EGFR mAbs first, followed by incubation with the Alexa Flour® 488-conjugated secondary goat anti-mouse IgG antibodies (red). Cell nuclei were stained by DAPI (blue). Scale bar: 25 μm
The uptake and internalization of immunocerasomes by A431, DU145, and HL-60 cells were examined by direct imaging analysis of the cells (Fig. 3a) and spectrophotometric analysis of the cell lysates (Fig. 3b). Consistent with our previous study on the endocytosis of cerasomes by DU145 cells [11], a direct imaging analysis showed that cerasomes were primarily internalized in the cytosol and some cerasomes may enter cell nuclei (Fig. 3a). The spectrophotometric analysis showed that a similar amount of cerasomes entered A431, DU145, and HL-60 cells and were internalized during the time period of 3 hr (Fig. 3b). Previously, we showed that the cellular uptake and internalization of cerasomes in DU145 cells are largely mediated by clathrin-dependent endocytosis [11]. It is likely that clathrin-dependent endocytosis is also responsible for the cellular uptake and internalization of cerasomes in A431 and HL-60 cells. This explains why all the three cell lines display a comparable level of cellular uptake of cerasomes during the same period of time.
Fig. 3.
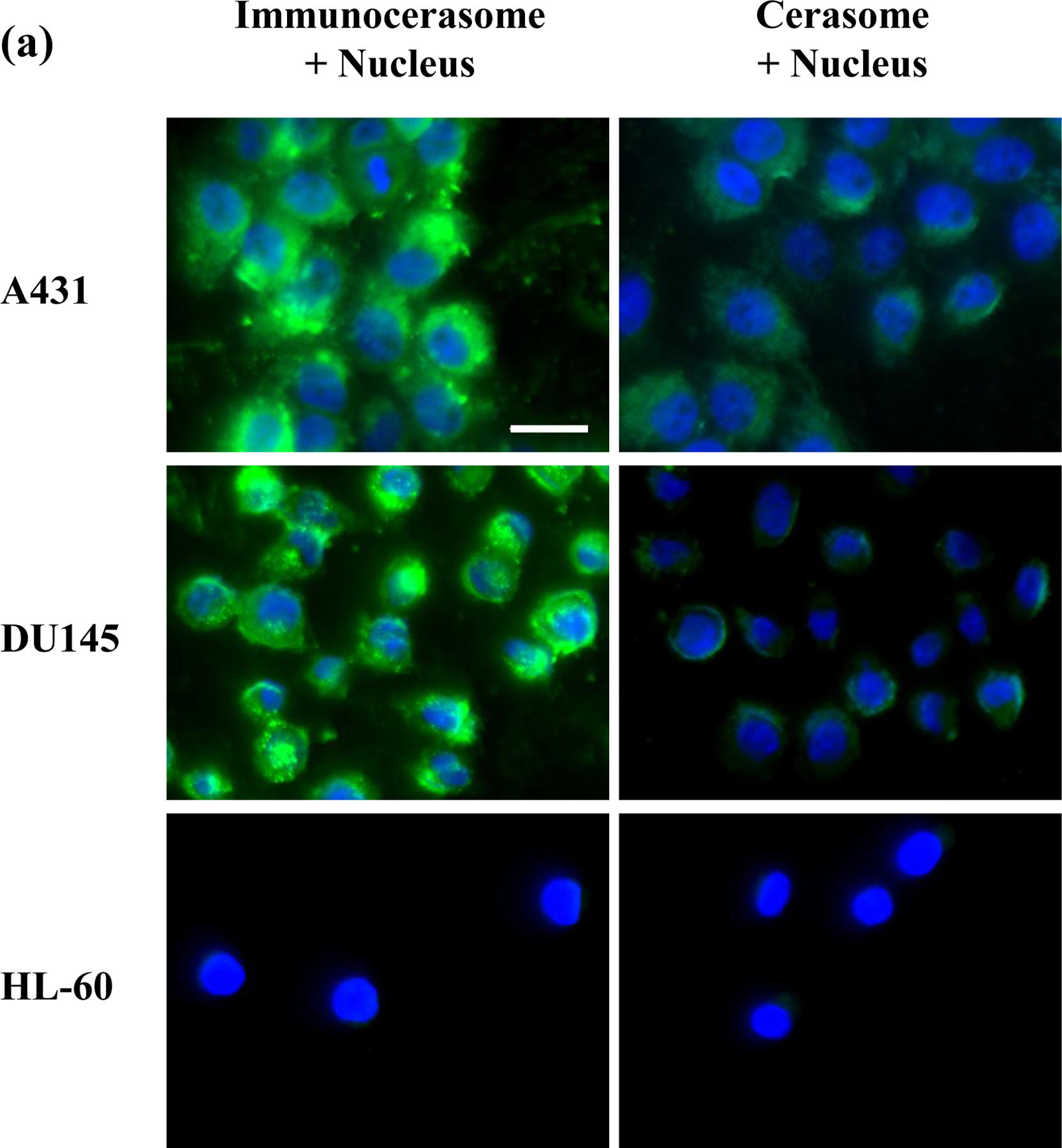
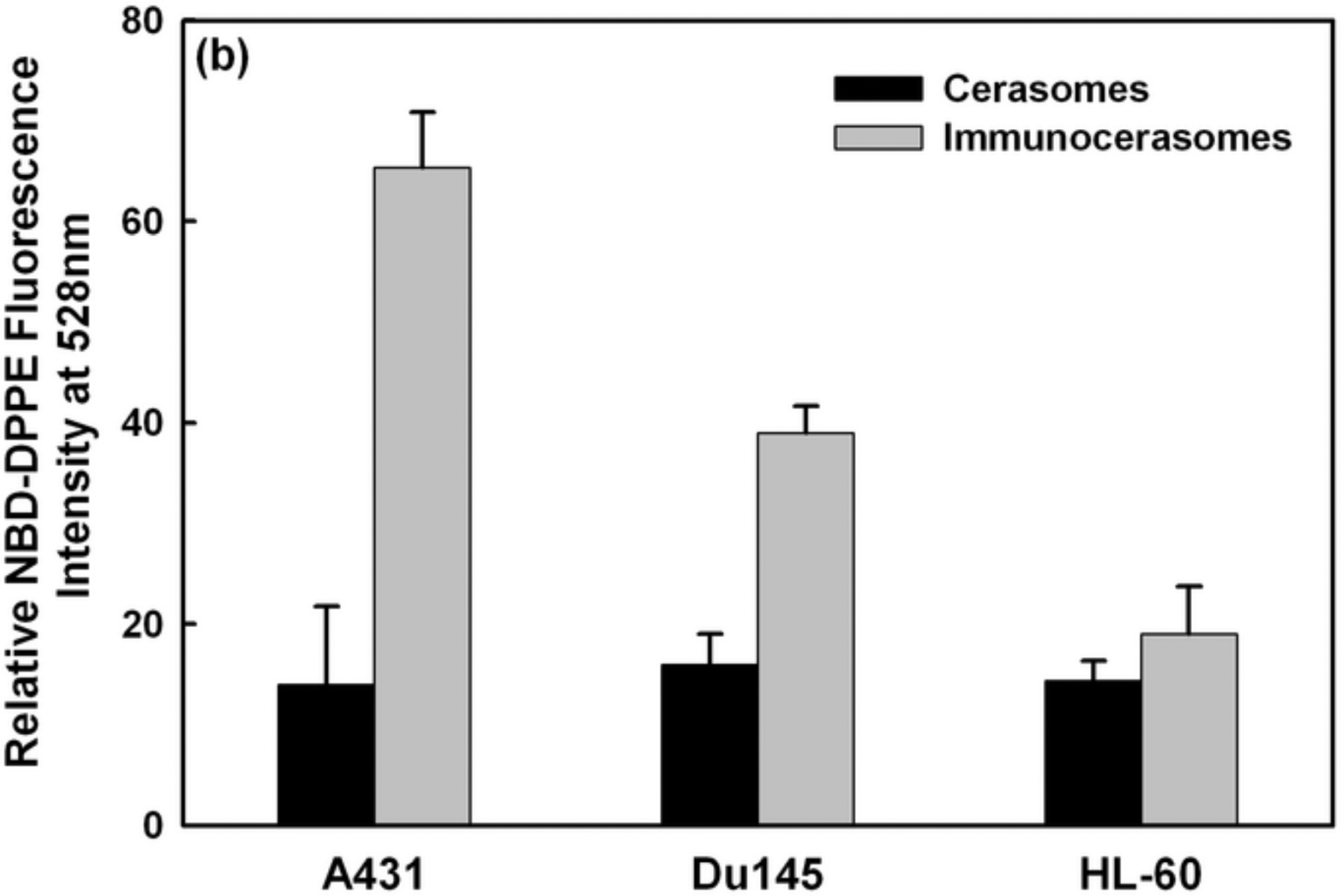
Fluorescent imaging analysis (a) and spectrophotometric analysis (b) of the cellular uptake and internalization of immunocerasomes in comparison to the cerasome control. A431, DU145, and HL-60 cells were incubated with immunocerasomes or cerasomes at a lipid concentration of 185 μM for 3 hr. (a) The cells were fixed and stained against nucleus by DAPI (blue), while immunocerasomes and cerasomes were labeled using NBD-DPPE (green). Scale bar: 25 μm (b) The relative amount of internalized immunocerasomes or cerasomes was measured by the NBD-DPPE fluorescence intensity of the cell lysates. The averages and errors were calculated from measurement of three replicate specimens. The p values were calculated to be 0.00059 for the endocytosis of cerasomes vs. immunocerasomes in A431 cells, 0.00031 in DU145 cells, and 0.117 in HL-60 cells, respectively.
Unlike the cerasome controls, the receptor-mediated endocytosis of immunocerasomes strongly correlates to the membrane expression of EGFR by cancer cells. As A431 cells express the highest level of EGFR among the three cell lines, the fluorescent imaging analysis revealed the largest increase in the cellular uptake and internalization of immunocerasomes by the cells (Fig. 3a). A strong increase in the cellular uptake and internalization of immunocerasomes was also observed in DU145 cells, as the cells over-express EGFR. As a negative control, HL-60 cells marginally express EGFR, and no noticeable changes in the endocytosis of immunocerasomes by the cells were observed from the direct imaging analysis. A quantitative spectrophotometric analysis of the lyses of A431 cells shows a 4.6-fold increase in cellular uptake and internalization (p < 0.001) when the nanovesicles are conjugated with anti-EGFR mAb (Fig. 3b). In DU145 cells, the conjugation of cerasomes with anti-EGFR mAb increases the cellular uptake of the lipid nanovesicles by 2.4 times in comparison to the cerasome control (p < 0.001). In HL-60 cells, a moderate increase of 32% in cellular uptake is statistically insignificant (p = 0.117) when cerasomes are conjugated with anti-EGFR mAbs.
