

Abstract
Background & objectives:
Oral squamous cell carcinoma is one of the most lethal forms of cancer, and its aetiology has been attributed to both genetic and epigenetic factors working in liaison to contribute to the disease. Epigenetic changes especially DNA methylation is involved in the activation or repression of gene functions. The aim of this study was to investigate the DNA methylation pattern and expression profiling of the promoter regions of FMS-related tyrosine kinase 3 (FLT3), erythrocyte membrane protein band 4.1-like 3 (EPB41L3) and stratifin (SFN) genes in oral cancer within the Khasi and Jaintia tribal population of Meghalaya in North East India.
Methods:
Quantitative methylation analyses of the selected genes were carried out by MassARRAY platform System, and the relative expression profiling was carried out by real-time polymerase chain reaction.
Results:
Quantitative methylation results indicated that the level of methylation was significantly higher (hypermethylated) for FLT3 and EPB41L3 and significantly lower (hypomethylated) for SFN in tumour tissues as compared to the adjacent paired normal tissue. Expression profiling was in concurrence with the methylation data whereby hypermethylated genes showed low mRNA level and vice versa for the hypomethylated gene.
Interpretation & conclusions:
The findings show that hyper- and hypomethylation of the selected genes play a potential role in oral carcinogenesis in the selected Khasi and Jaintia tribal population of Meghalaya. The methylation status of these genes has not been reported in oral cancer, so these genes may serve as promising biomarkers for oral cancer diagnosis as well as in disease monitoring.
Keywords: DNA methylation, ethnic population, gene expression, hypermethylation, hypomethylation, oral cancer
Oral squamous cell carcinoma (OSCC) or oral cancer is the sixth most common form1 and one of the most widespread and highly aggressive types of cancer2. It has an annual estimated incidence of over 500,000 cases worldwide1, and in India, as per the Global Cancer Statistics 2018 the incidence of new cases of cancer of the lip and oral cavity is about 119,9923. A case study carried out in India in 2010 on cancer mortality rate revealed that the cancer of the oral cavity was the third most common cancer among women, with the highest incidence of mortality among men. The study also showed that the highest incidence of cancer occur mainly in the northeastern parts of India4. As per the Cancer Atlas of India, Meghalaya has a high rate of cancer of the oral cavity, oropharynx, hypopharynx and oesophageal cancer5. In Meghalaya, a retrospective study carried out in Shillong Civil Hospital from 2007 to 2011 on the prevalence of head and neck cancer within the State showed that oropharyngeal and oral cancer were the most common in this region6.
The high rate of oral cancer in Meghalaya has been attributed to the lifestyle habits of the population. The involvement of betel quid has been shown in several types of cancer including oral cancer7,8.
The development of oral cancer involves many factors working together in synergy to bring about alterations in two large groups of genes, viz., oncogenes and tumour suppressor genes (TSGs). These alterations bring about molecular changes in both genetic and epigenetic levels leading to the diseased condition9. Epigenetic modifications such as DNA methylation (hyper- and hypomethylation) of a specific gene(s) is known to be one of the factors involved in the development and progression of oral cancer10. DNA hypermethylation of promoter region(s) of specific TSGs is a critical step in oral carcinogenesis11, and several genes hypermethylated in OSCC are known to cover a wide range of cellular processes2. Global genomic hypomethylation of repetitive DNA elements and oncogenes is a common feature in many malignancies, and their activation has been known to trigger genomic instability12.
A whole epigenomic study carried out by us revealed 45 differentially methylated genes13, and the present study was focused on the study of the methylation pattern of three selected genes, viz., FMS-related tyrosine kinase 3 (FLT3), erythrocyte membrane protein band 4.1-like 3 (EPB41L3) and stratifin (SFN) from those 45 genes, in patients with oral cancer in the Khasi and Jaintia tribal population of Meghalaya, India. Several studies have shown a disparity in the level of DNA methylation of specific functional gene and their altered expression based on race and ethnicity14,15,16, the present study may provide an insight on how promoter-associated CpG island methylation of specific functional genes is involved in the aetiology of oral cancer within this population.
