

Summary
Although antibodies are routinely used to label and isolate a desired cell type from a more complex mixture of cells, via either fluorescence-activated cell sorting (FACS) or magnetic-activated cell sorting (MACS), such antibody labeling is not easily reversible. We describe a FACS and MACS compatible method to reversibly label and purify cells using aptamers. Magnetic beads loaded with the EGFR-binding antagonistic aptamer E07 specifically isolated EGFR-expressing cells, and pure, label-free cells were recovered via treatment with an “antidote” oligonucleotide complementary to the aptamer. Additionally, while FACS sorting cells with E07 or EGFR antibody yielded EGFR(+) cells with impeded EGFR signaling, stripping off the aptamer via antidote treatment restored receptor function, returning cells to their native state which was not possible with the antibody. The ability to reversibly label or isolate cells without compromising their function is a valuable, versatile tool with important implications for both the lab and clinic.
Keywords: aptamer, FACS, MACS, antidote, cell purification, cell isolation
Graphical Abstract
eTOC Blurb
Cell purification methods rely upon poorly reversible antibody labeling of desired cells or upon depletion of unwanted cells. Powell Gray et al. demonstrate that aptamers can be used to positively purify cells and, unlike antibodies, yield cells in their native state using antidote-mediated reversal of aptamer cell-surface binding.
Introduction
Selective purification of cells is routinely performed and critical for numerous clinical and basic research applications. Clinically, CD34+ cell enrichment is required for the 45,000+ hematopoietic stem cell transplantations performed annually (Champlin et al. 2000; Handgretinger et al. 1998) (reviewed in Copelan et al. 2006; Sykes and Nikolic 2005), and CD8+ cell purification required for the rapidly growing field of adoptive T-cell therapy (Heslop et al. 1996; Mackensen et al. 2006) (reviewed in June 2007; Riddell and Greenberg 1995). Popular basic research purification schemes such as fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS) traditionally rely on antibodies for selective labeling and subsequent isolation of a target cell population (Bonner et al. 1972; Miltenyi et al., 1990, reviewed in Mattanovich et al. 2006; Herzenberg et al. 2002; Tomlinson et al. 2012). However, antibody-based cell labeling is difficult to reverse, reducing the utility of isolated cells in downstream applications. The inherent immunogenicity of antibodies strongly contraindicates their use in purification of cellular therapeutics, and antibodies that bind antagonistically to receptors disrupt native signaling pathways, potentially compromising the usefulness of isolated cells (Hwang and Foote 2005). Strategies to circumvent these well-recognized issues include negative depletion, in which undesired cells are labeled and discarded, and the use of antibodies engineered to lack the immunogenic Fc region (Locatelli et al. 2013) (reviewed in Handgretinger 2012; Holliger and Hudson 2005). However, neither of these approaches guarantee a pure population of functional and uninhibited cells for basic research or clinical use.
Recently a dielectrophoresis-based method was described which allows for separation of cells without the use of labels such as antibodies (Faraghat et al. 2017). However, there is still the need for a reversible method to specifically isolate cell populations based on selective receptor expression. A highly promising and alternative approach is to use aptamers as selective cell labels. Aptamers are innately non-immunogenic, single-stranded nucleic acid ligands that share the highly selective nature of antibodies yet are functionally reversible using complementary nucleic acid-based antidotes and nucleic acid-binding polymers (Oney et al. 2009). In thousands of patients, antidotes have been demonstrated to inactivate therapeutic aptamers targeting soluble coagulation proteins within minutes (Dyke et al. 2006; Chan et al. 2008).
Both RNA and DNA aptamers specific for extracellular proteins have been immobilized on a variety of surfaces to isolate cancer cells (reviewed in Zamay et al. 2017), demonstrating the utility of aptamers as affinity-reagents for cell capture. In particular, the well-characterized epidermal growth factor receptor (EGFR)-antagonizing aptamer E07 (Li et al. 2011; Avutu 2010) has been attached to nanotextured substrates (Wan et al. 2012) and microfluidic biochips (Wang et al. 2012; Wan et al. 2010; Wan et al. 2012; Wan et al. 2011) to bind and isolate EGFR-expressing cancer cells; significantly, the E07 aptamer more specifically and efficiently captured EGFR-expressing cells than an EGFR antibody (Wan et al. 2010). While both an accelerated flow rate (Wang et al. 2012) and a long E07-specific anti-sense RNA strand (Wan et al. 2012) have been used in an effort to subsequently strip bound cancer cells off of E07-biochips, neither technique attempted to isolate and purify EGFR-expressing cells out of a complex mixture of cells or validated that purified cells were returned to their native state following purification.
In theory, an aptamer-antidote system could be used to selectively label and purify cells before subsequently removing aptamer-based labels from the purified cells, restoring them to their native state. To thoroughly evaluate the utility of an aptamer-antidote system to selectively label and purify cells before restoring them to their native state, we sought to generate both fluorescent dye-aptamer conjugates and aptamer-magnetic bead conjugates to selectively label and purify cells, via FACS or MACS, respectively, before utilizing a matched antidote to remove the aptamer label and return the cells to their native state (Fig. 1). Focusing on a truncated version of the EGFR-antagonizing E07 aptamer, we developed an aptamer-antidote system capable of specifically isolating and purifying EGFR-expressing cells out of both a mixture of EGFR(+) and EGFR(−) cells and out of the more complex cellular mixture of blood. MACS purification of EGFR(+) cells using E07 aptamer-magnetic beads and subsequent treatment with a matched antidote effectively reversed E07 binding giving a pure population of ligand-free EGFR(+) cells.
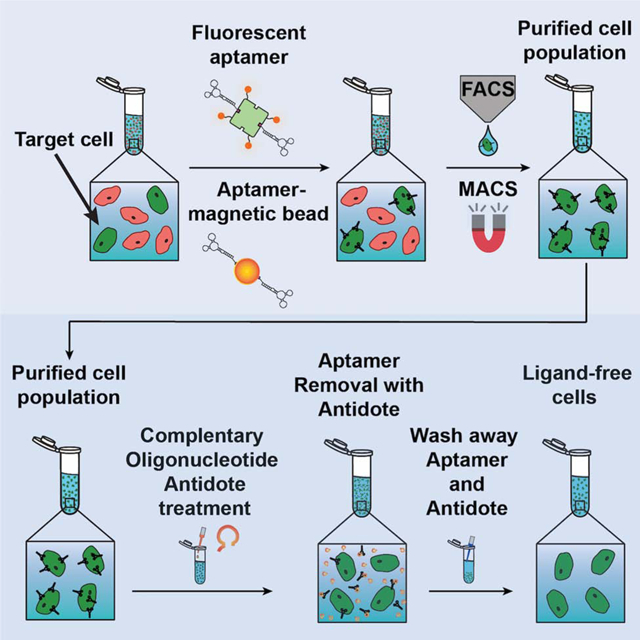
Figure 1. Overview of sorting and isolating aptamer-labeled target cells followed by antidote treatment to remove the aptamer and generate ligand-free cells.
Target cells of an initially heterogenous population are selectively labeled with a fluorescent dye tagged-aptamer (A) or with aptamer-magnetic beads (B) and then isolated using a conventionally antibody-based purification scheme such as FACS or MACS, respectively. However, the utility of purified target cells may be compromised as residual sorting ligand on purified cells can interfere with their native functions or elicit an immune response. Unlike antibody-based labels, treatment of aptamer-labeled cells with matched antidotes allows for ligand removal, returning cells to a ligand-free, native functional state.
Most importantly, we confirmed that antidote treatment effectively removes E07 from purified cells, returning the cells to their native state and restoring normal EGFR function. The E07-antidote pair serve as a sort of “molecular switch” allowing for cell staining that can be turned “on” via aptamer binding, turned “off” with antidote treatment and then turned “on” again via restaining with aptamer. Additionally, as the E07 aptamer blocks EGFR signaling by preventing the native epithelial growth factor (EGF) ligand from binding to EGFR, antidote-mediated removal of E07 from the cells frees EGFR to once again bind and respond to EGF, restoring normal signaling. Thus, using an E07 aptamer-antidote pair, we developed a method to specifically isolate and purify a desired cell population, resulting in label-free cells that are restored to their native state making them more amenable to study and utilize.
Results
Development of a 2’ O-Methyl RNA Antidote that Reverses E07 Binding and Function
The specificity and efficiency of the E07 aptamer for binding EGFR(+) cells makes it an attractive ligand for the isolation of receptor positive cells. Additionally, a truncated version of E07 eases synthesis while retaining EGFR binding and targeting (Avutu 2010). To utilize the truncated E07 aptamer as a reversible ligand capable of capturing and then releasing a pure population of EGFR(+) cells, we sought to develop an antidote oligonucleotide capable of annealing to E07 and disrupting its binding and function.
By walking down the E07 aptamer sequence, we rationally designed a series of 15 nucleotide long, single-stranded DNA antidote candidates complementary to different regions of the aptamer (Fig. 2A). To easily visualize aptamer targeting via flow cytometry, truncated E07 was chemically synthesized with a 5’ C6-thiol modifier and conjugated to a maleimide-Alexa Fluor 488 (AF488) dye (predicted structure, Fig. S1A). The A431 epidermoid carcinoma cell line that highly expresses EGFR (Masui et al. 1993; Schmidt-Ullrich et al. 1997; Sako et al. 2010) was then incubated with AF488-E07 +/− a 50-fold excess of one of the DNA antidotes 1–17 (A1-A17), washed to remove unbound AF488-E07 and antidote and then analyzed by flow cytometry. For the antidote screen, as well as all subsequent E07-binding and cell isolation experiments, salmon sperm DNA was added to block nonspecific oligonucleotide binding and subsequent internalization and verify that all aptamer targeting was specific. The most effective antidotes greatly reduced cell fluorescence by neutralizing AF488-E07 binding to cells (Fig. 2B; representative histograms shown in Fig. S2), although treatment at 37°C was necessary for the antidotes to function well; at 4°C even a 1000-fold excess of the DNA antidotes only minimally neutralized AF488-E07 binding (Fig. S3) likely due to better nucleation between the antidotes and the E07 aptamer at 37°C.
Figure 2. Development of a 2’ O-Methyl RNA Antidote that Reverses E07 Binding.

(A) Regions of the E07 sequence targeted by 15-base DNA antidote candidates. (B) A screening assay to identify the DNA antidote that best blocks E07 binding revealed a number of potent antidotes, including DNA Antidote 9 (A9). A431 cells were stained with AF488-E07 (or control aptamer AF488-C36) +/− a 50-fold excess of each of the DNA antidote candidates and analyzed by flow cytometry. The mean fluorescence intensity (MFI) of every sample was divided by the MFI of cells treated with AF488-E07 alone. Note that all antidotes are abbreviated as “A” and then the antidote number, such that Antidote 9 = A9. (C) Schematic of the E07 predicted structure with the antidote A9 target region illustrated via green dots. (D–E) After staining cells with 100nM of AF488-E07 at 4°C, cells were exposed to antidote at 37°C before returning the cells to 4°C for washing and flow cytometry analysis. Shown are representative flow cytometry analyses. (D) Cells were exposed to a 250-fold excess of antidote for either 2 min or 10 min at 37°C. Both DNA-based A9 and its 2’OMe RNA analog mA9, but not a scrambled-sequence control 2’OMe RNA antidote (sA9), enhanced removal of cell-bound AF488-E07 over time as analyzed by flow cytometry. (E) Cells were exposed to increasing concentrations of DNA A9 or 2’OMe mA9 antidotes for 10 min at 37°C. Both the DNA A9 and 2’OMe RNA mA9 antidotes were similarly effective at removing cell-bound E07 after incubation at 37°C for 10 min at concentrations ≥ 5uM (≥ 50-fold excess of antidote), but the 2’OMe RNA version was more potent at lower concentrations. (n=3; representative flow data of ≥ 104 gated events shown) See also Figure S1A, Figure S2 and Figure S3.