Comparison of the spectrophotometric and fluorescent imaging analyses reveals an interesting observation: the former suggests the cellular uptake of cerasomes or immunocerasomes in HL-60 cells is comparable to that of cerasomes in A431 and DU145 cells, but the latter indicates little cellular uptake of the lipid nanovesicles in HL-60 cells, in comparison to that of cerasomes in A431 and Du145 cells. This is probably because adhesive A431 and DU145 cells remain flat when cultured on a substrate and the lipid nanovesicles that are internalized in the cytosols can be readily visualized in the focal plan near the substrate surface. In contrast, HL-60 cells are non-adhesive and remain rounded when collected on a substrate. As a result, only a small fraction of the cytosols can be visualized, thereby underestimating the cellular uptake of the lipid nanovesicles using the fluorescent imaging technique.
The direct imaging and spectrophotometric analyses show that the amount of immunocerasomes internalized by cancer cells strongly correlates to the level of their membrane expression of EGFR. The higher the level of EGFR the cells express, the more immunocerasomes the cells internalize. The present study thus confirms that anti-EGFR mAbs were successfully conjugated to immunocerasomes via maleimide-thiol coupling and that the anti-EGFR mAbs conjugated on immunocerasomes retained their receptor-binding capability.
3.3. Selective anti-proliferative effects of immunocerasomes on A431 cells
The overgrowth of cancer cells of epidermal origin is often mediated by various growth factors, including epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). Binding of these growth factors to EGFR leads to autophosphorylation of receptor tyrosine kinase and activation of downstream signaling cascades for cell proliferation, differentiation, and survival [26]. Monoclonal antibodies such as anti-EGFR mAbs can block the binding of EGFR to its cognate ligands and suppress the proliferation of some types of cancer cells that overexpress EGFR [26]. The anti-proliferative effects of anti-EGFR mAbs, however, can be compromised by chemical modification [21,22]. Herein, we examine if anti-EGFR mAbs inhibit the proliferation of epidermal A431 and non-epidermal DU145 cells, which both overexpress EGFR, and if the chemical conjugation of anti-EGFR mAbs to immunocerasomes affects their anti-proliferative effects. In this process, the cytocompatibility of cerasomes was examined first. As shown in Fig.4, A431 and DU145 cells that were incubated with cerasomes displayed viability comparable to the untreated controls when the lipid concentration of cerasomes was in the range of 0.048 to 480 μM. This suggests that cerasomes are cytocompatible and that any subsequently observed anti-proliferative effects of immunocerasomes are induced by anti-EGFR mAbs.