Material & Methods
Surgically resected tumour and adjacent paired normal samples (at least 2 cm from the site of the primary tumour) were collected from the department of ENT, North Eastern Indira Gandhi Regional Institute of Health and Medical Sciences (NEIGRIHMS), Shillong, Meghalaya, India, from April 2013 to February 2014. Samples were collected from the ethnic tribal patients of Meghalaya i.e., the Khasis and Jaintias. The samples were flash-frozen in liquid nitrogen and stored at −80°C until further requirement. The tumour tissues were histopathologically confirmed for cancer cells. All participants provided written informed consent. The research protocol was approved by the Institutional Ethics Committee of North Eastern Hill University, Shillong, Meghalaya (IEC/13412).
DNA extraction and MassARRAY: A total of 17 pairs of tumour and paired adjacent normal tissues were used for methylation profiling by MassARRAY. Quantitative methylation MassArray EpiTyper (EpiTYPER® Agena Bioscience, CA, USA)13 was carried out for the selected genes, viz., FLT3, EPB41L3 and SFN for methylation analysis of its promoter region-associated CpG island. This bisulphite treatment-based method for detection and quantitation of DNA methylation uses base-specific cleavage and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) to differentiate between methylated and non-methylated DNA. All primers were designed using MethPrimer17 within the identified CpG islands (Table I). The details of procedure have been described elsewhere13.
Table I.
List of primers for the selected sets of genes used in the MassARRAY® EpiTYPER
| Amplicon name | Primer sequence | Amplicon size (bp) | Amplicon location relative to TSS | Total number of CpGs | |
|---|---|---|---|---|---|
| FLT3_CpG_1 | F | TATTTTTAAGAGAGTTATTTGTAG | 541 | −37 to +503 bp | 75 |
| R | AAAAAAAATCCTTAACCACCTAAC | ||||
| EPB41L3_CpG_1 | F | TTTATGTAATTGTTTTGAAGTATTG | 288 | −199 to +88 bp | 32 |
| R | TTACCTAAAATCAACAAAAAACCC | ||||
| SFN_CpG_2 | F | GGATATGGTAGTTTTTATGAAAGG | 259 | +111 to +369 bp | 15 |
| R | ATAACTATCCAACAAACCCAACAC |
TSS, transcription start site; FLT3, FMS-related tyrosine kinase 3; EPB41L3, erythrocyte membrane protein band 4.1-like 3; SFN, stratifin; bp, base pair; F, forward; R, reverse
RNA extraction and RT-PCR: RT-PCR was carried out for all three genes (FLT3, EPB41L3 and SFN) using β-actin as an endogenous control. Total RNA was extracted from tumour and paired adjacent normal tissues as described eleswhere13. mRNA sequence for the selected genes was retrieved from PubMed gene database (https://www.ncbi.nlm.nih.gov/pubmed/), and gene-specific primers were designed using Primer3web v4.0.0 (http://primer3.ut.ee/)1 and synthesized by Integrated DNA Technologies, Iowa, USA (Table II). Quantitative RT-PCR was performed and relative expression was calculated using the comparative delta Ct method13.
Table II.
List of primers for quantitative real-time polymerase chain reaction
| Gene | Primer sequence | Amplicon size (bp) | |
|---|---|---|---|
| EPB41L3 (NM_012307.3) | F | AGCAGTAAACTCTCTCGGTCT | 112 |
| R | AGCGTTTCTCTACATCACAGG | ||
| FLT3 (NM_004119.2) | F | TCAGTGGCAAGAAACGACAC | 90 |
| R | TCCCTTTTCTACGATGGTAACC | ||
| SFN (NM_006142.3) | F | TTGTGGCTGAGAACTGGACA | 113 |
| R | TGCTTTCCCTCAATCTCGGT | ||
| ACTB (NM_001101) | F | TCTACAATGAGCTGCGTGTG | 110 |
| R | GGTCTCAAACATGATCTGGGT |
ACTB, β-actin
Statistical analysis: Kolmogorov-Smirnov test was performed over the raw data of methylation and expression assay to check whether the data are normally distributed or not. Data were not normally distributed and hence required a non-parametric statistical test. Taking into consideration the small sample size, the result of normal distribution and paired nature of the sample, the Wilcoxon signed rank test for paired analysis was chosen for data analysis. All statistical analysis was carried out in SPSS v20.0 (IBM Corp., Armonk, NY, USA).