A number of potent antidotes emerged from the antidote screen, and the potent antidote A9 was chosen for downstream studies (Fig. 2B–C). The abilities of the DNA antidote A9, its 2’ O-Methyl (2’OMe) RNA analog (mA9) and a sequence-scrambled 2’OMe RNA control antidote (sA9) to reverse AF488-E07 staining over time were then evaluated. To prevent aptamer internalization and ensure that all bound aptamer was available to reverse, cells were maintained at 4°C and only incubated at 37°C for short periods of time with antidote present. Cells were stained with 100nM of AF488-E07 at 4°C, maintained at 4°C while washing away unbound aptamer and antidotes, and then pulsed with a 250-fold excess of antidotes A9, mA9 or sA9 for 2 min or 10 min at 37°C before returning the cells to 4°C for washing and flow cytometry analysis. At 37°C, E07-specific antidotes A9 and mA9 both stripped similar amounts of bound AF488-E07 from cells over time, reducing AF488-E07 staining to the background levels seen with the control non-targeting aptamer AF488-C36 (AF488 conjugate of C36, cntrl.36 (Wilner et al. 2012)) when incubated with the aptamer-cell complexes for 10 min (Fig. 2D). As expected, the scrambled control antidote sA9 did not engender removal of AF488-E07 relative to a no antidote treatment control. While A9 and mA9 performed similarly at the relatively high concentration of 25 μM (250-fold excess of antidote) after a 10 min incubation at 37°C, mA9 was more potent at lower concentrations (< 5 μM, < 50-fold excess) (Fig. 2E), likely due to the higher stability of 2’OMe RNA complexes versus DNA complexes; consequently, mA9 was utilized in all subsequent experiments.
E07-Aptamer Loaded Magnetic Beads + A Matched Antidote Reversibly Bind and Isolate a Pure Population of EGFR(+) Cells Out of a Cell Mixture
To determine if the E07 aptamer-antidote pair can be used to both specifically isolate EGFR(+) cells out of a cell mixture and then release a pure population of EGFR(+) cells, E07 was synthesized with a 5’ biotin linker (bE07, Fig. S1C) and complexed with streptavidin (SA)-magnetic beads. For specific tracking of EGFR(+) cells, A431 cells were labeled with the fluorescent dye, DiI. When analyzed via flow cytometry, the larger EGFR(+) DiI-labeled A431 cells gave a side scatter (SSC) vs forward scatter (FSC) profile distinct from that of the smaller EGFR(−) Jurkat cells (Fig. 3E–G). As expected, the DiI-labeled A431 cells were positive for DiI staining (Fig. 3E) and the unlabeled Jurkat cells negative for DiI staining (Fig. 3F). A 50–50 mixture of DiI-A431s and Jurkats gave an expected near 50–50 split in DiI staining, with 45.4% of the cells staining DiI(+) and 54.6% of the cells staining DiI(−) (Fig. 3G).
Figure 3. E07-Aptamer Loaded Magnetic Beads + A Matched Antidote Reversibly Bind and Isolate a Pure Population of EGFR(+) Cells Out of a Cell Mixture.
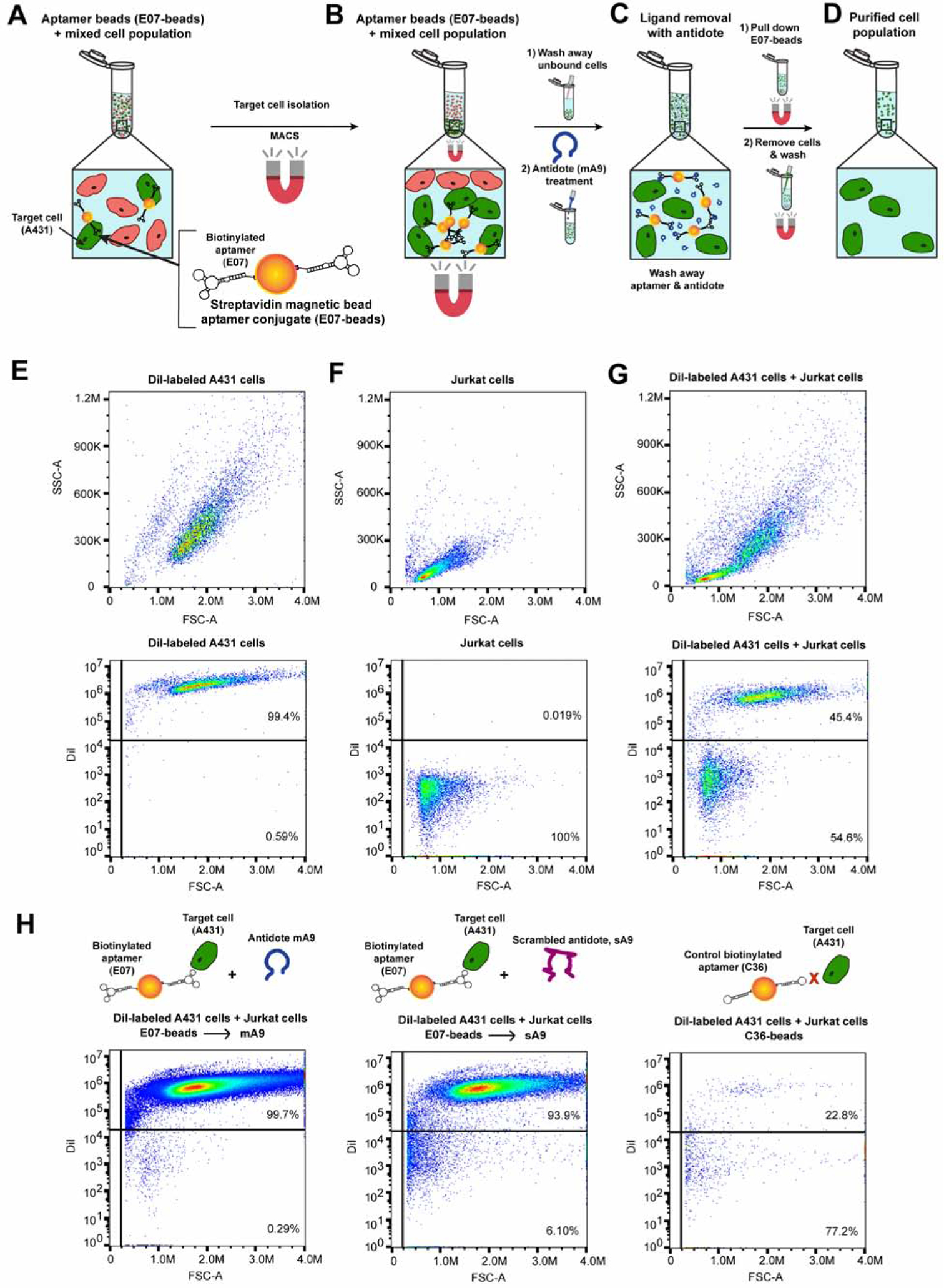
(A–D) Overview of E07 aptamer-antidote MACS purification of EGFR(+) cells out of a cell mixture. (A) Biotinylated-E07 was attached to SA-magnetic beads and the E07-beads incubated with a mixture of ~50% EGFR(+)A431 cells and ~50% EGFR(−) Jurkat cells. (B) MACS purification was used to specifically isolate E07-bead bound cells, allowing unbound cells to be washed away. (C) Subsequent treatment with 100-fold excess antidote mA9 stripped the E07-magnetic beads from isolated cells, and (D) additional magnetic-based pull down of the aptamer-antidote neutralized beads left a pure population of isolated cells in the supernatant. (E) EGFR(+) A431 cells were labeled with DiI dye, and dye incorporation verified by flow cytometry. (F) EGFR(−) Jurkat cells gave a SSC vs. FSC profile unique from that of the EGFR(+) A431 cells and were negative for the DiI dye. (G) Flow cytometry analysis of the ~50% A431 ~50% Jurkat cell mixture showed SSC vs. FSC profiles consistent with the presence of both cell types and a breakdown of ~50% DiI(+) staining and ~50% DiI(−) staining. (H) Treatment with E07-beads plus the specific mA9 antidote recovered a >99% pure population of DiI-labeled EGFR(+) cells, as evidenced by flow cytometry analysis of recovered cells. While treatment with E07-beads plus the scrambled control antidote sA9 also recovered a fairly pure population of DiI-labeled EGFR(+) cells, far fewer cells were recovered with the sA9 antidote verses the specific mA9 antidote (see Table S1 for cell recovery numbers). Control aptamer C36-beads also isolated very few cells, and the majority of the non-specific recovered cells were DiI(−) EGFR(−) Jurkat cells. (n=3; representative flow data of ≥ 104 gated events shown) See also Figure S1C and Table S1.
E07-beads were then incubated with the mixture of ~50% EGFR(+) DiI-labeled A431 cells and ~50% EGFR(−) Jurkat cells for 30 min at 4°C to ensure that the E07-beads only bound EGFR-expressing cells without internalizing into the cells (Fig. 3A). While maintaining the samples at 4°C, E07-bead-cell complexes were separated from the cell mixture via MACS (Fig. 3B), followed by extensive washing to remove unbound cells. The E07-bead-cell complexes were then resuspended in 100-fold excess mA9 to reverse aptamer binding and release the cells (Fig. 3C); as a 50-fold excess of antidote was shown to be sufficient for reversal of AF488-aptamer under these same conditions (Fig. 2E), the antidote concentration was doubled to ensure complete aptamer reversal and account for the multimeric display of the aptamer on the magnetic beads, which may require a greater excess of antidote to reverse. Subsequent magnetic separation of the E07-beads allowed for cell recovery in the supernatant and the isolation of a pure population of EGFR(+) cells (Fig. 3D). Flow cytometry analysis of the cells recovered from E07-beads treated with mA9 antidote resulted in a >99% pure population of DiI-labeled cells, indicating that only A431 cells were recovered (Fig. 3H). Analysis of the cells recovered from E07-beads treated with the scrambled control antidote (scrambled version of mA9), sA9, also resulted in a mostly pure (~94%) population of DiI-labeled A431 cells (Fig. 3H), but the scrambled antidote did not efficiently recover cells; treatment with the specific mA9 antidote led to the recovery of ~6-fold more cells than treatment with the control sA9 antidote, with a recovery of ~13% of the EGFR(+) A431 cells for the mA9 antidote (Table S1). Notably, very few cells were recovered from the control aptamer C36-beads (Table S1), and of the few cells that were collected, ~77% of them were the EGFR(−) Jurkat cells (Fig. 3H). Thus the combination of E07-magnetic beads and a matched antidote effectively isolates a pure population of EGFR(+) cells from a mixture of EGFR(+) and (−) cells.
E07-Aptamer Loaded Magnetic Beads + A Matched Antidote Reversibly Bind and Isolate a Pure Population of EGFR(+) Cells Out of Blood
To demonstrate that the E07 aptamer-antidote pair can also specifically isolate and release EGFR(+) cells out of a more complex and clinically relevant cellular mixture, human blood was spiked with DiI-labeled EGFR(+) A431 cells at a concentration equal to 5% of the leuokycte population. This concentration of A431 cells was chosen to closely model the isolation of a specific subset of leukocytes, such as CD4+ or CD8+ T-cells or B cells, all of which fall in the range of 1–20% of total leukocytes. When analyzed via flow cytometry, the EGFR(+) DiI-labeled A431 cells stained positive for DiI fluorescence while the blood was DiI negative (Fig. 4E–F).
Figure 4. E07-Aptamer Loaded Magnetic Beads + A Matched Antidote Reversibly Bind and Isolate a Pure Population of EGFR(+) Cells Out of Blood.