Fig. 4.
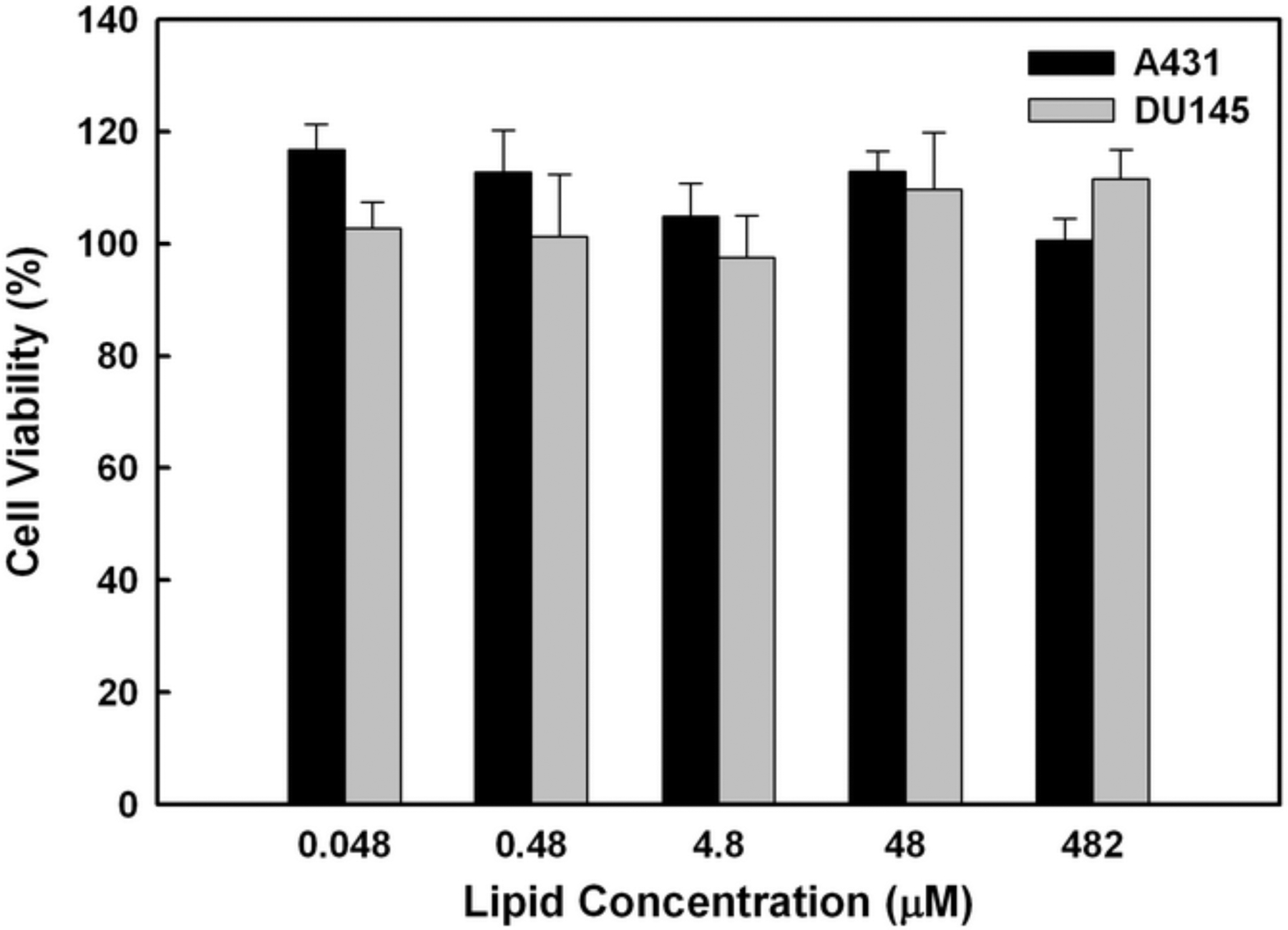
Cytocompatibility of cerasomes. A431 and DU145 cells were incubated with cerasomes at various concentrations for 48 hr, and then analyzed using the MTT assay. Results were represented as the percentage of viable cells in comparison to the untreated control. The averages and standard deviation were calculated from measurement of five replicate specimens.
The effects of free anti-EGFR mAbs and anti-EGFR-conjugated immunocerasomes on the proliferation of A431 and DU145 cells were analyzed using the MTT or Live/Dead assay. In A431 epidermoid carcinoma cells, no detectable anti-proliferative effects were observed when the cells were incubated with free anti-EGFR mAbs or immunocerasomes conjugated with anti-EGFR mAbs at an antibody concentration of 1 nM or lower (Fig. 5a). However, free anti-EGFR mAbs or immunocerasomes conjugated with anti-EGFR mAbs at nanomolar doses inhibited the proliferation of the epidermal A431 cells. When the antibody concentration was 11.6 nM, the viability of A431 cells dropped to about 86% (p = 0.049 for free anti-EGFR and p = 0.047 for immunocerasomes, respectively). A further increase in antibody concentration up to 100 nM did not lead to a lower survivability of A431 cells. Unlike A431 cells, no inhibitory effects on the proliferation of DU145 cells were observed when the cells were treated with free anti-EGFR mAbs or with immunocerasomes conjugated with anti-EGFR mAbs in the concentration range of 0.01 ~ 100 nM.
Fig. 5.