Results
Methylation level of FLT3, EPB41L3 and SFN: MassARRAY platform was used for the methylation analysis of FLT3, EPB41L3 and SFN. Quantitative methylation analyses were primarily focused on the promoter region of the gene. Hence, one amplicon for each genes was identified taking into consideration SNPs, repeats, a maximum number of CpGs and their relative position with respect to the transcription start site (TSS).
The methylation level of the each amplicon for the three genes showed a significant difference in the average level of methylation in tumour samples as compared to that of adjacent normal tissue. FLT3 amplicon with 75 CpG sites analysed showed a significantly higher level of average methylation across 46 CpG sites (P <0.05, P<0.01 and P<0.001, n=17) in tumour sample as compared to that of adjacent paired normal tissue (Fig. 1). Twenty three sites could not be detected and six sites were not significant. Similarly, EPB41L3 amplicon with 32 CpG sites showed a significantly higher level of average methylation across 18 CpG sites (P<0.01 and P<0.001, n=17) in tumour sample as compared to that of adjacent paired normal tissues (Fig. 2). Fourteen sites could not be detected. The higher level of DNA methylation in tumour sample as compared to that of paired adjacent normal tissue gives indicated that the CpGs within the amplicon of FLT3 and EPB41L3 were hypermethylated.
Fig. 1.

Average methylation level of each CpG site in FMS related tyrosine kinase 3 (FLT3) in tumour and adjacent normal tissues (n=17) detected and undetected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis. P*<0.05, **<0.01, ***<0.001 compared to normal. Data are shown as mean±standard error of mean.
Fig. 2.
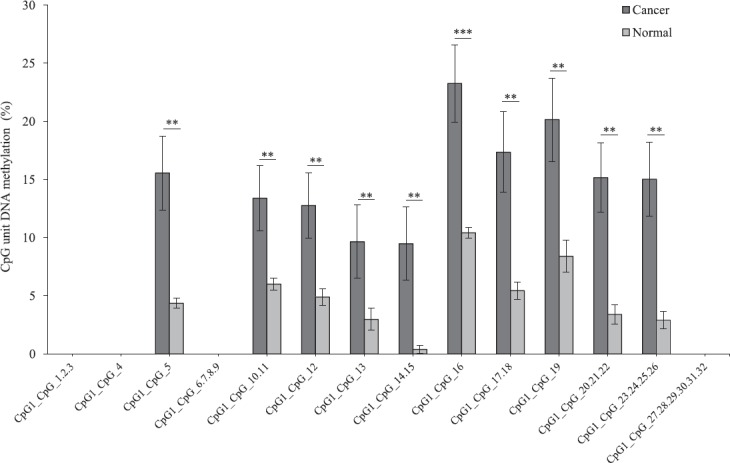
Average methylation level of each CpG site in erythrocyte membrane protein band 4.1-like 3 (EPB41L3) in tumour and adjacent normal tissues (n=17) detected and not detected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis. P*<0.05, **<0.01, ***<0.001 compared to normal. Data are shown as mean±standard error of mean.
In the case of SFN, of the 15 CpG sites analysed, the level of average methylation across 10 CpG sites was significantly lower in tumour sample as compared to that of adjacent paired normal tissues (P<0.05, n=17). Two sites could not be detected and three sites were NS (Fig. 3). This suggested that CpGs within the amplicon of SFN were hypomethylated.
Fig. 3.

Average methylation level of each CpG site in stratifin (SFN) in tumour and adjacent normal tissues (n=17) detected and undetected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis. *P<0.05 compared to normal. Data are shown as mean±standard error of mean.