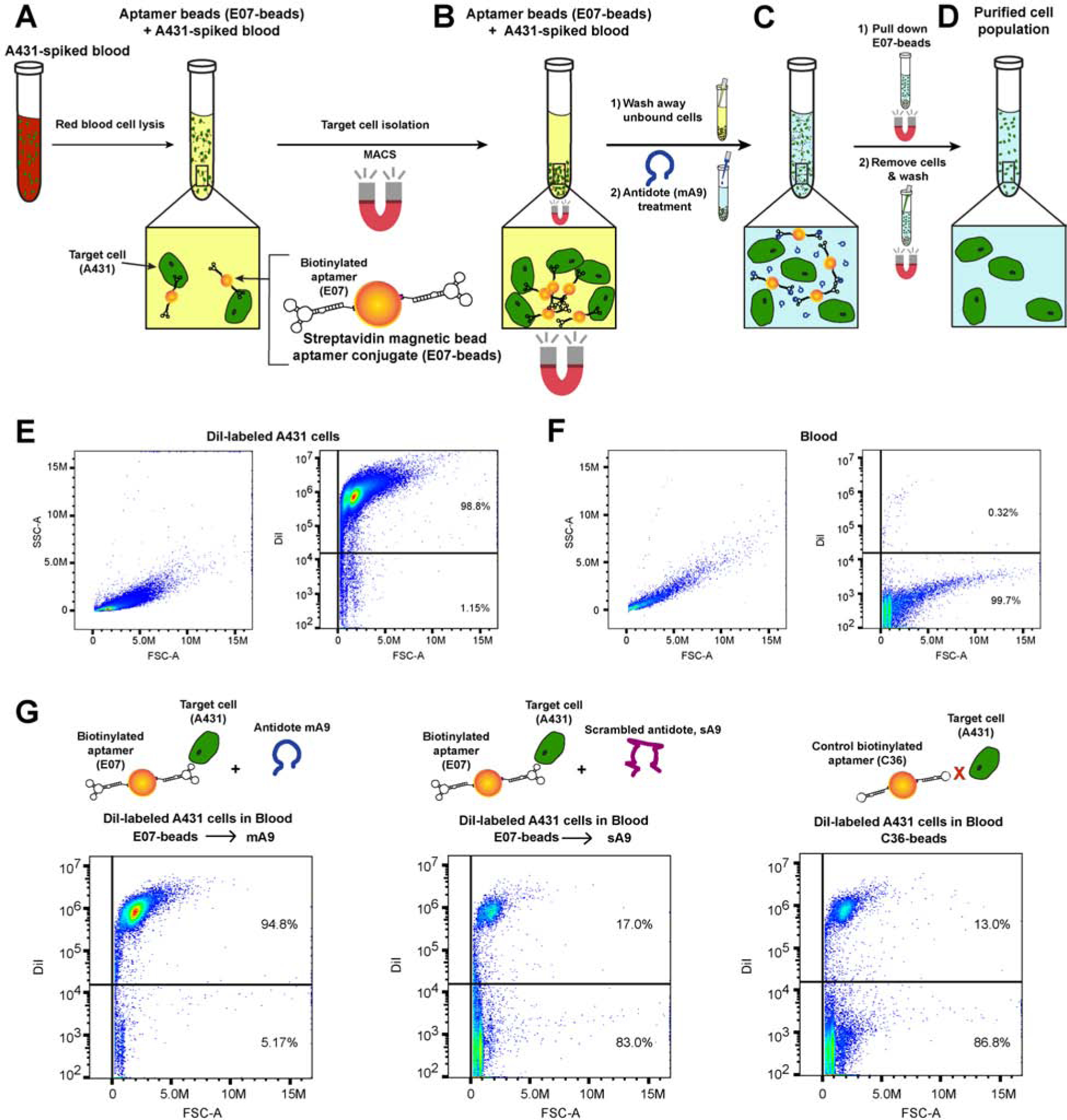
(A–D) Overview of E07 aptamer-antidote MACS purification of EGFR(+) cells out of blood. (A) A431 cells were spiked into human blood and red blood cells lysed prior to the addition of biotinylated-E07 attached to SA-magnetic beads. (B) MACS purification was used to specifically isolate E07-bead bound cells, allowing unbound cells to be washed away. (C) Subsequent treatment with 100-fold excess antidote mA9 stripped the E07-magnetic beads from isolated cells, and (D) additional magnetic-based pull down of the aptamer-antidote neutralized beads left a pure population of isolated cells in the supernatant. € EGFR(+) A431 cells were labeled with DiI dye, and dye incorporation verified by flow cytometry. (F) Blood gave a SSC vs. FSC profile unique from that of the EGFR(+) A431 cells and was negative for the DiI dye. (G) Treatment with E07-beads plus the specific mA9 antidote recovered a ~95% pure population of DiI-labeled EGFR(+) cells, as evidenced by flow cytometry analysis of recovered cells. See also Table S2 and Figure S4.
After red blood cell lysis, the A431 spiked blood sample was incubated with E07-magnetic beads (5’ biotin E07-SA-magnetic beads) (Fig. 4A) following a similar protocol to that used for the isolation of A431 cells out of the A431/Jurkat cell mixture. While maintaining the samples at 4°C to prevent aptamer internalization, E07-loaded beads were incubated with the spiked blood for 30 min prior to MACS separation of the E07-bead-cell complexes (Fig. 4B) and extensive washing to remove unbound cells. To reverse aptamer binding and release any bound cells, 100-fold excess antidote m9 was added to the E07-bead-cell complexes (Fig. 4C). Subsequent magnetic separation of the E07-beads allowed for cell recovery in the supernatant and the isolation of a pure population of EGFR(+) cells (Fig. 4D).
The combination of E07-beads plus antidote mA9 isolated a pure population of EGFR(+) A431 cells out of A431-spiked blood, with flow cytometry analysis showing ~95% positive staining for the DiI dye incorporated into the A431 cells (Fig. 4G). Additionally, almost a third of the EGFR(+) A431 cells were recovered from the blood (Table S2). As expected, very few cells were recovered from either the E07-beads treated with control antidote sA9 or from the control aptamer C36-beads (Fig. 4G), with both controls recovering < 2% of the A431 cells (Table S2). Increasing the initial number of EGFR(+) A431 spiked into blood to an amount equal to the number of leuokyctess also allowed for specific recovery of the EGFR(+) A431 cells. The E07-beads in combination with the mA9 antidote isolated a 99.9% pure population of EGFR(+) A431 cells from blood doped with the higher concentration of A431 cells (Fig. S4). While the combination of E07-beads with control antidote sA9 also isolated a ~93% pure population of A431 cells from the blood doped with a higher concentration of A431 cells, 3-fold fewer cells were recovered via incubation with the control antidote compared to the specific m9 antidote (Table S2). Thus E07 aptamer-magnetic beads along with a matched antidote allow for MACS isolation and recovery of a pure population of EGFR(+) cells from human blood at physiologically relevant concentrations.
The E07 Aptamer-Antidote Pair Can Be Used to Reversibly Stain and Restain EGFR(+) Cells, Creating a Molecular Sorting Switch
To validate the ability of an aptamer - antidote pair to fully reverse aptamer binding, stripping the aptamer off of the cellular receptor and leaving behind a native receptor, the E07 aptamer – mA9 antidote pair were used to stain, sort, and subsequently restain cells. Such a method would allow the flexibility to serially sort a cell population for multiple biomarkers using a single fluorophore or fluorophores with overlapping spectral profiles. Additionally, a reversible cell-staining ligand could serve as a molecular switch that can be toggled on and off. To demonstrate that this concept could be achieved, E07 was transcribed with a 5’ biotin to allow for multimerization and provide a better comparison with antibodies, which are typically bivalent. The 5’ end of E07 was also extended by an 8 nucleotide “tail” (biotin-tailed-E07, btE07) (Fig. S1B) to ensure the aptamer structure was maintained and limit steric effects that might impact aptamer function upon complexing with SA. btE07 was multimerized by labeling with AF488-SA at a ratio designed to complex 2 aptamers per SA-dye. EGFR(+) A431 cells were then stained with AF488-SA-E07 (Fig. 5A), sorted for E07/AF488+ signal and then destained with or without mA9 (Fig. 5B) generating a pool of ligand-free unlabeled cells (Fig. 5C) that were subsequently restained with btE07 labeled with the fluorophore AF647-SA (AF647-SA-E07) (Fig. 5D). Small aliquots were saved at each of these steps for subsequent analysis by flow cytometry (Fig. 5E–G). Successful antidote reversal of the AF488-SA-E07 stain should strip E07 from EGFR on the cell surface turning the molecular switch “off” and leaving the receptor available to turn “on” again by restaining with AF647-SA-E07. AF488-SA-E07 staining did not saturate all EGFR binding sites, allowing subsequent cell staining with AF647-SA-E07 without loss of AF488-SA-E07 signal (Fig. 5E, G). Conversely, antidote mA9 treatment switched “off” the EGFR staining by removing ~ 90% of the initial aptamer AF488 stain (Fig. 5F). Subsequent labeling with AF647-SA-E07 turned back “on” the EGFR switch, staining antidote-treated cells with ~ 80% more AF647 compared to the control, non-antidote treated cells (Fig. 5G). As expected, this method is not applicable to antibody labels due to their limited reversibility (Fig. S5 A).
Figure 5. The E07 Aptamer-Antidote Pair Can Be Used to Reversibly Stain and Restain EGFR(+) Cells, Creating a Molecular Sorting Switch.
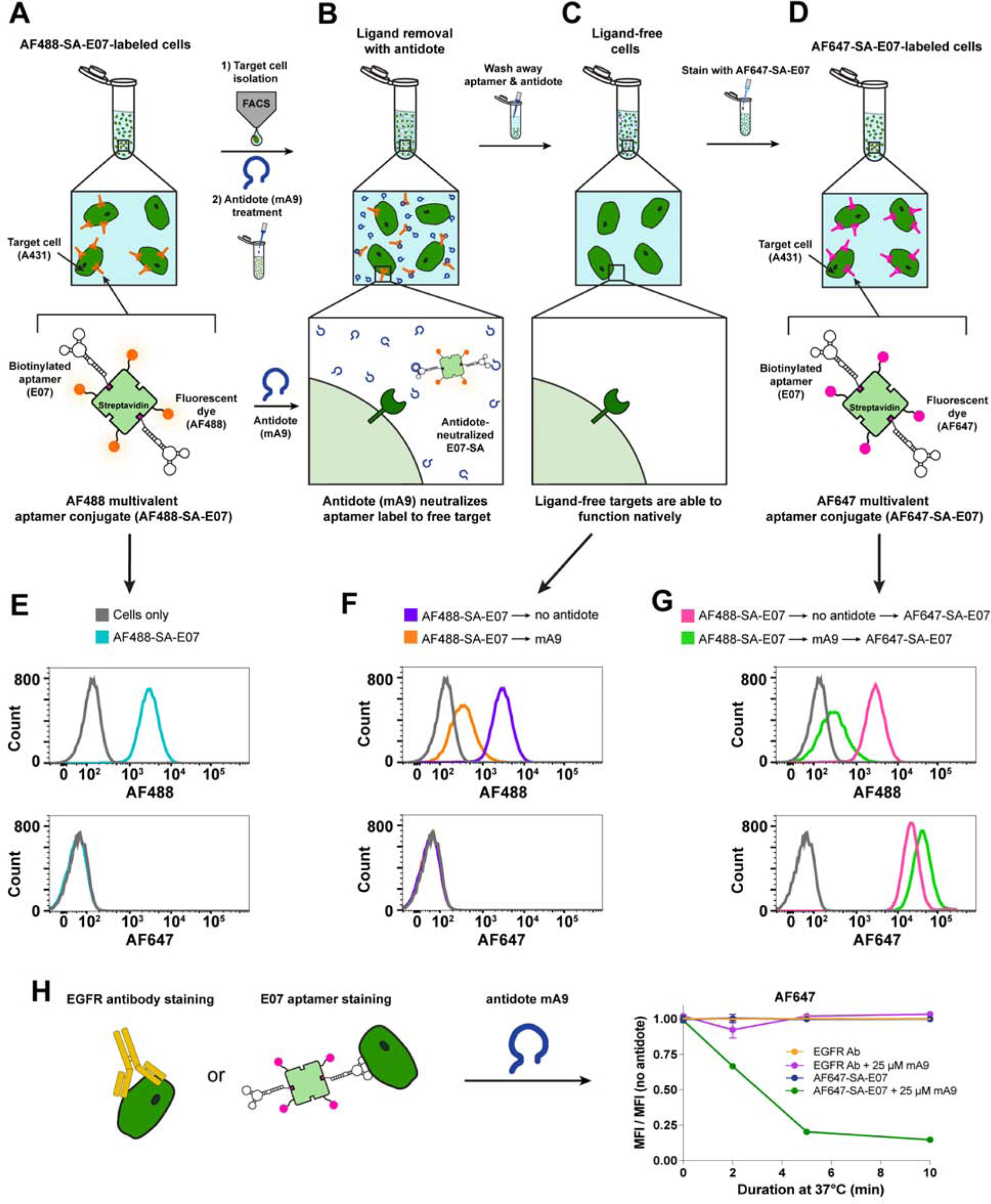
(A–D) Overview of AF488-SA-E07 aptamer-staining and FACS sorting of EGFR(+) cells followed by antidote treatment to remove the E07 stain and leave the cells free for additional staining with a different E07 label. (A) EGFR(+) A431 cells were stained with the green-fluorescent AF488-SA-E07, sorted for E07/AF488+ signal and then (B) destained with antidote mA9 to (C) generate a pool of ligand-free unlabeled cells that could be (D) subsequently restained with E07 labeled with the far-red-fluorophore AF647-SA. (E–G) Small aliquots were saved at each of these steps for subsequent analysis by flow cytometry. (E) AF488-SA-E07 bound and stained EGFR(+) A431 cells, allowing for FACS sorting based on AF488 signal. (F) mA9 antidote treatment of sorted AF488(+)/EGFR(+) cells removed ~ 90% of the AF488-SA-E07 stain. (G) The resultant label-free cells were restained with AF647-SA-E07, generating ~80% more AF647 signal compared to the cells that were not antidote destained. (H) mA9 enhanced removal of AF488-SA-E07 over time but did not affect the EGFR-binding antibody ICR10 as evidenced by flow cytometry analysis of stained A431 cells. A431 cells were labeled with a 1:2000 dilution of antibody ICR10 or with 100nM AF488-SA-E07 at 4°C, and then incubated with 250-fold excess of mA9 antidote for different lengths of time at 37°C. Shown is the mean fluorescence intensity (MFI) of the antibody or aptamer signal after antidote treatment normalized to the MFI of the respective treatment without antidote treatment. (n=3 for all studies; representative curves shown, all flow data shows ≥ 104 gated events) See also Figure S1B and Figure S5.
