


Abstract
In order to discover new antibiotics with improved activity and selectivity, we created a reliable in vitro reporter system to detect trans-translation activity, the main mechanism for recycling ribosomes stalled on problematic messenger RNA (mRNA) in bacteria. This system is based on an engineered tmRNA variant that reassembles the green fluorescent protein (GFP) when trans-translation is active. Our system is adapted for high-throughput screening of chemical compounds by fluorescence.
INTRODUCTION
During translation, ribosomes translate the messenger RNA (mRNA) and synthesize the nascent polypeptides until they encounter a stop codon on the mRNA. However, ribosomes regularly stall on the 3′ end of mRNA, particularly when termination codon is missing (1). In bacteria, trans-translation is the main rescue mechanism for clearing these trapped ribosomes (2,3). This process is driven by transfer-messenger RNA, or tmRNA (Figure 1A), along with its partner small protein B (SmpB) (4). tmRNA has similar properties to both transfer RNA (tRNA) and mRNA (5), is encoded by the ssrA gene, and is aminoacylated with an alanine. When ribosomes reach the 3′ end of non-stop mRNA, the ribosome decoding site (A site) becomes vacant. The tmRNA–SmpB complex associates with the EF-Tu elongation factor and recognizes the stalled ribosome since SmpB’s C-terminal tail enters the vacant mRNA path (6). Transpeptidation then occurs between the stalled nascent polypeptide and alanyl-tmRNA. The tRNA-like domain (TLD) of tmRNA then moves to the P site, and translation resumes on tmRNA’s internal mRNA-like domain (MLD). The stop codon present in the MLD facilitates termination of trans-translation. What is ingenious in this whole process is that tmRNA’s short internal open reading frame (AANDENYALAA in E. coli) encodes a specific sequence that is recognized by proteases and is followed by a termination codon. This allows degradation of the incomplete peptide after its release. The ribosomes are then recycled and the problematic non-stop mRNA is degraded by RNase R (7,8). Trans-translation is an appealing target for new antibiotic molecules because: (i) there is no trans-translation in eukaryotes, (ii) it is essential for either the survival or virulence of many pathogenic bacteria (9,10) and (iii) in cases where the deletion is not lethal, it induces hypersensitive phenotypes thus making antibiotics more efficient (11).
Figure 1.
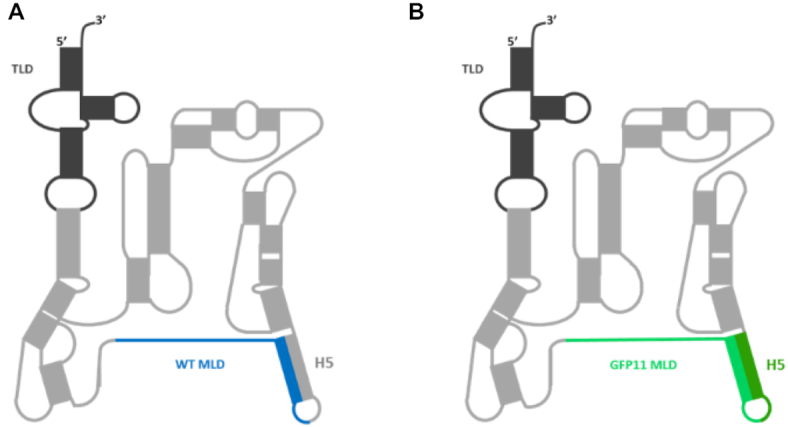
Secondary structure of tmRNA. (A) Wild-type Escherichia coli tmRNA. The transfer-like domain (TLD) is dark grey and the messenger-like domain (MLD) is blue (ANDENYALAA). (B) Mutated tmRNAGFP11 with an engineered MLD encoding the eleventh GFP domain (i.e. beta-strand) in green (ARDHMVLHEYVNAAGIT). Dark green compensatory mutations maintain the base-pairing interactions of helix H5.
In vivo trans-translation reporter systems were recently developed but they are specific to E. coli and dependent on other degradation or rescuing pathways of the cells (12,13). To date, several kinds of cell-free trans-translation systems have been developed. However, they are mainly based on the use of radio-labelled elements and/or purification of the trans-translated products by SDS/PAGE or HPLC (12,14–16), impairing their use for a wide screening of antibiotics in pathogenic bacteria. To explore the targeting of trans-translation for development of novel antibiotics, we designed and set up an effective in vitro fluorescent reporter system adapted to high throughput screening.