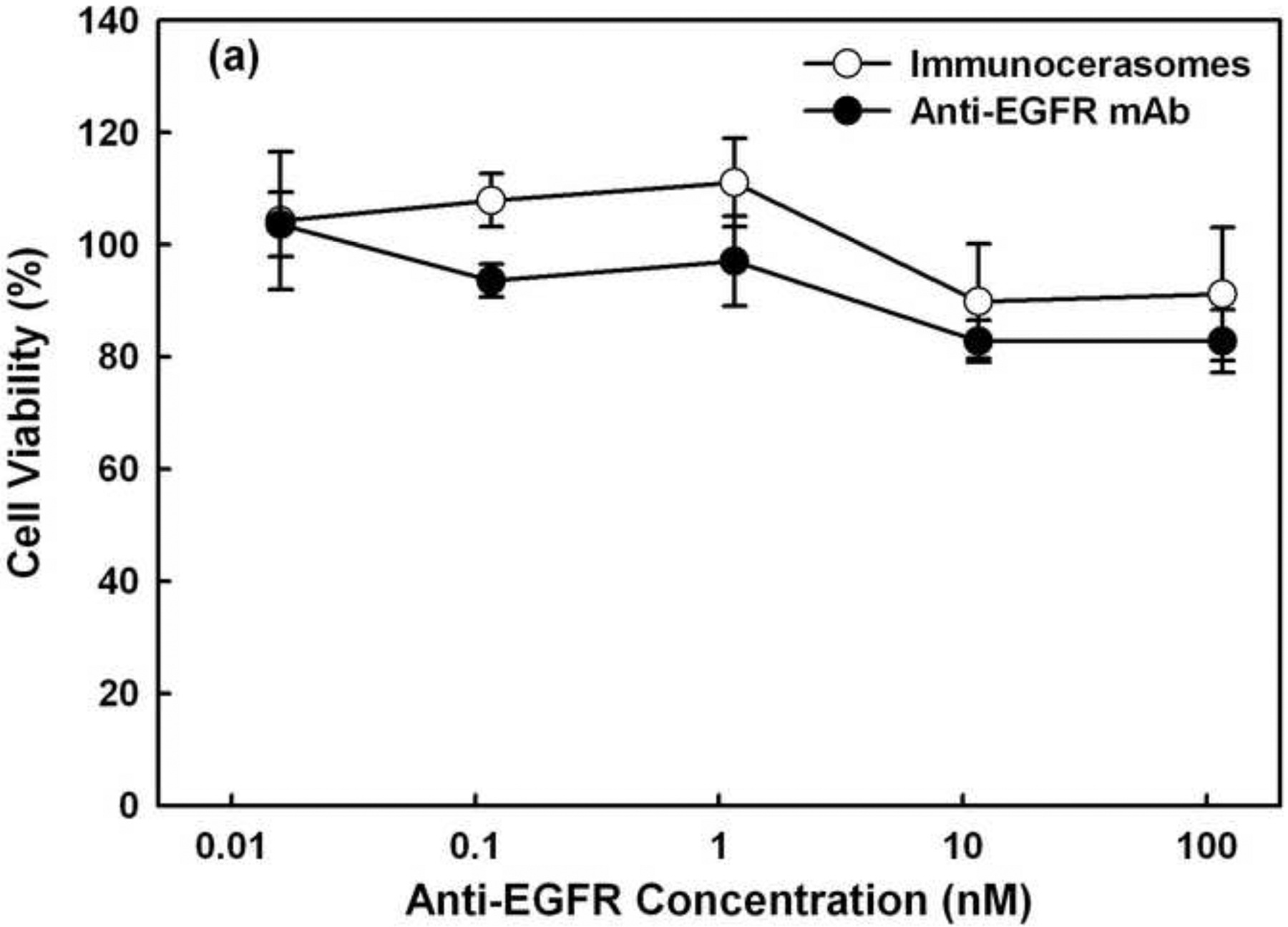
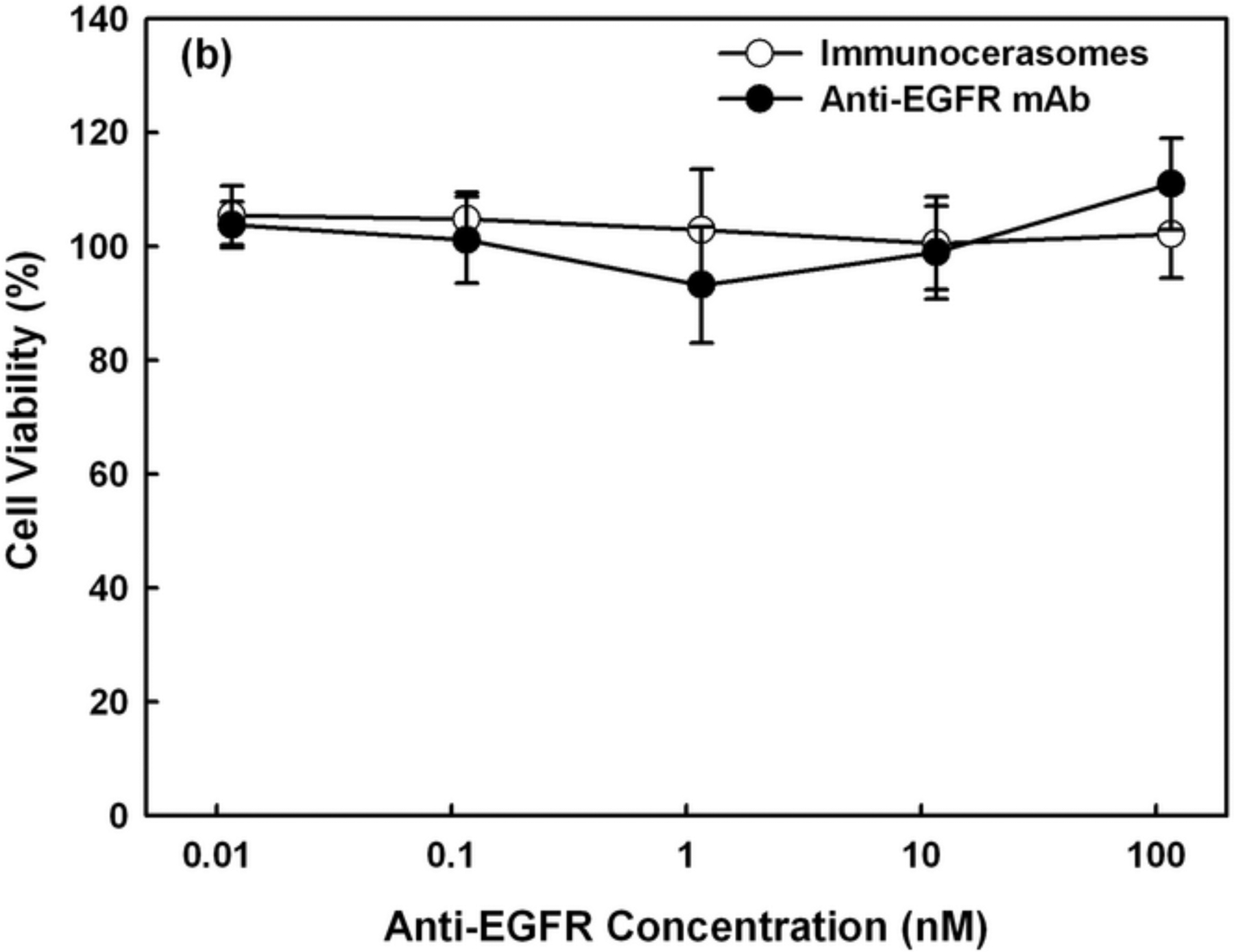
Anti-proliferative effects of free anti-EGFR mAbs and anti-EGFR-conjugated immunocerasomes on (a) A431 cells and (b) DU145 cells in serum-enriched medium. The cancer cells were incubated with free anti-EGFR mAbs or immunocerasomes that are conjugated with anti-EGFR mAbs at various antibody concentrations for 48 hr, and then examined using the MTT or Live/Dead assay. The averages and standard deviation were calculated from measurement of five replicate specimens.
