


Abstract
Ribosomal protein S1 plays important roles in the translation initiation step of many Escherichia coli mRNAs, particularly those with weak Shine-Dalgarno sequences or structured 5′ UTRs, in addition to a variety of cellular processes beyond the ribosome. In all cases, the RNA-binding activity of S1 is a central feature of its function. While sequence determinants of S1 affinity and many elements of the interactions of S1 with simple secondary structures are known, mechanistic details of the protein's interactions with RNAs of more complex secondary and tertiary structure are less understood. Here, we investigate the interaction of S1 with the well-characterized H-type pseudoknot of a class-I translational preQ1 riboswitch as a highly structured RNA model whose conformation and structural dynamics can be tuned by the addition of ligands of varying binding affinity, particularly preQ1, guanine, and 2,6-diaminopurine. Combining biochemical and single molecule fluorescence approaches, we show that S1 preferentially interacts with the less folded form of the pseudoknot and promotes a dynamic, partially unfolded conformation. The ability of S1 to unfold the RNA is inversely correlated with the structural stability of the pseudoknot. These mechanistic insights delineate the scope and limitations of S1-chaperoned unfolding of structured RNAs.
INTRODUCTION
Ribosomal protein S1 has a well-established role in translation by the Escherichia coli ribosome wherein it facilitates the binding of many mRNAs, particularly those with weak Shine-Dalgarno (SD) sequences and those of highly structured 5′ untranslated regions (UTRs), by the 30S subunit (1–4). Protein S1 therefore is required for cell growth and viability (3,5). Despite its loose association with the ribosome, S1 is also known to have other cellular activities (6), including roles in trans-translation (7), transcriptional cycling (8), stimulation of T4 endoribonuclease RegB (9,10) and as a subunit of Qβ replicase (11–14). In each of these functions, the RNA binding activity of S1 is essential, yet still poorly understood.
S1 is a large protein composed of six imperfect repeats of an RNA binding domain known as an OB-fold (15) (Figure 1A). The first two N-terminal domain repeats are involved in binding of the ribosome (16) and thus indispensable for cell survival (5,17). They are also implicated in binding of both Qβ replicase (12,14,18) and tmRNA (19). The, C-terminal domains, and in particular the central three domain repeats are thought to bind mRNA, with the strongest evidence supporting the involvement of domains 3 and 4 (17,20,21). Structural studies have also shown that the C-terminal domains of S1, particularly domains 4 and 6, are involved in ribosome inactivation and hibernation under stress by mediating 70S ribosome dimerization (22). Lastly, domains 5 and 6 were found to be most important for the ability of S1 to stimulate transcription (8).
Figure 1.

Tte riboswitch as a model pseudoknot to study RNA-S1 interactions. (A) Diagram of protein S1 highlighting some of the known activities of the OB-fold domains. (B) Secondary structure of the Tte riboswitch pseudoknot showing a subset of the key tertiary interactions in the Leontis-Westhof nomenclature (59). Annotations in square brackets or parenthesis indicate modifications made to the RNA for fluorescent EMSA or smFRET experiments, respectively. (C) Simplified representation of the pseudoknot's secondary structure domains. (D) Chemical structures of ligands preQ1 (7-aminomethyl-7-deazaguanine), guanine (Gua), 2,6-diaminopurine (DAP) and adenine (Ade).
S1 can unwind double-stranded RNA (dsRNA) (23,24) and, while it was once thought that this unwinding activity was not needed for its role in translation (21), a growing body of work suggests otherwise (1,5,25). For instance, the translation initiation region of the rpsO mRNA contains a pseudoknot, the unfolding of which is promoted by S1 in vitro (5). Furthermore, even when the strength of the SD sequence is enhanced in rpsO mRNA, the unfolding (but not binding) of the mRNA, which is required for subsequent formation of the translation initiation complex, is strongly impaired when the 30S subunit is depleted of S1 (5).
Despite the functional importance of S1, studies of its mechanism in binding and unfolding RNA are still relatively few. Here, we have investigated the mechanistic details of the interaction between RNA and S1 using the well-characterized, highly structured pseudoknot from the translational Thermoanaerobacter tengcongensis (Tte) preQ1 riboswitch as a model RNA structural motif. Pseudoknots are a common type of mixed secondary and tertiary structure motif found in a wide variety of RNAs, often directly involved in their diverse functions (26) such as riboswitch-mediated regulation of translation initiation. The class-I preQ1 riboswitch, which has undergone extensive structural characterization by our lab and others (27–30), contains an aptamer domain comprised of a small, well-defined H-type pseudoknot that senses intracellular metabolites for gene regulatory effect (Figure 1B and C). A number of ligands and their corresponding affinities are known for this riboswitch, including preQ1, guanine (Gua) and 2,6-diaminopurine (DAP) (31) (Figure 1D). These ligands provide a convenient handle through which the stability of the pseudoknot can be easily modulated while maintaining the same global fold, without the need for making sequence-level changes that may have less predictable effects on the structure, or fundamentally change the nature of the interaction under study. For example, an RNA helix can be made more stable simply by changing the GC content, which however, could become problematic when studying phenomena that exhibit sequence dependence, as is the case here given the well-known preference of S1 for A/U-rich sequences (2).
We demonstrate that S1 binds the preQ1 riboswitch, wherein the binding interaction strongly depends on the structural stability of the pseudoknot, which we modulated by using ligands of different affinity that stabilize the folded RNA to varying extents. We show that S1 has considerable interactions with the pseudoknot that is folded (but still dynamic) in the absence of ligand, and that S1 destabilizes its secondary and tertiary structure; this is also the case in the presence of ligands that only moderately increase the folded RNA’s stability. In contrast, S1 cannot bind to the compact, stably folded form of the pseudoknot promoted by binding of a high-affinity ligand. These observations are further supported by single molecule fluorescence microscopy, where changes in conformation of the pseudoknot are monitored by changes in intramolecular single molecule fluorescence resonance energy transfer (smFRET) between two fluorophores that report on the RNA’s pseudoknot fold. Analysis of the smFRET data uncovered three distinct patterns of dynamic RNA folding behavior, two of which are strongly correlated with the presence of protein S1, showing that binding of S1 significantly unfolds the pseudoknot to form a dynamic RNA-protein complex.
MATERIALS AND METHODS
Expression and purification of E. coli ribosomal protein S1
A plasmid vector containing the rpsA gene, encoding the E. coli ribosomal protein S1, with a cleavable N-terminal His-tag was prepared by mutagenesis from the ASKA(–) clone JW0894 (National BioResource Project—E. coli at National Institute of Genetics) (32). Additional details for mutagenesis are provided in Supplementary Information. The final plasmid, pCA24N_6xHis_TEV_rpsA, is available through Addgene (www.addgene.org).
pCA24N_6xHis-TEV_rpsA was expressed in the BLR(DE3) strain of E. coli using conditions loosely based on those described by Lancaster et al. (33). A detailed description of the purification protocol appears in Supplementary Information. Briefly, 1 L of LB-Miller broth containing 68 μg/mL chloramphenicol was inoculated 1:500 from a saturated overnight culture and grown with shaking at 37°C, induced with 1 mM IPTG at an OD600 ∼0.6, and harvested 2 h post-induction. All subsequent steps were performed at 4°C or on ice. The cell pellet was lysed using a microfluidizer in 30 ml of buffer B (15 mM Tris–HCl [pH 7.05 at 25°C], 30 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol, 0.1 mM PMSF), and clarified by centrifugation. The clarified lysate was combined with 5 ml of Ni-NTA Agarose resin (Qiagen, 30210) pre-equilibrated in buffer B and incubated for ∼2.5 h. The resin was washed with 25 ml of buffer C (15 mM Tris–HCl [pH 7.05 at 25°C], 30 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol, 10 mM imidazole [pH 8.0]) with 500 mM NaCl to reduce the amount of co-purifying RNA, and then washed again with 25 ml of buffer C to remove excess Na+. Bound protein was eluted using buffer D (15 mM Tris–HCl [pH 7.05 at 25°C], 30 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol, 250 mM imidazole [pH 8.0]). Fractions containing significant amounts of 6× His-TEV-S1 were pooled and the concentration of 6× His-TEV-S1 was estimated from the A280 of the solution (ϵ280 = 48 930 M−1 cm−1, ExPASy ProtParam, Swiss Institute of Bioinformatics). The N-terminal His-tag was cleaved using TEV protease during overnight dialysis into buffer E (15 mM Tris–HCl [pH 7.05 at 25°C], 5 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol). The cleaved His-tag and TEV protease were separated from the S1 protein using 5 ml of Ni-NTA Agarose resin (Qiagen) pre-equilibrated in buffer E, and the S1-containing flow-through was loaded onto a 5 ml Q Sepharose Fast Flow anion exchange column (GE Healthcare, 17-0510-01), pre-equilibrated with buffer E. The S1 protein was eluted using a step-wise gradient of buffer F (15 mM Tris–HCl [pH 7.05 at 25°C], 600 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol) in buffer E. S1-containing fractions were pooled, concentrated using a centrifugal filtration device, and dialyzed into protein storage buffer A(10) (25 mM Tris–HCl [pH 7.05 at 22°C], 100 mM NH4Cl, 10 mM MgCl2, 10% [v/v] glycerol, 6 mM β-mercaptoethanol). The S1 concentration was measured after dialysis (ϵ280 = 47 440 M−1 cm−1), then snap frozen in aliquots and stored at –80°C. This final protein solution had a measured 260/280 absorbance ratio of 0.74, suggesting the absence of any co-purifying nucleic acid. Protein aliquots were removed for use and thawed on ice; aliquots were kept for up to one week stored at −20°C and then discarded.
Preparation of DNA templates for in vitro transcription
DNA oligonucleotides (5′-CCC TTG TTT TGT TAA CTG GGG TTA CTG CGA CCC AGG ACC TAT AGT GAG TCG TAT TAA ATT-3′; 5′-AAT TTA ATA CGA CTC ACT ATA GG-3′) designed to give a partially double-stranded template for transcription of the wild-type Tte pseudoknot were gel purified prior to use. Additional details of the gel purification protocol are provided in Supplementary Information.
Site-directed mutagenesis of the pUC19_TTE1564 plasmid (Addgene ID 61000), which carries the wild-type pseudoknot sequence (34), was performed to generate the P2-deletion mutant. The mutagenesis reaction was performed according to the procedure described in the Supplementary Information, using the mutagenesis primer 5′-AAC AAA ATG CTC ACC TGG GTT CGC CCA GTT AAC AAA ACA AGG-3′, with slight modifications (Supplementary Information). The resulting plasmid carrying the desired mutation (pUC19_TTE1564_UUCG-loop) is available through Addgene.
The template for in vitro transcription of the P2-deletion mutant was prepared by PCR from the pUC19_TTE1564_UUCG-loop plasmid using PAGE-purified DNA primers 5′-TTT CCC AGT CAC GAC GTT-3′ and 5′-GGG CAC AAA ATT ACC TC-3′. PCR cycling and purification conditions are provided in Supplementary Information.