MATERIALS AND METHODS
Plasmid construction
We obtained the gene that encodes for the mature tmRNAGFP11 by primer-directed mutagenesis based on a pGEMEX-tmRNA plasmid (17) template using primers #1 and #2 (Supplementary Table S1). The specific ANDENYALAA sequence recognized by proteases was replaced by ARDHMVLHEYVNAAGIT (which contains the first conserved alanine of native tmRNA, underlined, plus the eleventh domain (i.e. beta-strand) of the ‘superfolder’ GFP called ‘sfGFP’). We also added compensatory mutations in order to preserve the H5 helix (see Figure 1B and sequence Supplementary Table S2). The fragment obtained was then inserted between the pGEMEX plasmid's NdeI and HindIII sites to generate pGEMEX-mtmRNAGFP11. We then used this first plasmid to construct pBstNav-mtmRNAGFP11 and pUC19mtmRNAGFP11 plasmids for in vitro and in vivo production, respectively. We also amplified the mature tmRNAGFP11 sequence (see Nucleotide Sequences) using primers #3 and #4 on the pGEMEX-mtmRNAGFP11 plasmid. The resulting sequences were cloned into the pBstNav vector between the EcoRI and PstI restriction sites. Meanwhile, the same tmRNAGFP11 sequence was amplified with primers #5 and #6 (Supplementary Table S1) and cloned between HindIII and BamHI restriction sites on the pUC19 vector, thus generating the pUC19mtmRNAGFP11 plasmid.
The smpB gene was amplified from the E. coli genome using primers #10 and #11 (Supplementary Table S1) and inserted into the pET-22b(+) vector (AmpR) between the NdeI and XhoI restriction sites. The resulting pF1275 plasmid was verified by sequencing, and allows for C-terminal his-tag SmpB protein production under control of the T7 promoter.
His-tagged alanyl-tRNA synthetase protein (AlaRS) was produced using the plasmid pQE30 containing his-tagged alaS gene (18).
Protein purification
SmpB: His-tagged SmpB proteins were expressed from the pF1275 vector under the control of a T7 promoter in BL21(DE3)ΔssrA cells (19). Cultures were made in lysogeny broth (LB) at 30°C supplemented with ampicillin (100 μg/ml) and kanamycin (50 μg/ml). Protein expression was induced in the exponential phase (OD600 nm 0.6) with 0.1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) overnight at 16°C. Cells were centrifuged, washed, then resuspended in lysis buffer (HEPES–KOH 50 mM, KCl 200 mM, imidazole 20 mM and DTT 1 mM, pH 7.5). Cell lysis was performed using a French press. The lysate was centrifuged, the supernatant was filtrated (0.2 μm) and injected on a Ni-NTA sepharose column (HisTrap FF, GE Healthcare) previously equilibrated with the lysing buffer. The column was washed with 100 ml lysis buffer and 50 ml washing buffer (HEPES–KOH 50 mM, KCl 200 mM, NH4Cl 1 M, imidazole 20 mM and DTT 1 mM pH 7.5) before elution with 500 mM imidazole. Finally, an Amicon 10 kDa system was used to concentrate the fractions containing pure SmpB, changing the buffer to a concentration buffer (HEPES–KOH 50 mM, KCl 100 mM, glycerol 10% and DTT 1 mM, pH 7.5).
AlaRS: His-tagged alanyl-tRNA synthetase (AlaRS) protein was purified similarly to SmpB. A final concentration of 1 mM IPTG was used for 4 h at 37°C to induce protein production. The buffers used to concentrate and dialyze the protein in the Amicon 100 kDa purification process were as follows: lysis buffer (NaH2PO4/Na2HPO4 50 mM, NaCl 500 mM, imidazole 10 mM and glycerol 10%, pH 7.4); washing buffer (NaH2PO4/Na2HPO4 50 mM, NaCl 500 mM, imidazole 30 mM, glycerol 10%, pH 7.4); elution buffer (NaH2PO4/Na2HPO4 50 mM, NaCl 500 mM, imidazole 500 mM and glycerol 10%, pH 7.4); and concentration buffer (Tris–HCl 60 mM, MgCl2 10 mM, glycerol 50% and DTT 1 mM, pH 7.5).
In vivo purification of tmRNAGFP11
Mutated tmRNAGFP11 was produced in vivo in a JM101tr E. coli strain. After phenol chloroform extraction, tmRNAGFP11 was purified in native conditions as previously described (20). The RNA molecule was first separated from the RNA pool in two steps using RESOURCE Q and MONO Q columns (GE Healthcare), both pre-equilibrated with a buffer (KH2PO4/K2HPO4 20 mM and EDTA 1 mM, pH 6.5), and using NaCl for elution. Sample purity was then polished on a GE Superdex 200 in a buffer solution (KH2PO4/K2HPO4 20 mM, 150 mM NaCl and 2 mM EDTA, pH 6.5). The tmRNAGFP11 eluted as monomers.
In vitro production of tmRNAGFP11
Mutated tmRNAGFP11 was transcribed in vitro from the pUC19mtmRNAGFP11 plasmid. To generate the CCACCA 3′ end needed for aminoacylation, the plasmid (10 μg) was digested by the BsmBI restriction enzyme, then the DNA was purified using phenol/chloroform. The purified digested plasmid was precipitated and the resulting pellet resuspended overnight in 40 μl nuclease-free water. We used a MEGAscript T7 transcription kit (ThermoFisher Scientific) to produce the tmRNAGFP11 before its purification using a MEGAclear kit (Ambion/Life Technologies).