Since both A431 and DU145 cells overexpress EGFR, the cell viability study suggests that anti-proliferative effects of anti-EGFR mAbs depend on the types of cancer cells. Specifically, anti-EGFR mAbs suppress the proliferation of epidermal A431 cells but not non-epidermal DU145 cells. The selective antitumor activities of anti-EGFR mAbs have been reported in early studies on nude mice [27]. More recent work has indentified the therapeutic effects of anti-EGFR mAbs on other cancers such as colorectal cancer [28]. The present study shows that anti-EGFR mAbs do not inhibit the in vitro proliferation of DU145 prostate cancer cells. This, however, does not compromise the potential of EGFR as a promising target for selective delivery of therapeutic and/or imaging agents to DU145 cells. Results from the current study are consistent with an early report that anti-EGFR mAb at nanomolar doses is required to inhibit the in vitro proliferation of epidermal cancer cells [29]. Within experimental errors, free anti-EGFR mAbs and immunocerasomes containing anti-EGFR mAbs at nanomolar doses display no significant difference in suppressing the proliferation of A431 cells. Further, neither free anti-EGFR mAbs nor immunocerasomes suppress the proliferation of DU145 cells. The present study indicates that the conjugation of anti-EGFR mAbs to immunocerasomes via maleimide-thiol coupling chemistry does not change antibody functions, including the abilities to bind to EGFR and to inhibit the proliferation of cancer cells of epidermal origin.
In studies presented in Figs. 5a and 5b, the cancer cells were cultured in serum-enriched medium. An early study shows a more pronounced anti-proliferative effect of free anti-EGFR mAbs on epidermal A431 cells that are cultured in serum-free medium [29]. To examine the influences of serum on the anti-proliferative activities of anti-EGFR mAbs and anti-EGFR-conjugated immunocerasomes on A431 cells, the cells were also cultured in serum-free medium and the cell viability was presented in Fig. 6. When A431 cells were cultured with free anti-EGFR mAbs or immunocerasomes conjugated with anti-EGFR mAbs at nanomolar doses (e.g., 10 to 100 nm), the viability of the cancer cells dropped to about 60%. It has been reported that serum proteins such as albumin [30] and insulin-like growth factor-II [31] can promote the proliferation of cancer cells. It is likely that serum proteins and growth factors containing in serum partially counterbalance the anti-proliferative effects of anti-EGFR mAbs on A431 cells. Further, serum proteins and growth factors may bind to other receptors and opsonized by the cells. They may stimulate the proliferation of A431 cells but not suppress the endocytosis of immunocerasomes via EFGR. To examine this effect, the cellular uptake of immunocerasomes by A431 cells was analyzed in serum-free and serum-enriched media, and compared to the cerasome control.
Fig. 6.
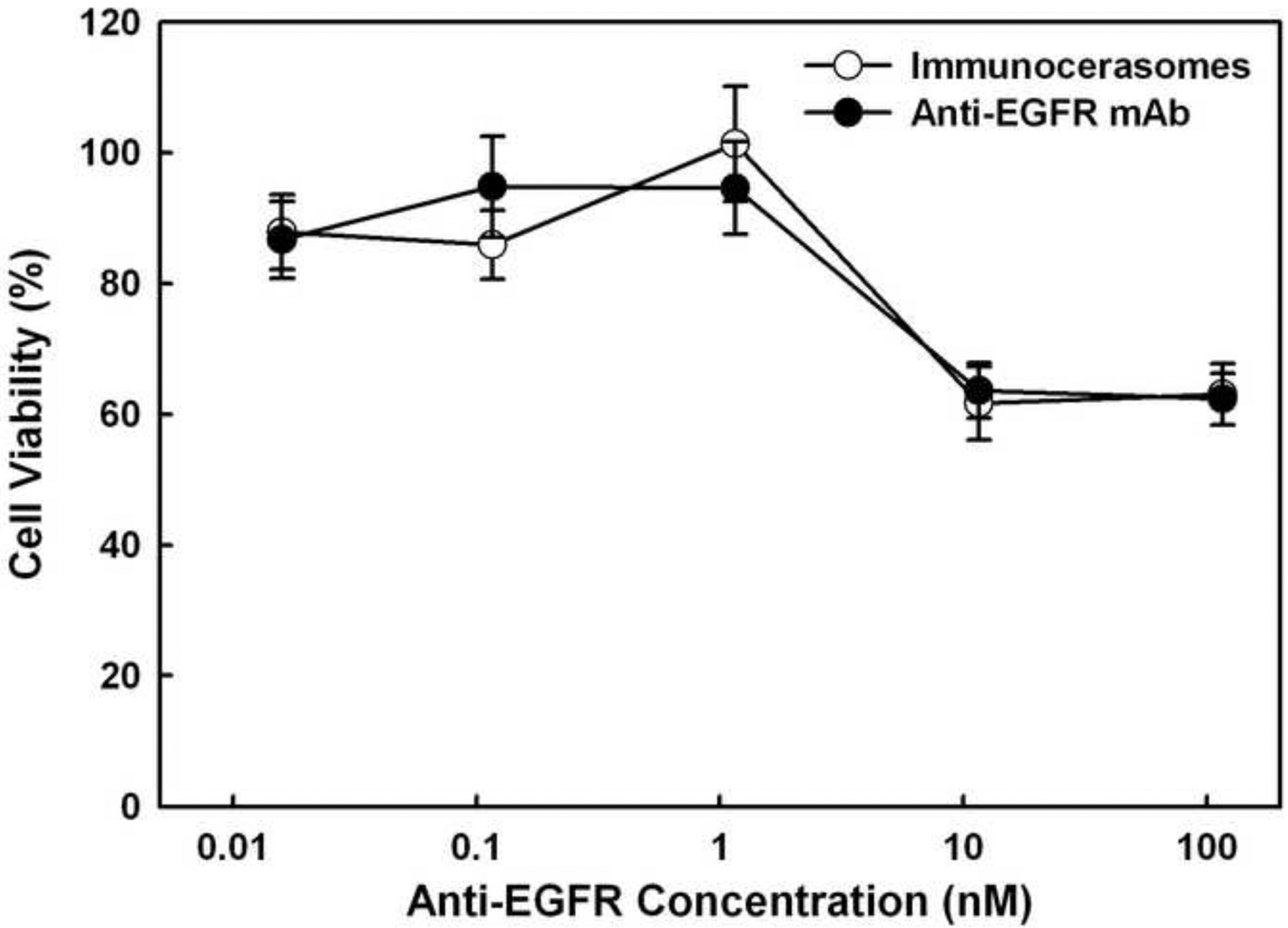
Anti-proliferative effects of free anti-EGFR mAbs and anti-EGFR-conjugated immunocerasomes on A431 cells in serum-free medium. The cancer cells were incubated with free anti-EGFR mAbs or immunocerasomes that are conjugated with anti-EGFR mAbs at various antibody concentrations for 48 hr, and then examined using the Live/Dead assay. The averages and standard deviation were calculated from measurement of five replicate specimens.