RNA preparation for EMSA and melting curve studies
RNA pseudoknots were generated by in vitro transcription using T7 RNA polymerase. The wild-type Tte riboswitch pseudoknot was transcribed from the partially double-stranded oligonucleotide template described above. The complete sequence of the wild-type pseudoknot used in this study, with the exception of smFRET experiments (see below), is 5′-GGU CCU GGG UCG CAG UAA CCC CAG UUA ACA AAA CAA GGG-3′.
The P2-deletion mutant construct was similarly prepared by in vitro transcription, using the PCR product described above as the template. The sequence of the P2-deletion mutant is 5′-GGG CAG UGA GCA ACA AAA UGC UCA CCU GGG uuc gCC CAG UUA ACA AAA CAA GGG AGG UAA UUU UGU GCC C-3′, where lower case indicates the site of mutation. Additional details of the in vitro transcription reaction conditions and purification schemes are provided in Supplementary Information.
3′ Fluorophore labeling of RNA
RNAs prepared by transcription as described above were labeled with a Cy3 fluorophore at their 3′ end following a method described previously by Willkomm and Hartmann (35) with several modifications. Detailed protocols for labeling and purification are provided in Supplementary Information. In this labeling method, the 3′ end of the RNA is first oxidized using sodium (meta) periodate (Fluka, 71859) on ice, and then precipitated. The oxidized RNA was then coupled with a hydrazide derivative of the fluorophore Cy3 (GE Healthcare, PA13120), and then purified using a combination of ethanol precipitation and G-50 desalting spin columns. The final concentration of RNA in the recovered solution was determined spectrophotometrically using the extinction coefficient ϵ260 = 485 437 M−1 cm−1 for the RNA and ϵ550 = 150 000 M−1 cm−1 for Cy3. The labeling efficiency was determined to be ∼60%. The contribution of dye to the absorbance at 260 nm was accounted for as follows: A260, RNA = A260 – 0.08 × A550.
Ligand preparation
The preQ1 ligand used in this study was synthesized as described previously (36) and was generously provided by Prof. George Garcia at the University of Michigan. The concentrations of the preQ1 stock solutions were measured by UV–vis spectrophotometry using a Nanodrop2000 spectrophotometer. Sparingly soluble guanine (Gua) (Sigma, G6779), 2,6-diaminopurine (DAP) (Sigma, 247847) and adenine (Ade) (Sigma, A8626) were dissolved in milliQ water by vortexing and then sonicating for ∼20 min. Final concentrations were measured by UV–vis spectrophotometry using the following molar extinction coefficients: preQ1 at 256 nm (11 200 M−1 cm−1), Gua at 243 nm (10 700 M−1 cm−1), DAP at 280 nm (9050 M−1 cm−1), Ade at 261 nm (13 400 M−1 cm−1).
Electrophoretic mobility shift assays
Binding of S1 to the pseudoknot was assessed using electrophoretic mobility shift assays (EMSA), using conditions adapted from McGinness et al. (17). Assay conditions were converted to a minigel format (84 mm × 74 mm × 1.0 mm), using a Mini-PROTEAN® Tetra Cell apparatus (Bio-Rad). All solutions containing fluorophore-labeled RNA were protected from light. For titration experiments serial dilutions of S1 were prepared similarly in buffer A(10) (25 mM Tris–HCl [pH 7.05 at 22°C], 100 mM NH4Cl, 10 mM MgCl2, 10% [v/v] glycerol, 6 mM β-mercaptoethanol). A typical binding reaction was prepared as follows: 1 μl of 0.1 μM of 3′ Cy3-labeled RNA (0.1 pmol total) was combined with 6 μl of 1.67× binding buffer (16.7 mM Tris–HCl [pH 7.5 at 22°C], 167 mM NH4Cl, 8.3% (v/v) glycerol, 1.67 mM DTT) and 1 μl of milliQ water in a 0.2 ml PCR tube. The mixture was refolded by heating in a 90°C copper bead bath for 3 min and allowed to cool for at least 15 min at room temperature (refolded RNA solution). 1 μl each of 1 mg/ml BSA and 10× S1 dilution series were then added to the refolded RNA, mixed thoroughly, and incubated at room temperature for 30 min. For ligand titration experiments (preQ1, DAP, Gua), ligands were serially diluted in milliQ water at 10× the desired final concentration. 1 ul of 10× ligand dilution series was then combined with 1 μl of 0.1 μM of 3′ Cy3-labeled RNA and 6 μl of 1.67× binding buffer and refolded similarly. Then 1 μl each of 1 mg/ml BSA and buffer A(10) were added to the refolded RNA, mixed thoroughly and incubated at room temperature for 30 min. The buffer composition in the final reactions (excluding RNA, preQ1, adenine, and S1) was 12.5 mM Tris–HCl, 125 mM NH4Cl, 6% (v/v) glycerol, 100 μg/ml BSA and 1 mM MgCl2 in a final volume of 10 μl. In S1 competition experiments, 1 μl each of 1 mg/mL BSA and 2.5 μM S1 in buffer A(10) were added to the refolded RNA, mixed thoroughly, and incubated at room temperature for 30 min. Following the 30 min incubation, reactions were mixed with 10 μl of cold loading buffer (10 mM Tris–HCl [pH 7.5 at 22°C], 100 mM NH4Cl, 100 μg/mL BSA, 60% [v/v] glycerol, 0.03% [w/v] bromophenol blue, 1 mM DTT) and placed on ice. Samples were electrophoresed at 4°C, 12% native polyacrylamide gels in 1× TGE (25 mM Tris base, 190 mM glycine, 1 mM EDTA) that had been pre-run for ∼1 h at 50 V, and then run at 200 V until the bromophenol blue band was at the bottom edge of the gel (∼90 min for 12% gels). Gels were scanned in the glass plates on a Typhoon™ 9410 Variable Mode Imager (GE Healthcare) operating in Fluorescence mode, with 532 nm laser excitation, default emission filter set for Cy3 (580 BP 30) and 50 μm pixel size. The PMT voltage was adjusted to maximize signal without saturating the detector (typically 720–760 V). The fractions of bound and unbound RNA in resulting images were quantified in ImageQuant v5.2 (Molecular Dynamics).
5′ Radiolabeling of RNA
5′ Radiolabeling of RNA was achieved following a protocol from New England Biolabs with several modifications. Briefly, 2.5 μl (∼100 pmol) of RNA was combined with 1 μl (5 units) of Antarctic phosphatase (NEB, M0289), 2 μl of 10× Antarctic Phosphatase Reaction Buffer and 14.5 μl of milliQ water. The mixture was then incubated at 37°C for 30 min. After incubation, excess metal ions (Mg2+ and Zn2+) present in the reaction buffer were quenched by adding 0.264 μl of 125 mM EDTA to prevent metal induced degradation and the reaction was stopped by heat-inactivation at 80°C for 2 min. Then, 5 μl (∼25 pmol) of the dephosphorylated RNA was combined with 2 μl of [γ-32P] ATP (MP Biomedicals 0135001U; ∼6000 Ci/mmol, 10 mCi/ml), 1 μl of 10× polynucleotide kinase (PNK) buffer, 1 μl (20 units) of T4 PNK (NEB, M0201) and 1 μl milliQ water. The reaction mixture was incubated in 37°C for 30 min. The radiolabeled RNA was then purified twice with mini Quick Spin RNA Columns (Roche Diagnostics, 11814427001) to remove excess [γ-32P] ATP.
Binding assay for the estimation of dissociation equilibrium constants
Dissociation constants were determined by native EMSA experiments with [γ-32P] ATP end-labeled Tte riboswitch. For S1 binding experiments serial dilutions of S1 were prepared in buffer A(10). A typical binding reaction was prepared as follows: 0.5 μl of 100 times diluted radio-labeled RNA (trace amount) was combined with 6 μl of 1.67× binding buffer (16.7 mM Tris–HCl [pH 7.5 at 22°C], 167 mM NH4Cl, 8.3% (v/v) glycerol, 1.67 mM DTT) and 1.5 μl of milliQ water in a 0.2 ml PCR tube. The mixture was refolded by heating in a 90°C copper bead bath for 3 min and allowed to cool for at least 15 min at room temperature (refolded RNA solution). 1 μl each of 1 mg/ml BSA and 10× S1 dilution series were then added to the refolded RNA, mixed thoroughly, and incubated at room temperature for ∼20 min. Following the incubation, reactions were mixed with 10 μl of cold loading buffer (10 mM Tris–HCl [pH 7.5 at 22°C], 100 mM NH4Cl, 100 μg/mL BSA, 60% [v/v] glycerol, 0.03% [w/v] bromophenol blue, 1 mM DTT) and placed on ice. The samples were electrophoresed at 4°C on a 12% native polyacrylamide gel (18 cm × 14.5 cm × 1.5 mm) in 1× TGE that had been prerun for ∼1 h at 100 V, and run with sample at 18 mA for 3 h. The fraction of RNA bound was quantified by using an Amersham Typhoon PhosphorImager (GE lifesciences) and analyzed with ImageQuant software v5.2. The equilibrium dissociation constants for the two complexes were determined using a two-site binding model (37) where the fraction of RNA bound in complex 1 and 2 were fitted simultaneously using equations (1) and (2).
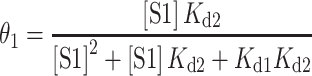 |
(1) |
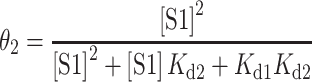 |
(2) |
Melting curve studies
Because Tris-based buffers are poorly suited for melting curve studies, melting experiments were performed using sodium phosphate as the buffering salt (38,39). Melting experiments were performed on a Beckman DU® 640B spectrophotometer fitted with a high performance temperature controller unit, transport accessory and Tm six-cell holder. A typical sample for melting curve analysis was prepared as follows: a solution of 0.6 μM RNA (no fluorophores) in 10 mM sodium phosphate (pH 7.0 at 22°C), 100 mM NH4Cl, and 1 mM MgCl2 was refolded by heating for 3 min in a 90°C copper bead bath, then transferred to a 70°C copper bead bath for 3 min, then transferred to a 60°C heating block for 3 min, and finally allowed to cool to room temperature over ∼1 h in the same heating block for RNA. 325 μl of refolded RNA solution was carefully transferred to each cuvette (Beckman, 523878) and tightly stoppered. The instrument was blanked with buffer without RNA, and absorbance at 260 nm was monitored with a 0.5 s read averaging time with the absorbance 340 or 320 nm used for background correction. Cuvettes were allowed to equilibrate in the instrument at 10°C for 15 min before the start of the discontinuous heating ramp. The cuvette holder was purged with a gentle of nitrogen during portions of the ramp <20°C to prevent condensation on the cuvette. The temperature was increased at a rate of 1°C/min between 10 and 22°C with a reading made every 1°C, then at a rate of 0.5°C/min between 22 and 75°C with a reading made every 0.5°C, then at a rate of 1°C/min between 75 and 95°C with a reading made every 1°C. For experiments in the presence of saturating ligands, 0.6 μM RNA was refolded with 3× molar excess of ligands over RNA in the same buffer and returned to the cuvette. The same temperature ramp program was then run. Analysis of the melting curve data was performed using custom scripts written in Matlab. For additional details of this analysis, please see Supplementary Information.