DNA template and oligonucleotide production
For trans-translation assays the nonstop GFP1-10 sequence was produced by PCR using primers #7 and #8 (with pETGFP 1–10 vector as a template). Similarly, sfalaGFP (the superfolder GFP with the additional conserved alanine between the sfGFP1–10 and sfGFP11 domains, contained in the tmRNAGFP11 sequence) was amplified using primers #7 and #9 from the same template, both of which have a T7 promoter upstream from their coding sequences. The resulting PCR products were purified using QIAquick® PCR Purification Kit (Quiagen). Antisense oligonucleotides (Supplementary Table S1) were ordered from Eurofins company.
Translation and trans-translation assays
In vitro translation was performed as recommended by New England Biolabs (NEB) using PURExpress®In Vitro Protein Synthesis Kit. 250 ng of PCR product were added to the reaction to produce the sfalaGFP. Translation reactions were incubated for 3 h and trans-translation reactions for 4 h in a thermocycler at 37°C. The trans-translation assays were performed using the same protein synthesis kit. PURExpress® 70S ribosomes were first stalled by adding 250 ng of purified PCR product encoded for nonstop sfGFP1-10 under control of the T7 promoter (sequence Supplementary Table S2), for 15 min at 37°C in a thermocycler. During this step, to neutralize any native tmRNA that might be present in the kit, 5 μM oligonucleotide antisense anti-tmRNA were added (sequence A, Supplementary Table S1). Then, stalled ribosomes were pooled with 50 pmol tmRNAGFP11 and 100 pmol SmpB before a 4 h incubation. Note that either trans-translation assays with in vivo or in vitro purified tmRNAGFP11 works in a similar way. The same is true with an aminoacylated tmRNAGFP11. In that case, 50 pmol tmRNAGFP11 was aminoacylated with 50 pmol SmpB, 75 pmol AlaRS, 2.5 mM ATP and 30 μM alanine during 30 min at 37°C. The aminoacylated tmRNAGFP11 was then added to the stalled PURExpress® ribosomes supplemented by 50 other pmol of SmpB (100 pmol final quantity).
Trans-translation inhibition assays were performed by adding the different molecule inhibitors together with stalled ribosomes and before addition of tmRNAGFP11. Various concentrations of KKL-35 compound (1–400 μM) and PA-1 aptamer (2.5–20 μM) were tested. KKL-35 was provided by Dr Mickael Jean, Inserm COSS U1242, Rennes, France). Its antimicrobial activity was controlled by CMI measurements on E. coli BW25113ΔtolC in parallel of each trans-translation assay (13). The peptide Aptamer PA-1 (GGVTFLVNTYPNGVQSRAGG-NH2) was ordered from ProteoGenix (Schiltingheim, France). Because KKL-35 and PA-1 were conserved in DMSO, the trans-translation reactions as well as the controls contain 2.86% final concentration of DMSO.
Fluorescence analysis
After incubations at 37°C, translation or trans-translation reaction volumes were adjusted to 150 μl and put it into cuvettes for fluorescence measurement using a LS-55 fluorescence spectrometer (PerkinElmer). The translated or trans-translated sfalaGFP showed a fluorescence intensity of 510 nm when using an excitation of 485 nm. The photomultiplicator was programmed at 650 V for translation assays and at 775 V for trans-translation assays.
Trans-translation radioactive assay
In vitro trans-translation experiments were performed as described above, using the PURExpress kit, but with a different PCR product encoding for a non-stop DHFR. This PCR product was amplified from the plasmid template supplied by NEB using primers #9 (Supplementary Table S1) and ‘dhfr-ns reverse’ (12). Visualization of translated and trans-translated non-stop DHFR was made possible by the addition of [35S]-methionine, according to manufacturer's instructions (PURExpress, NEB). After 4 h incubation at 37°C, reactions were separated on 15% (w/v) SDS polyacrylamide gels, and visualized by exposure to a phosphor screen (Typhoon FLA 9500, GE Healthcare). Radioactive bands intensities were calculated using ImageJ software. Trans-translation efficiencies were calculated as the percentage of tagged DHFR over total translated DHFR (tagged + non-tagged), after background signals subtraction.
Validation of the assay by screening a new set of oxadiazole compounds
Assays were performed using PURExpress®In Vitro Protein Synthesis Kit supplemented with 6.25 ng/μl of purified PCR product encoding for nonstop sfGFP1-10, 1.25 μM of tmRNAGFP11, 2.5 μM of SmpB, 5 μM of antisense A, 2% DMSO (vehicle and neutral control) and 2 μM of antisense B (scale reference control). In order to optimize the reaction in 96-well plates format, all the components of the master-mix were added at the same time. Assays were then assembled in three steps. First, 400 nl of control or compound (100 μM final) were transferred in PCR microtubes. Next, 19.6 μl of master-mix were added. Finally, the tubes were vortexed and 19 μl of each reactional volume were transferred in qPCR microplates. After 300 min incubation at 37°C, fluorescence was measured by using the PCR system Step One plus (Applied Biosystems). The assays were validated statistically by determining the Z’ factor (calculated using the formula: Z’ = 1 – [(3σneutral control + 3σscale reference control)/(μneutral control – μscale reference control)]) (21).