3.4. Serum effects on the endocytosis of cerasomes and immunocerasomes
As shown in Fig. 7, the study of the serum effects on the cellular uptake and internalization of cerasomes and immunocerasomes by A431 cells reveals a few interesting phenomena. First, in comparison to the cerasome control, a reduction of 41.5 ± 9.2% was observed in the cellular uptake and internalization of immunocerasomes when the culture medium was serum-free. Because anti-EGFR mAbs are highly selective to EGFR, the endocytosis of anti-EGFR-conjugated immunocerasomes by A431 cells is primarily mediated by EGFR. Upon binding, antibody-EGFR complexes and thus immunocerasomes migrate to clathrin-coated pits for endocytosis [32,33]. In comparison to immunocerasomes, cerasomes can also bind to other receptors and the clathrin-dependent endocytosis of cerasomes may be improved. Additionally, anti-EGFR mAbs bind to inactive EGFR, promoting receptor internalization and subsequent degradation without receptor phosphorylation and activation [34]. Degradation of the internalized receptor reduces the surface availability of EGFR, slowing down the receptor-mediated endocytosis of immunocerasomes.
Fig. 7.
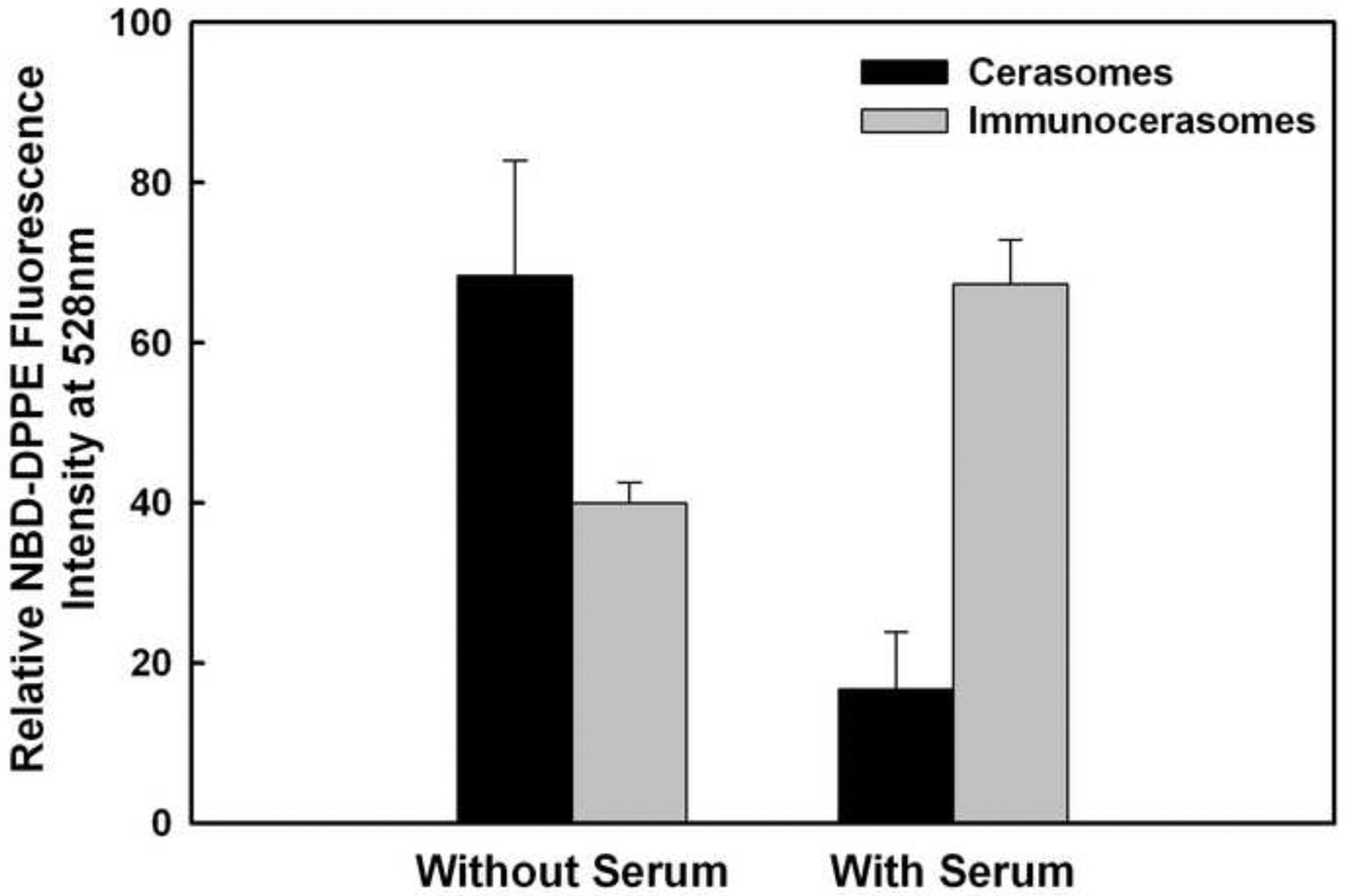
Serum effects on the cellular uptake of cerasomes and immunocerasomes by A431 cells. The cells were incubated with immunocerasomes or cerasomes at a lipid concentration of 185 μM for 3 hr. The relative amount of internalized immunocerasomes or cerasomes was measured by the NBD-DPPE fluorescence intensity of the cell lysates. The averages and standard deviation were calculated from measurement of three replicate specimens. The p values were calculated to be 0.03630 for the endocytosis of cerasomes vs. immunocerasomes in the serum-free medium, 0.00044 for the endocytosis of cerasomes vs. immunocerasomes in the serum-enriched medium 0.00610 for the endocytosis of cerasomes in serum-free vs. serum-enriched media, and 0.00277 for the endocytosis of immunocerasomes in serum-free vs. serum-enriched media, respectively.