To further validate btE07 as a reversible alternative to antibodies, A431 cells were stained with either AF647-SA-E07 or with an EGFR-binding antibody (ICR10) and then treated with or without mA9 to destain. AF647-SA-E07 and ICR10 each exhibited robust cell-staining and stability at 37°C in the absence of antidote (Fig. 5H). In contrast, treatment with 25 μM mA9 readily removed bound AF647-SA-E07 but did not remove bound ICR10 as antibodies are not sensitive to antidote treatment. Importantly, the gentle nature of this sequential method maintained cell viability (Fig. S5 B–C). Thus, successful antidote reversal of E07 aptamer staining restores cells to their native state, presenting the option of restaining the cells, and creating a molecular switch that can be easily switched from “on” to “off” to back “on” again.
The E07 Aptamer is an Antidote-Reversible Cell-Sorting Ligand that Isolates and Then Returns EGFR+ Cells to Their Native State Making Them Responsive to EGF
The ability of reversible cell-ligands to return cells to a native functional state was next explored by using mA9 to rescue EGFR function on cells stained with either AF488-SA-E07 or with an EGFR antibody (D1D4J). This approach was assessed by incubating cells with EGF, a native agonist for EGFR that, upon binding, promotes phosphorylation of the receptor’s intracellular domain, consequently initiating downstream signaling that modulates cellular function. E07 and D1D4J are both EGFR antagonists that block EGF-induced stimulation, reducing phosphorylation. Antidote removal of AF488-SA-E07 from stained cells should thus restore EGFR phosphorylation to native levels, indicating functional recovery of cells previously crippled due to the presence of the EGFR-targeting ligand that impedes EGF binding (Fig. 6A). This restoration was evaluated using quantitative western blots that probed for total and phosphorylated EGFR (pEGFR) and allowed determination of relative phosphorylation levels (pEGFR/total EGFR) of cells in different treatment groups. For unsorted (mock sorted) and sorted samples, both AF488-SA-E07 and D1D4J-stained cells exhibited significantly reduced stimulation relative to the positive control of native cells stimulated with EGF (Fig. 6B–D). However, treatment of AF488-SA-E07-stained cells with 5 μM mA9 rescued EGF stimulation to native levels, indicating a complete functional recovery of the previously blocked receptor (Fig. 6C–D). In contrast, EGFR Ab D1D4J binding was irreversible under the same conditions and resulted in permanently crippled EGFR signaling and function. To further examine the potential reversibility of the antibody, cells were stained with EGFR Ab D1D4J for 30 min at 4°C and then incubated with a 10-fold excess of the EGFR extracellular domain for either 10 min or 6 hrs at 37°C (Fig. S6). Unlike E07 aptamer reversibility with an antidote, a 10 min incubation with excess EGFR extracellular domain did not reverse the DAD4J Ab stain, and even a 6 hr incubation with the EGFR extracellular domain only minimally reversed the Ab binding.
Figure 6. The E07 Aptamer is an Antidote-Reversible Cell-Sorting Ligand that Isolates and Then Returns EGFR+ Cells to Their Native State Making Them Responsive to EGF.
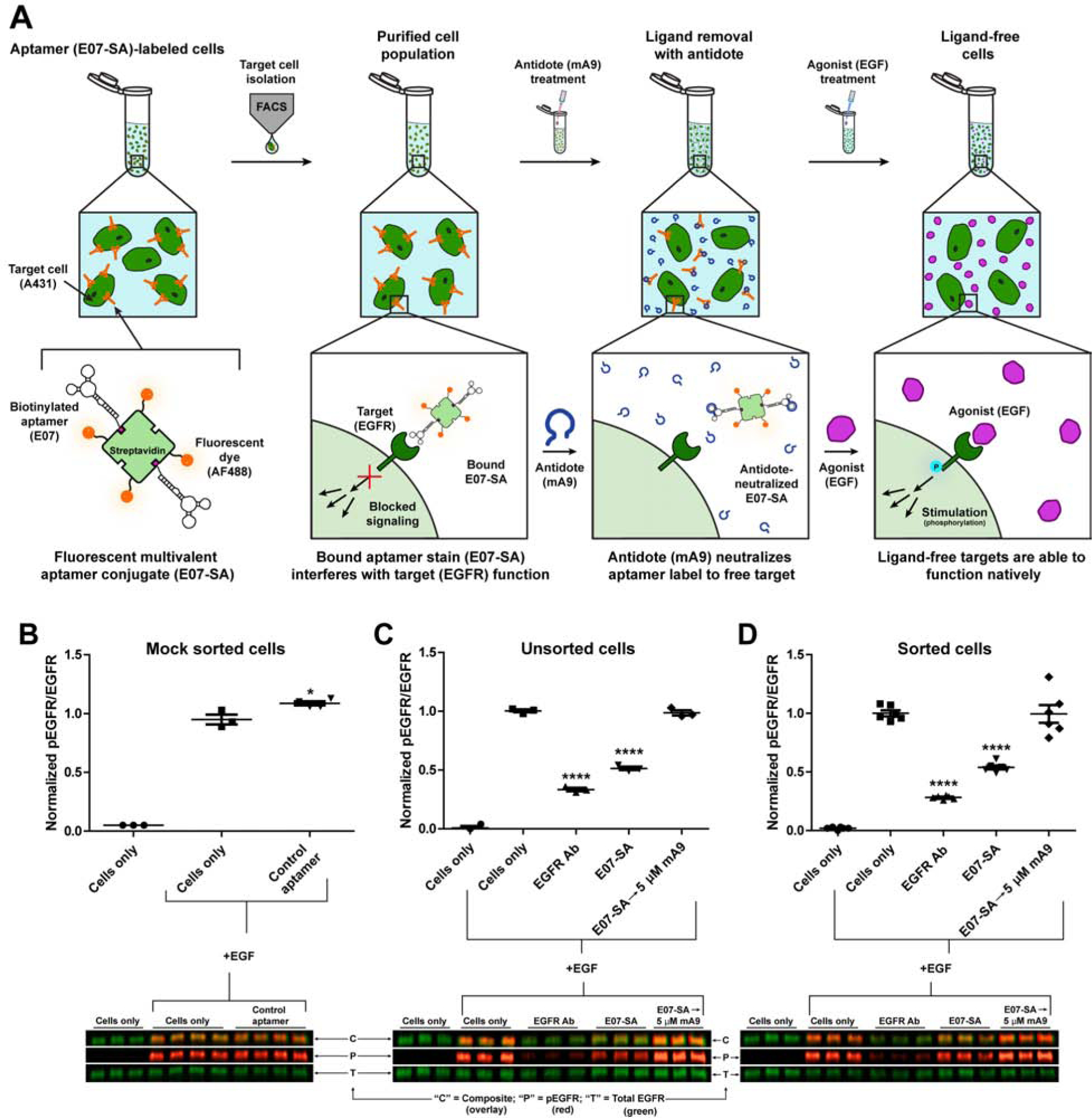
(A) Overview of AF488-SA-E07 aptamer-staining and FACS sorting of EGFR(+) cells followed by antidote treatment to remove the E07 stain and leave the cells ligand-free, reversing the EGFR antagonist effects of the E07 aptamer and restoring native signaling. EGFR(+) A431 cells were stained with AF488-SA-E07 and sorted for E07/AF488+ signal generating a pool of cells with aptamer-neutralized EGFR signaling. Destaining with antidote mA9 generated a pool of ligand-free unlabeled cells with restored EGFR signaling pathways. (B–D) Quantitative western blots of A431 cells probed for both phosphorylated EGFR (pEGFR; red) and total EGFR (green) after stimulation with EGF (indicated by “+EGF”). Cells were sorted for EGFR expression after staining with either 500nM AF488-SA-E07 or with a 1:50 dilution of the EGFR-antagonizing antibody (D1D4J). Cells alone or cells stained with the control C36 aptamer were mock sorted by running them through the cell sorter. While EGF stimulated EGFR phosphorylation in both unstained cells and cells stained with the control C36 aptamer, incubating/staining both unsorted (C) and sorted (D) cells with either AF488-SA-E07 or an EGFR-antagonizing antibody impedes stimulation of EGFR by EGF, indicating compromised receptor function. Subsequent treatment with mA9 (5 mM) for 5 min at 37°C efficiently removed AF488-SA-E07 to restore EGFR stimulation to native wild type levels. In contrast, irreversibly bound antibody permanently inhibited stimulation of EGFFR by EGF (n ≥ 3). One-way ANOVAs followed by Tukey-Kramer post-hoc tests were used to determine significance as indicated by * (p = 0.012) and **** (p < 0.0001). Data are mean ± SEM. See also Figure S6.
The ability to reversibly label cells without compromising their viability or function is a valuable, versatile tool with important implications for both the lab and clinic. Here, we demonstrate that multivalent aptamers can be used to sort cells in place of antibodies while also retaining the aptamer-exclusive benefit of rapid and efficient antidote-mediated reversibility. This methodology serves as an adaptable platform for other aptamer-antidote pairs that should prove useful for a myriad of research and clinical applications.
Discussion
Many basic research applications, as well as a growing number of clinical applications, rely on the isolation and purification of specific cell types. Antibodies selective for specific cell types are standardly used to label and isolate the desired cells via FACS or MACS. However, isolated cells retain the antibody label. Thus, isolation of ligand-free cells requires utilizing antibodies specific for unwanted cell types and sequentially depleting these cells until the desired cells remain. To selectively target and purify label-free cells, we developed an aptamer-based purification method. Aptamers can be used to label cells, purify them via either FACS or MACS, and then be stripped from the cells via treatment with specific complementary oligonucleotide “antidotes”, leaving behind a purified pool of label-free cells in their native state.
As a proof of principle for this technique, we focused on the well-characterized EGFR-antagonizing E07 aptamer to develop an aptamer-antidote system capable of specifically isolating and purifying EGFR-expressing cells out of a more complex cellular mixture. After screening a series of short complementary nucleic acid strands as potential antidotes, we identified an antidote, mA9, capable of effectively reversing E07 binding and activity. E07-fluorescent dye conjugates and E07-coated magnetic beads labeled and purified EGFR-expressing cells via both FACS and MACS, and subsequent treatment with the matched antidote mA9 removed the aptamer label to generate a pure population of label-free cells, returning the cells to their native state (Figure 1). E07-magnetic beads were able to capture EGFR(+) A431 cells out of both a mixture of EGFR(+) and EGFR(−) cells as well as out of a complex mixture of blood spiked with A431 cells. Addition of antidote mA9 to the cell-E07-magnetic bead complexes generated a ~95–99% pure population of label-free EGFR(+) cells, as evidenced by flow cytometry analysis of recovered cells.