RESULTS AND DISCUSSION
To allow for the monitoring of trans-translation via the fluorescence levels of a reconstituted green fluorescent protein (GFP) we developed an assay based on reassembling an active ‘superfolder’ GFP (sfGFP) after tmRNA tagging. Indeed, semi-synthetic green fluorescent proteins, such as the well-folded sfGFP (22), are made up of eleven domains that can be assembled by adding a short fragment to a truncated protein. This split GFP method (23) is based on a break between the tenth and eleventh beta-strands of GFP, after which the two resulting moieties, GFP1-10 and GFP11, are easily reassembled (24). The GFP1-10 fragment is not fluorescent on its own, but recovers complete fluorescence when complemented by GFP11, a short peptide made of 16 amino acids only. Their combination results in chromophore maturation within the reconstituted functional GFP. Rather than simply being added by self-complementation as occurs in split GFP (25,26), in our approach the missing domain is added covalently to a non-stop sfGFP1-10 through a manipulated trans-translation process. To do this, we engineered a mature tmRNA variant in which the MLD encoding for the sequence specifically recognized by proteases was replaced by the 11th domain sfGFP11 sequence (ARDHMVLHEYVNAAGIT, Figure 1B) followed by a termination codon. The original nucleotides upstream from the resume codon and the alanine resume codon (underline in the above sequence) were kept, since they both play instrumental roles in re-registration of the reading frame tagging (27). To minimize disruption of the tmRNA structure and conserve its helix H5 base-pairing interactions, we also engineered compensatory mutations (dark green H5, Figure 1B). We named the resulting modified tmRNA ‘tmRNAGFP11’ (sequence in Supplementary Table S2). We then took advantage of the reconstituted cell-free protein synthesis system, PURExpress® (New England Biolabs), to evaluate the tagging activity of tmRNA in vitro (28) (see Supporting information). This kit contains all of the E. coli components necessary for both transcription and translation. To transform this kit into a fluorescent in vitro trans-translation system, we added the components needed for trans-translation: tmRNAGFP11; SmpB; and a non-stop DNA sequence encoding the first ten sfGFP domains to stall ribosomes (sequence in Supplementary Table S2). Scheme 1 depicts our approach, beginning with the ribosome stalled on a non-stop mRNA (step 2, Scheme 1). The complex made of alanyl-tmRNAGFP11 and SmpB is added to the reaction. The complex is accommodated on the ribosome, and the translation restarts on the tmRNAGFP11 MLD (steps 3 and 4, Scheme 1). As a consequence, the eleventh domain is attached to sfGFP1-10 by trans-translation. A functional sfalaGFP is released and the two ribosomal subunits recycled (step 5, Scheme 1).
Scheme 1.

Reassembling GFP by in vitro trans-translation. 1: Canonical translation of sfGFP1-10 mRNA lacking a stop codon (red cross). 2: Due to the absence of termination signal, ribosome is stalled. Two tRNAs are in the P (yellow) and E (purple) sites, and the A site is empty. 3: The tmRNAGFP11-SmpB complex binds to the stalled ribosome, and SmpB’s C-terminal tail recognizes the empty A site. 4: The translation restarts thanks to the tmRNAGFP11 MLD, which encodes the missing eleventh domain of the sfGFP (green tmRNA section) and therefore adds this beta-strand to the incomplete sfGFP1-10. 5: The process ends when the tmRNA stop codon (red star) is reached. The complete sfGFP is released and becomes fluorescent. The 50S and 30S subunits are dissociated to be reused, and the tmRNAGFP11–SmpB complex is recycled.
We first assessed the effectiveness of transcription and translation of our in vitro fluorescent reporter system by ensuring that neither the PURExpress kit components alone, nor with the sfGFP1-10 first ten translated domains displayed any basal fluorescence (not shown). The next step was to ensure that the PURExpress system allows synthesis of a functional (i.e. fluorescent) sfGFP, even with an additional alanine between the sfGFP1-10 and sfGFP11 domains (‘sfalaGFP’). The results show a strong fluorescent signal for production of sfalaGFP (sequence in Supplementary Table S2), indicating that the template DNA was well-transcribed and well-translated, and that the fluorescent protein was functional (Table 1, line 1). The fluorescence was reduced by five times in the presence of 100μM chloramphenicol, a strong inhibitor of protein synthesis (Table 1, line 2)
Table 1.