Second, a reduction of 75.6 ± 5.7% in the cellular uptake of cerasomes by A431 cells resulted when cell culture medium was enriched with fetal bovine serum. Clathrin-dependent endocytosis, by which cerasomes are internalized, has also been identified as a prominent mechanism for the cellular entry of essential nutrients and nanoparticles [35]. Therefore, serum proteins, such as serum albumin, and insulin-like growth factors, likely compete with cerasomes for entrance to the cells via clathrin-coated vesicles [36–39]. Due to their small sizes and high mobility, serum proteins may possess an enhanced ability to bind to a variety of receptors and to be internalized via clathrin-dependent endocytosis, in comparison to cerasomes. Previously, Johnstone et al. reported that surface-associated serum proteins may change the properties of the PEG that reduces the cellular uptake of PEG-bearing liposomes [40]. In the current study, PEG is included in the fabrication of cerasomes and immunocerasomes to improve the colloidal stabilities of the nanovesicles. It is possible that serum proteins may adversely affect the non-specific interactions between PEG-bearing cerasomes and A431 cells and the subsequent uptake of cerasomes by the cells. In contrast, immunocerasomes that are conjugated with high-affinity anti-EGFR mAbs selectively bind to the EGF receptors of A431 cells. Serum proteins cannot compete with immunocerasomes for binding to EGFR and opsonized via EGFR-mediated endocytosis. As a result, the cellular uptake and internalization of immunocerasomes by A431 cells does not decrease in serum-enriched medium, in comparison to the serum-free controls.
Last, a moderate increase of 68.3 ± 3.2% was observed in the endocytosis of immunocerasomes when the cells were cultured in serum-enriched medium than in serum-free medium (p = 0.00277). A speculation is that this phenomenon may be due to the serum-enhanced recycling and surface availability of EGFR. When EGFR binds to anti-EGFR mAbs, the receptors are internalized and degraded [34]. When EGFR binds to other ligands, the receptors may be internalized and partially recycled. Since serum proteins can stimulate a variety of cellular activities, the cycling of internalized, non-degraded EGFR may be more improved in serum-enriched medium than in serum-free medium, leading to the enhanced endocytosis of immunocerasomes.
4. Conclusion
Organic-inorganic hybrid liposomal immunocerasomes were synthesized as nanocarriers for the selective delivery of therapeutic/imaging agents to cancer cells. Specifically, organic-inorganic liposomal cerasomes were prepared using an ethanol injection approach, incorporated with fluorescent lipid NBD-DPPE for imaging analysis, and conjugated with anti-EGFR mAbs via maleimide-thiol coupling chemistry, creating immunocerasomes. About 60 ± 13 anti-EGFR mAbs were conjugated on each lipid nanovesicle. Selective targeting of immunocerasomes to cancer cells was examined in A431 cells that expressed an abnormally high level of EGFR, DU145 cells that overexpressed EGFR, and HL-60 that marginally expressed EGFR. In comparison to the cerasome controls, the receptor-mediated endocytosis of immunocerasomes was increased by 4.6-fold in A431 cells, by 2.4-fold in DU145 cells, and only marginally in HL-60 cells. The endocytosis study revealed a strong correlation between the cellular uptake and internalization of anti-EGFR-conjugated immunocerasomes and the membrane expression of EGFR by cancer cells. Further, free anti-EGFR mAbs and immunocerasomes conjugated with anti-EGFR mAbs at nanomolar doses moderately inhibited the proliferation of epidermal A431 cells but not non-epidermal DU145 cells, although both cells overexpressed EGFR. Last, the presence of serum in a cell culture medium increased the endocytosis of immunocerasomes but greatly decreased the cellular uptake of cerasomes by A431 cells. It is believed that serum proteins may effectively compete with cerasomes for non-specific binding to various receptors but not with immunocerasomes for specific, high-affinity binding to EGFR for receptor-mediated endocytosis. In addition, nutrient provided by serum proteins may stimulate a variety of cell activities such as the recycling of internalized EGFR, leading to the enhanced endocytosis of immunocerasomes by A431 cells. Taken together, the present study demonstrated that anti-EGFR mAbs can be successfully conjugated to immunocerasomes and that the antibody functions, including the ability to bind to EGFR and inhibit the proliferation of A431 cells, are retained in immunocerasomes.
Supplementary Material
Table I.
Key parameters of immunocerasome preparation
| Sulfhydryl groups per antibody | Antibody/lipid molar ratio | Antibody conjugation efficiency | Most probable size of vesicles | Number of antibodies on a vesicle |
|---|---|---|---|---|
| 23 ± 5 | 1:1,385 | 36.52 ± 1.54% | 140 nm | 60 ± 13 |
Anti-EGFR antibodies were conjugated to highly stable organic-inorganic cerasomes, creating immunocerasomes.
Cellular uptake and internalization of immunocerasomes were examined in epidermoid carcinoma, prostate carcinoma, and leukemia cells.
Selective delivery of immunocerasomes to the cells was directly correlated to the membrane expression of EGFR by the cells.
Immunocerasomes displayed selective anti-proliferative effects on epidermoid carcinoma cells.
Acknowledgment
This work was supported by the US National Institutes of Health (R21EB009160) and National Science Foundation (CMMI0855890), and National Science Foundation of China (2007AA03Z316, 30970829).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary data on cell surface EGFR expression on NIH-3T3 and DU145 cells.