Expression analysis by RT-PCR: Quantitative real-time PCR of FLT3, EPB41L3 and SFN were carried out on 12 paired samples. The expression of FLT3 and EPB41L3 showed a significant reduction in the level of mRNA expression in tumour sample as compared to that of adjacent paired normal tissues (P<0.01). There was an approximate 6-fold and 4-fold decrease in the level of mRNA expression relative to FLT3 and EPB41L3, respectively (Fig. 4). SFN, on the other hand, showed a significant increase in the level of mRNA expression in the tumour sample as compared to that of adjacent paired normal tissues (P<0.01, n=12). A 21-fold increase in the level of mRNA expression relative to SFN was observed. The level of mRNA expression seen in FLT3 and EPB41L3 was in conjunction with the methylation data as hypermethylation usually decreases the level of expression. Similarly, SFN level of mRNA expression was in concurrence with the methylation data as hypomethylation usually increases the level of expression (Fig. 4).
Fig. 4.

Average mRNA expression of FMS-related tyrosine kinase 3 (FLT3), erythrocyte membrane protein band 4.1 like-3 (EBP41L3) and stratifin (SFN). Real-time polymerase chain reaction was performed to identify the mRNA expression of all three genes in tumour and adjacent paired normal tissues (n=12). **P<0.01 compared to normal. Data are shown as mean±standard error of mean.
Discussion
Promoter hyper- or hypomethylation for genes, especially around the TSS has been known to play a critical role in cancer development, especially at an early stage of tumour development13. These early alterations lead to either loss of function of TSGs or gain in function of oncogenic genes and elements. Altered gene function results in loss of cell cycle control, up- or downregulation of gene transcription and altered cell-cell and cell-substratum interaction. The three genes (FLT3, EPB41L3 and SFN) showed their role with reference to cell function, and alteration in their functionality by methylation pattern may give rise to altered gene expressions and abnormal development.
FLT3 belongs to the receptor tyrosine kinase family and is important for the normal development of the haematopoietic and immune systems18. Many studies on FLT3 have been focused on the somatic alteration of the gene consisting of an internal tandem duplication of the JM (juxtamembrane) domain coding sequence in acute myeloid leukemia19. Studies carried out on the status of DNA methylation profiling of FLT3 gene and cancer showed a higher level of DNA methylation within the FLT3 gene in oesophageal cancer20 and pancreatic cancer21, respectively, indicating that the gene was hypermethylated. Tan et al21 reported a decrease in the level of mRNA expression in pancreatic cancer2, and treatment on HCC cell lines by 5-aza-dC showed re-expression of the methylated genes22. Underexpression of the FLT3 genes may contribute to the lack of sensitivity in certain tyrosine kinase target drugs, such as imatinib23. Kuo et al20 have reported that FLT3 may function as tumour suppressor-like genes in oesophageal squamous cell carcinoma (ESCC) tumourigenesis as hypermethylation of the gene showed an association with poor survival outcome. No literature was available for the methylation status of this gene in oral cancer.
The EPB41L3 gene encodes for an adhesion protein belonging to the 4.1 family of membrane-associated proteins that regulates cell growth9 and cell adhesion24. EPB41L3 promoter hypermethylation has been attributed to the cause of ovarian cancer25, cervical cancer24, diffuse gliomas9 and prostate cancer26. Hypermethylation of the EPB41L3 promoter is a common mechanism by which the gene is downregulated during tumour development and re-expression of EPB41L3 has been known to induce extensive apoptotic cell death25. EPB41L3 may play an important role in suppressing metastasis by regulating the proper arrangements of actin stress fibres and increasing the cell motility associated with metastatic behaviour27. Because of this feature, EPB41L3 may be considered as a tumour suppressor in ESCC27 and a candidate ovarian cancer-suppressor gene25. A study carried out by Perez-Janices et al9, showed that the expression of EPB41L3 in gliomas was low or completely absent in cases where the promoter region of the gene was hypermethylated. Several studies also showed that treatment of cell lines by 5-azadC restored transcription of the EPB41L3 gene9,24.
SFN belongs to the 14-3-3 family of acidic polypeptides. Capable of binding to more than 100 functionally diverse cellular proteins, these can play significant roles in different cellular functions such as cell cycle regulation, signal transduction, apoptosis, malignant transformation and cytoskeleton organization28. Overexpression of SFN has been reported in lung cancer29 and pancreatic cancer21. The overexpression of the SFN gene has been attributed to hypomethylation, as confirmed by Sato et al30 in pancreatic cancer.