Fluorescence intensities in PURExpress® kit
| Translation | Trans-translation | |||
|---|---|---|---|---|
| Neutral control | Scale reference control (+ Chloramphenicol) | Neutral control | Scale reference control (+ antisense B) | |
| Fluorescence (A.U.) | 520619.2 ± 36674.79 | 10338.30 ± 2027.69 | 206805.93 ± 11323.28 | 4233.22 ± 1046.49 |
We then moved on to test trans-translation by using non-stop DNA sequence encoding the first ten sfGFP domains, tmRNAGFP11 and SmpB. The strongest fluorescent signal was recovered after 4 h (negative control vs trans-translation, Figure 2A), confirming that the in vitro system developed in this study is efficient and might be useful to investigate trans-translation inhibitors. To avoid a possible competition between the endogenous tmRNA already present in the kit and our tmRNAGFP11, we also added an antisense oligonucleotide targeting wild-type tmRNA (WT tmRNA) only (antisense A, Supplementary Table S1). Indeed, the addition of this antisense increases the fluorescent signal by almost 60% (trans-translation vs antisense A, Figure 2A). The level of tagged GFP fluorescence is 2.5 times lower relative to full sfalaGFP (Table 1, compare lines 1 and 3).
Figure 2.

Quantification of in vitro trans-translation in the presence of inhibitors by fluorescence and radioactivity assays. (A) Fluorescent assay. The ‘negative control’ contains all trans-translation components except tmRNAGFP11; ‘trans-translation’: same as negative control with tmRNAGFP11; ‘Antisense A’ and ‘Antisense A and B’: same as ‘trans-translation’ respectively with an antisense targeting WT tmRNA or an antisense targeting tmRNAGFP11 (Supplementary Table S1). (B) Radioactive assay. Trans-translation reactions were performed as in fluorescent assays but using a non-stop DHFR instead of truncated GFP and [35S]-methionine (see methods). The non-stop DHFR is indicated by a black arrow and the lower-mobility trans-translated DHFR proteins bands are indicated by a black or a green star, depending on their tagging by WT tmRNA or tmRNAGFP11, respectively. (C) In vitro translation assays. Effect of SmpB peptide aptamer PA-1 on in vitro translation tested at final 2.5–20 μM concentrations. For experiments, the PURExpress® kit basal fluorescence was subtracted from the measurements and the results are then normalized with respect to the translation control (symbolized with the black dotted line). A t-test was performed using that control hypothetical value. Bars represent standard deviation of at least three independent experiments. ‘ns’ is for non-significant (D) In vitro trans-translation assays. Peptide aptamer was added at the same concentration than in translation assays. For these experiments, measurements were normalized with respect to the trans-translation control (‘Antisense A’) without any compounds (symbolized with the black dotted line). A t-test was performed using that control hypothetical value. P-values indicates significant differences compared to the control. Bars represent standard deviation of at least three independent experiments.
To confirm that inhibition of trans-translation is correlated to a loss of fluorescence signal, we first designed an antisense oligonucleotide to inhibit tmRNAGFP11 activity (antisense B, Supplementary Table S1). Its addition decreases fluorescence intensities by more than 95% (antisense A and B, Figure 2A). This demonstrates the strong correlation between trans-translation and fluorescence rates (Table 1, line 4). Note that trans-translation assays were performed using tmRNAGFP11 purified either in vivo or in vitro, and with or without a prior tmRNAGFP11 aminoacylation step, with no relevant differences (data not shown).
In order to compare this new assay with previously described methods, we performed the same in vitro trans-translation assays using radioactivity (15) (see Materials and Methods). We used both tmRNAGFP11 and WT tmRNA, in order to compare their trans-translation efficiencies (Figure 2B). The bands corresponding to the tagged DHFR were confirmed by adding specific antisense oligonucleotides (Figure 2B, lines 3 and 6). Addition of 50 pmol (as in the GFP assay) WT tmRNA to the reaction led to a total tagging activity of 49% (Figure 2B, line 2). However, it turns out that endogenous WT tmRNA from the PURExpress kit has a significant activity of 31% (Figure 2B, line 1). Therefore, after subtraction of this signal, the tagging activity corresponding to the addition of 50 pmol WT tmRNA is of 18% only. Same experiments were then performed using 50 pmol tmRNAGFP11. An antisense oligonucleotide was used to target endogenous WT tmRNA and avoid competition. Since tmRNAGFP11 adds a larger tag than WT tmRNA, its activity was directly measured by calculating the intensity of the upper band (Figure 2B, line 4). It reaches 21% (Figure 2B, line 5), highlighting the similar efficiency of tmRNAGFP11 and WT tmRNA.
Finally, peptide aptamer PA-1 (sequence GGVTFLVNTYPNGVQSRAGG) known to knock down SmpB (29,30) was then used as trans-translation inhibitor. To avoid compounds that inhibit any step necessary for fluorescence (e.g. transcription, translation, or GPF folding) to be scored as positive and result in false positive hits, trans-translation assays are always preceded by a translation assay. As expected, excesses of PA-1 did not exhibit any activity on transcription or translation (Figure 2C). Consequently, we evaluated the anti-trans-translation activity of this molecule (Figure 2D). Contrary to what we observed for translation, the peptide aptamer strongly inhibits trans-translation even at the smallest concentration (which represent a 1:1 ratio SmpB:Aptamer), according to in vivo inhibitions assays reported by Liu et al., in 2016 and 2017 (29,30). Additionally, we also tested the chemical oxadiazole compound KKL-35 (12) whose in vivo activity against trans-translation was recently called into question (13,31). A minor activity was observed and for concentrations higher or equal to 10 μM with no dose-dependent effect (Supplementary Figure S1). This moderate effect, taken together with in vivo trans-translation assays experiments (13,31), suggests that KKL-35 have indeed other in vivo targets besides trans-translation. The same order of magnitude results were obtained using tmRNAGFP11 in radioactive assays (Figure 2 and Supplementary Figures S1 and S3), suggesting that the sensitivity of the fluorescent assay is similar to the radioactive one, with the advantage of making rapid and non-radioactive screening experiments possible.