E07 aptamer-antidote based MACS purification recovered close to 15% of the EGFR(+) cells present in the EGFR(+) A431 and EGFR(−) Jurkat cell mixtures and up to 33% of the EGFR(+) A431 spiked into blood. Notably, reducing the number of EGFR(+) A431 cells spiked into blood from an amount equal to the number of leuokyctess to 5% of the leuokycte population, increased A431 cell recovery and reduced nonspecific recovery of A431 cells. For the blood samples spiked with A431s at a concentration equal to the leuokycte population, we recovered ~12% of the A431 cells. However, for the blood samples spiked with A431s at a concentration equal to 5% of the leuokycte population, we recovered 33% of the A431 cells. These results suggest the ratio of EGFR(+) cells per aptamer-beads/antidote can be optimized to increase specificity and yield. We also expect that moving to a magnetic column purification platform would further increase yield by reducing washing losses seen with a magnet pulldown in a tube. Additionally, although other 2’F pyrimidine modified aptamers have been shown to be stable in human serum for 4–10 hours, suggesting that the E07 aptamer used in these studies should be stable throughout the entire purification procedure, using a fully 2’OMe modified aptamer could expand the utility of this technique by extending the serum stability to a predicted half-life of at least 20 days (Kratschmer and Levy 2017).
While further optimization of this technique is expected to yield even higher recovery levels, the ability to specifically tag cells for separation and subsequently generate such a pure population of label-free cells remains a major advantage compared to existing methods. Additionally, as MACS purification is usually used to deplete out unwanted cell types, rather than to isolate a desired cell type, it is difficult to compare the percent recovery for aptamer-antidote MACS purification to the recovery of traditional antibody-based MACS purification. Moreover, the ability to specifically tag cells for separation and subsequently generate such a pure population of label-free cells remains unique. Traditional antibody-MACS purification is not always sufficient to generate a pure population of a desired cell type (Fong et al. 2009) or requires extensive optimization to effectively separate out different cell types (Sutermaster and Darling 2019). While antibody-based FACS purification can also be used to deplete unwanted cells, ~70% of all cells are lost during the process (Sutermaster and Darling 2019). By contrast, as our aptamer-antidote method is based upon specific labeling and isolation of a particular cell type as opposed to the removal of undesired cells, unwanted cells are washed away leaving behind only a pure population of labeled cells that can be subsequently isolated via antidote treatment.
A similar approach was recently described by Kacherovsky et. al. using CD8 aptamer-magnetic beads for MACS-based isolation of CD8+ T cells followed by incubation with a complementary antidote to dissociate the cells from the aptamer-beads (Kacherovsky et al. 2019). Unfortunately, while the authors looked at broad phenotypic responses after aptamer removal, they did not specifically look at the function of the CD8 receptor after aptamer removal to verify that receptor signaling is restored to a native state. Moreover, the design of the CD8 aptamer reversal method makes it impossible to determine whether the antidote actually contributed to release of the aptamer from the cells. After incubation with the antidote, the solution used to wash and isolate cells contained 5 mM EDTA, a concentration of EDTA frequently used to dissociate adherent cells from plates, suggesting that the EDTA wash itself may have contributed to cell dissociation from the beads. Additionally, as EDTA chelates divalent cations which are often necessary for proper folding of aptamers, EDTA washes may cause an aptamer to unfold and release from its target. Since neither a control antidote nor a control including EDTA washes without antidote treatment were included in the CD8 T-cell purifications, unfortunately it is impossible to distinguish whether the cells released from the aptamer due to the antidote incubation or due to EDTA cation chelation. Thus it is currently unclear whether this approach actually depends upon antidote-based reversal of the aptamer. Nevertheless, we have observed that antidote oligonucleotides can reverse the binding of aptamers to a variety of proteins in vitro, in animals and in patients (Dyke et al. 2006; Rusconi et al. 2002; Rusconi et al. 2004; Bompiani et al. 2012; Soule et al. 2016; Powell Gray et al. 2018; Gunaratne et al. 2018; Nimjee et al. 2019; Nimjee et al. 2017). Thus, we anticipate that the aptamer-antidote purification method we describe herein should be broadly applicable to other aptamers.
Importantly, antidote treatment returned cells stained and sorted based upon EGFR(+)/E07(+) signal to a label-free state, allowing for restaining with a different E07-label, generating a “molecular sorting switch” that can be easily toggled between the “on” and “off” states. Such a method will also allow the flexibility to serially sort a cell population for multiple biomarkers using a single fluorophore or fluorophores with overlapping spectral profiles. As the E07 aptamer also blocks EGFR signaling by preventing binding of the native ligand EGF, antidote-mediated removal of E07 from purified cells restores responsiveness to EGF and native EGFR signaling. Thus, using an E07 aptamer-antidote pair, we developed a method to specifically isolate and purify a desired cell population, resulting in label-free cells that are restored to their native state.
As this aptamer-antidote purification method utilizes either the well-established techniques of FACS or MACS, it should serve as an adaptable platform for use with other aptamer-antidote pairs. Numerous cell-binding aptamers already exist in the literature (see aptamers referenced in review (Yan and Levy 2018)), and we demonstrated both in this study as well as in previous work(42), that antidotes can be readily designed against cell binding aptamers. Additionally, aptamers can be selected against other cellular targets via either Systematic Evolution of Ligands by EXponential enrichement (SELEX) (Ellington and Szostak 1990; Tuerk and Gold 1990), where a library of oligonucleotides is selected for binding to a target protein, and/or Cell-SELEX(reviewed in (Shangguan et al. 2015)) or Cell-Inernalization SELEX (method in (Yan and Levy 2014)), which use a cell, instead of a protein, as the target of interest. We therefore expect this technique to translate to the isolation of a variety of different cell types for both laboratory and clinical use. This may include isolation of various immune cell populations for basic research applications as well as isolation of CD8+ T-cells to generate CAR T-cells for cancer therapy or CD34+ stem cells for hematopoietic stem-cell transplantation. Thus, this methodology serves as an adaptable platform for other aptamer-antidote pairs that are able to suit a myriad of research and clinical applications.
Significance
Current methods of specific cell purification rely upon either poorly reversible antibody labeling of desired cell types or upon successive depletion of unwanted cells. Here, we demonstrate that aptamers can be used to label and purify cells in place of antibodies while also retaining the aptamer-exclusive benefit of antidote-mediated reversibility. Cells can be labeled with aptamers, purified using the standard techniques of FACS or MACS, and the aptamer label removed via treatment with a complementary antidote oligonucleotide, returning the cells to their native, label-free state. Using this method, we demonstrated that a ~99% pure population of target cells can be isolated out of various complex cell mixtures. Significantly, antidote removal of aptamer tags generates label-free cells, reversing aptamer effects on the cells and returning cells to their native state. Additionally, the aptamer-antidote pair can be used as a sort of molecular sorting switch to toggle between aptamer labeled “on” states and unlabeled “off” states. This methodology serves as an adaptable platform for other aptamer-antidote pairs and is easily translatable and scalable by replacing antibodies with aptamers in standard FACS and MACS protocols. Thus, we developed a technique to reversibly label and isolate cells without compromising their function that has broad implications for both basic science and clinical use.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce Sullenger (bruce.sullenger@duke.edu). The sequences of all oligonucleotides generated in this study are listed in the Key Resources Table and Table S3 and may therefore be synthesized, transcribed, or purchased from oligonucleotide vendors.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-EGFR Antibody 1F4 | Cell Signaling Technologies | Cat# 2239 |
| Rabbit monoclonal anti-EGFR Antibody D1D4J (PE Conjugate) | Cell Signaling Technologies | Cat# 48685 |
| Rat monoclonal anti-EGFR Antibody ICR10 (FITC Conjugate) | Abcam | Cat# ab11400 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Phosphate Buffered Saline (DPBS) with MgCl2 and CaCl2 | Sigma | Cat# D8662–500ML |
| Dulbecco’s Phosphate Buffered Saline (DPBS) without MgCl2 or CaCl2 | Gibco | Cat# 14190–144 |
| Fetal Bovine Serum (not heat-inactivated) | Gibco | Cat# 16000–044 |
| Heat-Inactivated Fetal Bovine Serum | Gibco | Cat# 10082–147 |
| High Glucose Dulbecco’s Modified Eagle Medium | Sigma | Cat# D6429–500ML |
| RPMI 1640 | Gibco | Cat# A10491 |
| Salmon Testes DNA, Sodium Salt | Millipore Sigma | Cat# 262012–1GM |
| Human EGFR protein extracellular domain | G&P Biosciences | Cat# FCL0169 |
| Gibco EGF Recombinant Human Protein | ThermoFisher | Cat# PHG0315 |
| eBioscience 10x RBC Lysis Buffer | ThermoFisher | Cat# 00-4300-54 |
| 5’-Biotin-G-Monophosphate | TriLink Biotechnologies | Cat# N-6003 |
| 5’-Thiol-C-6 Disulfide Modifier CED phosphoramidite | ChemGenes | Cat# CLP-8506 |
| Biotin (BB) CED phosphoramidite | ChemGenes | Cat# CLP-1517 |
| 5’-DMT-2’-TOM-ribo Adenosine (n-acetyl) OP | ChemGenes | Cat# ANP-3201 |
| 5’-DMT-2’-TOM-ribo Guanosine (n-acetyl) OP | ChemGenes | Cat# ANP-3203 |
| 2’-Fluoro-2’-deoxy Cytidine (n-ac) CED phosphoramidite | ChemGenes | Cat# ANP-9152 |
| 2’-Fluoro-2’-deoxy Uridine CED phosphoramidite | ChemGenes | Cat# ANP-9154 |
| Triethylamine Acetate (TEAA) 2.0M, HPLC grade | Glen Research | Cat# 60-4110-52 |
| Acetonitrile for HPLC, gradient grade, ≥99.9% | Sigma Aldrich | Cat# 34851–4×4L |
| Critical Commercial Assays | ||
| Streptavidin, Alexa Fluor™ 647 Conjugate | ThermoFisher Scientific | Cat# S21374 |
| Streptavidin, Alexa Fluor™ 488 Conjugate | ThermoFisher Scientific | Cat# S32354 |
| Vybrant™ DiI cell-labeling solution | ThermoFisher Scientific | Cat# V-22885 |
| LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34975 |
| Experimental Models: Cell Lines | ||
| A431 cells | Duke Cell Culture Facility | n/a |
| Jurkat cells | Duke Cell Culture Facility | n/a |
| Oligonucleotides | ||
| Biotin E07 (RNA) 5’-Biotin linker-GGA fCGG AfUfU fUAA fUfCG fCfCG fUAG AAA GfCA fUGfU fCAA AGfC fCGG AAfC fCGfU fCfC idT-3’ |