Our study results were in concurrence with other published studies whereby promoter hyper- and hypomethylation of the functional genes in OSCC were shown to lead to their altered gene expressions. Our study also showed involvement of new and novel genes involved in OSCC, and this might further provide an additional insight on the existing molecular mechanism involved in the occurrence, development and progression of the disease.
Acknowledgment
Authors acknowledge Shri Shaiphyrnai Marnagar (Project Assisstant-DBT project) for providing support in laboratory maintenance, Dr Prashant Singh and the team at the Genomics Shared Resources, Rosewell Park Cancer Institute, Buffalo, New York, for providing all the technical support required for carrying out the MassArray EpiTyper experiment.
Footnotes
Financial support & sponsorship: Authors acknowledge the funding support provided by the Department of Biotechnology (DBT), New Delhi, in the form of financial support (BT/PR5546/MED/12/560/2012 dated 09/09/2013 awarded to the last author (SG) and Council of Scientific & Industrial Research (CSIR), New Delhi, for providing Senior Research Fellowship (CSIR-SRF) to the first author (SK)
Conflicts of Interest: None
References
- 1.Lingen M, Pinto A, Mendes R, Franchini R, Czerninski R, Tilakaratne W, et al. Genetics/epigenetics of oral premalignancy: Current status and future research. Oral Dis. 2011;17:7–22. doi: 10.1111/j.1601-0825.2011.01789.x. [DOI] [PubMed] [Google Scholar]
- 2.Mascolo M, Siano M, Ilardi G, Russo D, Merolla F, de Rosa G, et al. Epigenetic disregulation in oral cancer. Int J Mol Sci. 2012;13:2331–53. doi: 10.3390/ijms13022331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 4.Dikshit R, Gupta PC, Ramasundarahettige C, Gajalakshmi V, Aleksandrowicz L, Badwe R, et al. Cancer mortality in India: A nationally representative survey. Lancet. 2012;379:1807–16. doi: 10.1016/S0140-6736(12)60358-4. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Atlas India. [accessed on December 12, 2017]. Available from: http://www.canceratlasindia.org/
- 6.Shunyu NB, Syiemlieh J. Prevalence of Head and Neck Cancer in the State of Meghalaya : Hospital-based Study. Int J Head Neck Surg. 2013;4:1–5. [Google Scholar]
- 7.Wary KK, Sharan RN. Aqueous extract of betel-nut of North-East India induces DNA-strand breaks and enhances rate of cell proliferation in vitro. J Cancer Res Clin Oncol. 1988;114:579–82. doi: 10.1007/BF00398180. [DOI] [PubMed] [Google Scholar]
- 8.Gasche J, Goel A. Epigenetic mechanisms in oral carcinogenesis. Futur Oncol. 2013;8:1407–25. doi: 10.2217/fon.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Janices N, Blanco-Luquin I, Tuñón MT, Barba-Ramos E, Ibáñez B, Zazpe-Cenoz I, et al. EPB41L3, TSP-1 and RASSF2 as new clinically relevant prognostic biomarkers in diffuse gliomas. Oncotarget. 2015;6:368–80. doi: 10.18632/oncotarget.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, et al. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res. 2002;8:3164–71. [PubMed] [Google Scholar]
- 11.Viet CT, Schmidt BL. Methylation array analysis of preoperative and postoperative salivaDNA in oral cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17:3603–11. doi: 10.1158/1055-9965.EPI-08-0507. [DOI] [PubMed] [Google Scholar]
- 12.Ting Hsiung D, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. Global DNA-methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16:108–14. doi: 10.1158/1055-9965.EPI-06-0636. [DOI] [PubMed] [Google Scholar]
- 13.Khongsti S, Lamare FA, Shunyu NB, Ghosh S, Maitra A, Ghosh S. Whole genome DNA methylation profiling of oral cancer in ethnic population of Meghalaya, North East India reveals novel genes. Genomics. 2017;2:112–23. doi: 10.1016/j.ygeno.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Piyathilake CJ, Bell WC, Jones J, Henao OL, Heimburger DC, Niveleau A, et al. Pattern of nonspecific (or global) DNA methylation in oral carcinogenesis. Head Neck. 2005;27:1061–7. doi: 10.1002/hed.20288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song MA, Brasky TM, Marian C, Weng DY, Taslim C, Dumitrescu RG, et al. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics. 2015;10:1177–87. doi: 10.1080/15592294.2015.1121362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011;6:623–9. doi: 10.4161/epi.6.5.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 18.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17:1738–52. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- 19.Noguera NI, Brecci a M, Divona M, Diverio D, Costa V, De Santis S, et al. Alterations of the FLT3 gene in acute promyelocytic leukemia: Association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia. 2002;16:2185–9. doi: 10.1038/sj.leu.2402723. [DOI] [PubMed] [Google Scholar]
- 20.Kuo IY, Chang JM, Jiang SS, Chen CH, Chang IS, Sheu BS, et al. Prognostic CpG methylation biomarkers identified by methylation array in esophageal squamous cell carcinoma patients. Int J Med Sci. 2014;11:779–87. doi: 10.7150/ijms.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan AC, Jimeno A, Lin SH, Wheelhouse J, Chan F, Solomon A, et al. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol Oncol. 2009;3:425–38. doi: 10.1016/j.molonc.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin SH, Kim BH, Jang JJ, Suh KS, Kang GH. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array. J Korean Med Sci. 2010;25:1152–9. doi: 10.3346/jkms.2010.25.8.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YC, Huang RL, Huang YK, Liao YP, Su PH, Wang HC, et al. Methylomics analysis identifies epigenetically silenced genes and implies an activation of β-catenin signaling in cervical cancer. Int J Cancer. 2014;135:117–27. doi: 10.1002/ijc.28658. [DOI] [PubMed] [Google Scholar]
- 24.Guerrero-Setas D, Pérez-Janices N, Blanco-Fernandez L, Ojer A, Cambra K, Berdasco M, et al. RASSF2 hypermethylation is present and related to shorter survival in squamous cervical cancer. Mod Pathol. 2013;26:1111–22. doi: 10.1038/modpathol.2013.32. [DOI] [PubMed] [Google Scholar]
- 25.Dafou D, Grun B, Sinclair J, Lawrenson K, Benjamin EC, Hogdall E, et al. Microcell-mediated chromosome transfer identifies EPB41L3 as a functional suppressor of epithelial ovarian cancers. Vol. 12. Neoplasia Neoplasia Press, Inc; 2010. pp. 579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulz WA, Alexa A, Jung V, Hader C, Hoffmann MJ, Yamanaka M, et al. Factor interaction analysis for chromosome 8 and DNA methylation alterations highlights innate immune response suppression and cytoskeletal changes in prostate cancer. Mol Cancer. 2007;6:1–16. doi: 10.1186/1476-4598-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng R, Huang JP, Li XF, Xiong WB, Wu G, Jiang ZJ, et al. Epb41l3 suppresses esophageal squamous cell carcinoma invasion and inhibits MMP2 and MMP9 expression. Cell Biochem Funct. 2016;34:133–41. doi: 10.1002/cbf.3170. [DOI] [PubMed] [Google Scholar]
- 28.Shiba-Ishii A, Noguchi M. Aberrant stratifin overexpression is regulated by tumor-associated CpG demethylation in lung adenocarcinoma. Am J Pathol. 2012;180:1653–62. doi: 10.1016/j.ajpath.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Shiba-Ishii A, Kano J, Morishita Y, Sato Y, Minami Y, Noguchi M. High expression of stratifin is a universal abnormality during the course of malignant progression of early-stage lung adenocarcinoma. Int J Cancer. 2011;129:2445–53. doi: 10.1002/ijc.25907. [DOI] [PubMed] [Google Scholar]
- 30.Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma frequent hypomethylation of multiple genes overexpressed in pancreatic. Cancer Res. 2003;63:4158–66. [PubMed] [Google Scholar]