Therefore, in order to perform screening assays, we first optimized the current fluorescent assay in 96-well plates format (see Materials and Methods) and carried out kinetic experiments to follow fluorescence over time. The maximum signal was recovered after ∼300 min (Figure 3A). These conditions were then used to assess the suitability of the assay to screen a small library of compounds. To date, 1,3,4-oxadiazole compounds have been identified as the most promising inhibitors of trans-translation (12). However, due to multiple cellular targets in bacteria, it is difficult to compare the direct effects of the oxadiazole variations on trans-translation in vivo, making the current in vitro assay necessary. In order to improve their potential anti-trans-translation activity, we recently designed and synthesized a new series of oxadiazole derivatives (33 and Supplementary Table S3) that were submitted to the present assay. We first performed a translation assay that confirmed that none of the compounds inhibits any step (e.g. transcription, translation, or GPF folding) necessary for fluorescence (Supplementary Figure S2). On the other hand, the thirteen molecules behave differently regarding trans-translation (Figure 3B). Most of the molecules displayed the same moderate effects as KKL-35, the parent compound, while CT1-83 inhibited trans-translation much more strongly, displaying an inhibitory effect of 26% at concentration of 100 μM. This indicates that replacement of the benzene of the chloro-aryl moiety of KKL-35 by a pyridine group in CT1-83 affects the specificity of the molecule towards trans-translation, information that can be used to make future improvements. The activity of CT1-83 against trans-translation was confirmed using the same conditions in a radioactive assay, despite at a lower level (Supplementary Figure S3). Finally, and most surprisingly, CT1-69 caused a slight increase in trans-translation rates, certainly due to an activation of another step necessary for fluorescence (Supplementary Figure S2). This suggests that the nitrogen atoms from the heterocyclic ring are necessary for targeting trans-translation. The accuracy of the test was confirmed through Z′ factor calculation (21) (Figure 3B). The results show low plate-to-plate and well-to-well variation as evidenced by a high Z′ factor of 0.82, indicating that the assay is robust enough for high throughput screening (32).
Figure 3.
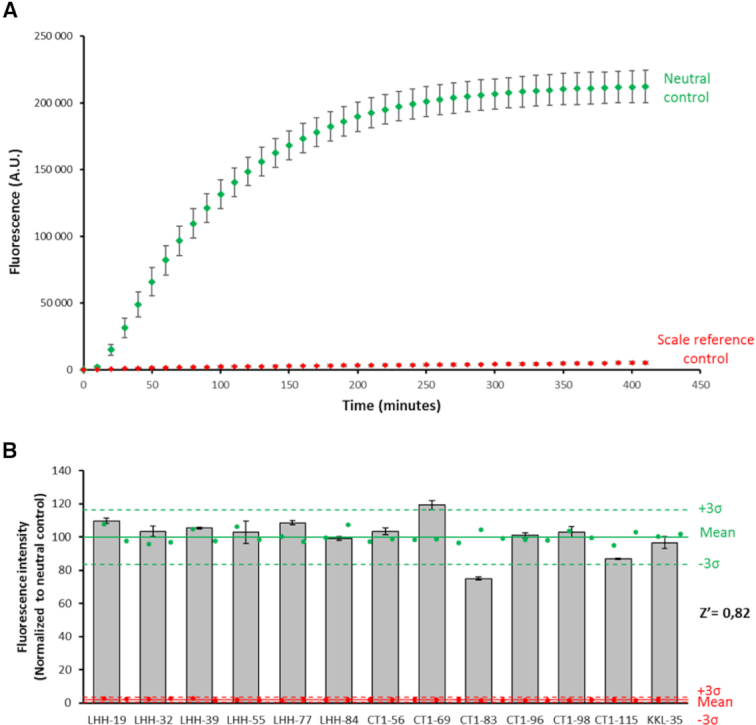
Fluorescent trans-translation in microplates for HTS. (A) Trans-translation kinetics over time in microplates for HTS. The fluorescent increase is directly linked to trans-translation. The neutral control contains all necessary components for trans-translation (see methods). The scale reference control is the same as the neutral control but in the presence of 2 μM antisense B (Supplementary Table S1). The five experiments have been performed during five different days (n = 6 or 5 or 3 wells per group per plate). (B) Screening of a small library of compounds. The anti-trans-translation activity of the compounds at 100 μM concentration was measured in 96-well qPCR plates. Neutral controls are green circles (n = 28), scale reference controls are red circles (n = 27). The solid green or red lines represent the mean fluorescence, the dashed green or red lines represent 3SD of respectively neutral and scare reference controls and tested compounds are grey bars (n = 3). The results were shown as mean ± standard deviation and normalized to neutral control (%) neutral control = (fluorescence compounds condition/mean fluorescence neutral control) × 100. Compounds that inhibited the assay to >3 SD were identified as positive hits. The average Z′ factor is 0.82.