References
- [1].Maeda H, in Weber G (Eds.), Advances in Enzyme Regulation, Vol 41 (2001), p 189–207. [DOI] [PubMed] [Google Scholar]
- [2].Gordon KB, Tajuddin A, Guitart J, Kuzel TM, Eramo LR, and Vonroenn J, Cancer 75 (1995): 2169–2173. [DOI] [PubMed] [Google Scholar]
- [3].Laginha K, Mumbengegwi D, and Allen T, BAB-Biomembranes 1711 (2005): 25–32. [DOI] [PubMed] [Google Scholar]
- [4].Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang QQ, Storm G, Molema G, Lu PY, Scaria PV, and Woodle MC, Nucleic Acids Research 32 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang XQ, Lam R, Xu XY, Chow EK, Kim HJ, and Ho D, Advanced Materials 23 (2011) 4770. [DOI] [PubMed] [Google Scholar]
- [6].Yu CC, Hu Y, Duan JH, Yuan W, Wang C, Xu HY, and Yang XD, Plos One 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berry JD and Gaudet RG, New Biotechnology 28 (2011): 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, and Dallaire BK, Journal of Clinical Oncology 16 (1998): 2825–2833. [DOI] [PubMed] [Google Scholar]
- [9].Mendelsohn J and Baselga J, Journal of Clinical Oncology 21 (2003): 2787–2799. [DOI] [PubMed] [Google Scholar]
- [10].Kawamoto T, Sato JD, Le A, Polikoff J, Sato GH, and Mendelsohn J, Proc Natl Acad Sci U S A 80 (1983): 2787–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Leung S, Zha Z, Teng W, Cohn C, Dai Z, and Wu X, Soft Matter 8 (2012): 5756–5764. [Google Scholar]
- [12].Torchilin VP, Nat Rev Drug Discov 4 (2005): 145–160. [DOI] [PubMed] [Google Scholar]
- [13].Ishihara K, Nomura H, Mihara T, Kurita K, Iwasaki Y, and Nakabayashi N, Journal of Biomedical Materials Research 39 (1998): 323–330. [DOI] [PubMed] [Google Scholar]
- [14].Ishihara K and Takai M, Journal of the Royal Society Interface 6 (2009): S279–S291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Weingart J, Vabbbilisetty P, and Sun XL, Advances in Colloid and Interface Science 197-198 (2013): 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cashion MP, Long TE, Accounts of Chemical Research 42 (2009): 1016–1025. [DOI] [PubMed] [Google Scholar]
- [17].Matsui K, Sando S, Sera T, Aoyama Y, Sasaki Y, Komatsu T, Terashima T, and Kikuchi J, Journal of the American Chemical Society 128 (2006): 3114–3115. [DOI] [PubMed] [Google Scholar]
- [18].Katagiri K, Hashizume M, Ariga K, Terashima T, and Kikuchi J, Chemistry-a European Journal 13 (2007): 5272–5281. [DOI] [PubMed] [Google Scholar]
- [19].Cao Z, Ma Y, Yue XL, Li SZ, Dai ZF, and Kikuchi J, Chemical Communications 46 (2010): 5265–5267. [DOI] [PubMed] [Google Scholar]
- [20].Ethier SP, Semin Radiat Oncol 12 (2002): 3–10. [DOI] [PubMed] [Google Scholar]
- [21].Fan Z, Masui H, Altas I, and Mendelsohn J, Cancer Res 53 (1993): 4322–4328. [PubMed] [Google Scholar]
- [22].Park JW, Hong K, Carter P, Asgari H, Guo LY, Keller GA, Wirth C, Shalaby R, Kotts C, Wood WI, Papahadjopoulos D, and Benz CC, P Natl Acad Sci USA 92 (1995): 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Banerjee R, Tyagi P, Li S, and Huang L, International Journal of Cancer 112 (2004): 693–700. [DOI] [PubMed] [Google Scholar]
- [24].Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P, Benz CC, and Papahadjopoulos D, Biochemistry-Us 36 (1997): 66–75. [DOI] [PubMed] [Google Scholar]
- [25].El Sheikh SS, Domin J, Abel P, Stamp G, and Lalani EN, Neoplasia 6 (2004): 846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herbst RS, Int J Radiat Oncol Biol Phys 59 (2004): 21–26. [DOI] [PubMed] [Google Scholar]
- [27].Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, and Mendelsohn J, Cancer Res 44 (1984): 1002–1007. [PubMed] [Google Scholar]
- [28].Zhang W, Gordon M, and Lenz HJ, Annals of Medicine 38 (2006): 545–551. [DOI] [PubMed] [Google Scholar]
- [29].Kawamoto T, Mendelsohn J, Le A, Sato GH, Lazar CS, and Gill GN, J Biol Chem 259 (1984): 7761–7766. [PubMed] [Google Scholar]
- [30].Francis GL, Cytotechnology 62 (2010): 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Guo N, Ye JJ, Liang SJ, Mineo R, Li SL, Giannini S, Plymate SR, Sikes RA, and Fujita-Yamaguchi Y, Growth Hormone & Igf Research 13 (2003): 44–53. [DOI] [PubMed] [Google Scholar]
- [32].Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, and Burgess AW, Exp Cell Res 284 (2003): 31–53. [DOI] [PubMed] [Google Scholar]
- [33].Sigismund S, Argenzio E, Tosoni D, Cavallaro E, Polo S, and Di Fiore PP, Dev Cell 15 (2008): 209–219. [DOI] [PubMed] [Google Scholar]
- [34].Martinelli E, De Palma R, Orditura M, De Vita F, and Ciardiello F, Clinical and Experimental Immunology 158 (2009): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sahay G, Alakhova DY, and Kabanov AV, Journal of Controlled Release 145 (2010): 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mukherjee S, Ghosh RN, and Maxfield FR, Physiol Rev 77 (1997): 759–803. [DOI] [PubMed] [Google Scholar]
- [37].Aleksic T, Chitnis MM, Perestenko OV, Gao S, Thomas PH, Turner GD, Protheroe AS, Howarth M, and Macaulay VM, Cancer Res 70 (2010): 6412–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen YW, Boyartchuk V, and Lewis BC, Neoplasia 11 (2009): 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yumoto R, Nishikawa H, Okamoto M, Katayama H, Nagai J, and Takano M, Am J Physiol Lung Cell Mol Physiol 290 (2006): L946–L955. [DOI] [PubMed] [Google Scholar]
- [40].Johnstone SA, Masin D, Mayer L, and Bally MB, Biochim Biophys Acta 1513 (2001): 25–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.