In-gel FRET electrophoretic mobility shift assay
Structural changes in the doubly fluorophore-labeled RNA upon S1 binding were assayed using a similar EMSA to that described above, with slight differences. The preparation of the doubly fluorophore-labeled Tte pseudoknot was the same as for our smFRET experiments, the Methods section of which describes it. The doubly labeled Tte riboswitch was gel purified on a 20% urea–PAGE gel to rigorously remove RNA that was labeled with only DY547, as well as residual free Cy5 fluorophore that is not covalently attached to the RNA. This additional step is required for this assay to simplify the interpretation of in-gel FRET. A solution of 1 μM RNA in milliQ water was refolded by heating for 2 min in a 70°C copper bead bath then allowed to cool to room temperature over 20 min. The binding reactions were assembled with 1 μl of 1 μM RNA (1 pmol total), 1 μl of 10× S1 dilution series, and buffer in a final volume of 10 μl; the buffer composition in the final reactions (excluding RNA and S1) was 12.5 mM Tris–HCl, 125 mM NH4Cl, 6% (v/v) glycerol, 100 μg/ml BSA and 1 mM MgCl2. Note that a higher concentration of RNA is used in these reactions compared to those described above. The assembled binding reactions were incubated at room temperature for 50 min, after which loading buffer was added as described above. The samples were electrophoresed at 4°C on a 12% native polyacrylamide gel (18 cm × 14.5 cm × 1.5 mm) in 1X TGE that had been prerun for ∼3 h at 100 V, and then run at 18 mA for 3 h. Reference lanes containing the RNA pseudoknot labeled with only DY547 and a doubly Cy5 end-labeled DNA strand (5′-Cy5-CTT TAC CAC AAG GAT GTG-Cy5-3′) were included for later use in correction of background and cross-talk between fluorescence channels. The gels were scanned in the glass plates on a Typhoon™ 9410 Variable Mode Imager (GE Healthcare) operating in Fluorescence mode. For in-gel FRET, the gel was imaged first with 532 nm laser excitation and the default emission filter set for Cy3 (580 BP 30) and PMT voltage of 580 (channel 1). The gel was subsequently imaged upon 532 nm laser excitation with the default emission filter set for Cy5 (670 BP 30) and a PMT voltage of 650 V (channel 2). Cross-talk between channels and background was corrected in FluorSep v2.2 (Molecular Dynamics) using automatic fluorochrome separation as described in the software's manual; the bands in the RNA-DY547 only and Cy5-DNA reference lanes were each boxed separately as references for channels 1 and 2, respectively. The approximate fractions of RNA in each band as a function of S1 concentration were calculated from a Cy5-only scan, in which Cy5 is directly excited (633 nm laser, 670 BP 30 filter set, PMT voltage of 550), in ImageQuant v5.2 (Molecular Dynamics).
In-line probing analysis
In-line probing analyses with radiolabeled Tte riboswitch were performed following a method previously established by Roth et al. (31) with minor modifications. For each in-line probing reaction, 1 μl (2.5 pmol, ∼6000 cpm) of radiolabeled Tte riboswitch aptamer was combined with 1 μl of 0.5 M Tris–HCl of pH 8.3, 1 μl of 1 M KCl and 4 μl of milliQ water. The mixture was then refolded by heating in a 90°C copper bead bath for 3 min and allowed to cool for at least 15 min at room temperature. 1 μl each of 10 mM MgCl2, 1 mg/ml BSA and A(10) buffer were then added to the refolded RNA to a final volume of 10 ul, mixed thoroughly, and incubated at room temperature for ∼45 h. For experiments performed in the presence of S1, 1 μl each of 10 mM MgCl2, 1 mg/ml BSA and 10× S1 in A(10) buffer (20 μM S1) were added to the refolded RNA to attain a final S1 concentration of 2 μM. For experiments in the presence of preQ1, 1.8 μl of 16.3 μM preQ1 was combined with radiolabeled RNA before refolding to attain a final preQ1 concentration of 3 μM. A recombinant RNase inhibitor (RNaseOUT, ThermoFisher Scientific) was included in a small subset of reactions at 100 units per reaction where indicated to rule out the presence of residual nuclease activity in the S1 protein preparation (Supplementary Figure S6). After incubation, the reactions were stopped by adding 2× formamide gel loading buffer containing 95% formamide, 18 mM EDTA, 0.025% each of SDS, xylene cyanol and bromophenol blue. Resulting RNA fragments were then resolved on a 15% denaturing polyacrylamide gel in 0.5X TBE that had been pre-run at 35 W (constant power) for ∼0.5 h prior to loading, and then run at 35 W for ∼80 min. After electrophoresis, one of the glass plates was removed, leaving the gel adhered to the second glass plate. The gel and plate were wrapped in plastic cling wrap and imaged using a storage phosphor screen (∼90 min exposure) and Typhoon 9410 Variable Mode Imager (GE Healthcare Life Sciences). Gel images were quantified using ImageQuant v 5.2 (Molecular Dynamics). The intensity of each band was determined by dividing the intensity of that band by total intensity of the lane.
RNase A protection assay
To investigate the changes in secondary structure of the Tte pseudoknot, we performed an RNase A protection assay with 5′ radiolabeled pseudoknot. For each reaction, 1 μl (∼2.5 pmol) of radiolabeled Tte pseudoknot was combined with 6 μl of 1.67× buffer (16.7 mM Tris–HCl [pH 7.5 at 22°C], 167 mM NH4Cl, 8.3% (v/v) glycerol, 1.67 mM DTT) and 1 μl of ligand (or H2O for ligand condition). The mixture was refolded by heating in a 70°C copper bead bath for 3 min and allowed to cool for at least 15 min at room temperature (refolded RNA solution). 1 μl each of 1 mg/ml BSA and 2 μM S1 in buffer A(10) were then added to the refolded RNA, mixed thoroughly and incubated at room temperature for 20 min. Finally, 1 μl of 0.01 μg/ml RNase A (Roche, 10109142001) was added to each reaction and incubated for 2 min at room temperature. The reaction was then stopped quickly by adding 10 μl of 2× formamide gel loading buffer and placing on ice. Resulting RNA fragments were then resolved on a 15% denaturing polyacrylamide gel as described above, except with a run time of ∼60 min. After electrophoresis, the gel was imaged using a storage phosphor screen as descried above.
smFRET experiments
The doubly fluorophore-labeled RNA pseudoknot used for single molecule fluorescence experiments (Figure 1B) is identical to that used in previous studies by Suddala and Rinaldi et al. (30); the sequence of this construct is 5′-biotin-UCA CCU GGG UCG CAG (U-Cy5)AA CCC CAG UUA ACA AAA CAA GG G-DY547-3′. The Tte construct was purchased from Dharmacon Inc. (Fayette, CO) with a 5′ biotin modification, 3′ DY547 label, and a 5-aminoallyl-uridine (5 NU) modification at position U12 for later functionalization with Cy5. The synthesized RNA was HPLC purified by the manufacturer. Upon receipt, the 2′ACE protecting groups were removed according to the manufacturer-provided protocol: RNA was dissolved in deprotection buffer (100 mM acetic acid, adjusted to pH 8.3 with TEMED), incubated at 60°C for 2 h, and then brought to dryness in a vacuum centrifuge. The labeling with Cy5 was achieved by adding one pack of Cy5-NHS ester (GE Healthcare) dissolved in 30 μl of DMSO to ∼3.4 nmol RNA in a total reaction volume of 50 μl containing 0.1 M sodium bicarbonate buffer (pH 8.7) and incubating for 4 h at room temperature in the dark. Excess dye was removed using a NAP-5 gel filtration column (GE Healthcare), followed by ethanol precipitation. The doubly labeled RNA was dissolved in deionized water and used for smFRET experiments.
To reduce non-specific binding of protein that results in high background fluorescence, quartz slide surfaces for single molecule experiments were passivated with a mixture of PEG/biotin-PEG as described previously using established protocols (40). Microfluidic channels (∼80–100 μl capacity) were assembled using the passivated slides and coverslips (40,41), and the biotin-PEG reacted with 0.2 mg/mL streptavidin in T50 buffer (10 mM Tris–HCl [pH 8.0 at 2°C], 50 mM NaCl) for 10 min, then washed again with T50. A typical sample was prepared as follows: 6.25 nM Tte (smFRET) RNA was refolded by heating in a 90°C copper bead bath for 2 min with 2 μl of 100 mM Tris–HCl (pH 7.5 at 22°C), 0.5 μl of 4 M NH4Cl and 2 μl of 50% (v/v) glycerol in a total volume of 16 μl. The RNA in solution was refolded by allowing it to cool for at least 15 min at room temperature, after which 2 μl of 1 mg/ml BSA and 2 μl of S1 protein storage buffer A(10) (Expression and purification of E. coli ribosomal protein S1 above). The composition of the resulting 20 μl solution was 5 nM RNA, 12.5 mM Tris–HCl, 125 mM NH4Cl, 6% (v/v) glycerol, 100 μg/ml BSA, and 1 mM MgCl2. The refolded sample was then diluted to a final RNA concentration of 25 pM in buffer I (12.5 mM Tris–HCl, 125 mM NH4Cl, 6% (v/v) glycerol, 100 μg/ml BSA, 1 mM MgCl2). This diluted RNA solution was applied to the slide and allowed to incubate for 10 min. Excess, unbound RNA was washed by flowing at least 100 μl of buffer I. Finally, at least 100 μl of buffer I as well as an oxygen scavenging system, consisting of 5 mM protocatechuic acid, 50 nM protocatechuate-3,4-dioxygenase (to slow photobleaching) (42) and 4 mM Trolox, to reduce photoblinking (43), was applied to the slide and allowed to equilibrate for 5 min before imaging on a prism-TIRF microscope, as described previously (30). In previous work we extensively tested OSS cocktails for RNase contamination and found our PCD/PCA system to be RNase-free (44). Fluorescence emission from single molecules excited with a 532 nm diode laser was recorded at 10 frames per second (integration time of 100 ms) in mj2 format using an intensified CCD camera (I-Pentamax, Princeton Instruments) using a custom acquisition script written in Matlab. Movie files were converted from mj2 to pma format and fluorescence-over-time traces extracted using IDL (Exelis Visual Information Solutions). Alternatively, fluorescence time traces were extracted from the mj2 files directly using custom Matlab scripts (The MathWorks). Genuine fluorescence time traces were selected manually and analyzed using custom Matlab (The MathWorks) scripts as described previously (30). FRET distribution histograms were built by combining the apparent FRET efficiencies observed in 100 frames from each trace in a given condition. The resulting histograms were fit with Gaussian function using OriginLab 9 (OriginLab Corporation).
To measure binding affinity of ligands (Gua and DAP) by smFRET, we performed single molecule experiments with varying ligand concentration ranging from 0 to 5 μM for Gua, and 0 to 500 μM for DAP. For these experiments, at least 100 μl of solution containing exact concentration of ligand as well as an oxygen scavenging system was flowed on the slide and allowed to equilibrate for 5 min before imaging. The proportion of molecules in the high-FRET state was determined using the area under the curve after Gaussian fitting of the smFRET histogram, and the % high-FRET state as a function of ligand concentration was fit using Hill equation in OrginLab to determine the apparent ligand affinity (K1/2).
Rate constant analysis from smFRET data was performed as described previously (30), with minor modifications. Notably, the error associated with the measured rate constants was estimated by bootstrapping using a custom Matlab script. Additional details for rate constant calculation and error estimation are provided in Supplementary Information.
Single-molecule measurement of Cy5-labeled S1 binding to DY547-labeled Tte pseudoknot
To observe real time binding and dissociation of protein S1 to the Tte pseudoknot, we labeled the primary amine (–NH2) group of lysine residues of S1 nonspecifically by following a method described previously by Koh et al. (45). The labeling reaction was performed with a ∼10-fold excess of dye to protein S1, at a final S1 concentration of 8 μM to achieve a final dye to protein ratio close to 1:1. In brief, a single Cy5-NHS ester dye packet (GE Healthcare, PA25001) containing ∼344 nmol of dye was dissolved in 50 μl anhydrous DMSO. In a typical reaction, 13 μl of stock S1 was mixed with 0.5 μl of stock dye solution, 3 μl of 1 M sodium bicarbonate buffer, pH 8.5, and 33.5 ul of milli-Q water. Reactions were protected from light and incubated at room temperature for 30 min with agitation. The excess dye was removed by passing the reaction mixture twice through a Micro Bio-Spin P-6 gel spin column (Bio-Rad, 7326221). To minimize non-specific adsorption of labeled S1 on the PEG-coated slide containing immobilized Tte pseudoknot (labeled with a 3′-DY547), we further passivated the slide surface by incubating with 10 mg/ml BSA for ∼15 min before flowing the final imaging solution containing Cy5 labeled S1. Co-localization of the labeled S1 protein and RNA was monitored via TIRF microscopy with simultaneous excitation using 532 nm and 640 nm lasers.
Single molecule cluster analysis (SiMCAn) of smFRET traces
To identify and visualize the various behavioral patterns in the smFRET trace data, single molecule cluster analysis (SiMCAn) was performed in MATLAB as described previously (46), with minor modifications. Additional details are provided in Supplementary Information.
Data availability
The various Matlab scripts, input data, and resulting analysis files associated with this work are available in DeepBlue from the University of Michigan library (https://deepblue.lib.umich.edu/data).
RESULTS
The Tte pseudoknot as a structured RNA model for studying S1 binding
We first assessed the ability of S1 to bind to the Tte preQ1 pseudoknot using an electrophoretic mobility gel shift assay (EMSA) employing the 3′ Cy3 fluorophore-labeled RNA. A representative gel in Figure 2A shows the change in RNA migration on the native gel in the presence of increasing concentrations of S1. The free RNA migrates as two bands, corresponding to the pre-folded (slower migrating) and fully folded (faster migrating) forms identified previously (30). The pre-folded form represents an ensemble of conformations in which the A-rich L3 adopts only transient interactions with the P1–L1 stem–loop, whereas the fully folded form consists of conformations that are more compact as a result of a docked L3 and more fully formed P2 stem (30).
Figure 2.
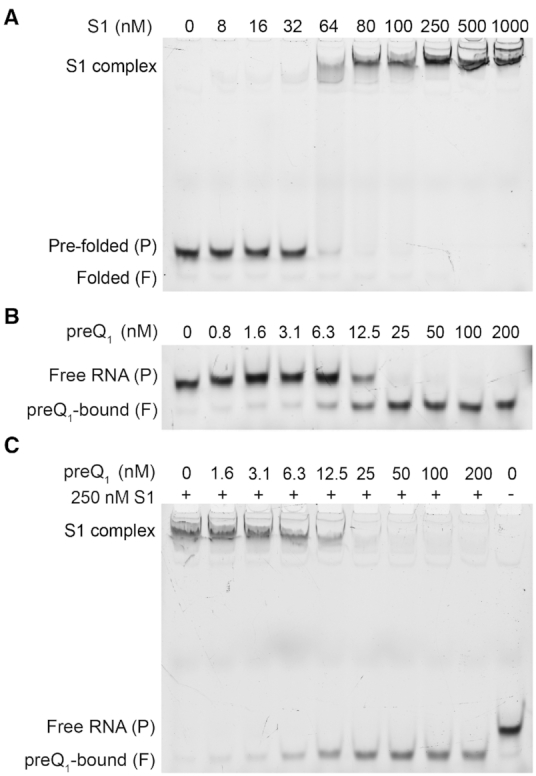
S1 binds to the Tte pseudoknot as a function of pseudoknot stability. (A) An EMSA titration of the 3′-Cy3-labeled Tte pseudoknot with increasing concentration of S1 shows clear shift in mobility, indicating the formation of an RNA-protein complex. For the free pseudoknot, two bands are observed due to the presence of two pseudoknot conformations which were earlier referred to as the folded (F, faster migrating) and pre-folded (P, slower migrating) forms. (B) A titration with high-affinity ligand preQ1 shows the formation of a compact, faster moving preQ1-bound (folded, F) species. (C) EMSA titration as in (B) except in the presence of 250 nM S1. Note that at sub-saturating concentrations of ligand, the less compact pre-folded (P) form of the pseudoknot is preferentially bound by S1. The RNA concentration was 10 nM in all lanes in panel A, B and C.
In contrast, in the presence of S1 we observed the appearance of a new shifted band that migrates much slower than the free RNA, indicating binding of S1 to the RNA pseudoknot to form a slow-moving RNA–protein complex. At ∼250 nM S1, the free RNA bands completely disappear so that we observed a single band for the slow-migrating S1 complex. While these results clearly show that S1 is capable of binding to the RNA pseudoknot, the relatively high concentrations of RNA and protein necessary for detection by fluorescence used in this assay are not suitable for an estimation of the dissociation constant (Kd). Instead, we performed a similar EMSA assay with trace amounts of 5′ radiolabeled Tte pseudoknot (Supplementary Figure S1). With this more sensitive EMSA, we similarly see two bands for the pre- and fully folded forms of the RNA. In addition, this EMSA exhibited two RNA-S1 complexes at the highest S1 concentrations. The formation of the second, even slower migrating complex at concentrations of >250 nM S1 (Supplementary Figure S1A; Complex 2) is likely due to binding of a second protein S1 to the already formed RNA–S1 complex, enabled in part through protein-protein interactions and the much higher relative concentration ratio of S1 to RNA than in the fluorescence-based EMSA. From the radioactive assay, the dissociation equilibrium constants for these two RNA-S1 complexes can be estimated using a two-site binding model (37) as Kd1 = 120 ± 23 nM and Kd2 = 440 ± 80 nM for Complexes 1 and 2, respectively (Supplementary Figure S1B). These results are consistent with earlier observations of two S1 molecules binding to one molecule of tmRNA with apparent binding constants of ∼90 nM and ∼300 nM (47), and establish the Tte preQ1 pseudoknot as a suitable structured RNA model for studying ribosomal protein S1.
The structural stability of the Tte pseudoknot can be tuned with ligands of different affinity
The aptamer domain of the Tte riboswitch binds a variety of ligands with varying affinity, which in turn considerably changes the stability of its structure. PreQ1 as the cognate ligand is known to possess high affinity for the pseudoknot and greatly stabilizes the fully folded conformation of the pseudoknot (31). An EMSA using the fluorescently labeled RNA in the presence of increasing concentrations of preQ1 converts the ligand-free, pre-folded form into the ligand-bound, fully folded form, characterized by a more compact conformation arising with a sharp inflection point at a stoichiometry of ∼1:1, demonstrating that the ligand binds the pseudoknot stoichiometrically and with high affinity (Figure 2B).
To understand how this ligand-induced conformational stabilization of the pseudoknot affects S1 binding, we tested if S1 can bind the RNA in the presence of preQ1. Figure 2C demonstrates that the amount of Tte riboswitch shifted into the S1-bound complex decreases when the RNA is pre-incubated with increasing concentrations of preQ1; consequently, only a single, fast-moving, ligand-bound band was observed at the highest preQ1 concentration of 200 nM (Figure 2C). This finding indicates that S1 cannot bind to the stably folded preQ1-bound Tte pseudoknot. In addition, under conditions where ligand is sub-saturating (e.g. compare 12.5 nM preQ1 condition in Figure 2B and C) no significant band was observed for the ligand-free RNA, suggesting that S1 instead preferentially binds this less compact, only partially folded RNA (Supplementary Figure S2).
To further investigate ligand-induced conformational stability of the RNA, we next examined two additional known near-cognate ligands: guanine (Gua) and 2,6-diaminopurine (DAP). We first determined the dissociation constants for these two ligands using an smFRET approach as described previously (30), where ligand-induced changes in pseudoknot conformation can be monitored via FRET between a donor DY547 attached to the 3′ terminus of the RNA pseudoknot and acceptor Cy5 coupled to U12 (Figure 1B). High-FRET and mid-FRET levels are observed for the folded and pre-folded pseudoknot conformations, respectively, and the apparent half-titration values K1/2 for ligand binding can be determined by monitoring changes in the equilibrium position between high- and mid-FRET as a function of ligand concentration. We found the K1/2 for Gua and DAP to be >10 and >1000-fold weaker, respectively, than that of preQ1 (Figure 3A, B), consistent with previous reports of weaker binding affinities of these ligands to the related B. subtilis preQ1 riboswitch (31). Accordingly, very high excesses of Gua or DAP over RNA were required to convert the pseudoknot into the fully folded form, and no sharp inflection points were observed upon addition of these two ligands as analyzed by fluorescent EMSA, in contrast to preQ1 (Figures 3C and Supplementary Figure S3A, B).
Figure 3.

The stability of the Tte pseudoknot can be monitored by using ligands of different affinity. (A, B) The percentage of high FRET state determined from smFRET experiments was plotted as a function of ligand concentration. The data were fit with a non-cooperative binding isotherm and the respective apparent K1/2 values are indicated for guanine (Gua, A) and 2,6-diaminopurine (DAP, B). The error is reported as the standard error of the fit from Origin. (C) Quantification of the fraction of ligand-bound RNA in the presence of increasing concentrations of Gua, DAP and adenine (Ade). (D) Melting curve analysis monitored at 260 nm absorbance indicates that melting point of P2 stem increases in the presence of ligands with increasing binding affinities. Tm1 and Tm2 represent the melting temperatures of P2 and P1 stem, respectively. (E, F) EMSA titrations as in (C) except in the presence of 250 nM S1, for Gua (E) and DAP (F).
To better understand the correlation between ligand binding affinity and the resulting degree of RNA structural stabilization, the relative stabilities of the free and ligand-bound pseudoknots were assessed by melting curve analysis (Figure 3D). In the absence of ligand, the Tte riboswitch showed two clear melting transitions at ∼50°C and ∼74°C. To assign these two transitions in the melting curve more confidently, we generated a pseudoknot variant lacking the P2 helix by replacing the corresponding portion of the pseudoknot sequence with the well-characterized UNCG tetraloop. As a result, we observed only a single transition >70°C, as expected for the melting of a highly stable tetraloop-capped hairpin structure (48) (Supplementary Figure S4A, B). This finding allowed the first, lower temperature transition to be confidently assigned to melting of the P2 stem, and the second, higher temperature transition to melting of P1. In the presence of ligands, no significant change in the melting temperature of P1 (∼74°C) was observed, however, the melting temperature of P2 was observed to increase from DAP (∼53°C) to Gua (56°C) to preQ1 (67°C), indicating that helix P2 is stabilized to an increasing extent (Figure 3D). In the case of preQ1, the melting transition of P2 largely overlaps with that of P1, suggesting a more uniform and cooperative unfolding in the presence of that ligand. In contrast, in the case of DAP the melting transition for P2 is more well defined than for the ligand-free RNA without significantly increasing the melting temperature, suggesting that the ligand may be able to organize but not significantly stabilize P2. These results suggest that the relative stabilities of the ligand-bound pseudoknot follows a decreasing trend of preQ1 > Gua ≫ DAP.
To ask how well ligands having lower affinity for the Tte riboswitch compete with S1, we next tested the binding of S1 to the Tte riboswitch in the presence of the less stabilizing Gua and DAP ligands. Significantly, we observed that S1 can strongly interact with the less stably folded, weaker ligand bound RNA pseudoknots, as indicated by the observation of noticeable bands for the S1-bound RNAs even in the presence of high concentrations of Gua and DAP (Figure 3E, F). In contrast, adenine, which is not a near-cognate ligand for the preQ1 riboswitch and does not induce a conformational change in the RNA pseudoknot (31), had no effect on S1 binding to the RNA (Supplementary Figure S3C, 5). Taken together, these results demonstrate that binding of S1 to the Tte pseudoknot is correlated inversely with pseudoknot stability and can be tuned by using ligands of different affinity for the riboswitch.
S1 binding significantly unfolds and alters the conformation of the pseudoknot
To better characterize the structure of the S1-bound RNA species, an S1 titration EMSA was performed using a doubly-fluorophore labeled pseudoknot (Figure 4A) (30). Consistent with our previous results, in the absence of S1 the Tte riboswitch is present in two forms: the faster migrating, more compact, fully folded form (high FRET, or red), and the slower migrating, relatively less compact, i.e. pre-folded, form (mid FRET, or yellow). In lanes containing S1, the in-gel FRET efficiency demonstrates that the RNA in complex with S1 exhibits lower FRET (i.e. a green shift), indicative of greater inter-fluorophore distance. Although the in-gel FRET efficiencies cannot be used to calculate exact physical distances, a lower apparent FRET efficiency of the S1-bound complex indicates that the RNA is unfolded to a greater extent than it is in either of its free forms.
Figure 4.

S1 significantly alters the pseudoknot conformation. (A) Titration of the doubly fluorophore labeled Tte pseudoknot with S1. The RNA concentration was 100 nM in all lanes. The S1-bound form of the pseudoknot exhibits lower apparent FRET efficiency (green pseudocolor), indicative of greater inter-fluorophore distance than the undocked (mid-FRET, yellow) or docked (high FRET, red) forms of the free RNA. Both forms of the free RNA migrate slightly faster when labeled with only one fluorophore (DY547 reference lane, far right). A Cy5-DNA reference lane used for cross-talk correction is omitted for clarity. (B) In-line probing analysis of the 5′-32P-labeled Tte pseudoknot in the presence of S1 reveals some decreased and some increased spontaneous cleavage sites, suggesting destabilization of the pseudoknot. In contrast, the cleavage pattern of the preQ1-bound Tte pseudoknot in the absence and presence of S1 shows very little difference. The cleavage products were resolved on a denaturing 15% polyacrylamide gel at 35 W for 1 h. NR, no reaction; T1, RNase T1 digestion ladder; –OH, alkaline hydrolysis ladder. (C) Differential in-line probing reactivity of the Tte pseudoknot in the presence of S1 reveals the sites of decreased and increased cleavages. Values are reported as the mean ± S.D. of n = 3 independent experiments. a.u., arbitrary units. (D) Differential in-line probing reactivity of the preQ1-bound Tte pseudoknot in the absence and presence of S1. (E) Differential in-line probing reactivity mapped onto the Tte pseudoknot secondary and tertiary structures, indicating sites of both decreased and increased cleavage (PDB ID: 3Q51).
To obtain more detail about the changes in RNA structure upon S1 binding, 5′-32P-labeled Tte riboswitch was subjected to in-line probing analysis with 1 mM Mg2+ in the absence and presence of 250 nM S1. Taking advantage of slow, spontaneous cleavage of phosphodiester linkages in the RNA backbone via intramolecular transesterification, in-line probing has been used extensively to investigate changes in tertiary structure of riboswitches in the absence and presence of their metabolite ligands (31). In general, spontaneous cleavage of highly structured RNA regions is low due to their less flexible, non-in-line conformation compared to unstructured, single-stranded regions that can more readily adopt an in-line orientation, which in turn leads to greater spontaneous backbone cleavage (49,50). The analysis of the 5′-radiolabeled Tte riboswitch pseudoknot in the absence of S1 indicates strong in-line cleavage at loop L1 (nucleotides C9 to U12) and flexible region of L3 (nucleotides U22, A26 and A31), while regions around the P1 helix (nucleotides C1 to G8 and A13 to G20) are relatively resistant to cleavage (Figure 4B), consistent with the known pseudoknot structure. Addition of protein S1 leads to a significant change in the structure of the pseudoknot, as evidenced by the different cleavage pattern of the pseudoknot. A major increase in cleavage activity near the P1 helix (nucleotides U-2 to G5, C16 to C18) and at C7, along with changes in cleavage activity in the P2 region (nucleotides C9, A10 and A31) (Supplementary Figure S6), suggests that the folded pseudoknot is destabilized. In addition, decreased cleavage in the L2 and L3 loop regions (U12, U22 and A26) suggests a further perturbation of the overall pseudoknot structure (Figure 4B, C). We note that the relative depletion of full-length RNA in the presence of S1 may lead to more than a single cleavage event of the same RNA molecule. Nevertheless, a titration with S1 shows the robustness of our in-line probing results (Supplementary Figure S6).
Given the strong competition of the cognate ligand with S1 binding, the Tte riboswitch structure was also studied in the presence of both S1 and a saturating concentration of 200 nM preQ1. As might be expected, the pseudoknot structure was strongly stabilized upon addition of preQ1, as evidenced by greatly reduced in-line probing activity overall, and a cleavage profile largely consistent with that of the preQ1-bound pseudoknot and a dramatic reduction of in-line probing activity signatures associated with the S1-bound pseudoknot (Figure 4B, D). This observation suggests that S1 cannot significantly interact with the preQ1-bound pseudoknot, consistent with our previous fluorescence-EMSA results. One notable exception appears to be the very 5′ end of the RNA (U-2 to C1), a single-stranded region that S1 apparently can still access (Figure 4B, D). These observations are further supported by an RNase A probing assay. RNase A preferentially cleaves single-stranded RNA at C and U positions on a much shorter reaction timescale (2 min) than in-line probing, and the strong increase in cleavage activity at C1 strongly supports an increase in single-stranded character in this region upon the addition of S1 (Supplementary Figure S7). In the presence of saturating concentrations of ligands (preQ1, Gua and DAP), the cleavage activity at C1 significantly decreases again.
Sites affected most significantly by S1 alone were mapped onto the known secondary and tertiary structure of the Tte pseudoknot (Figure 4E), highlighting regions with increased and decreased in-line probing reactivity. Taken together, our results show that S1 destabilizes the P1 helix, shifts the conformations of specific base pairs of P2, and stabilizes (or protects) L3, but only in the absence of saturating concentrations of competing preQ1.
smFRET analysis demonstrates that S1 interferes with 3′ tail docking of the RNA
To gain insight into how S1 binding alters the conformational dynamics of the RNA, we performed smFRET experiments using the FRET-labeled pseudoknot (Figure 1B) to monitor 3′ tail docking and formation of the P2 helix as established by our in-gel FRET experiments. The pseudoknot additionally was immobilized on a quartz slide using a 5′ biotin for observation with a prism-based total internal reflection fluorescence (TIRF) microscope (Figure 5A). Analysis of individual fluorescence time traces showed transitions between a high-FRET (∼0.96) state and a relatively short-lived, mid-FRET (∼0.82) state (Figure 5B); these states were previously assigned to the folded conformation of the pseudoknot where the P2 stem is fully base paired (i.e. docked), and the pre-folded conformation where the P2 stem is only partially formed (undocked), respectively (30).
Figure 5.

smFRET analysis reveals S1-mediated unfolding of the Tte pseudoknot. (A) Schematic depiction of our smFRET experimental setup. The doubly labeled Tte pseudoknot was immobilized on a PEG-coated quartz slide via a biotin-streptavidin linkage and imaged by TIRF microscopy. (B) Representative smFRET trace (top) for the pseudoknot alone showing fluctuations between mid-FRET (pre-folded; P) and high-FRET (folded; F) states. The cyan line represents the idealized FRET trace from Hidden Markov modeling (HMM). An smFRET histogram (bottom) constructed from the FRET values observed in the first 100 frames from N single molecule traces reveals the two major FRET states fitted with Gaussian distributions. (C) Representative smFRET trace (top) and corresponding histogram (bottom) as in (B) of the Tte pseudoknot in the presence of 250 nM S1, showing the dynamic nature of the S1-bound unfolded (U) pseudoknot. The addition of S1 introduces a third, low-FRET state in the smFRET histogram, corresponding to RNA pseudoknot conformations that are unfolded to an even greater extent. (D, E) Transition occupancy density plots (TODPs) illustrating the most common FRET transitions (left panels) and corresponding mean rate constants (right panels) in the absence (D) and presence (E) of S1.
Incubation with 250 nM S1 protein favors the undocked conformations of the pseudoknot, as indicated by the observation of higher occupancy of the mid-FRET state in the smFRET histogram; the value of this mid-FRET state is also shifted slightly lower to ∼0.77. The fraction of high-FRET state (folded conformation) was determined from the area under the high-FRET peak of smFRET histograms based on over 100 molecules and was found to decrease from 27% in the absence of S1 to 19% in the presence of S1 (Figure 5B, C). In addition, we observed that the S1-bound pseudoknot visits a short-lived, more fully unfolded (undocked) ensemble of conformations, evidenced by the brief transitions to FRET values below 0.5 in the representative trace and the appearance of a broad, much lower FRET state (∼0.45) in the population-averaged smFRET histogram (Figure 5C). However, frequent transitions to the folded, high-FRET conformation provide evidence for reversible dissociation of S1 from the pseudoknot. The reversibility of binding was further tested using a single molecule assay with Cy5-labeled S1 where on average one lysine per protein molecule was labeled nonspecifically. 3′-DY547-labeled Tte pseudoknot molecules were immobilized on a quartz slide at low density. The Cy5-labeled S1 was then flowed onto the slide and imaged by TIRF microscopy. Binding will thus be characterized by DY547 and Cy5 signals co-localized within a diffraction-limited spot (Supplementary Figure S8A). While this approach led to significant non-specific surface binding of the labeled S1 and possibly some heterogeneity in its folding and RNA binding properties, as expected, the resulting Cy5 intensity trajectories revealed repetitive binding events of S1 to single Tte pseudoknot molecules (Supplementary Figure S8B).
To investigate the dynamic nature of these S1-dependent RNA conformational changes, we fit the FRET traces using a two- or three-state Hidden Markov Model (HMM) in the QuB software suite as described previously (51), generating idealized FRET traces. These HMMs in turn allowed us to generate transitional occupancy density plots (TODPs) that display the fraction of molecules exhibiting transitions between specific initial and final FRET states (51). TODPs from the free-RNA traces underscore the existence of a large fraction of static high-FRET (∼0.96) states along with a small fraction of dynamic states showing reversible transitions between folded and pre-folded conformations (FRET states of ∼0.96 and ∼0.8, respectively) (Figure 5D, left panel). In contrast, upon addition of 250 nM protein S1, molecules with static behavior disappeared entirely while interstate transitions involving a total of three FRET states (0.96, 0.77 and 0.45) emerged (Figure 5E, left panel), further supporting the notion that S1 binding destabilizes the folded conformation of the pseudoknot.
From the HMM idealizations we also estimated the rate constants for docking (kP→F) and undocking (kF→P) of the nucleotides that form the 3′ side of the P2 stem (Figure 5D, E right panels; Supplementary Figure S9). kP→F decreases from 1.1 s−1 in the absence of S1 to 0.7 s−1 in the presence of S1. In contrast, the value of kF→P remains almost unchanged upon addition of S1 (0.51 s−1) compared to its absence (0.56 s−1), suggesting that stabilization of the pseudoknot in the pre-folded conformations arises almost entirely from S1 interfering with 3′ tail docking.
Ligand-induced stabilization counteracts S1-mediated unfolding
We next examined how interactions between S1 and the Tte riboswitch pseudoknot change as a function of increasing pseudoknot stability, brought about by ligand binding. As expected, preQ1 significantly stabilizes the folded conformation of the Tte pseudoknot, indicated by a significant static high-FRET signal in the presence of 200 nM preQ1 (Figure 6A). Consistent with our earlier observations that S1 does not efficiently compete with preQ1 binding to the riboswitch, conformational dynamics of the preQ1-bound pseudoknot remain unaltered in the presence of 250 nM S1 (Figure 6B). Importantly, the same behavior was observed regardless of whether the RNA was first incubated with preQ1 and then followed by a buffer containing S1 and ligand, or when the RNA was incubated first with S1, and then buffer containing both S1 and preQ1 was introduced (data not shown). In contrast, in the presence of saturating (5 μM) Gua and 250 nM S1, smFRET traces of Gua-bound pseudoknot molecules showed increasing transitions to the mid-FRET state of ∼0.77, leading to lower occupancy of the high-FRET state (37%) compared to Gua-only bound pseudoknot (62%, Figure 6C, D). The increasing emergence of such molecules with dynamic (off-diagonal) state transitions in the TODP suggests that S1 can transiently interact with the pseudoknot stabilized by Gua. The least-stabilizing DAP-bound pseudoknot was even more destabilized by S1 binding, as evidenced by the now pronounced transitions to the mid-FRET states (Figure 6E, F and Supplementary Figure S10, 11). The presence of a larger fraction of molecules with dynamic behavior observed in the TODP further underscores the ability of S1 to unfold the DAP-bound pseudoknot considerably more than the preQ1- or Gua-bound riboswitches, inversely correlating with ligand affinity.
Figure 6.

Ligand modulated structural stability of the pseudoknot determines S1-mediated unfolding. Representative smFRET traces, corresponding histograms and TODP plots for the ligand-bound pseudoknots in the absence (A, C, E) and presence (B, D, F) of 250 nM S1. Ligand concentrations were 200 nM preQ1 (A, B), 5 μM Gua (C, D) and 500 μM of DAP (E, F). The folded pseudoknot conformation is significantly stabilized in the presence of saturating concentrations of ligand. However, S1 can unfold the pseudoknot in the presence of weak ligands (Gua or DAP).
Cluster analysis reveals distinct conformational dynamic behaviors associated with S1-RNA interactions
Because smFRET enables direct observation of complex behaviors (52), such as the transitions between multiple distinct conformational states that we observed for the RNA pseudoknot in the presence of S1, the data can be challenging to analyze and interpret. As described above, we observed that the interaction of the RNA pseudoknot with S1 changes upon the addition of ligands that stabilize the folded RNA structure to varying extents. In particular, the fully folded (high-FRET) and pre-folded (mid-FRET) states are always present, however, the resulting FRET transitions between conformational states of the RNA are more similar for some ligands than others.
To better understand the interplay between the ligand-induced stabilization of the folded RNA and protein S1, we performed single-molecule clustering analysis (SiMCAn), a method developed to identify in unbiased fashion unique and shared patterns of dynamic conformational behavior across large numbers of smFRET traces (46). Given the enhanced conformational dynamics of the RNA in the presence of S1, indicated by the wide range of observed FRET values (Figure 5C), we first idealized the FRET traces from all conditions using vbFRET (53), allowing for up to five FRET states to be fit. As done previously (46), idealized FRET values were then re-binned into ten uniformly spaced bins to facilitate analysis. Notably, in a TODP the information on which transitions occur is preserved, but information on which transitions are observed together in the same molecule trace is not. By contrast, SiMCAn is able to identify these patterns of transition behavior and thus preserves this information in algorithmic fashion, which otherwise can only be gained from cumbersome and somewhat subjective visual inspection of the traces and thus may easily be missed.
A first round of clustering was performed to identify common patterns of transitions between different conformational (i.e., FRET) states of the RNA (indicated by transitions), using the combined data from all experimental conditions (RNA alone, RNA + S1, RNA + preQ1, RNA + Gua, RNA + DAP, RNA + preQ1 + S1, RNA + S1 + preQ1, RNA + Gua + S1, RNA + DAP + S1). Of the 1160 total single molecule traces included in the analysis, 631 were classified into seven static clusters with a single characteristic FRET state. The remaining 529 traces with dynamic transitions between FRET states were clustered based on the FRET Similarity Matrix (FSM) of each molecule, which takes into account the number and type of transitions between FRET states for a given molecule, as well as the time spent in each FRET state (46). The hierarchical tree resulting from the clustering of these dynamic molecules (Figure 7A) was trimmed so as to maximize the differences between clusters, while simultaneously maximizing the similarity of molecules within a given cluster (Materials and Methods, Supplementary Figure S12). This strategy allowed us to quickly identify three patterns of behavior or clusters (denoted by colored branches), where molecules within a given cluster have similar FSMs and thus exhibit a common pattern of FRET transition probabilities. These clusters can be loosely described by the FRET states that the molecules occupy and transition between most frequently (Figure 7B, C), namely high-FRET states (0.95–0.85, H), mid- to high-FRET states (0.75–0.95, M1), and mid-range FRET states (0.85–0.65, M2). Notably, all clusters include traces that occupy lower FRET states.
Figure 7.

Single molecule cluster analysis identifies classes of dynamic behavior as a function of RNA stability. (A) Dendrogram of dynamic traces resulting from hierarchical clustering analysis to identify patterns of trace behavior. Of the 1160 total molecules included in the analysis, only the 529 dynamic molecules are shown in the dendrogram. The dashed line indicates the threshold of three clusters used to describe the data. Dynamic clusters were assigned labels loosely based on the predominant FRET ranges observed: M2 (mid-FRET 2, teal), M1 (mid-FRET 1, purple), H (high-FRET, orange). (B) Occupancy heat maps for traces in each of the dynamic clusters shown in (A). Each trace is represented by a single row in the heat map, and is colored according to the fraction of the total observation time spent in each of the 10 binned FRET states (Occupancy). N, number of single molecule traces in the cluster. (C) Representative trace segments from each of the dynamic clusters. For each dynamic cluster, 25 s of idealized FRET trace data are shown for five individual traces, separated by magenta bars. The first trace shown (colored segment) is taken from the trace closest to the cluster centroid, and the remaining four segments are randomly chosen from other traces in the cluster. The gray arrowhead represents the point at which photobleaching of the fluorophores occurred. (D) Clustergram from second-round clustering showing the relationship between patterns of static and dynamic trace behavior identified in first-round clustering (rows) and experimental condition (columns). Each experimental condition is represented by a heat map, colored according to the fraction of traces from that condition that were grouped into each of the static or dynamic first-round clusters. White vertical lines indicate experimental conditions that group together based on similar distributions of the observed patterns of trace behaviors. * indicates the experimental condition where S1 was introduced before the addition of the preQ1 ligand.
First-round clustering was performed without regard to experimental condition, yet the three types of dynamic behavior (H, M1, M2) are not equally represented across each experimental condition. To more easily visualize how these behaviors relate to the presence of the RNA-stabilizing ligands and S1, as well as integrate information about time traces in static clusters that exhibited a single, stable conformation, a second round of hierarchical clustering was performed based on the cluster's abundance profile (i.e., the fraction of molecules from an experimental condition in a given static or dynamic behavior cluster), as well as by experimental condition. This allows experimental conditions with common behavioral profiles to be more easily identified. The resulting clustergram (Figure 7D) consists of a heat map showing the fractional distribution of molecules in each cluster, where behavior clusters are grouped according to similarities in their abundance profile across conditions (row dendrogram), whereas experimental conditions are grouped according to the clusters into which their traces are classified (column dendrogram).
Examining this clustergram, it is immediately apparent that in the absence of ligand or protein S1, RNA pseudoknot molecules exhibit a variety of behaviors and conformational states, with the H and static 0.95 FRET (S-0.95) clusters, corresponding to conformations with a predominantly folded P2 stem, being the most highly represented. Reassuringly, this profile closely reflects what one would expect given the predominant features observed in the TODP for this condition (Figure 5D). A significant fraction (∼20%) of the traces from the RNA-alone condition cluster into M1 and M2, underscoring that the pseudoknot is inherently conformationally dynamic. The fraction of molecules clustering into M1 and M2 increases upon addition of S1 (∼45%), concurrent with the dramatic loss of molecules from the S-0.95 cluster, suggesting that while the M2 dynamic behavior is not exclusive to RNA-S1 interaction, it is far more prevalent in the presence of S1. The RNA+S1 condition also appears as its own branch in the condition dendrogram, highlighting the unique conformational behavior of the RNA induced by S1.
In the presence of ligands that stabilize the pseudoknot structure to varying degrees (preQ1 > Gua ≫ DAP), the fraction of molecules in the S-0.95, S-0.85 and H clusters increases, accompanied by a loss of molecules from the M1 and M2 clusters, indicating that the pseudoknot becomes less conformationally dynamic, and instead adopts conformations with a more fully folded P2 stem. Somewhat surprisingly, the fraction of molecules in the S-0.95 condition is highest in the presence of DAP, despite DAP being the most weakly binding and least stabilizing (i.e., conferring the smallest increase in melting temperature, Figure 3D) of the ligands tested. This increase in S-0.95 in the presence of DAP is accompanied by a slight decrease in S-0.85 compared to the other stabilizing ligands, providing evidence for slight conformational changes in P2 when accommodating DAP in the ligand binding pocket.
For the most strongly stabilizing ligand, preQ1, the addition of S1 makes little difference in the observed conformational behavior of the RNA, irrespective of whether the RNA is incubated with S1 before the addition of preQ1 (Figure 7D, denoted with *) or when added after ligand. This finding is also reflected in the fact that the +preQ1 –/+ S1 conditions all cluster together, indicative of their similar behavior. The ability of preQ1 to counteract the effects of S1 despite pre-incubation with protein suggests that the interactions between S1 and the RNA are transient in nature and that the strongly binding preQ1 ligand is able to displace S1.
In contrast, when S1 is added to RNA that has been pre-incubated with the less stabilizing ligands DAP and Gua, more dynamic and conformationally dynamic RNA behavior is observed, reflected by an increased occupancy of the M1 and M2 clusters, similar to RNA alone. This finding suggests that S1 prevents molecules from adopting a static high-FRET state where the P2 stem is fully docked and that S1 can possibly convert molecules in the S-0.95 FRET to the mid-FRET dynamic behaviors (M1, M2), but that there is also an upper limit to RNA structural stability that S1 is able to unfold.
DISCUSSION
In this work, using the well-characterized pseudoknot of the Tte riboswitch as a model, we have studied the mechanistic details of ribosomal protein S1 binding to a highly structured, yet conformationally dynamic, RNA motif where the folded conformation can sequester the first two nucleotides of the SD sequence through the formation of P2 helix (Figure 1). Our results reveal that S1 considerably interacts with the dynamic Tte pseudoknot, interfering with P2 helix docking, an interaction that is disfavored when P2 is stabilized by binding of cognate, but less so near-cognate, ligands (Figures 2 and 3). In-line probing studies suggest that S1 preferentially interacts with the single-stranded regions in the pseudoknot, apparent also from the observation in EMSA experiments that S1 does not bind to the faster migrating form of the RNA pseudoknot adopted in the presence of ligand, instead gel-shifting only the less compact form (Figure 2C). Furthermore, S1 binding to the pseudoknot appears reversible and thus binding of ligand, which requires (re)formation of the ligand binding pocket, results in stabilization of P2 and disfavors the S1-RNA interaction (Figure 2C). Taken together, these insights lead to a model wherein protein S1 preferentially binds the unfolded or pre-folded, more single-stranded conformations of the RNA pseudoknot, partially blocking 3′ tail docking. A thermodynamically stably binding ligand can counteract this effect by accessing its binding pocket and closing the pseudoknot so that S1 is more likely to dissociate, resulting in the coupled binding and folding equilibria depicted in Figure 8.
Figure 8.

Model of S1-mediated unfolding of the Tte-pseudoknot. S1 preferentially binds to the pre-folded conformation (EFRET = 0.82) of the Tte riboswitch pseudoknot and stabilizes a partially unfolded conformation (EFRET = 0.77). The S1 bound pseudoknot can also transiently form a more completely unfolded conformation (EFRET = 0.45) that folds quickly to either the pre-folded conformation or folded conformation (EFRET = 0.96). The ability of S1 to unfold the pseudoknot strongly depends on the stability of the pseudoknot, which can be altered by using ligands of varying affinity to the pseudoknot. S1 cannot interact with the strong ligand-bound, stably-folded pseudoknot, but can significantly interact with and unfold the weak ligand-bound, weakly-folded pseudoknot.
Our work is generally consistent with recent studies of S1-mediated unfolding of RNA pseudoknots (5,20). Initial binding of S1 is known to occur through A/U-rich single-stranded regions of an RNA, which are either freely available for binding or transiently accessible through the reversible nature of RNA secondary structure folding. This initial binding destabilizes local secondary and tertiary RNA structure and leads to further pseudoknot unfolding in a multistep process (5,20). Accordingly, we find that the thermodynamic stability of the Tte RNA pseudoknot plays an important role in the S1–RNA interaction, as transient unfolding is required to initiate the binding of S1. In our study, we were able to conveniently modulate this stability by exploiting (near-)cognate ligands of varying affinity.
Upon binding to the pseudoknot, S1 significantly alters the RNA structure as evidenced by a decrease in FRET for the S1-bound form of the pseudoknot labeled with a FRET pair that reports on docking of the P2 helix (Figure 4A). In addition, S1-induced structural destabilization of the pseudoknot is supported by the significant increase in in-line probing activity of the P1 and P2 helices with a concomitant decrease around the A/U-rich single-stranded L3 region (Figure 4B). In light of the well-established preference of S1 for unstructured A/U-rich sequences, a likely candidate for the S1 binding site within these pseudoknots is the sequence element 5′ -UUA ACA AAA CAA-3′, comprised of the A/U-rich L3 loop and three nucleotides of the 3′ half of P2 (Figure 4E). This initial interaction then disrupts the long-range interactions between L3 and the P1 helix, which in turn increases the dynamics within P1. In addition, S1 binding extends L3 away from the stacked helical body of the pseudoknot and slows docking of the 3′ tail to form helix P2, as directly observed here by smFRET (Figures 5–7). This pathway is in good agreement with a recent study by Qu et al. (20), wherein optical tweezers were employed to study the ability of S1 to unwind a long RNA helix. The authors determined that S1 acts as a chaperone that passively unwinds the RNA duplex by binding to its terminal base pairs and rendering them transiently single-stranded, aided by thermal breathing and thus preventing reannealing. In the Tte pseudoknot, the resulting undocking of the 3′ tail ‘unties’ the pseudoknot, resulting in a more elongated conformation, and in doing so necessarily exposes sequences that were previously base paired, albeit transiently (Figure 8). This kind of RNA remodeling into a generally more elongated conformation by S1 provides clues into how the protein facilitates translation initiation consistent with recent work suggesting a role for S1 in coupling transcription with translation in E. coli, where S1 forms part of the interface between RNA polymerase and the small ribosomal subunit to help direct the nascent mRNA from the polymerase onto the ribosome (54).
Our smFRET results provide further, direct insight into the mechanism of S1-mediated RNA unfolding. As seen previously (30), the Tte pseudoknot adopts pre-folded (mid FRET, EFRET ∼0.82) and fully folded (high FRET, EFRET ∼0.96) conformations that exhibit inter-state transitions (Figure 5B). Upon binding, S1 opens the less-compact, pre-folded pseudoknot conformation to adopt a lower FRET value (EFRET ∼0.77) and increases the conformational dynamics of the pseudoknot, as indicated by the smFRET histograms and TODPs (Figure 5C, E). We were able to gain additional insights from applying SiMCAn, an analysis that uses a model-free clustering algorithm and is agnostic to the experimental conditions so that it can identify molecular processes across different datasets that may otherwise have stayed hidden (46). An increase in occupancy of the dynamic clusters H, M1, M2 similarly suggests that S1 induces conformational dynamics in the RNA (Figure 7D), thus exerting its unfolding effect, consistent with the findings by Qu et al. (20), suggesting that passive unwinding of RNA is facilitated by thermal breathing. Earlier studies reported that the unfolding activity of E. coli S1 arises from a structural rearrangement of its domains upon binding to RNA (55). A recent NMR study by Qureshi et al. reported that, in V. vulnificus S1, OB-fold domains D3 and D4 provide the RNA binding platform while RNA chaperone activity is related to the conformational dynamics of domain D5 (56), supporting the picture of coupled RNA and protein folding invoked in our model (Figure 8).
Our smFRET data show that the S1-bound pseudoknot also occasionally visits a more completely unfolded conformation (EFRET ∼0.45), where the pseudoknot transiently adopts significant single-stranded character (Figure 5C, E). This observation may be of particular importance for S1′s role in translation initiation, where it dynamically allows the ribosome to access the translation initiation region of an mRNA (5). However, fully unfolding an RNA structure requires expending a significant amount of free energy, leading to an overall decreased translation rate (57), consistent with our single molecule results indicating that the less single-stranded pre-folded state is favored more than the completely undocked conformation of the pseudoknot. This notion is also reflected in the fact that S1 results in mostly increases in the M1 and M2 clusters, in which FRET is generally >0.7, as opposed to static low FRET (< 0.65) clusters, which are observed only at low abundance.
Consistent with this interplay between RNA and protein dynamics, our results additionally indicate that the unfolding capability of protein S1 strongly depends on the structural stability of the pseudoknot. The observation of largely static (cluster) behavior of the single molecule traces of the preQ1-bound pseudoknot in the presence of S1 suggests that the protein, similar to many other RNA chaperones that act in a largely sequence-unspecific manner and without the use of external energy sources (58), has negligible thermodynamic power to unfold a stable RNA structure. In contrast, S1 can considerably interact with and unfold the weakly stabilized, Gua- or DAP-bound pseudoknots since they retain significant conformational dynamics, allowing S1 to interact with transiently emerging single-stranded regions. These mechanistic insights help define the scope and limitations of how protein S1 can modulate the conformational stability of highly structured 5′ UTRs of mRNAs to facilitate their assembly with the translation initiation complex.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. G. Garcia for the generous gift of preQ1 and Prof. J. Maddock for the original ASKA clones used to prepare the pCA24N_6xHis-TEV_rpsA plasmid.