Synthesized for this paper. Kratschmer et al. 2018 Oligo (without biotin linker) is 1 nucleotide truncate of minimized version of E07 (original full-length E07 - Li et al. 2011; initial E07 truncate – Avutu 2010) |
n/a |
| Biotin C36 (RNA) 5’-Biotin linker-GGfC GfUA GfUG AfUfU AfUG AAfU fCGfU GfUG fCfUA AfUA fCAfC GfCfC-3’ |
Synthesized for this paper. Oligo (without biotin linker) from Wilner et al. 2012 |
n/a |
| S-S-C6-E07 (RNA) 5’-S-S-C6-GGA fCGG AfUfU fUAA fUfCG fCfCG fUAG AAA GfCA fUGfU fCAA AGfC fCGG AAfC fCGfU fCfC idT-3’ |
Synthesized for this paper. Kratschmer et al. 2018 Oligo (without thiol linker) is 1 nucleotide truncate of minimized version of E07 (original full-length E07 - Li et al. 2011; initial E07 truncate – Avutu 2010) |
n/a |
| S-S-C6-C36 (RNA) 5’- S-S-C6-GGfC GfUA GfUG AfUfU AfUG AAfU fCGfU GfUG fCfUA AfUA fCAfC GfCfC-3’ |
Synthesized for this paper. Oligo (without biotin linker) from Wilner et al. 2012 |
n/a |
| btE07 forward primer (DNA) 5’-GAT AAT ACG ACT CAC TAT AGG GAT TTA GGA CGG ATT TAA TCG CCG TAG AA-3’ |
Purchased from Integrated DNA Technologies (IDT) | n/a |
| btE07 reverse primer (DNA) 5’-GGA CGG TTC CGG CTT TGA CAT GCT TTC TAC GGC GAT TAA ATC CGT CCT AAA TCC C-3’ |
Purchased from Integrated DNA Technologies (IDT) | n/a |
| btE07 (RNA) 5’-GGG AfUfU fUAG GAfC GGA fUfUfU AAfU fCGfC fCGfU AGA AAG fCAfU GfUfC AAA GfCfC GGA AfCfC GfUfC fC-3’ |
Transcribed for this paper. | n/a |
| btC36 forward primer (DNA) 5’-GAT AAT ACG ACT CAC TAT AGG AAA ATA GGC GTA GTG ATT ATG AAT CGT-3’ |
Purchased from Integrated DNA Technologies (IDT) | n/a |
| btC36 reverse primer (DNA) 5’-GGC GTG TAT TAG CAC ACG ATT CAT AAT CAC TAC GCC TAT TTT CC-3’ |
Purchased from Integrated DNA Technologies (IDT) | n/a |
| btC36 (RNA) 5’-GGA AAA fUAG GfCG fUAG fUGA fUfUA fUGA AfUfC GfUG fUGfC fUAA fUAfC AfCG fCfC-3’ |
Transcribed for this paper. | n/a |
| See Table S3 for antidote oligonucleotides | ||
| Software | ||
| FlowJo 10.4.1 to 10.6.1 | FlowJo LLC | https://www.flowjo.com |
| GraphPad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageLab Software | BioRad | http://www.bio-rad.com/en-us/product/image-lab-software |
| M-Fold Software | Zuker 2003 | http://unafold.rna.albany.edu/?q=mfold/download-mfold |
| Other | ||
| BD Vacutainer® Sodium Citrate Tubes | BD Biosciences | Cat#369714 |
| Dynabeads™ MyOne™ Streptavidin C1 Beads | Invitrogen | Cat# 65001 |
| DynaMag™-2 Magnet | ThermoFisher | Cat# 12321D |
fC = 2’Fluoro cytidine; fU = 2’Fluoro guanosine; idT = inverted deoxythymidine; mA = 2’OMethyl adenine; mC = 2’OMethyl cytidine; mG = 2’OMethyl guanosine; mU = 2’OMethyl uridine
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
A431 female epidermoid carcinoma cells and Jurkat male T lymphocyte cells sourced from the American Type Culture Collection (ATCC) were obtained through the Duke Cell Culture Facility. A431 cells were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) containing L-glutamine, sodium pyruvate, and sodium bicarbonate with 4.5 g/L glucose supplemented with 10% non-heat inactivated fetal bovine serum (FBS) (Gibco) per ATCC recommendations. For fluorescent dye-SA-aptamer flow cytometry experiments, 100 U/mL penicillin and 100μg/mL streptomycin were also added to the cell culture media. Jurkat cells were cultured in RPMI 1640 with 10% heat-inactivated FBS (Gibco). Cells were cultured in a 5% CO2 humidified incubator at 37°C. A431 cells were pass aged at 70–90% confluency using 0.25% trypsin-EDTA (Gibco) and split 1:2 – 1:4 into new tissue culture flasks. Jurkat cells were passaged every 4 to 7 days to keep cells at a concentration of 2–8 × 105 cells/mL.
METHOD DETAILS
Buffers, reagents and experimental samples were maintained at 4°C either on ice, in a cold room or in refrigerated centrifuges during all procedures unless otherwise stated. Care was taken to minimize ambient light exposure of fluorescent samples. Media supplemented only with 1% bovine serum albumin (“Media+”) was used for staining, antidote solutions, and washes unless otherwise noted. All phosphate-buffered saline (PBS+/+) contained Ca2+ and Mg2+ unless otherwise noted.
Chemical Synthesis of Aptamers
The E07 aptamer, non-specific control C36 aptamer, antidote mA9 and scrambled control antidote sA9 were synthesized by solid phase synthesis on an Expedite 8909 DNA synthesizer (Biolytic Lab Performance, Fremont, CA) or MerMade 12 (Bioautomation, Irving, TX) as previously described (Wilner et al. 2012). Briefly, all aptamers were synthesized on inverted dT CPG columns with 2’fluoro (2’F) pyrimidines and 2’OH purines and the antidotes synthesized with 2’OMe amidites. Synthesis reagents were purchased from Glen Research (Sterling, VA) and Chemgenes (Wilmington, MA). Aptamers used for 5’ labeling with Alexa Fluor dyes were synthesized bearing a 5’ thiol using a C6 S-S phosphoramidite. Aptamers that were labeled with a 5’ biotin linker were synthesized bearing a 5’ biotin phosphoramidite. Aptamer sequences are provided in the methods. The aptamers and antidotes were reversed-phase HPLC purified using a linear gradient of acetonitrile in 0.1 M TEAA, pH 7.5, on a 10 × 50 mm Xbridge C18 column (Waters) at 65°C.
Aptamer-Dye Conjugation
To prepare for dye conjugation, aptamers bearing a 5’ thiol C6 S-S linker were reduced to give a free thiol by heating at 70°C for 3 minutes in 0.1 M TEAA with 500 mM TCEP and then incubating at room temperature for 57 minutes. After confirming thiol reduction by HPLC, TCEP was removed by buffer exchanging into PBS (without Ca2+and Mg2+) + 2mM EDTA using Amicon Ultra 10kD spin columns (Millipore, Billerica, MA). The reduced aptamers were then 5’ labeled with Alexa Fluor 488 (AF488) or Alexa Fluor 647 (AF647) using the thiol-reactive AF488-C5-maleimide or AF647-C2-maleimide dyes (ThermoFisher Scientific). The dyes were dissolved in DMSO to a concentration of 20 mM and added to the reduced aptamers at 5–10-fold molar excess of dye:drug. Labeling efficiency was determined by analytical HPLC and routinely proceeded to 99% purity. Free dye was removed by washing labeled aptamers with 10 mM Tris-EDTA pH 7.5 (TE) in Amicon Ultra 10kD spin columns. The concentration of dye-labeled aptamer was quantitated using a NanoDrop spectrophotometer (ThermoFisher Scientific).
Screen for DNA antidotes that block E07 cell binding at 37°C
Experiments utilized cells at 75–90% confluency that had been plated 48–72 hours in advance. Trypsin was used to harvest cells and immediately neutralized with growth media upon cell detachment. Cells were washed once with Media+, counted with a hemocytometer, and then 5×105 cells per sample were partitioned into chilled 96 well plates. Staining was performed by resuspending cells in 70–100uL of the appropriate aptamer-staining solution (5 million cells per mL). All aptamers were folded in PBS+/+ via denaturation at 65°C for 5 minutes followed by passive cooling to ambient temperature for 3 minutes prior to staining. Cells were stained with 100 nM of AF488-E07 or control aptamer AF488-C36 in DMEM+ with 1 mg/mL salmon sperm DNA +/− 50-fold excess of DNA antidotes at 37°C for 30min. After staining, cells were washed with once with 100 uL Media+ and analzyed for AF488 fluorescence on a CytoFLEX (Beckman-Coulter).
Screen for DNA antidotes that block E07 cell binding at 4°C
Experiments utilized cells at 75–90% confluency that had been plated 48–72 hours in advance. Trypsin was used to harvest cells and immediately neutralized with growth media upon cell detachment. Cells were washed once with Media+, counted with a hemocytometer, and then 5×105 cells per sample were partitioned into chilled 1.5 mL microcentrifuge tubes. Staining was performed by resuspending cells in 100uL of the appropriate aptamer-staining solution (5 million cells per mL). All aptamers were folded in PBS+/+ via denaturation at 65°C for 5 minutes followed by passive cooling to ambient temperature for 3 minutes prior to staining. Cells were stained with 100 nM of AF488-E07 or control aptamer AF488-C36 in DMEM+ with 1 mg/mL salmon sperm DNA +/− 1000-fold excess of DNA antidotes at 4°C for 30min. After staining, cells were washed with once with 500 uL Media+ and analzyed for AF488 fluorescence on BD FACSCalibur or BD FACSCanto II flow cytometers (BD Biosciences).
Screen for antidote removal of E07 cell staining
All aptamers were folded in PBS+/+ via denaturation at 65°C for 5 minutes followed by passive cooling to ambient temperature for 3 minutes. Cells were stained with 100 nM of AF488-E07 or control aptamer AF488-C36 in DMEM+ with 1 mg/mL salmon sperm DNA at 4°C for 30min. After washing, samples were resuspended +/− antidotes in DMEM+ and incubated for either 2 min or 10 min at 37°C before returning the cells to 4°C for washing and flow cytometry analysis of AF488 fluorescence on a CytoFLEX (Beckman-Coulter) or BD FACSCalibur or FACSCanto II (BD Biosciences).
Immobilization of aptamers on magnetic beads
5’ Biotinylated, synthesized aptamers were folded by incubation in PBS+/+ for 5 minutes at 65°C followed by passive cooling to room tempera ture for 3 minutes. Beads were vortexed for 30 seconds and washed with 1 mL of SB1 buffer (40mM HEPES, 125mM NaCl, 5mM KCl, 1mM MgCl, 1mM CaCl, .05% Tween 20) per 32uL of beads before washing 3 additional times with SB1 buffer at a volume equal to the bead volume. Note that all bead washes involved pulling down the beads with a magnet. The beads were then resuspended to a final concentration of 2.5 μg/μL in SB1 buffer with 1 mg/mL salmon sperm DNA. Folded aptamers (12.5pmol per 40μL of beads) were incubated with the beads for 15 minutes at room temperature with gentle rotation before removing the solution and washing 3 times with SB1 buffer with 1 mg/mL salmon sperm DNA (washing with a volume of 50 μL wash buffer per 40 μL of beads). Washed aptamer-beads were resuspended at a bead concentration of 2.5 μg/μL in DMEM+ with 1 mg/mL salmon sperm DNA.
Aptamer-MACS recovery of cells from a cell mixture
A431 cells were labeled with DiI dye by incubating cells in serum-free DMEM with 50 mg/mL DiI (ThermoFisher) at a concentration of 1×106cells/mL for 20 minutes at 37°C, then washing three times with serum-free DMEM. DiI-labeled cells were resuspended in Media+ with 1mg/mL salmon sperm DNA at a concentration of 4×107cells/mL and incubated with the beads (1 ×107cells per 40 μL beads) at 4°C with gentle rotation for 30 minutes. The beads and cells were maintained at 4°C and pulled down via MACS bef ore 3 additional magnetic pull downs to wash with 500 μL of Media+ per 40 μL bead sample. Washed beads were resuspended in Media+ at a bead concentration of 2.5 μg/μL. 100-fold molar excess of the specific antidote mA9 or scrambled antidote sA9 was added to detach cells from the beads by incubating at 37°C with gentle rotation for 10 minutes. Isolated cells were then recovered from the supernatant after magnetic pull down of the beads. The beads were magnetically pulled down 3 additional times and washed with Media+ (500 μL of Media+ per 40 μL bead sample), and the supernatant from each wash combined with the initial collected supernatant. Recovered cell samples were resuspended in 260 μL of flow buffer (PBS + 1% BSA) and split into 4 different 65uL aliquots into a 96-well flow plate. The cells in each well were counted and analyzed using a CytoFLEX flow cytometer (Beckman-Coulter). A431 recovery was determined by counting the number of Di positive cell events per sample.