To summarize, we created a reliable, easy-to-use in vitro fluorescent system that can be used to measure trans-translation efficiency. In addition to its inherent rapidity, it is also optimized for HTS of chemical compounds in multi-well microplates using a microplate fluorimeter. Miniaturized assays can be handled by reducing volumes further to few nanoliters to lower the average screening costs. Adapting this system to specific multiresistant pathogenic bacteria will be the next step for the development of narrow-spectrum antibiotics. Towards this end we will adapt this system by using tmRNAGFP11, SmpB and ribosomes from the six ESKAPE bacterial pathogens commonly associated with antimicrobial resistance (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.).
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Stéphanie Cabantous (Institute of Pharmacology and Structural Biology, Toulouse) for providing the pETGFP 1–10 vector and Mickael Jean for providing KKL-35 and other oxadiazole compounds. The authors also are particularly grateful to Emmanuel Giudice, Daniel Boujard and Romain Gibeaux for their precious advices and Juliana Berland for insightful comments on the manuscript.
Author contributions: Charlotte Guyomar (C.G.) and Marion Thépaut (M.T.) performed the experiments and analysis. C.G., M.T. and Renan Goude (R.Go.) measured fluorescence, R.Go. participated to clonings, Sylvie Nonin-Lecomte produced and purified tmRNAs in vivo. Agnès Méreau, C.G. and M.T. performed radioactivity assays experiments. C.G. and R.Gi. wrote the paper. R.Gi. supervised the project. All authors have given approval to the final version of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale pour la Recherche, the Direction Générale de l’Armement [ANR-14-ASTR-0001]; Agence Nationale pour la Recherche under the frame of the Joint Programming Initiative on Antimicrobial Resistance (JPIAMR) Project named ‘Ribotarget – Development of novel ribosome-targeting antibiotics’; SATT Ouest-Valorisation [DV 2552]; Charlotte Guyomar is supported by Direction Générale de l’Armement and Université de Rennes 1 for PhD financial support. Funding for open access charge: CNRS.
Conflict of interest statement. Charlotte Guyomar and Reynald Gillet are co-inventors of the system described here (patent application #EP/2018/063780).
REFERENCES
- 1. Giudice E., Gillet R.. Bacterial trans-translation: from functions to applications. Reviews in Cell Biology and Molecular Medicine. 2014; American Cancer Society; 1–33. [Google Scholar]
- 2. Giudice E., Macé K., Gillet R.. Trans-translation exposed: understanding the structures and functions of tmRNA-SmpB. Front. Microbiol. 2014; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Himeno H., Kurita D., Muto A.. tmRNA-mediated trans-translation as the major ribosome rescue system in a bacterial cell. Front. Genet. 2014; 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karzai A.W., Susskind M.M., Sauer R.T.. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA). EMBO J. 1999; 18:3793–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muto A., Ushida C., Himeno H.. A bacterial RNA that functions as both a tRNA and an mRNA. Trends Biochem. Sci. 1998; 23:25–29. [DOI] [PubMed] [Google Scholar]
- 6. Neubauer C., Gillet R., Kelley A.C., Ramakrishnan V.. Decoding in the absence of a codon by tmRNA and SmpB in the ribosome. Science. 2012; 335:1366–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards J., Mehta P., Karzai A.W.. RNase R degrades non-stop mRNAs selectively in an SmpB-tmRNA-dependent manner. Mol. Microbiol. 2006; 62:1700–1712. [DOI] [PubMed] [Google Scholar]
- 8. Domingues S., Moreira R.N., Andrade J.M., dos Santos R.F., Bárria C., Viegas S.C., Arraiano C.M.. The role of RNase R in trans-translation and ribosomal quality control. Biochimie. 2015; 114:113–118. [DOI] [PubMed] [Google Scholar]
- 9. Huang C., Wolfgang M.C., Withey J., Koomey M., Friedman D.I.. Charged tmRNA but not tmRNA-mediated proteolysis is essential for Neisseria gonorrhoeae viability. EMBO J. 2000; 19:1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Julio S.M., Heithoff D.M., Mahan M.J.. ssrA (tmRNA) plays a role in Salmonella enterica Serovar Typhimurium Pathogenesis. J. Bacteriol. 2000; 182:1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de la Cruz J., Vioque A.. Increased sensitivity to protein synthesis inhibitors in cells lacking tmRNA. RNA. 2001; 7:1708–1716. [PMC free article] [PubMed] [Google Scholar]
