


Abstract
Many organisms exchange small RNAs (sRNAs) during their interactions, that can target or bolster defense strategies in host–pathogen systems. Current sRNA-Seq technology can determine the sRNAs present in any symbiotic system, but there are very few bioinformatic tools available to interpret the results. We show that one of the biggest challenges comes from sequences that map equally well to the genomes of both interacting organisms. This arises due to the small size of the sRNAs compared to large genomes, and because a large portion of sequenced sRNAs come from genomic regions that encode highly conserved miRNAs, rRNAs or tRNAs. Here, we present strategies to disentangle sRNA-Seq data from samples of communicating organisms, developed using diverse plant and animal species that are known to receive or exchange RNA with their symbionts. We show that sequence assembly, both de novo and genome-guided, can be used for these sRNA-Seq data, greatly reducing the ambiguity of mapping reads. Even confidently mapped sequences can be misleading, so we further demonstrate the use of differential expression strategies to determine true parasite-derived sRNAs within host cells. We validate our methods on new experiments designed to probe the nature of the extracellular vesicle sRNAs from the parasitic nematode Heligmosomoides bakeri that get into mouse intestinal epithelial cells.
INTRODUCTION
Organisms do not live in isolation. The wonderful diversity and complexity in life arises in part due to the contacts that living beings have with their peers. Symbioses can have positive or negative consequences to one or both of the interacting partners. These interactions are not only obvious at the macroscopic level, but molecular exchanges underlie many of them. Molecules moving between organisms of different species include antibiotics, toxins, volatiles, sugars, amino acids, amongst many others.
RNA is a molecule of incredible functional versatility, participating in central cellular processes as messenger, transfer and ribosomal RNA, but also in complex regulatory layers, from bacterial riboswitches to eukaryotic microRNAs (miRNAs). Yet, RNA has historically been regarded as an unsuitable molecule for exchanging signals between cells or organisms due to its instability, even though it was proposed as an extracellular communicator several decades ago (1).
Recent advances in sequencing technology have allowed researchers to measure RNAs with unprecedented sensitivity, leading to the surprising discovery that many small RNAs (sRNAs), including miRNAs, are extracellular components of human bodily fluids including blood, tears and maternal milk (2–4). These extracellular RNAs can be protected from degradation through binding to proteins like Argonaute and/or encapsulation within extracellular vesicles (EVs) (5). Even so, a report that miRNAs from plant food sources could be detected in the mammalian bloodstream was quite surprising (6). These so-called ‘xenomiRs’ have been hotly debated, with a slight consensus arising that miRNAs detected after passing through the vertebrate digestive tract are probably contaminations or other molecular errors coming from index swapping during Illumina library preparation (7–10).
A key discovery came when Botrytis cinerea, a fungal plant pathogen, was shown to secrete sRNAs that traffic into plant cells to help block the host defense response (11). Since then, we and others have shown that sRNAs are detected in material exchanged between a large variety of pathogens and their hosts (12–18). The parasitic nematode Heligmosomoides bakeri secretes sRNAs inside EVs into the host gut environment, modulating the immune response of mice (12). The parasitic plant Cuscuta campestris produces miRNAs that travel into the host tissue eliciting a functional silencing response in Arabidopsis (14). Plants can also deliver their own sRNAs to strike back at their pathogens (15,16). According to some recent reports, RNA exchange may even occur between the different domains of life: the bacterium Salmonella uses the host Argonaute to produce miRNA-like RNA fragments that increase its survival (19), and mammalian miRNAs present in the gut can be internalized into bacteria and affect their growth thereby shaping the microbiota (20). Although there has been more focus in the literature on RNA released from pathogens, RNAs are also being exchanged in other types of symbioses (20–24). Although most of the focus in this area has been on very small (∼22 nt) RNA species that can act through RNA interference mechanisms, it is clear that larger RNAs can also travel between cells. For example, full length Y-RNA like molecules (∼70 nt in length) are abundant in the extracellular vesicles of Heligmosomoides bakeri and can enter host cells (12). Mammalian EVs also contain messenger RNAs (mRNAs) that can shuttle between mammalian cells and even be translated in the recipient cell (25). Recent work has shown that fungal EVs contain mRNAs that can be translated in vitro (26); however there are no data yet to show that imported parasite mRNAs make functional proteins inside of host cells. Sequencing technologies are at a state where detecting RNAs of different sizes, from all sorts of biological material, even single cells, is accessible to most research groups. Analysis of RNA sequencing data from interacting organisms began a few years ago, with ‘Dual RNA-Seq’ experiments that focused on transcriptional analyses of bacterial pathogen–host systems (27,28). To successfully perform these experiments, several technical aspects were addressed to account for highly abundant rRNA or tRNA from phylogenetically heterogeneous samples, the lack of poly-A tails in bacteria and scenarios where one of the organisms was present in very small relative amounts. In contrast, bioinformatic analyses of these results are generally straightforward, since 100–150 nt sequences (the most common read-length of current Illumina sequencers) can usually be easily assigned to the correct position, in the correct genome of origin.
Dealing with smaller RNAs, such as eukaryotic sRNAs (∼20–30 nt), presents completely different challenges. Removal of full length rRNA and tRNA, or poly-A selection is not required, since a size-selection step prior to, or after library generation will enrich for the RNA population of interest. On the other hand, bioinformatic analyses can be challenging since very short sequences can map perfectly to a large genome just by chance. Furthermore, short sequences can map to multiple locations, leading to uncertainty that is sometimes solved by discarding these sequences. Some sequences can also genuinely arise from different species. Ancient miRNAs, as well as highly conserved rRNA/tRNA fragments can share up to 100% identity between phylogenetically diverse organisms like nematodes and mammals. On the other hand, new miRNAs are constantly evolving, and they have been proposed as phylogenetic markers (29). Taking advantage of this idea, miRTrace was developed to predict the taxonomic diversity in any sRNA-Seq sample or detect the origin of cross-species contamination (30). Yet because it relies on a database of clade-specific miRNAs, it cannot classify sequences that have not been curated and does not account for the other categories of sRNAs in samples.
There is increasing interest in studying the sRNAs that are naturally exchanged between organisms. We initially reported, using standard library preparation techniques, that EVs secreted by the parasitic nematode H. bakeri mostly contain microRNAs (12). Recently we discovered that 5′ triphosphate small interfering RNAs (siRNAs) derived from repetitive elements are in fact the most dominant cargo (31). This is quite interesting, since the sRNAs secreted by B. cinerea that impair plant defense responses derive from transposable elements (11). It is possible that various pathogens use repetitive elements of their genome to efficiently explore a wide range of sequences to interfere with their hosts. There are no available methods to confidently detect and quantify these kinds of sRNAs within the cells or tissues of another organism. Here we describe the development of methods to detect, quantify, and characterize sRNAs that can move between different species.
We downloaded available data from experiments designed to probe inter-organismal communication mediated by sRNAs between three eukaryotic parasites and their hosts. We also included a symbiosis model of nodulating bacteria. To further increase our dataset diversity, we designed new experiments to discover which of the siRNAs in H. bakeri EVs actually get into mouse host cells. We detail the difficulties of analysing all of these kinds of experiments, and propose a series of strategies to solve them. One of the biggest challenges arises from the sRNAs which can confidently map to the genomes of both interacting species. We show that this ambiguity, as expected, depends on the length of the sRNA, the size of the genomes, and their phylogenetic relationship. We next demonstrate how sequence assembly of the raw sRNA-Seq data extends the length of many sRNAs and reduces the ambiguity problem. Finally, we show how differential expression analysis, in combination with sRNA assembly, and proper experimental designs, can be leveraged to confidently detect and quantify the sRNAs that move between even closely related species.
MATERIALS AND METHODS
Selected experiments and reference genomes
The list of host–symbiont species used in this work is shown in Table 1. Further information of the sRNA-Seq data processed from these experiments is included in Supplementary Table S1. The reference genomes used are described in Supplementary Table S2. To facilitate finding ambiguous reads across both genomes, a combined reference was produced by concatenating the sequences from both genomes for each experiment. In cases where rRNA was missing, these were manually added as an extra contig. A two-word label was added to all fasta headers to differentiate parasite form host genome sequences. All genome files were indexed using Bowtie-1.2.2 (32).
Table 1.
Small RNA sequencing datasets of interacting organisms
| Host | Symbiont | Tissue or condition | Data availability | Reference |
|---|---|---|---|---|
| Arabidopsis thaliana | Botrytis cinerea | Rosette leaves: 24, 48 and 72 h after infection | Sequence Read Archive: SRP019801. Samples: SRX252403, SRX252404, SRX252405 | (11) |
| Arabidopsis thaliana | Cuscuta campestris | Arabidopsis stems 4 cm above a Cuscuta haustorium | Sequence Read Archive: SRP118832. Samples: SRX3214812, SRX3214813 | (14) |
| Meriones unguiculatus | Litomosoides sigmodontis | Serum from infected gerbils | GEO: GSE112949. Samples: GSM3091975, GSM3091976, GSM3091977, GSM3091978, GSM3091979 | (50) |
| Mus musculus | Heligmosomoides bakeri | MODE-K cell line: 4 and 24 h after adding EVs | GEO: GSE124506. Samples: GSM3535462, GSM3535463, GSM3535464, GSM3535468, GSM3535469, GSM3535470 | This work |
| Glycine max | Bradyrhizobium japonicum | 10 and 20 days nodule | Sequence Read Archive: SRP164711. Samples: SRR7986783, SRR7986788 | (24) |
H. bakeri life cycle and EV isolation
CBA × C57BL/6 F1 (CBF1) mice were infected with 400 L3 infective-stage H. bakeri larvae by gavage and adult nematodes were collected from the small intestine 14 days post infection. The nematodes were washed and maintained in serum-free media in vitro as described previously (31). To collect H. bakeri EVs, culture media from the adult worms were collected from 24–92 h post-harvest from the mouse (the first 24 h of culture media was excluded to reduce host contaminants). Eggs were removed by spinning at 400 g and the supernatant was then filtered through 0.22 mm syringe filter (Millipore) followed by ultracentrifugation at 100 000 g for 2 h in polyallomer tubes at 4 °C in an SW40 rotor (Beckman Coulter). Pelleted material was washed two times in filtered PBS at 100 000 g for 2 h and re-suspended in PBS. The pelleted H. bakeri EVs, were quantified by Qubit Protein Assay Kit (Thermo Fisher), on a Qubit 3.0.
MODE-K uptake assays
MODE-K cells were kindly provided by Dominique Kaiserlian (INSERM) and were maintained as previously described (33). Uptake experiments were carried out with 2.5 ug EVs per 50 000 cells for 4 and 24 h time points, in a 37 °C, 5% CO2 incubator. Cells that were not incubated with H. bakeri EVs were treated as controls for the two-time points. Cells were washed twice in PBS before RNA extraction with a miRNeasy mini kit (Qiagen), according to manufacturer's instructions. The RNA Integrity Number (RIN) was tested with the Agilent RNA 6000 Pico Kit on an Agilent 2100 Bioanalyzer. Three biological replicates were included for each of the samples.
Small RNA library prep and sequencing
Total RNA was treated with RNA 5′ Polyphosphatase (Epicenter) following manufacturer's instructions, before library preparation. Libraries for sRNA sequencing were prepared using the CleanTag sRNA library prep kit according to manufacturer's instructions. Although there are other sRNA library prep methods that can reduce the quantification bias due to adapter ligation, e.g. (34), in our hands and due to the small amount of starting material, CleanTag kits yield much higher signal:background (fewer adapter–dimers) for extracellular material. For all samples, 1:2 dilutions of both adapters were used with 18 amplification cycles (TriLink BioTechnologies). Libraries of 140–170 bp in length were size-selected and sequenced on an Illumina HiSeq 2500 in high-output mode with v4 chemistry and 50 bp SE reads, by Edinburgh Genomics at the University of Edinburgh (Edinburgh, UK). This insert size was chosen to focus on the small interfering guides of exWAGO, the only Argonaute protein detected within Heligmosomoides EVs. We know that these siRNAs are mainly 23–24nt and are one of the main small RNA components of EVs (31).
Processing of small RNA-Seq reads
The quality of all sRNA-Seq reads from each library was inspected using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw reads were then cleaned and trimmed to remove 3′ adapter using reaper (35) with the following parameters: -geom no-bc, -mr-tabu 14/2/1, -3p-global 12/2/1, -3p-prefix 8/2/1, -3p-head-to-tail 1, -nnn-check 3/5, -polya 5 -qqq-check 35/10, -tri 35. Trimmed reads smaller than 18nt were discarded. When needed, reads were collapsed into individual sequences with counts, using tally (35). One replicate of the MODE-K control cells (incubated for 24 h without treatment) was an outlier according to PCA analysis, did not have a clear peak of mouse miRNAs (suggesting degraded RNA), and was excluded from further analyses.
Calculations of host, symbiont and ambiguous reads
Each library was aligned to the separate host and symbiont genomes using Bowtie-1.2.2 (32) and requiring perfect end-to-end hits (-v 0). Each read was classified as: host if it only hit the host genome, symbiont if it only hit the symbiont reference and ambiguous if it hit both genomes. The resulting length distributions of trimmed reads (classified as host, symbiont or ambiguous) for all experiments are shown in Supplementary Figure S1.
Shared k-mers between genomes
To calculate the fraction of shared k-mers (s) of length 12–30 in two random genomes of fixed sizes we used the following equation:
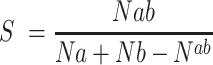 |
where Na and Nb are the number of k-mers in random genomes a and b, respectively. Nab corresponds to the number of shared k-mers in both genomes. The values of Na, Nb and Nab were calculated using the theoretical approach given by (36).
The fractions of k-mers between sizes 12–30 that are shared between each pair of real genomes were calculated using Jellyfish 2.2.10 (37).
Genome-guided sRNA assembly
To perform genome-guided sRNA assembly, ShortStack 3.8.2 (38) was used with parameters favoring smaller clusters: a minimum coverage of one read, requiring 0 mismatches, using unique-mapping reads as guide to assign multi-mapping reads (mmap: u), a padding value of 1, reporting all bowtie alignments (bowtie_m: ‘all’), and a ranmax value of 5000 to avoid losing reads mapping to multiple sites. The default bowtie cores and sorting memory values were also increased to improve processing time. Reads were aligned to the concatenated host and symbiont reference genomes described above.
De novo assembly of sRNA-Seq
To evaluate de novo assembly of sRNA reads, six popular RNA-Seq de novo assemblers were selected: Oases (39), rnaSpades (40), SOAPdeNovo (41), Tadpole (https://jgi.doe.gov/data-and-tools/bbtools/), TransAbyss (42) and Trinity (43). These assemblers were also evaluated using only their first ‘k-mer extension’ step: (a) rnaSpades ‘–only-assembler’, Trans-AbySS ‘–stage contigs’ and Trinity ‘–no_run_chrysalis’; (b) the equivalent for Oases was to use contigs generated by velvetg, while for SOAPdenovo-Trans the .contig output file was used; (c) Tadpole is a simple assembler that only performs k-mer extension. Additional parameters for each configuration are available in Supplementary Table S3. All the generated contigs were post-processed as follows: (i) all reads used to generate the assembly were aligned back to the contigs using Bowtie-1.2.2 (-v 0) and (ii) using the BAM files from these alignments, contig edges that did not have any reads mapping to them were trimmed back. All contigs were then mapped to the reference genomes to decide if they were host or symbiont.
Disambiguation of host–symbiont mixed samples
To try to disambiguate reads that mapped equally well to both genomes, we used the assembled de novo contigs or genome-guided clusters. Clusters are assembled directly on a specific genome, so by definition they are not ambiguous. Contigs are assembled in the absence of a genome, but since they are longer than reads, they will be less ambiguous (e.g. see Figure 1). So we mapped the contigs to the genomes, first with Bowtie-1.2.2 (32) to find perfect hits, and the remainder with a more relaxed setting using Bowtie2–2.3.3 (44) (allowing indels and mismatches). For contigs that mapped imperfectly to both genomes, the alignment with fewer mismatches was selected (XM:i<N> SAM optional field). We then mapped all reads to contigs or clusters. For simplicity the following will refer to contigs only.
Figure 1.

Factors that influence the number of ambiguous k-mers between pairs of genomes. X-axes represent the k-mer size and Y-axes the fraction of shared or ambiguous k-mers. (A) Random genomes of sizes equivalent to those of A. thaliana and B. cinerea. (B) Fixed A. thaliana genome, compared to full B. cinerea genome or a sample corresponding to 50% or 10% of the complete genome. (C) All genomes were subsampled to the size of the smallest, that of B. japonicum. (D) Real fractions of ambiguous k-mers in each pair of complete genomes. Insets correspond to a zoomed in area of k-mer sizes 18–23.
We classified the mapped reads into three groups: those that mapped to multiple contigs (multi-mapping reads), reads that mapped uniquely to one contig (uniquely-mapping reads) and reads that did not align to any contig. This is now a problem similar to assigning multi-mapping reads to transcript isoforms. Tools such as ERANGE (45), a method developed for CAGE (46), RSEM (47) and ShortStack (48) use the information in uniquely mapping reads to ‘rescue’ reads that map to multiple transcripts. The core idea is that the proportion of uniquely-mapping reads is a good estimate for the proportion of multi-mapping reads produced from each transcript. In our implementation we first summed the counts of all the uniquely-mapping reads for each contig across all libraries, producing global uniquely-mapping counts. In order to only use the most informative contigs with high global uniquely-mapping counts, we selected the top 0.2% (we evaluated different cutoffs, and ∼90% of the multi-mapping reads can be assigned with this cutoff). We then distributed the counts of the multi-mapping reads that mapped to these contigs, proportional to the global uniquely-mapping counts. Reads that mapped to other contigs, as well as those that mapped to ambiguous contigs or did not map, remained ambiguous. The R code with our implementation of these steps is available in the repository: https://github.com/ObedRamirez/Disentangling-sRNA-Seq.
Differential expression analysis
To perform differential expression analysis, a matrix was first built for individual sequences using all distinct reads in the libraries to be compared. In this matrix, rows represent individual sequences and columns represent libraries. Each cell represents the times a sequence was found in a given library. A similar procedure was done to obtain matrices for contigs and clusters, except that their counts were obtained by mapping each library to FASTA files of the contigs or clusters, and multi-mapping reads were disambiguated as described in the previous section.
Differential expression analyses were performed using the edgeR package (49). The sRNA elements (individual sequences, de novo assembled contigs or genome-guided clusters) with low expression were filtered out: only those with more than one count per million in at least two libraries were kept. The MODE-K vesicle-treated libraries were compared to the control untreated MODE-K libraries, regardless of the incubation time (4 and 24 h). To determine differential expression, a generalized linear model (GLM) likelihood ratio test was used, always fixing a common dispersion value of 1.626, which was estimated using the unassembled reads. False discovery rates (FDR) were calculated, and sequences that mapped to the symbiont and had an FDR < 0.1 and a positive log fold-change were considered symbiont sequences according to differential expression. The R scripts we used to perform these analyses are also available in the repository: https://github.com/ObedRamirez/Disentangling-sRNA-Seq.
Defining sRNA classes by length and first nucleotide
For the H. bakeri experiments, the first nucleotide and length of each sequence mapping to the de novo assembled contigs and genome-guided clusters was calculated using custom R scripts and the Rsamtools package. Reads beginning with a Guanine and between 21–24 nucleotides were classified as ‘22G’. Reads beginning with a Thymine and between 21–24 nucleotides were classified as ‘22U’. These criteria were defined by observing the properties of the pure EV and MODE-K libraries (Supplementary Figure S2).
Expression comparison with H. bakeri EV libraries
To compare the expression of the nematode sequences detected in MODE-K libraries with pure EV nematode libraries, we mapped EV reads to Up-regulated (Up) symbiont unassembled reads, or to all reads assigned to our Up contigs or clusters using Bowtie-1.2.2. This approach was chosen to achieve a fairer comparison between assembled and unassembled reads.
RESULTS AND DISCUSSION
A diverse selection of species that exchange small RNAs
To build a foundation for bioinformatically probing cross-species RNA communication, we selected five phylogenetically diverse pairs of interacting organisms from publications with available sRNA-Seq data, and where small RNAs were proposed as mediators of cross-species communication (Table 1). These were the model plant Arabidopsis thaliana infected by a fungus (Botrytis cinerea) (11) or a parasitic plant (Cuscuta campestris) (14), and the mongolian gerbil (Meriones unguiculatus) infected by a filarial parasite (Litomosoides sigmodontis) (50). Given our interest in parasitic nematodes and their secreted extracellular vesicles (EVs) (12,31), we designed new experiments to explore the sRNA guides of exWAGO present in EVs from Heligmosomoides bakeri that get internalized by host cells, using sRNA-Seq of a mouse intestinal epithelial cell line. Finally, we also included soybean (Glycine max) nodules, containing the bacterium (Bradyrhizobium japonicum) that helps its host by fixing nitrogen (24). The full list of sRNA-Seq samples available from these experiments are described in Supplementary Table S1.
Since the field of cross-species communication by RNA is still young, these represent some of the only real-world scenarios of symbiotic or parasitic models that have been examined with sRNA-Seq. The biological material sampled in each case is diverse: infected stems or leaves in the case of Arabidopsis, serum from infected gerbils, a cell culture for our nematode-mouse model, and nodules from soybean. The amount of RNA present from the two organisms within the samples can also be quite different. Botrytis spores are used to infect Arabidopsis leaves, from which RNA is extracted after the necrotrophic fungus has grown and invaded the tissue of its host. At the time of sampling the parasite had overgrown a small portion of the leaf. The Cuscuta experiment includes different types of samples (Supplementary Table S1), and for our main analysis we selected those from Arabidopsis stems ∼4 cm above the parasite haustorium. The rodent pathogens release extracellular RNA to the host environment and the parasites are not themselves present in the collected material. The soybean samples are from nodule tissue at two time points, so they include the bacterial cells growing within. We expected the rodent samples in particular to be akin to a ‘needle in a haystack’ problem, with very small amounts of parasite sRNA amongst a very large amount of host RNA. In contrast, the plant samples are expected to contain a mixed population of sRNAs from both species at more comparable levels. The Botrytis and soybean samples include cells from both organisms, instead of extracellular material. Nevertheless, the Botrytis experiment represents one of the first and most cited publications regarding RNA communication between species, and the soybean-bacteria experiment provides an interesting contrast to the eukaryotic parasites.
Determining the amount of host, symbiont and ambiguous reads in sRNA datasets
As a first step to identify the genome of origin of sRNAs involved in cross-species communication, we prepared a combined reference genome for each pair of interacting species (see Methods, and Supplementary Table S2). We then focused on sRNA reads of at least 18 nucleotides that map with 100% identity to the corresponding combined reference. These mapped reads are then divided into three categories: (i) host (if they only map to the host portion of the reference), (ii) symbiont (if they only map to the symbiont) and (iii) ambiguous (if they map at least once to the host and at least once to the symbiont). With this partitioning, different experiments yield varying proportions of host, symbiont and ambiguous reads (Supplementary Figures S1 and S3). Although in principle sRNAs of any size could be exchanged and be functionally important, the experiments we analyzed included a size-fractionation step to enrich sequences between 20nt and 30nt. Nevertheless, a range of sequences of up to 50nt (the maximum read length) remain in most experiments, as can be appreciated in Supplementary Figure S1.
The Arabidopsis + Botrytis libraries show between ∼4–7% of symbiont reads, with ambiguous reads accounting for ∼1–7%. The Arabidopsis + Cuscuta libraries show ∼4% of symbiont and up to ∼55% of ambiguous reads. The infected gerbil serum had between ∼1–3% of symbiont reads but only ∼0.5% of ambiguous reads. The MODE-K cells treated with extracellular vesicles from H. bakeri yielded the lowest amount of symbiont reads: 0.4–0.9%. In this case, the symbiont reads are clearly outnumbered by the ambiguous ones, with ∼4–6% being assigned to this category. Finally, the soybean libraries contain ∼9–19% bacterial reads and only ∼0.1–0.2% ambiguous reads. These results highlight the difficulty in correctly identifying the origin of all the sRNAs. Whilst one approach would be to discard the ambiguous reads we could be throwing away an important amount of sequencing information that may include bonafide RNA molecules involved in cross-species communication.
Ambiguity in host–symbiont sRNA-Seq reads is influenced by read length, genome size and phylogenetic distance
We next wanted to explore the factors that lead to ambiguous reads in our host–symbiont models. Similar problems have been approached before, showing that read length, genome size and phylogenetic relationships are important (36). We describe these factors for our models, using ‘k-mers’ (nucleotide words of length k) as a proxy for reads (Figure 1).
Read length
Intuitively, it is more likely that a small k-mer will be present in two genomes compared to a longer k-mer. To illustrate this, we define two random genomes of the same size as A. thaliana and B. cinerea, and calculated the fraction of shared k-mers of different sizes (36). The shared k-mers between these two random genomes decrease rapidly as k increases. For instance, almost 80% of k-mers of length 12 are shared, but when considering k-mers of length 18, <0.3% are shared (Figure 1A).
Genome size
The size of each genome determines the maximum number of distinct k-mers that it contains (Supplementary Figure S4). A smaller genome will have fewer distinct k-mers, and so the number of shared k-mers it can have with another genome is also expected to be smaller. To highlight this property, we took the real A. thaliana genome, but sampled decreasing fractions of the B. cinerea genome (100%, 50% and 10%) to visualize how the number of shared k-mers changes. As expected, smaller Botrytis genomes share a smaller percent of k-mers of any length (Figure 1B).
Phylogenetic distance
Real genomes are not random concatenations of nucleotides, but are related through shared ancestry. Thus, the phylogenetic distance between two genomes should also influence the number of shared k-mers and therefore our ability to distinguish sRNAs that might map to both. If we imagine two genomes that have just begun to diverge, almost all k-mers will be shared. To quantify the effect of phylogenetic separation, but ignoring the effect of genome size which we described above, we fixed the smallest of the genomes under consideration (B. japonicum) and randomly down-sampled each of the other eight genomes to this size.
The effect of phylogenetic distance is small but noticeable (Figure 1C). In particular A. thaliana shares more k-mers with another plant (C. campestris) than with a fungus (B. cinerea), and the smallest number of shared k-mers are between a plant (G. max) and a bacteria (B. japonicum). While both pairs of animal genomes are expected to be similarly related (rodents and nematodes), H. bakeri shares fewer k-mers with mouse than L. sigmodontis with the gerbil. This can be explained since H. bakeri has a particularly large genome (∼700 Mb, compared to ∼65 Mb for L. sigmodontis), that is full of repetitive elements many of which are unique to this species (31). A random sample of the H. bakeri genome will thus include more k-mers from these repetitive elements. This helps explain the smaller fraction of shared k-mers than expected due to phylogeny, and highlights an extra contributing factor: genome composition and complexity, which we will not explore further in this work.
It is thus not possible to predict the exact number of ambiguous k-mers between two species just based on their genome size and phylogenetic distance, but if the genomes are available it can be efficiently calculated using tools like Jellyfish (37). By doing so, we can see that H. bakeri and M. musculus show the highest level of ambiguous k-mers, while G. max and B. japonicum show the lowest (Figure 1D). These are the biggest and smallest pairs of genomes, respectively, indicating that genome size is a major factor driving these differences. But at longer k-mers the two plant genomes are the pair with the highest ambiguity. This is due to their close phylogeny (both species are eudicotyledons, a clade of flowering plants). In all the eukaryotic pairs, the ambiguous k-mers between real genomes, at larger k-mer sizes, become much higher than expected exclusively by genome size, reflecting the contribution of shared ancestry (Supplementary Figure S5).
From these results, there are two important things to note for our purposes: (i) even for k-mers the size of biologically important molecules like microRNAs (∼21 nucleotides), there is always a fraction that will be shared identically between two genomes and (ii) if we could increase the length of any sequence, even by one or two nucleotides, the probability that it will be shared between genomes substantially decreases.
High levels of ambiguity in host–symbiont sRNA-Seq reads is caused by conserved sequences like ribosomal, transfer and microRNAs
The levels of ambiguity in our real sRNA-Seq data are much higher than predicted by the fractions of k-mers shared between pairs of genomes. For instance, 52–55% of all 18–50nt reads from the A. thaliana and C. campestris interaction are ambiguous (Supplementary Figure S3), while only 1.8% of k-mers of size 18 are shared between the genomes (Figure 1D). This is a consequence of sRNA-Seq reads not being produced randomly across the genome, and indicates that many come from regions with a higher-than-average level of conservation. This is not surprising since conserved classes of RNA, like ribosomal RNA, are always sequenced to some extent. So, from what regions are our sRNA-Seq reads being produced, particularly the ambiguous ones? We sought to answer this, focusing on the A. thaliana and C. campestris interaction where the problem of ambiguous reads is most apparent (Supplementary Figure S3).
We extracted all the ambiguous reads from libraries of A. thaliana stems 4 cm above the C. campestris primary haustorium (average of 1 260 368 from the two replicates) and tabulated them by length (Figure 2). The length distribution suggests slightly degraded RNA, with a tendency towards reads of shorter lengths (Figure 2A). Plants usually show a peak of 21nt enriched with miRNAs, and a peak of 24nt enriched with siRNAs that target transposable elements, and these are not as clearly defined here as in some of the other samples from the same experiment (Supplementary Figure S6). We then traced where all the ambiguous reads mapped in A. thaliana, which is better annotated, and classified them according to the annotation of this genome (Figure 2B). Most ambiguous reads map to rRNA (91.6%), followed by those that map to miRNA at the expected ∼21nt (3.2%), and tRNA (2.6%). This represents a clear enrichment for rRNA, miRNA and tRNA, since together they occupy ∼0.05% of the Arabidopsis genome, while comprising 97.5% of the ambiguous reads. Only 0.47% of the ambiguous reads map to other annotations, including exons, introns, transposable elements, pseudogenes and other non-coding RNA (in total occupying 75.3% of the genome), while the remaining 2% map to unannotated intergenic regions, which in total occupy 24.5% of the genome. Many plant miRNAs are highly conserved (51), so it is not surprising that a large fraction of the 21nt ambiguous reads coincide with conserved and highly-expressed miRNAs like MIR159, MIR319a and MIR396a. Ribosomal reads are more evenly distributed across all read lengths, suggesting that their presence is caused by low levels of fragmentation of highly abundant RNA molecules. Ribosomal, transfer RNA and miRNA contribution is also the main explanation for the ambiguous reads in libraries collected from the Cuscuta stem above the primary haustorium, and from Arabidopsis stems with a C. campestris haustorium attached (Supplementary Figure S6).
Figure 2.

Genomic origin of ambiguous reads from libraries of A. thaliana stems 4cm above a C. campestris haustorium. Each bar represents the sequenced reads of one size between 18 and 50 nucleotides. Bar height represents the actual number of reads (top) or the fraction of reads (bottom). (A) Mapping categories are: host (green), symbiont (blue) or ambiguous (purple). (B) Genomic annotation of ambiguous reads only: intergenic (light green), miRNA (yellow), rRNA (light purple), tRNA (red) or other annotation (orange).
With these results we can see that discarding sRNA-Seq reads that map to rRNA and tRNA annotations, which is a common practice, can lead to a substantial reduction of ambiguity. Yet, the ultimate goal of this work is to be able to detect RNA transfer between species and emerging literature suggests tRNA and rRNA fragments could be extracellular signaling molecules. For instance, tRNA fragments can be selectively packaged into extracellular vesicles and move between cells (52), while tRNA fragments in sperm can contribute to intergenerational inheritance (53). Furthermore, bacterial tRNA-derived sRNAs have been implicated in plant root nodulation (24).
Discarding conserved miRNA sequences would be even more problematic, since foreign miRNAs are known to benefit from hijacking existing regulatory networks. A Kaposi's sarcoma herpesvirus miRNA uses the same target site as the cellular miR-155 (54), while we have shown that nematode miR-100 and let-7, which are identical to their mouse counterparts, are present in secreted material during infection (12). During parasitism, novel miRNAs from Cuscuta were shown to enter and target host mRNAs (14), so conserved miRNAs could also be exchanged and functional. Therefore, there is a need to be able to track the origin of ambiguous sequences.
Even highly conserved miRNAs, tRNAs and rRNAs have point differences in some part of their sequence. For example, rRNAs contain variable regions that are leveraged during phylogenetic analyses, and the loops of miRNA hairpins tend to be poorly conserved. Due to the high depth of current sequencing technology, there will be overlapping reads with slightly different 5′ and 3′ ends, due to imperfect enzymatic processing or degradation. Depending on the length range being sequenced, reads from precursors before processing/degradation can also be present. In fact, the existence of reads from different parts of miRNA hairpin precursors, including both arms and the loop region, is the basis of popular prediction tools like miRDeep2 (55). Thus, as long as we are able to extend the conserved sequences into a less conserved portion, we should be able to disambiguate them. We therefore explore the possibility of using sRNA-Seq assembly to reduce ambiguity through extension of reads.
Assembly of sRNA-Seq reads
Most work on RNA sequence assembly has focused on producing full-length transcripts from mRNA-Seq data. There are many methods that work in a genome-guided fashion: first mapping reads to the genome, then assembling clusters (exons) and connecting them with rules based on splicing properties and sequencing depth, e.g. Cufflinks (56) and Stringtie (57). Analogous to these, there are some tools that cluster sRNA-Seq reads where they map to the genome, in order to predict sRNA-producing loci: segmentSeq (58), CoLIde (59), and ShortStack (38). ShortStack fits our needs quite well, since it analyses reference-aligned sRNA-Seq reads to cluster them in order to predict sRNA genes, which we shall refer to as genome-guided clusters from here on. So, we used ShortStack to perform a genome-guided sRNA assembly and quantification.
We were also particularly interested in finding out if we could deal with situations in which the genomes for the interacting organisms were not available, or were not of sufficient quality. In these cases, a de novo assembly approach is the only option. There has been a lot of development regarding de novo RNA-Seq assemblers. These tools do not require genome sequences, but rely instead on breaking down reads into k-mers, building a graph, and finding paths through the graph to build longer sequences. These RNA-Seq de novo assemblers are not designed for using on sRNA-Seq data. For example, k-mers of at least 25 nucleotides are usually used to improve the assembly quality, while some functional molecules in sRNA-Seq (e.g. miRNAs) are smaller than this size. In our case, though, we want to extend all sRNA sequences in order to capture sequence variation that can help us infer the correct genome of origin.
We tested six popular de novo transcriptome assemblers: Oases (39), rnaSPAdes (40), SOAPdenovo (41), Tadpole (https://jgi.doe.gov/data-and-tools/bbtools/), TransABySS (42) and Trinity (43). These programs first generate contigs by extending k-mers in a graph. This step produces short contigs that are later connected into full-length transcripts, but for our purpose of slightly extending sRNAs it could be sufficient, so we included the output of this ‘k-mer extension’ step as a standalone method when possible (see Materials and Methods). One of the most important parameters for all the assemblers is the k-mer size, which affected the number of reads that we could remap to the assembly (Supplementary Figure S7). The optimal k-mer was 19 for our sRNA-Seq datasets, except for the A. thaliana + B. cinerea data where 23 was slightly better.
The four assemblies generated with only the first ‘k-mer extension’ step (rnaSPAdes-only-assembler, Tadpole, Trans-ABySS-stage-contigs and Trinity-inchworm) performed quite differently than the full pipelines (Supplementary Figure S8). They generated a larger number of contigs (Supplementary Figure S8A), that were shorter (Supplementary Figure S8B), and mapped more often to the reference genomes (Supplementary Figure S8C) than the full transcriptome assemblers. Additionally, library re-mapping was higher than with the other evaluated assemblies (Supplementary Figure S8D). From these, Trinity-inchworm showed the highest library re-mapping in the majority of the evaluated datasets and is therefore used in our subsequent analyses.
Assembly reduces ambiguity of host–symbiont sRNA-Seq reads
To compare the amount of ambiguity between the original reads (unassembled), de novo contigs and genome-guided clusters, we first assigned contigs and clusters to their genome of origin (see Materials and Methods). We then mapped reads directly to the sequences of the contigs or clusters. The reads that mapped to more than one of these are ambiguous, but this problem is analogous to when reads map to different transcript isoforms or paralogous genes. Several tools, including ERANGE (45), a method developed for CAGE (46), RSEM (47) and ShortStack (48) assume that the proportion of reads that uniquely map to each isoform is a good proxy for the fraction of ambiguous reads that are produced from those isoforms. This logic is supported by independent simulations (47,48,60) as well as correlations with microarray and qPCR experiments (45,46,48). We reimplemented these ideas to use the number of uniquely-mapping reads to help distribute the reads that mapped equally well to more than one contig or cluster (see Materials and Methods).
With either type of assembly, many previously ambiguous reads can now be assigned to one of the two interacting genomes (Figure 3). The results vary by dataset, with the de novo contigs reducing more ambiguity than the genome-guided clusters in three out of five cases. It seems that de novo assembly benefits more from using longer sRNA reads (31–50nt) since during an initial assembly where these reads were excluded, genome-guided clusters outperformed de novo assembly (not shown). Nevertheless, both strategies outperform the baseline use of unassembled reads.
Figure 3.

Percent of ambiguous and symbiont reads before and after assembly. The name of the two interacting species is shown for each experiment above three bars. All 18–50nt reads were classified and the percent of each category were averaged across each experiment's samples. The first bar of each group represents unassembled reads, the second de novo contigs, the third genome-guided clusters. The Y-axes are independently zoomed and cut to highlight the percent of symbiont (blue) and ambiguous (purple) reads. Host reads (green) always represent the remainder of 100%.
These results show how the assembled versions of the sRNA-Seq data contain more reads that can be assigned to the interacting organisms, and less ambiguity, allowing researchers to use more information from their experiments. However, the assembled sequences could include reads from the wrong genome, due to errors during assembly. So, we ideally want an independent test for validating the origin of the assembled sequences mapped to the symbiont genome.
Differential expression analysis improves detection of parasite sRNAs
Ideally, parasite sRNAs should be present in those samples that were infected with the parasite, and be absent (no reads) in uninfected samples. Unfortunately, this does not perfectly hold due to problems like index-swapping during library preparation (61). Especially for situations when the parasite sRNAs can be present in very low numbers, a statistical framework is needed to determine which sRNAs are reliably present in the infected compared to uninfected samples. For this, we can use differential expression analysis, which also helps to confirm if our assembled sequences behave like parasite or host sequences.
We designed our new H. bakeri extracellular-vesicle (EV) experiment to be amenable to differential expression analysis. We collected RNA from six biological replicates of MODE-K intestinal epithelial cell cultures treated with H. bakeri EVs, and the corresponding untreated controls. Since we do not know the dynamics of import, or the stability of foreign sRNA once inside the cells, we performed RNA extraction for half our replicates at 4 h, and the other half at 24 h after treatment and following extensive washing of cells. We then mapped all the sRNA-Seq reads to our assembled contigs and clusters, and quantified their expression as above (see Methods). We also obtained the simple counts of each unique unassembled read for the baseline analysis. For these three types of count matrices, we performed the exact same steps of a differential expression analysis (see Materials and Methods). We also kept track of H. bakeri, M. musculus or ambiguous mapping status for reads, contigs and clusters and used this information when visualizing our results. This helps us determine which reads/contigs/clusters may actually come from the host genome, despite mapping perfectly and preferentially to the parasite genome.
The process of sequence assembly reduces ambiguity, but another advantage is that it reduces the number of statistical tests performed during differential expression analysis (there are fewer distinct contigs/clusters than unassembled reads), reducing a problem known in statistics as multiple-testing. In addition, if the reads are grouped correctly into real biological entities with a consistent expression pattern, we should get higher counts and increased statistical power.
Although we conservatively performed the differential expression analysis starting with all unassembled reads, contigs or clusters, we focused only on the subset that should contain the real parasite sequences: those that were assigned to the H. bakeri genome (parasite), and that were up-regulated in the EV-treated samples (Up). With these criteria, the parasite sequences we detected with each strategy included an average of 17 506 counts for the Up unassembled reads, 40 334 counts for the Up de novo contigs, and 42 092 counts for the Up genome-guided clusters (Supplementary Table S4 and Figure S9). These results show how the assemblies have increased the number of confidently detected parasitic sequences: the de novo contigs contain 2.3 times more counts, and the genome-guided clusters about 2.4 times more counts, compared to the unassembled reads.
Our mapping results (Figure 3) indicated that all sequences that map perfectly to the parasite genome represent genuine parasite sRNAs. Our differential expression results suggest that even sRNAs that appear to be correctly mapped can still be divided into those that are genuine parasite sRNAs (up-regulated in samples treated with parasite EVs), and those that more likely represent host sRNAs (similar expression levels in treated and control samples). Nevertheless, our differential expression analysis could be underpowered (due to a relatively small number of replicates and high biological variability) leading to false negative predictions. So, we next wanted to further validate these results.
Validation of differentially expressed parasitic sRNAs
A distinctive property of H. bakeri EV sRNAs is that the majority are 22–23 nucleotides in length and begin with a Guanine (31). This is in stark contrast to endogenous MODE-K sRNAs that are dominated by miRNAs of 22 nucleotides that begin with a Uracil (Supplementary Figure S1). We thus have a simple method to determine whether there is a signature in the reads associated with true parasite sRNAs: compare the first-nucleotide preference of our predictions. We first classified all sRNA reads according to starting nucleotide and length, defining three categories: 22G (enriched in parasite EVs), 22U (enriched in MODE-K) or other (see Methods). As a reference, libraries prepared from pure H. bakeri EVs contain 75% 22G and 1.4% 22U reads, while untreated MODE-K libraries contain 2.5% 22G and 43.8% 22U (Figure 4A). The assembled contigs and clusters that do not show evidence of differential expression (non-Up) have high fractions of 22U reads, similar to mouse MODE-K libraries (Figure 4B). This would suggest that some of the assembled sequences are actually chimeras, i.e. they have incorporated a large number of sequences that are really from the host. Unfortunately, we cannot rule out that some of these contain true parasite miRNA sequences that are diluted by the host content and remain as false negatives of our differential expression analysis. Nevertheless, the sRNAs that are significantly upregulated (Up) after treatment with parasite EVs are enriched with 22Gs, consistent with them being true parasitic sRNAs (Figure 4B). Our proposed strategies show that the assembled contigs and clusters allowed us to discover a larger number of true parasitic sequences (more upregulated counts and similar 22G enrichment), compared to the baseline analysis with unassembled reads. In general, our results show that considering mapping information alone can be misleading, and that a differential expression approach is always useful to separate parasite from host sequences.
Figure 4.

Evaluation of differential expression results. Reads were categorized as 22G (yellow), 22U (red), or other (grey) based on length and first-nucleotide. (A) sRNA profiles of control samples: purified H. bakeri Extracellular Vesicles (EVs) and untreated MODE-K cells. Bar height represents the fraction of all reads. (B) sRNA profiles of unassembled reads, de novo contigs and genome-guided clusters. For each of these sets, there are two bars, the first one represents differentially expressed up-regulated elements (Up) and the second, elements that lack evidence for differential expression in this direction (non-Up). Bar height represents the number of reads (top) or the fraction of reads (bottom) belonging to these categories. (C) Percent of reads from pure H. bakeri EV libraries, recovered during differential expression analysis of MODE-K cells treated with EVs. Each circle represents the total reads from EV libraries. The recovered fractions are indicated in blue. The numbers of H. bakeri differentially expressed elements are shown to the right, as well as total read counts (from B).
As a final validation of the parasite Up sequences that we detect inside host cells, we checked if they are also found in pure H. bakeri EV libraries. To do so, we first mapped all our pure H. bakeri EV reads to Up unassembled reads, or to all reads assigned to our Up contigs or clusters (see Materials and Methods). We do not expect to recover every sRNA read observed in EV libraries, since some EV sRNAs might not get into MODE-K cells, others might be turned over quickly or degraded, and others might not be detected due to insufficient sequencing depth. We reasoned, though, that the percent of recovered EV reads is an indication of how good the method is at recovering true parasite sRNAs within host cells. This analysis showed us that 561 Up unassembled reads correspond to 16.92% of the total reads in EV libraries, while 1118 Up contigs and 1186 Up clusters receive 27.81% and 29.74% of all EV reads, respectively (Figure 4C). These results again highlight the improvement achieved by both assembly strategies.
CONCLUSIONS
We are now realizing that the phenomenon of organisms exchanging RNA during their interactions is surprisingly widespread. These sRNAs can be produced and secreted by the cells of one organism, travel within extracellular vesicles, and perform regulatory functions when entering cells of a different species. We know very little about which kinds of RNAs can be secreted, which ones make it inside the cells of the receiving organism, and which have a functional role for the interacting organisms. We are just beginning to understand the potential functions and applications of this kind of RNA-based communication. Although the sequencing technology is at a state where we can begin to interrogate any pair of interacting species at unprecedented detail, there are no bioinformatic tools to correctly interpret the results. Before we can properly study the mechanisms and functions of RNA communication, we need to be able to correctly disentangle the sRNA-Seq data that is being acquired. We have shown here that the small size of sRNA-Seq sequences, and the large size of genomes, leads to many sequences mapping incorrectly or ambiguously to both interacting genomes (Figure 1). Even worse, many of the produced sRNAs that can be exchanged include sequences from highly conserved miRNAs, rRNA or tRNAs that are even more likely to map well to both genomes (Figure 2). We first showed that by performing sequence assembly of the sRNA-Seq data, we can reduce the problem of ambiguity, and assign more sequences to their correct genome of origin (Figure 3). Importantly, we revealed that mapping information can still be misleading, and we showed that differential expression analysis can be used to confidently detect parasitic sRNAs that have been internalized by host cells (Figure 4). The conclusion that mapping can be misleading can have profound implications for many projects, for example those that rely mainly on sRNA mapping to a foreign genome to infer the transfer of dietary miRNAs to the mammalian blood stream (6,62,63).
We designed new experiments to detect the parasitic EV sRNAs from H. bakeri that successfully enter a mouse epithelial cell line. With our assembly methods, we showed that up to 2% of the sRNA-Seq reads within treated MODE-K cells might come from the parasite. This is a substantial increase over the simple approach of mapping to the genomes and dividing perfect hits between parasite and host, which suggested that only 0.6% of the sRNA-Seq reads were parasitic (Figure 3). Nevertheless, we showed with differential expression that these numbers are probably inflated with sequences that are really from the host. The sRNAs that pass our differential expression filter have all the characteristics of true H. bakeri EV sequences: they show the expected length and first-nucleotide 22G preference (Figure 4A and B) and include almost twice the number of reads sequenced from independently purified EVs, compared to the approach using unassembled reads (Figure 4C).
There are still some caveats to the methods we propose. Highly conserved sequences from the host, like miRNAs, can be misincorporated into parasitic sequence assemblies. The magnitude of this problem will depend on the relative level of expression of the conserved sRNA from both organisms in the sequenced sample. In our nematode-mouse experiment, a few miRNAs that we know are present in purified EVs (e.g. let-7, miR-100) are naturally expressed in MODE-K cells. Assuming a ratio of 98% host sRNA to 2% parasite sRNA (Figure 3), even for equally expressed sRNAs the mouse copy should be almost 50 times more abundant than the nematode one. It is thus not surprising that some mouse sequences erroneously contribute to the nematode assemblies. In any case, we believe that there is still room for improving sRNA-Seq assembly strategies. Promisingly, programs for de novo RNA-Seq assembly can be used, with appropriate parameters, and yield results that are comparable with genome-guided sRNA-Seq cluster assembly.
We have come to appreciate the great advantage of designing experiments to study RNA communication with differential expression in mind. Ideally this implies sampling from the separate organisms, and from the interacting material, all with several biological replicates. We realise that this might be a limitation in some cases, due to cost, the availability of sufficient quantity of biological material (e.g. purified EVs) or even the possibility of obtaining certain samples (e.g. from an obligate intracellular parasite). Nevertheless, we would like to stress the importance of having biological replicates and controls of at least one of the interacting organisms, particularly for confidently detecting low-abundance sRNAs. Other steps can also improve the ability to experimentally detect these sRNAs, such as separately processing control and infected samples to reduce index-swapping and contamination between samples (61). Finally, there have been many advances in library prep methods that can reduce adapter ligation biases and improve identification and quantification of individual sequences (34,64,65).
Regardless of the experimental and bioinformatic approaches, there may always be sequences that are 100% identical between the interacting organisms. In organisms that can be grown separately and then allowed to interact, chemically modifying the nucleotides of one organism would allow one to experimentally confirm the origin of some of these sequences. The most interesting next steps, though, will be to focus on understanding the function of the exchanged RNAs. A lot of work has focused on small extracellular RNAs that are of miRNA-like length (∼20–24nt), with the assumption that they will behave as miRNAs when inside a different organism. Nevertheless, this is not the only mechanism by which foreign sRNAs can act, and the presence of longer sRNAs (e.g. yRNAs) and a variety of RNA-binding proteins associated with EVs (66), is a reminder that the field should keep an open mind.
We have recently shown that H. bakeri EV sRNAs are mainly 5′ triphosphate species that are bound to a non-conventional worm Argonaute, which is unlikely to function like a miRNA Argonaute (31). We now show that these parasite sRNA sequences are stably detected inside mouse cells and future experiments will focus on understanding what these foreign RNA messages are doing to the host.
DATA AVAILABILITY
The new sRNA-Seq data produced for this paper are available through NCBI’s GEO under accession GSE124506. The sRNA-Seq data from other publications is referenced in Table 1. The main scripts for the analyses presented in this paper are available in the repository: https://github.com/ObedRamirez/Disentangling-sRNA-Seq
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Araceli Fernández Cortés for support using the Mazorka HPC cluster at Langebio. We also want to acknowledge Pablo Manuel González de la Rosa for help with running sRNA annotation pipelines and Sujai Kumar for suggesting and testing Tadpole for assembling sRNA-Seq data.
Notes
Present address: Franklin Wang-Ngai Chow, Department of Microbiology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Human Frontiers Science Program (HFSP) [RGY0069 to A.H.B., C.A.-G.]; Consejo Nacional de Ciencia y Tecnología - México (CONACyT) [CB-284884 to C.A-G.]; J.R.B.-B. holds a CONACyT fellowship [434580]. Funding for open access charge: CONACYT [CB-284884].
Conflict of interest statement. None declared.
REFERENCES
- 1. Benner S.A. Extracellular ‘communicator RNA’. FEBS Lett. 1988; 233:225–228. [DOI] [PubMed] [Google Scholar]
- 2. Taylor D.D., Gercel-Taylor C.. The origin, function, and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front. Genet. 2013; 4:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X., Liang H., Zhang J., Zen K., Zhang C.-Y.. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012; 22:125–132. [DOI] [PubMed] [Google Scholar]
- 4. Hoy A.M., Buck A.H.. Extracellular small RNAs: what, where, why. Biochem. Soc. Trans. 2012; 40:886–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turchinovich A., Samatov T.R., Tonevitsky A.G., Burwinkel B.. Circulating miRNAs: cell-cell communication function. Front. Genet. 2013; 4:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang L., Hou D., Chen X., Li D., Zhu L., Zhang Y., Li J., Bian Z., Liang X., Cai X. et al.. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. 2012; 22:107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witwer K.W., McAlexander M.A., Queen S.E., Adams R.J.. Real-time quantitative PCR and droplet digital PCR for plant miRNAs in mammalian blood provide little evidence for general uptake of dietary miRNAs: limited evidence for general uptake of dietary plant xenomiRs. RNA Biol. 2013; 10:1080–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dickinson B., Zhang Y., Petrick J.S., Heck G., Ivashuta S., Marshall W.S.. Lack of detectable oral bioavailability of plant microRNAs after feeding in mice. Nat. Biotechnol. 2013; 31:965–967. [DOI] [PubMed] [Google Scholar]
- 9. Tosar J.P., Rovira C., Naya H., Cayota A.. Mining of public sequencing databases supports a non-dietary origin for putative foreign miRNAs: underestimated effects of contamination in NGS. RNA. 2014; 20:754–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Witwer K.W., Zhang C.-Y.. Diet-derived microRNAs: unicorn or silver bullet. Genes & Nutrition. 2017; 12:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weiberg A., Wang M., Lin F.-M., Zhao H., Zhang Z., Kaloshian I., Huang H.-D., Jin H.. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science. 2013; 342:118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buck A.H., Coakley G., Simbari F., McSorley H.J., Quintana J.F., Le Bihan T., Kumar S., Abreu-Goodger C., Lear M., Harcus Y. et al.. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 2014; 5:5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang M., Weiberg A., Lin F.-M., Thomma B.P.H.J., Huang H.-D., Jin H.. Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat Plants. 2016; 2:16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shahid S., Kim G., Johnson N.R., Wafula E., Wang F., Coruh C., Bernal-Galeano V., Phifer T., dePamphilis C.W., Westwood J.H. et al.. MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature. 2018; 553:82–85. [DOI] [PubMed] [Google Scholar]
- 15. Cai Q., Qiao L., Wang M., He B., Lin F.-M., Palmquist J., Huang S.-D., Jin H.. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science. 2018; 360:1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang T., Zhao Y.-L., Zhao J.-H., Wang S., Jin Y., Chen Z.-Q., Fang Y.-Y., Hua C.-L., Ding S.-W., Guo H.-S.. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nature Plants. 2016; 2:16153. [DOI] [PubMed] [Google Scholar]
- 17. Koeppen K., Hampton T.H., Jarek M., Scharfe M., Gerber S.A., Mielcarz D.W., Demers E.G., Dolben E.L., Hammond J.H., Hogan D.A., Stanton B.A.. A novel mechanism of host–pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016; 12:e1005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hou Y., Zhai Y., Feng L., Karimi H.Z., Rutter B.D., Zeng L., Choi D.S., Zhang B., Gu W., Chen X. et al.. A phytophthora effector suppresses trans-kingdom RNAi to promote disease susceptibility. Cell Host Microbe. 2019; 25:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gu H., Zhao C., Zhang T., Liang H., Wang X.-M., Pan Y., Chen X., Zhao Q., Li D., Liu F. et al.. Salmonella produce microRNA-like RNA fragment Sal-1 in the infected cells to facilitate intracellular survival. Sci. Rep. 2017; 7:2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu S., da Cunha A.P., Rezende R.M., Cialic R., Wei Z., Bry L., Comstock L.E., Gandhi R., Weiner H.L.. The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe. 2016; 19:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu K., Liu M., Fu Z., Zhou Z., Kong Y., Liang H., Lin Z., Luo J., Zheng H., Wan P. et al.. Plant microRNAs in larval food regulate honeybee caste development. PLos Genet. 2017; 13:e1006946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mayoral J.G., Hussain M., Joubert D.A., Iturbe-Ormaetxe I., O’Neill S.L., Asgari S.. Wolbachia small noncoding RNAs and their role in cross-kingdom communications. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:18721–18726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teng Y., Ren Y., Sayed M., Hu X., Lei C., Kumar A., Hutchins E., Mu J., Deng Z., Luo C. et al.. Plant-derived exosomal microRNAs shape the gut microbiota. Cell Host Microbe. 2018; 24:637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ren B., Wang X., Duan J., Ma J.. Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science. 2019; 365:919–922. [DOI] [PubMed] [Google Scholar]
- 25. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O.. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007; 9:654–659. [DOI] [PubMed] [Google Scholar]
- 26. Peres da Silva R., Longo L.G.V., Cunha J.P.C., Sobreira T.J.P., Rodrigues M.L., Faoro H., Goldenberg S., Alves L.R., Puccia R.. Comparison of the RNA content of extracellular vesicles derived from Paracoccidioides brasiliensisandParacoccidioides lutzii. Cells. 2019; 8:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Westermann A.J., Gorski S.A., Vogel J.. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012; 10:618–630. [DOI] [PubMed] [Google Scholar]
- 28. Westermann A.J., Förstner K.U., Amman F., Barquist L., Chao Y., Schulte L.N., Müller L., Reinhardt R., Stadler P.F., Vogel J.. Dual RNA-seq unveils noncoding RNA functions in host–pathogen interactions. Nature. 2016; 529:496–501. [DOI] [PubMed] [Google Scholar]
- 29. Tarver J.E., Sperling E.A., Nailor A., Heimberg A.M., Robinson J.M., King B.L., Pisani D., Donoghue P.C.J., Peterson K.J.. miRNAs: small genes with big potential in metazoan phylogenetics. Mol. Biol. Evol. 2013; 30:2369–2382. [DOI] [PubMed] [Google Scholar]
- 30. Kang W., Eldfjell Y., Fromm B., Estivill X., Biryukova I., Friedländer M.R.. miRTrace reveals the organismal origins of microRNA sequencing data. Genome Biol. 2018; 19:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chow F.W.-N., Koutsovoulos G., Ovando-Vázquez C., Neophytou K., Bermúdez-Barrientos J.R., Laetsch D.R., Robertson E., Kumar S., Claycomb J.M., Blaxter M. et al.. Secretion of an Argonaute protein by a parasitic nematode and the evolution of its siRNA guides. Nucleic Acids Res. 2019; 47:3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vidal K., Grosjean I., Evillard J.P., Gespach C., Kaiserlian D.. Immortalization of mouse intestinal epithelial cells by the SV40-large T gene. Phenotypic and immune characterization of the MODE-K cell line. J. Immunol. Methods. 1993; 166:63–73. [DOI] [PubMed] [Google Scholar]
- 34. Kim H., Kim J., Kim K., Chang H., You K., Kim V.N.. Bias-minimized quantification of microRNA reveals widespread alternative processing and 3′ end modification. Nucleic Acids Res. 2019; 47:2630–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davis M.P.A., van Dongen S., Abreu-Goodger C., Bartonicek N., Enright A.J.. Kraken: a set of tools for quality control and analysis of high-throughput sequence data. Methods. 2013; 63:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fofanov Y., Luo Y., Katili C., Wang J., Belosludtsev Y., Powdrill T., Belapurkar C., Fofanov V., Li T.-B., Chumakov S. et al.. How independent are the appearances of n-mers in different genomes. Bioinformatics. 2004; 20:2421–2428. [DOI] [PubMed] [Google Scholar]
- 37. Marçais G., Kingsford C.. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011; 27:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shahid S., Axtell M.J.. Identification and annotation of small RNA genes using ShortStack. Methods. 2014; 67:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schulz M.H., Zerbino D.R., Vingron M., Birney E.. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012; 28:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bushmanova E., Antipov D., Lapidus A., Przhibelskiy A.D.. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience. 2019; 8:giz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie Y., Wu G., Tang J., Luo R., Patterson J., Liu S., Huang W., He G., Gu S., Li S. et al.. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014; 30:1660–1666. [DOI] [PubMed] [Google Scholar]
- 42. Robertson G., Schein J., Chiu R., Corbett R., Field M., Jackman S.D., Mungall K., Lee S., Okada H.M., Qian J.Q. et al.. De novo assembly and analysis of RNA-seq data. Nat. Methods. 2010; 7:909–912. [DOI] [PubMed] [Google Scholar]
- 43. Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q. et al.. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011; 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B.. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628. [DOI] [PubMed] [Google Scholar]
- 46. Faulkner G.J., Forrest A.R.R., Chalk A.M., Schroder K., Hayashizaki Y., Carninci P., Hume D.A., Grimmond S.M.. A rescue strategy for multimapping short sequence tags refines surveys of transcriptional activity by CAGE. Genomics. 2008; 91:281–288. [DOI] [PubMed] [Google Scholar]
- 47. Li B., Ruotti V., Stewart R.M., Thomson J.A., Dewey C.N.. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010; 26:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson N.R., Yeoh J.M., Coruh C., Axtell M.J.. Improved placement of multi-mapping small RNAs. G3. 2016; 6:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quintana J.F., Kumar S., Ivens A., Chow F.W.N., Hoy A.M., Fulton A., Dickinson P., Martin C., Taylor M., Babayan S.A., Buck A.H.. Comparative analysis of small RNAs released by the filarial nematode Litomosoides sigmodontis in vitro and in vivo. PLoS Negl. Trop. Dis. 2019; 13:e0007811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chávez Montes R.A., de Fátima Rosas-Cárdenas F., De Paoli E., Accerbi M., Rymarquis L.A., Mahalingam G., Marsch-Martínez N., Meyers B.C., Green P.J., de Folter S.. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014; 5:3722. [DOI] [PubMed] [Google Scholar]
- 52. Chiou N.-T., Kageyama R., Ansel K.M.. Selective export into extracellular vesicles and function of tRNA fragments during T cell activation. Cell Rep. 2018; 25:3356–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Q., Yan M., Cao Z., Li X., Zhang Y., Shi J., Feng G.-H., Peng H., Zhang X., Zhang Y. et al.. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016; 351:397–400. [DOI] [PubMed] [Google Scholar]
- 54. Gottwein E., Mukherjee N., Sachse C., Frenzel C., Majoros W.H., Chi J.-T.A., Braich R., Manoharan M., Soutschek J., Ohler U. et al.. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007; 450:1096–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Friedländer M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N.. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012; 40:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hardcastle T.J., Kelly K.A., Baulcombe D.C.. Identifying small interfering RNA loci from high-throughput sequencing data. Bioinformatics. 2012; 28:457–463. [DOI] [PubMed] [Google Scholar]
- 59. Mohorianu I., Stocks M.B., Wood J., Dalmay T., Moulton V.. CoLIde: a bioinformatics tool for CO-expression-based small RNA Loci Identification using high-throughput sequencing data. RNA Biol. 2013; 10:1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lipson D., Speed T.P., Taub M.. Methods for allocating ambiguous short-reads. Commun. Inform. Syst. 2010; 10:69–82. [Google Scholar]
- 61. Griffiths J.A., Richard A.C., Bach K., Lun A.T.L., Marioni J.C.. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 2018; 9:2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang J., Hotz T., Broadnax L., Yarmarkovich M., Elbaz-Younes I., Hirschi K.D.. Anomalous uptake and circulatory characteristics of the plant-based small RNA MIR2911. Sci. Rep. 2016; 6:26834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang J., Elbaz-Younes I., Primo C., Murungi D., Hirschi K.D.. Intestinal permeability, digestive stability and oral bioavailability of dietary small RNAs. Sci. Rep. 2018; 8:10253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dard-Dascot C., Naquin D., d’Aubenton-Carafa Y., Alix K., Thermes C., van Dijk E.. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genomics. 2018; 19:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Giraldez M.D., Spengler R.M., Etheridge A., Godoy P.M., Barczak A.J., Srinivasan S., De Hoff P.L., Tanriverdi K., Courtright A., Lu S. et al.. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018; 36:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mateescu B., Kowal E.J.K., van Balkom B.W.M., Bartel S., Bhattacharyya S.N., Buzás E.I., Buck A.H., de Candia P., Chow F.W.N., Das S. et al.. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell. Vesicles. 2017; 6:1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The new sRNA-Seq data produced for this paper are available through NCBI’s GEO under accession GSE124506. The sRNA-Seq data from other publications is referenced in Table 1. The main scripts for the analyses presented in this paper are available in the repository: https://github.com/ObedRamirez/Disentangling-sRNA-Seq