Aptamer-MACS recovery of cells from blood using an A431 concentration equal to the WBC concentration
A431 cells were labeled with DiI dye by incubating cells in serum-free DMEM with 50 mg/mL DiI (ThermoFisher) at a concentration of 1×106cells/mL for 20 minutes at 37°C, then washing three times with serum-free DMEM. Human whole blood was collected according to a protocol approved by the institutional review board of the Duke University Medical Center. Blood was collected in sodium citrate-treated vacutainer tubes (BD Biosciences), and 650 μL of citrated blood was mixed with 5×106 DiI-stained A431 cells. The A431-spiked blood was centrifuged at 1200g for 10 minutes. After removing the plasma supernatant, the pellet was incubated with 2.6 mL red blood cell lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0.1 mM EDTA) per 266 μL of blood at 37°C with gentle rotat ion for 15 minutes. Samples were then centrifuged at 1000g for 10 minutes and washed two times with red blood cell (RBC) lysis buffer followed by one wash with Media+ before resuspending in 750 μL of Media+.
RBC lysed, A431-spiked blood samples were incubated with aptamer-beads (250 μL spiked-blood per 40 μL beads) at 4°C with gentle rotation for 30 minutes. The beads and blood samples were maintained at 4°C and pulled down via MACS before 3 additional magnetic pull downs to wash with 500 μL of Media+ per 40 μL bead sample. Washed beads were resuspended in Media+ at a bead concentration of 2.5 μg/μL. 100-fold molar excess of the specific antidote mA9 or scrambled antidote sA9 was added to detach cells from the beads by incubating at 37°C with gentle rotation for 10 minu tes. Isolated cells were then recovered from the supernatant after magnetic pull down of the beads. The beads were magnetically pulled down 3 additional times and washed with Media+ (500 μL of Media+ per 40 μL bead sample), and the supernatant from each wash combined to with the initial collected supernatant. Recovered cell samples were resuspended in 300 μL of flow buffer (PBS + 1% BSA) and split into 4 different 75uL aliquots into a 96-well flow plate. The cells in each well were counted and analzyed using a CytoFLEX flow cytometer (Beckman-Coulter). A431 recovery was determined by counting the number of Di positive cell events per sample.
Aptamer-MACS recovery of cells from blood using an A431 concentration equal to 5% of the WBC concentration
A431 cells were labeled with DiI dye by incubating cells in serum-free DMEM with 50 mg/mL DiI (ThermoFisher) at a concentration of 1×106cells/mL for 20 minutes at 37°C, then washing three times with serum-free DMEM. Human whole blood was collected according to a protocol approved by the institutional review board of the Duke University Medical Center. Blood was collected in sodium citrate-treated vacutainer tubes (BD Biosciences), and, for most samples, a Heska HemaTrue used to determine the CBC and get a WBC. DiI-stained A431 cells were added to the blood at a concentration equal to 5% of the determined WBC or at a concentration of ~375,000 cells/mL (5% of median WBC). A 1x dilution of the eBioscience 10x RBC Lysis Buffer was then used to lyse RBCs.
RBC lysed, A431-spiked blood samples were incubated with aptamer-beads (500 μL spiked-blood per 40 μL beads) at 4°C with gentle rotation for 30 minutes. The beads and blood samples were maintained at 4°C and pulled down via MACS before 3 additional magnetic pull downs to wash with 500 μL of Media+ per 40 μL bead sample. Washed beads were resuspended in Media+ at a bead concentration of 2.5 μg/μL. 100-fold molar excess of the specific antidote mA9 or scrambled antidote sA9 was added to detach cells from the beads by incubating at 37°C with gentle rotation for 10 minu tes. Isolated cells were then recovered from the supernatant after magnetic pull down of the beads. The beads were magnetically pulled down 3 additional times and washed with Media+ (1 mL of Media+ per 40 μL bead sample), and the supernatant from each wash combined to with the initial collected supernatant. Recovered cell samples were resuspended in 100 μL of flow buffer (PBS + 1% BSA), and the cells counted and analyzed using a CytoFLEX flow cytometer (Beckman-Coulter). A431 recovery was determined by counting the number of Di positive cell events per sample.
Biotinylated aptamer transcription
E07 and C36 with 5’-biotinylated eight-nucleotide tail extensions (btE07 or btC36) were transcribed to enable conjugation to AF488 or AF647-labeled streptavidin (AF488-SA or AF647-SA; Thermo Fisher). The nucleotide extension was designed to ensure that the predicted secondary structure of E07 (Mfold software) was not impacted (Fig. S1B). Double-stranded DNA (dsDNA) templates for both btE07 and btC36 containing T7 RNA polymerase (RNAP) promoters were generated by annealing purchased primers (Integrated DNA Technologies, IDT) and using Klenow Fragment (NEB) to fill in the single-stranded overhangs. The dsDNA templates for tbE07 and tbC36 were subsequently transcribed using Y639F mutant T7 RNAP along with 2’F pyrimidine and 2’OH purine nucleotides doped with a 10-fold molar excess of 5’-biotin-G-monophosphate (TriLink Biotechnologies). Aptamers were purified on denaturing 12% polyacrylamide gels, extracted overnight at 4°C int o TE, pH 7.5, desalted in Amicon Ultra 10kD spin columns (Millipore) and quantitated via absorbance at 260nm on a NanoDrop.
Aptamer-streptavidin complex preparation
Biotinylated-aptamer-SA-dye complexes were prepared by first folding btE07 or btC36 aptamers in PBS+/+ by denaturation at 65°C for 5 mi nutes followed by passive cooling to ambient temperature for 3 minutes. Folded btE07 or btC36 were then incubated with AF488-SA or AF647-SA at a 2:1 molar ratio (RNA:SA-dye) in PBS+/+ for 20 minutes at room temperature. The conjugates were next resuspended in DMEM+ with 1 mg/mL ssDNA and incubated for another 20 min.
Staining Cells with Dye-SA-Aptamers or EGFR Abs for FACS
Experiments utilized A431 cells at 75–90% confluency that had been plated 48–72 hours in advance. Trypsin was used to harvest cells and immediately neutralized with growth media upon cell detachment. Cells were washed once with Media+, counted with a hemocytometer, and then 5×105 cells per sample were partitioned into chilled 1.5 mL microcentrifuge tubes. Staining was performed by resuspending cells in 100μL of the appropriate antibody- or aptamer-staining solution (5 million cells per mL). Cells were stained with 500 nM of Alexa Fluor-SA-aptamer solutions in DMEM+ with 1 mg/mL salmon sperm DNA or with 1:50 dilutions of antibodies in Media+ with 1 mg/mL salmon sperm DNA and incubated for 30 min at 4°C. For flow cytometry studies, a FITC-conjugated ICR10 (Abcam) monoclonal EGFR antibody was used for staining. For EGFR functional assays involving FACS sorting followed by western blots, cells were stained with a PE-labeled version of the EGFR neutralizing antibody D1D4J (Cell Signaling Technologies). Cells were then washed once with Media+ and either analyzed by flow cytometry or subsequently used for FACS sorting.
Fluorescence activated cell sorting (FACS)
Alexa Fluor 488-SA-E07, Alexa Fluor 488-SA-C36 or EGFR Ab stained samples (or unstained samples for mock sorting) were filtered through 40 μm cell strainers, resuspended to 106 cells/mL and kept on ice prior to sorting. Sorting was performed at 30 psi and a rate of 5×103 cells per second on a BD DiVa (BD Biosciences) utilizing refrigeration to maintain both the sample and collection tubes at 4°C. Collection tubes contained 3 mL Media+ and approximately 1mL sheath fluid (PBS without Ca2+ or Mg2+) was added per 0.5 million sorted cells. Sorting-associated losses were assessed by independently analyzing samples immediately prior to and after sorting on a BD FACSCalibur or BD FACSCanto II (BD Biosciences).
Destaining FACS sorted cells
Stained cells maintained on ice were resuspended in 100 μL Media+ with or without 2’OMe RNA antidote mA9 or 2’OMe scrambled control antidote sA9 at indicated concentrations and incubated in a water bath at 37°C for durations up to 10 minutes. Immediately after destaining, samples were placed back on ice. Cells were then washed once with Media+ and either analyzed by flow cytometry or subsequently used in E07 restaining assays or EGF stimulation assays.
Cell viability assays
Viability of cells that had been stained, sorted and then destained was evaluated by using a LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (ThermoFisher) according to the instructions of the manufacturer. Treated samples were then analyzed by flow cytometry on a BD FACSCanto II flow cytometer (BD Biosciences). Unstained sorted cells that had been heated at 65°C for 20 minutes served as a positive (dead cell) control for gating.
Restaining Cells with Dye-SA-Aptamer Complexes
Sorted cells that had been destained with antidote, or control cells that were not destained, were resuspended in 100uL of 500nM AF647-SA-E07 (5 million cells per mL) in DMEM+ with 1 mg/mL salmon sperm DNA and incubated for 30 min at 4°C. Cells were then washed once with Media+ and analyzed by flow cytometry.
Flow cytometry analysis of stained cells
Alexa Fluor-SA-E07 or Ab stained cells reversed for flow cytometry analysis were resuspended in FACS buffer (PBS+/+ with 0.1% BSA) and analyzed on BD FACSCalibur or BD FACSCanto II flow cytometers (BD Biosciences).
EGF stimulation assays and western blotting
Mock sorted or sorted, destained cells were stimulated with 5 nM EGF in Media+ for 15 minutes on ice and then lysed by resuspension in radioimmuniprecipitation (RIPA) buffer containing phosphatase and protease inhibitor cocktails (ThermoFisher). Lysate was either used immediately or frozen at −80°C until use. The amoun t of protein in crude lysate was quantified using a bicinchronic acid (BCA) assay kit (ThermoFisher) according to the instructions of the manufacturer. Crude lysate samples containing equal amounts of protein were prepared in Laemmli sample buffer containing 10% β-mercaptoethanol, boiled for 5 minutes at 95°C and then run on 4–15% denaturing polyacrylamide gels (Bio-Rad) in Tris/glycine/SDS buffer at 300V. Electrophoresed samples were blotted onto low-fluoresence polyvinylidene (PVDF) membranes with the Trans-Blot Turbo Transfer System and Transfer Packs (Bio-Rad) using the default setting for transfer of high molecular weight proteins.
Membranes were blocked for 1 hour at room temperature with PBS containing 0.05% Tween 20 (PBST) and 5% bovine serum albumin (blocking buffer). Blocked membranes were incubated overnight at 4°C in blocking buffer containing 1:1000 dilutions of primary monoclonal antibodies for total EGFR (mouse anti-human; Cell Signaling Technologies 2239S) and pEGFR (rabbit anti-human; Cell Signaling Technologies 4407S). Membranes were next washed three times for 5 minutes with PBST on a rocker and then incubated with blocking buffer containing fluorophore-conjugated secondary antibodies for detection of total EGFR (1:10,000 dilution of goat anti-mouse AF546 conjugate; Life Technologies A11030) and pEGFR (1:20,000 dilution of goat anti-rabbit AF647 conjugate; Jackson Labs 111-605-144) in separate fluorescent channels without spectral overlap. Blots were washed three times for 5 minutes with PBST on a rocker and then imaged on a Chemidoc MP equipped with green and red LEDs that enabled fluorescent multiplexing (Bio- Rad). Bands corresponding to total EGFR and pEGFR were manually identified using rectangular volumes in Image Lab software (Bio-Rad), and the amount of each protein was taken as the background-adjusted intensity of each specified volume. The pEGFR/total EGFR ratio for each sample was normalized to the average ratio for stimulated, unstained cells, which served as the positive control for native, uninhibited stimulation.
Cell Viability Assays
Viability of unfixed cells that had been stained, sorted and then destained was evaluated by using a LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit according to the instructions of the manufacturer. Treated samples were then analyzed by flow cytometry. Unstained sorted cells that had been heated at 65°C for 20 min served as a positive (dead cell) control for gating.
EGFR Antibody Reversal with the EGFR extracellular domain
Antibody staining solutions contained a 1:50 dilution of PE-labeled EGFR neutralizing antibody D1D4J (Cell Signaling Technologies) in Media+. To stain cells, 100,000 A431 cells per sample were partitioned into chilled 1.5 mL microcentrifuge tubes and incubated with 100μL of antibody staining solutions +/− 10-fold excess EGFR extracellular domain for 30 min at 4°C. Cells were then placed at 37°C for 10min or 6hrs be fore washing once with 100μL of Media+ and analyzing by flow cytometry on a CytoFLEX (Beckman-Coulter).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed by one-way analysis of variance (ANOVA) and the Tukey-Kramer post hoc test in JMP software to establish significant differences between experimental conditions where appropriate, with significance set at p < 0.05.