Notes
Present address: May Daher, Department of Chemistry, University of Detroit Mercy, Detroit, MI 4822, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [GM062357, GM118524 and GM131922 to N.G.W.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. de Smit M.H., van Duin J.. Translational initiation on structured messengers. Another role for the Shine-Dalgarno interaction. J. Mol. Biol. 1994; 235:173–184. [DOI] [PubMed] [Google Scholar]
- 2. Komarova A.V., Tchufistova L.S., Supina E.V., Boni I.V.. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA. 2002; 8:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sorensen M.A., Fricke J., Pedersen S.. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo. J. Mol. Biol. 1998; 280:561–569. [DOI] [PubMed] [Google Scholar]
- 4. Tedin K., Resch A., Blasi U.. Requirements for ribosomal protein S1 for translation initiation of mRNAs with and without a 5′ leader sequence. Mol. Microbiol. 1997; 25:189–199. [DOI] [PubMed] [Google Scholar]
- 5. Duval M., Korepanov A., Fuchsbauer O., Fechter P., Haller A., Fabbretti A., Choulier L., Micura R., Klaholz B.P., Romby P. et al.. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013; 11:e1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hajnsdorf E., Boni I.V.. Multiple activities of RNA-binding proteins S1 and Hfq. Biochimie. 2012; 94:1544–1553. [DOI] [PubMed] [Google Scholar]
- 7. Saguy M., Gillet R., Skorski P., Hermann-Le Denmat S., Felden B.. Ribosomal protein S1 influences trans-translation in vitro and in vivo. Nucleic Acids Res. 2007; 35:2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sukhodolets M.V., Garges S., Adhya S.. Ribosomal protein S1 promotes transcriptional cycling. RNA. 2006; 12:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Durand S., Richard G., Bisaglia M., Laalami S., Bontems F., Uzan M.. Activation of RegB endoribonuclease by S1 ribosomal protein requires an 11 nt conserved sequence. Nucleic Acids Res. 2006; 34:6549–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lebars I., Hu R.M., Lallemand J.Y., Uzan M., Bontems F.. Role of the substrate conformation and of the S1 protein in the cleavage efficiency of the T4 endoribonuclease RegB. J. Biol. Chem. 2001; 276:13264–13272. [DOI] [PubMed] [Google Scholar]
- 11. Brown D., Gold L.. Template recognition by an RNA-dependent RNA polymerase: identification and characterization of two RNA binding sites on Qβ replicase. Biochemistry. 1995; 34:14765–14774. [DOI] [PubMed] [Google Scholar]
- 12. Takeshita D., Yamashita S., Tomita K.. Molecular insights into replication initiation by Qbeta replicase using ribosomal protein S1. Nucleic Acids Res. 2014; 42:10809–10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tomita K. Structures and functions of Qbeta replicase: translation factors beyond protein synthesis. Int. J. Mol. Sci. 2014; 15:15552–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vasilyev N.N., Kutlubaeva Z.S., Ugarov V.I., Chetverina H.V., Chetverin A.B.. Ribosomal protein S1 functions as a termination factor in RNA synthesis by Qbeta phage replicase. Nat. Commun. 2013; 4:1781. [DOI] [PubMed] [Google Scholar]
- 15. Salah P., Bisaglia M., Aliprandi P., Uzan M., Sizun C., Bontems F.. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009; 37:5578–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giorginis S., Subramanian A.R.. The major ribosome binding site of Escherichia coli ribosomal protein S1 is located in its N-terminal segment. J. Mol. Biol. 1980; 141:393–408. [DOI] [PubMed] [Google Scholar]
- 17. McGinness K.E., Sauer R.T.. Ribosomal protein S1 binds mRNA and tmRNA similarly but plays distinct roles in translation of these molecules. PNAS. 2004; 101:13454–13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gytz H., Mohr D., Seweryn P., Yoshimura Y., Kutlubaeva Z., Dolman F., Chelchessa B., Chetverin A.B., Mulder F.A., Brodersen D.E. et al.. Structural basis for RNA-genome recognition during bacteriophage Qbeta replication. Nucleic Acids Res. 2015; 43:10893–10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okada T., Wower I.K., Wower J., Zwieb C.W., Kimura M.. Contribution of the second OB fold of ribosomal protein S1 from Escherichia coli to the recognition of TmRNA. Biosci. Biotechnol. Biochem. 2004; 68:2319–2325. [DOI] [PubMed] [Google Scholar]
- 20. Qu X., Lancaster L., Noller H.F., Bustamante C., Tinoco I. Jr. Ribosomal protein S1 unwinds double-stranded RNA in multiple steps. PNAS. 2012; 109:14458–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Subramanian A.R. Structure and functions of ribosomal protein S1. Prog. Nucleic Acid Res. Mol. Biol. 1983; 28:101–142. [DOI] [PubMed] [Google Scholar]
- 22. Beckert B., Turk M., Czech A., Berninghausen O., Beckmann R., Ignatova Z., Plitzko J.M., Wilson D.N.. Structure of a hibernating 100S ribosome reveals an inactive conformation of the ribosomal protein S1. Nat. Microbiol. 2018; 3:1115–1121. [DOI] [PubMed] [Google Scholar]
- 23. Kolb A., Hermoso J.M., Thomas J.O., Szer W.. Nucleic acid helix-unwinding properties of ribosomal protein S1 and the role of S1 in mRNA binding to ribosomes. PNAS. 1977; 74:2379–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas J.O., Szer W.. RNA-helix-destabilizing proteins. Prog. Nucleic Acid Res. Mol. Biol. 1982; 27:157–187. [DOI] [PubMed] [Google Scholar]
- 25. Studer S.M., Joseph S.. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol. Cell. 2006; 22:105–115. [DOI] [PubMed] [Google Scholar]
- 26. Peselis A., Serganov A.. Structure and function of pseudoknots involved in gene expression control. Wiley Interdiscip. Rev. RNA. 2014; 5:803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eichhorn C.D., Kang M., Feigon J.. Structure and function of preQ1 riboswitches. Biochim. Biophys. Acta. 2014; 1839:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jenkins J.L., Krucinska J., McCarty R.M., Bandarian V., Wedekind J.E.. Comparison of a preQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation. J. Biol. Chem. 2011; 286:24626–24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spitale R.C., Torelli A.T., Krucinska J., Bandarian V., Wedekind J.E.. The structural basis for recognition of the PreQ0 metabolite by an unusually small riboswitch aptamer domain. J. Biol. Chem. 2009; 284:11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suddala K.C., Rinaldi A.J., Feng J., Mustoe A.M., Eichhorn C.D., Liberman J.A., Wedekind J.E., Al-Hashimi H.M., Brooks C.L. 3rd, Walter N.G.. Single transcriptional and translational preQ1 riboswitches adopt similar pre-folded ensembles that follow distinct folding pathways into the same ligand-bound structure. Nucleic Acids Res. 2013; 41:10462–10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roth A., Winkler W.C., Regulski E.E., Lee B.W., Lim J., Jona I., Barrick J.E., Ritwik A., Kim J.N., Welz R. et al.. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat. Struct. Mol. Biol. 2007; 14:308–317. [DOI] [PubMed] [Google Scholar]
- 32. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H.. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005; 12:291–299. [DOI] [PubMed] [Google Scholar]
- 33. Lancaster L., Noller H.F.. Involvement of 16S rRNA nucleotides G1338 and A1339 in discrimination of initiator tRNA. Mol. Cell. 2005; 20:623–632. [DOI] [PubMed] [Google Scholar]
- 34. Rinaldi A.J., Lund P.E., Blanco M.R., Walter N.G.. The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat. Commun. 2016; 7:8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Willkomm D.K., Hartmann R.K.. Handbook of RNA Biochemistry. 2008; Wiley-VCH Verlag GmbH; 86–94. [Google Scholar]
- 36. Roth A., Winkler W.C., Regulski E.E., Lee B.W.K., Lim J., Jona I., Barrick J.E., Ritwik A., Kim J.N., Welz R. et al.. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat. Struct. Mol. Biol. 2007; 14:308–317. [DOI] [PubMed] [Google Scholar]
- 37. Bevilacqua P.C., Cech T.R.. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996; 35:9983–9994. [DOI] [PubMed] [Google Scholar]
- 38. Mergny J.L., Lacroix L.. Analysis of thermal melting curves. Oligonucleotides. 2003; 13:515–537. [DOI] [PubMed] [Google Scholar]
- 39. Puglisi J.D., Tinoco I. Jr. Absorbance melting curves of RNA. Methods Enzymol. 1989; 180:304–325. [DOI] [PubMed] [Google Scholar]
- 40. Roy R., Hohng S., Ha T.. A practical guide to single-molecule FRET. Nat. Methods. 2008; 5:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joo C., Ha T.. Selvin PR, Ha T. Single-Molecule Techniques: A Laboratory Manual. 2008; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 42. Aitken C.E., Marshall R.A., Puglisi J.D.. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008; 94:1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rasnik I., McKinney S.A., Ha T.. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods. 2006; 3:891–893. [DOI] [PubMed] [Google Scholar]
- 44. Widom J.R., Nedialkov Y.A., Rai V., Hayes R.L., Brooks C.L. 3rd, Artsimovitch I., Walter N.G.. Ligand modulates cross-coupling between riboswitch folding and transcriptional pausing. Mol. Cell. 2018; 72:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koh H.R., Wang X., Myong S.. Visualizing repetitive diffusion activity of double-strand RNA binding proteins by single molecule fluorescence assays. Methods. 2016; 105:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blanco M.R., Martin J.S., Kahlscheuer M.L., Krishnan R., Abelson J., Laederach A., Walter N.G.. Single Molecule Cluster Analysis dissects splicing pathway conformational dynamics. Nat. Methods. 2015; 12:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bordeau V., Felden B.. Ribosomal protein S1 induces a conformational change of tmRNA; more than one protein S1 per molecule of tmRNA. Biochimie. 2002; 84:723–729. [DOI] [PubMed] [Google Scholar]
- 48. Blose J.M., Proctor D.J., Veeraraghavan N., Misra V.K., Bevilacqua P.C.. Contribution of the closing base pair to exceptional stability in RNA tetraloops: roles for molecular mimicry and electrostatic factors. J. Am. Chem. Soc. 2009; 131:8474–8484. [DOI] [PubMed] [Google Scholar]
- 49. Soukup G.A., Breaker R.R.. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999; 5:1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Strauss B., Nierth A., Singer M., Jaschke A.. Direct structural analysis of modified RNA by fluorescent in-line probing. Nucleic Acids Res. 2012; 40:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blanco M., Walter N.G.. Analysis of complex single-molecule FRET time trajectories. Methods Enzymol. 2010; 472:153–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jalihal A.P., Lund P.E., Walter N.G.. Coming together: RNAs and proteins assemble under the Single-Molecule fluorescence microscope. Cold Spring Harb. Perspect. Biol. 2019; 11:a032441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bronson J.E., Fei J.Y., Hofman J.M., Gonzalez R.L., Wiggins C.H.. Learning rates and states from biophysical time series: a bayesian approach to model selection and Single-Molecule FRET data. Biophys. J. 2009; 97:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Demo G., Rasouly A., Vasilyev N., Svetlov V., Loveland A.B., Diaz-Avalos R., Grigorieff N., Nudler E., Korostelev A.A.. Structure of RNA polymerase bound to ribosomal 30S subunit. Elife. 2017; 6:pii: e28560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aliprandi P., Sizun C., Perez J., Mareuil F., Caputo S., Leroy J.L., Odaert B., Laalami S., Uzan M., Bontems F.. S1 ribosomal protein functions in translation initiation and ribonuclease RegB activation are mediated by similar RNA-protein interactions: an NMR and SAXS analysis. J. Biol. Chem. 2008; 283:13289–13301. [DOI] [PubMed] [Google Scholar]
- 56. Qureshi N.S., Bains J.K., Sreeramulu S., Schwalbe H., Furtig B.. Conformational switch in the ribosomal protein S1 guides unfolding of structured RNAs for translation initiation. Nucleic Acids Res. 2018; 46:10917–10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Espah Borujeni A., Channarasappa A.S., Salis H.M.. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014; 42:2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Doetsch M., Schroeder R., Furtig B.. Transient RNA-protein interactions in RNA folding. FEBS J. 2011; 278:1634–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The various Matlab scripts, input data, and resulting analysis files associated with this work are available in DeepBlue from the University of Michigan library (https://deepblue.lib.umich.edu/data).