- 12. Ramadoss N.S., Alumasa J.N., Cheng L., Wang Y., Li S., Chambers B.S., Chang H., Chatterjee A.K., Brinker A., Engels I.H. et al.. Small molecule inhibitors of trans-translation have broad-spectrum antibiotic activity. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:10282–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Macé K., Demay F., Guyomar C., Georgeault S., Giudice E., Goude R., Trautwetter A., Ermel G., Blanco C., Gillet R.. A genetic tool to quantify trans-translation activity in vivo. J. Mol. Biol. 2017; 429:3617–3625. [DOI] [PubMed] [Google Scholar]
- 14. Himeno H., Kurita D., Muto A.. Mechanism of trans-translation revealed by in vitro studies. Front. Microbiol. 2014; 5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurita D., Muto A., Himeno H.. In vitro trans-translation assays. Methods Mol. Biol. Clifton NJ. 2012; 905:311–325. [DOI] [PubMed] [Google Scholar]
- 16. Ivanova N., Pavlov M.Y., Felden B., Ehrenberg M.. Ribosome rescue by tmRNA requires truncated mRNAs. J. Mol. Biol. 2004; 338:33–41. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi T., Konno T., Muto A., Himeno H.. Various effects of paromomycin on tmRNA-directed trans-Translation. J. Biol. Chem. 2003; 278:27672–27680. [DOI] [PubMed] [Google Scholar]
- 18. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T.. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001; 19:751–755. [DOI] [PubMed] [Google Scholar]
- 19. Cougot N., Molza A.-E., Delesques J., Giudice E., Cavalier A., Rolland J.-P., Ermel G., Blanco C., Thomas D., Gillet R.. Visualizing compaction of polysomes in bacteria. J. Mol. Biol. 2014; 426:377–388. [DOI] [PubMed] [Google Scholar]
- 20. Ranaei-Siadat E., Mérigoux C., Seijo B., Ponchon L., Saliou J.-M., Bernauer J., Sanglier-Cianférani S., Dardel F., Vachette P., Nonin-Lecomte S.. In vivo tmRNA protection by SmpB and pre-ribosome binding conformation in solution. RNA. 2014; 20:1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J.H., Chung T.D., Oldenburg K.R.. A Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999; 4:67–73. [DOI] [PubMed] [Google Scholar]
- 22. Cabantous S., Waldo G.S.. In vivo and in vitro protein solubility assays using split GFP. Nat. Methods. 2006; 3:845–854. [DOI] [PubMed] [Google Scholar]
- 23. Kent K.P., Childs W., Boxer S.G.. Deconstructing green fluorescent protein. J. Am. Chem. Soc. 2008; 130:9664–9665. [DOI] [PubMed] [Google Scholar]
- 24. Kamiyama D., Sekine S., Barsi-Rhyne B., Hu J., Chen B., Gilbert L.A., Ishikawa H., Leonetti M.D., Marshall W.F., Weissman J.S. et al.. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016; 7:11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cabantous S., Terwilliger T.C., Waldo G.S.. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005; 23:102–107. [DOI] [PubMed] [Google Scholar]
- 26. Miller K.E., Kim Y., Huh W.-K., Park H.-O.. Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome-wide interaction studies. J. Mol. Biol. 2015; 427:2039–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kapoor S., Samhita L., Varshney U.. Functional significance of an evolutionarily conserved alanine (GCA) resume codon in tmRNA in Escherichia coli. J. Bacteriol. 2011; 193:3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T.. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001; 19:751–755. [DOI] [PubMed] [Google Scholar]
- 29. Liu P., Chen Y., Wang D., Tang Y., Tang H., Song H., Sun Q., Zhang Y., Liu Z.. Genetic selection of peptide aptamers that interact and inhibit both small protein B and alternative ribosome-rescue factor A of Aeromonas veronii C4. Front. Microbiol. 2016; 7:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu P., Huang D., Hu X., Tang Y., Ma X., Yan R., Han Q., Guo J., Zhang Y., Sun Q. et al.. Targeting inhibition of SmpB by Peptide aptamer attenuates the virulence to protect zebrafish against Aeromonas veronii infection. Front. Microbiol. 2017; 8:1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brunel R., Descours G., Durieux I., Doublet P., Jarraud S., Charpentier X.. KKL-35 exhibits potent antibiotic activity against Legionella species independently of trans-Translation inhibition. Antimicrob. Agents Chemother. 2018; 62:e01459-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sinnett S.E., Sexton J.Z., Brenman J.E.. A high throughput assay for discovery of small molecules that bind AMP-activated protein kinase (AMPK). Curr. Chem. Genomics Transl. Med. 2013; 7:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tresse C., Radigue R., Gomes Von Borowski R., Thepaut M., Hanh Le H., Demay F., Georgeault S., Dhalluin A., Trautwetter A., Ermel G. et al.. Synthesis and evaluation of 1,3,4-oxadiazole derivatives for development as broad-spectrum antibiotics. Bioorg. Med. Chem. 2019; 27:115097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.