DATA AND SOFTWARE AVAILABILITY
This study did not generate datasets.
Supplementary Material
Highlights.
Aptamers can be used to reversibly label and purify cells via FACS or MACS.
EGFR aptamer-magnetic bead MACS isolates EGFR+ cells out of complex cell mixtures.
An oligonucleotide “antidote” strips off aptamers to recover label-free cells.
EGFR aptamer FACS sorting and antidote removal restores native receptor function.
Acknowledgements
We thank Matt Levy’s lab, formerly at Albert Einstein College of Medicine, for use of their Expedite 8909 DNA synthesizer for aptamer synthesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Duke University has applied for a patent on this technology and has optioned the intellectual property to the Duke spin-out company Upstream Therapeutics. Dr. Sullenger is a founder and shareholder in Upstream Therapeutics.
Supplemental Information
See attached Supporting Information file.
References
- Avutu V (2010) Avidity effects of MinE07, an anti-EGFR aptamer, on binding to A431 cells. The University of Texas at Austin Texas ScholarWorks, https://repositories.lib.utexas.edu/handle/2152/13407. [Google Scholar]
- Bompiani KM, Monroe DM, Church FC, and Sullenger BA (2012). A high affinity, antidote-controllable prothrombin and thrombin-binding RNA aptamer inhibits thrombin generation and thrombin activity. J. Thromb. Haemost 10, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner WA, Hulett HR, Sweet RG, and Herzenberg LA (1972). Fluorescence Activated Cell Sorting. Rev. Sci. Instrum 43, 404–409. [DOI] [PubMed] [Google Scholar]
- Champlin RE, Passweg JR, Zhang M-J, Rowlings PA, Pelz CJ, Atkinson KA, Barrett AJ, Chan J-Y, Drobyski WR, Gale RP, et al. (2000). T-cell depletion of bone marrow transplants for leukemia from donors other than HLA-identical siblings: advantage of T-cell antibodies with narrow specificities. Blood 95, 3996–4003. [PubMed] [Google Scholar]
- Chan MY, Cohen MG, Dyke CK Myles SK, Aberle LG, Lin M Walder J, Steinhubl SR, Gilchrist IC, Kleiman NS, et al. (2008). Phase 1b Randomized Study of Antidote-Controlled Modulation of Factor IXa Activity in Patients With Stable Coronary Artery Disease. Circulation 117, 2865–2874. [DOI] [PubMed] [Google Scholar]
- Copelan EA (2006). Hematopoietic stem-cell transplantation. NEJM 354, 1813–1826. [DOI] [PubMed] [Google Scholar]
- Dyke CK, Steinhubl SR, Kleiman NS, Cannon RO, Aberle LG, Lin M, Myles SK, Melloni C, Harrington RA, Alexander JH, et al. (2006). First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation 114, 2490–2497. [DOI] [PubMed] [Google Scholar]
- Ellington AD and Szostak JW (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Faraghat SA, Hoettges KF, Steinbach MK, van der Veen DR, Brackenbury WJ, Henslee EA, Labeed FH, and Hughes MP (2017). High-throughput, low-loss, low-cost, and label-free cell separation using electrophysiology-activated cell enrichment. PNAS 114, 4591–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CY, Peh GSL, Gauthaman K and Bongso A (2009). Separation of SSEA-4 and TRA-1–60 Labelled Undifferentiated Human Embryonic Stem Cells from A Heterogeneous Cell Population Using Magnetic-Activated Cell Sorting (MACS) and Fluorescence-Activated Cell Sorting (FACS). Stem Cell Rev. and Rep. 5, 72–80. [DOI] [PubMed] [Google Scholar]
- Gunaratne R, Kumar S, Frederiksen JW, Stayrook S, Lohrmann JL, Perry K, Bompiani KM, Chabata CV, Thalji NK, Ho MD, et al. (2018). Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat. Biotechnol 36, 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgretinger R (2012). Negative depletion of CD3+ and TcRαβ+ T cells. Curr. Opin. Hematol 19, 434–439. [DOI] [PubMed] [Google Scholar]
- Handgretinger R, Lang P, Schumm M, Taylor G, Neu S, Koscielnak E, Niethammer D, and Klingebiel T (1998). Isolation and transplantation of autologous peripheral CD34+ progenitor cells highly purified by magnetic-activated cell sorting. Bone Marrow Transplantat. 21, 987–993. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M, and Herzenberg LA (2002). The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin. Chem 48, 1819–1827. [PubMed] [Google Scholar]
- Heslop HE, Ng CYC, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, and Rooney CM (1996). Long–term restoration of immunity against Epstein–Barr virus infection by adoptive transfer of gene–modified virus–specific T lymphocytes. Nat. Med 2, 551–555. [DOI] [PubMed] [Google Scholar]
- Holliger P and Hudson PJ (2005). Engineered antibody fragments and the rise of single domains. Nat. Biotechnol 23, 1126–1136. [DOI] [PubMed] [Google Scholar]
- Hwang WYK and Foote J (2005). Immunogenicity of engineered antibodies. Methods 36, 3–10. [DOI] [PubMed] [Google Scholar]
- June CH (2007). Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest 117, 1466–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacherovsky N, Cardle II, Cheng EL, Yu JL, Baldwin ML, Salipante SJ, Jensen MC, and Pun SH (2019). Traceless aptamer-mediated isolation of CD8(+) T cells for chimeric antigen receptor T-cell therapy. Nat. Biomed. Eng 3, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratschmer C and Levy M (2017). Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid Ther. 27, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratschmer C and Levy M (2018) Targeted Delivery of Auristatin-Modified Toxins to Pancreatic Cancer Using Aptamers. Mol. Ther. Nucleic Acids 10, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Nguyen HH, Byrom M, and Ellington AD (2011). Inhibition of cell proliferation by an anti-EGFR aptamer. PLoS ONE 6, e20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli F, Bauquet A, Palumbo G, Moretta F, and Bertaina A (2013). Negative depletion of α/β+ T cells and of CD19+ B lymphocytes: A novel frontier to optimize the effect of innate immunity in HLA-mismatched hematopoietic stem cell transplantation. Immunol. Lett 155, 21–23. [DOI] [PubMed] [Google Scholar]
- Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, and Andreesen R (2006). Phase I Study of Adoptive T-Cell Therapy Using Antigen-Specific CD8+ T Cells for the Treatment of Patients With Metastatic Melanoma. J. Clin. Oncol 24, 5060–5069. [DOI] [PubMed] [Google Scholar]
- Masui H, Castro L, Mendelsohn J (1993) Consumption of EGF by A431 cells: evidence for receptor recycling. J. Cell Biol. 120, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattanovich D and Borth N (2006). Applications of cell sorting in biotechnology. Microb. Cell Fact. 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltenyi S, Müller W, Weichel W, and Radbruch A (1990). High gradient magnetic cell separation with MACS. Cytometry 11, 231–238. [DOI] [PubMed] [Google Scholar]
- Nimjee SM, Dornbos D 3rd, Pitoc GA, Wheeler DG, Layzer JM, Venetos N, Huttinger A, Talentino SE, Musgrave NJ, Moody H, et al. (2019). Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol. Ther 27, 1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimjee SM, White RR, Becker RC, and Sullenger BA (2017). Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol 57, 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oney S, Lam RT, Bompiani KM, Blake CM, Quick G, Heidel JD, Liu JY, Mack BC, Davis ME, Leong KW, and Sullenger BA (2009) Development of universal antidotes to control aptamer activity. Nat. Med 15, 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR and Greenberg PD (1995). Principles for Adoptive T Cell Therapy of Human Viral Diseases. Annu. Rev. Immunol 13, 545–586. [DOI] [PubMed] [Google Scholar]
- Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G Jr., Scardino E, Fay WP, and Sullenger BA (2004) Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol 22, 1423–1428. [DOI] [PubMed] [Google Scholar]
- Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TA, Monroe D, and Sullenger BA (2002). RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419, 90–94. [DOI] [PubMed] [Google Scholar]
- Sako Y, Minoghchi S, Yanagida T (2000). Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2, 168–172. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich RK, Mikkelsen RB, Dent P, Todd DG, Valerie K, Kavanagh BD, Contessa JN, Rorrer WK, Chen PB (1997). Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene 15, 1191–1197. [DOI] [PubMed] [Google Scholar]
- Shangguan D, Bing T and Zhang N (2015). Cell-SELEX: Aptamer Selection Against Whole Cells. In Aptamers Selected by Cell-SELEX for Theranostics, Tan W and Fang X, eds. (Springer; ), pp. 13–33. [Google Scholar]
- Soule EE, Bompiani KM, Woodruff RS, and Sullenger BA, (2016). Targeting Two Coagulation Cascade Proteases with a Bivalent Aptamer Yields a Potent and Antidote-Controllable Anticoagulant. Nucleic Acid Ther. 26, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell Gray B, Kelly L, Ahrens DP, Barry AP, Kratschmer C, Levy M, and Sullenger BA (2018) Tunable cytotoxic aptamer-drug conjugates for the treatment of prostate cancer. PNAS 115, 4761–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutermaster BA and Darling EM (2019). Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Sci. Rep 9, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M and Nikolic B (2005). Treatment of severe autoimmune disease by stem-cell transplantation. Nature 435, 620–627. [DOI] [PubMed] [Google Scholar]
- Tomlinson MJ, Tomlinson S, Yang XB, and Kirkham J (2012). Cell separation: Terminology and practical considerations. J. Tissue Eng. 4, 204173141247269–204173141247214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C and Gold L (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Wan Y, Kim Y-t., Li N, Cho SK, Bachoo R, Ellington AD, and Iqbal SM (2010). Surface-Immobilized Aptamers for Cancer Cell Isolation and Microscopic Cytology. Cancer Res. 70, 9371–9380. [DOI] [PubMed] [Google Scholar]
- Wan Y, Liu Y, Allen PB, Asghar W, Mahmood MAI, Tan J, Duhon H, Kim Y-t., Avutu V, and Iqbal SM (2012). Capture, isolation and release of cancer cells with aptamer-functionalized glass bead array. Lab Chip 12, 4693–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Mahmood MAI, Li N, Allen PB, Kim Y-t., Bachoo R, Avutu V, and Iqbal SM (2012) Nanotextured substrates with immobilized aptamers for cancer cell isolation and cytology. Cancer 118, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Tan J, Asghar W, Kim Y-t., Liu Y, and Iqbal SM (2011). Velocity Effect on Aptamer-Based Circulating Tumor Cell Isolation in Microfluidic Devices. J. Phys. Chem. B 115, 13891–13896. [DOI] [PubMed] [Google Scholar]
- Wang L, Zheng Q, Zhang Q, Xu H, Tong J, Zhu C, and Wan Y (2012). Detection of single tumor cell resistance with aptamer biochip. Oncol. Lett 4, 935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilner SE, Wengerter B, Maier K, de Lourdes Borba Magalhães M, Del Amo DS, Pai S, Opazo F, Rizzoli SO, Yan A, and Levy M (2012). An RNA Alternative to Human Transferrin: A New Tool for Targeting Human Cells. Mol. Ther. Nucleic Acids 1, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan A and Levy M (2014). Cell Internalization SELEX: In Vitro Selection for Molecules That Internalize into Cells. Methods Mol. Biol 1103, 241–265. [DOI] [PubMed] [Google Scholar]
- Yan AC and Levy M (2018). Aptamer-Mediated Delivery and Cell-Targeting Aptamers: Room for Improvement. Nucleic Acid Therapeutics 28, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamay AS, Zamay GS, Kolovskaya OS, Zamay TN and Berezovski MV (2017). Aptamer-Based Methods for Detection of Circulating Tumor Cells and Their Potential for Personalized Diagnostics. Adv. Exp. Med. Biol 994, 67–81. [DOI] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acid Ther. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets.