


Abstract
UVA-induced mutagenesis was investigated in human pol eta-deficient (XP-V) cells through whole-exome sequencing. In UVA-irradiated cells, the increase in the mutation frequency in deficient cells included a remarkable contribution of C>T transitions, mainly at potential pyrimidine dimer sites. A strong contribution of C>A transversions, potentially due to oxidized bases, was also observed in non-irradiated XP-V cells, indicating that basal mutagenesis caused by oxidative stress may be related to internal tumours in XP-V patients. The low levels of mutations involving T induced by UVA indicate that pol eta is not responsible for correctly replicating T-containing pyrimidine dimers, a phenomenon known as the ‘A-rule’. Moreover, the mutation signature profile of UVA-irradiated XP-V cells is highly similar to the human skin cancer profile, revealing how studies involving cells deficient in DNA damage processing may be useful to understand the mechanisms of environmentally induced carcinogenesis.
INTRODUCTION
Ultraviolet (UV) light-induced cellular DNA damage and its relation to mutagenesis have been extensively demonstrated. However, mutation studies have mostly been limited to single genes such as HPRT (1–3), ATP1A1 (NaK-ATPase) (4) and TP53 (5) or to other genes carried in vectors derived from viruses (6,7). Although these studies have clarified many questions about the mutagenicity of UV light, they have many limitations, such as the clustering of information from various samples and the use of genes that are under selection (such as the HPRT or TP53 gene) (8). In the last decade, the use of next-generation sequencing (NGS), specifically whole-genome and whole-exome sequencing, has provided a simple and efficient diagnosis method for patients with Mendelian disorders (9) and tumour mutation signatures (10,11). This technology has also made it possible to identify the effects of well-known mutagens, potential DNA damage processes, failure of DNA repair mechanisms, and mutations responsible for the development of distinct tumours (12).
Exposure to sunlight is the leading cause of human skin cancer, due mostly to UV radiation-induced DNA damage. UV light is usually divided into three wavelength ranges: UVC (200–280 nm), UVB (280–320 nm) and UVA (320–400 nm) (13). UVA light is an important environmental mutagen and corresponds to more than 95% of UV light that reaches the Earth's surface (14). Although UVB energy is greater, UVA light can penetrate deeper into the skin, causing DNA damage (14). Therefore, the original causes of various types of skin cancer might be at least partly associated with UVA irradiation (13).
It is well established that solar UVA light produces different types of DNA damage (15). The two most critical types of UVA-induced photolesions are cyclobutane pyrimidine dimers (CPDs) (16–19) and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxoG) (20,21). However, the contribution of pyrimidine dimers or oxidized bases to sunlight-induced mutagenesis and skin cancer remains a matter of debate (14,22).
Different types of DNA damage are responsible for the induction of different types of mutations; thus, ‘mutational signatures’ can help to identify the processes involved in mutagenesis as well as the original type of lesion that caused such mutations. The UVB signature is defined as C>T transitions due to mispairing of A with C, formed mainly at pyrimidine dimer sites (1,2). This signature has been discussed as a characteristic of UV radiation in general and not specifically of UVB (15), since it has also been observed in UVA-irradiated cells (2,23). When in syn configuration, 8-oxoG may result in the erroneous pairing of an A, which can produce a G>T (or C>A) transversion (24), leading to a specific mutagenic replication process (25).
Cells exhibit several DNA repair mechanisms that address different types of DNA damage. Oxidized bases, for example, are commonly repaired by specific glycosylases via base excision repair (BER). On the other hand, to repair pyrimidine dimers (such as CPDs), which cause bulky distortions in DNA, the cell mainly recruits the nucleotide excision repair (NER) pathway (26). These lesions may also be tolerated by translesion synthesis (TLS) DNA polymerases, preventing cell death due to DNA synthesis blockage.
DNA polymerase η (pol eta) replicates injured DNA in skin cells, suppressing the formation of skin cancer by reducing UV-induced mutagenesis (27,28). Mutations in pol eta result in a recessive hereditary disorder known as Xeroderma Pigmentosum Variant (XP-V). XP-V cells are NER proficient but are slightly more sensitive to UV irradiation, including UVA light (29,30). In the absence of pol eta, dimers are replicated less efficiently and with less fidelity by other TLS polymerases, such as polymerase ι (pol iota), resulting in the UV-induced hypermutation phenotype of XP-V cells (31,32). It has been proposed that non-instructional lesions, such as abasic sites or pyrimidine dimers, may be replicated by TLS polymerases through the preferential insertion of an A, known as the ‘A-rule’, which would reduce mutations at T-containing dimers (33). In vitro experiments indicate that pol eta can also replicate 8-oxoG and insert an A or C opposite the lesion, whether in syn or anti configuration (34,35), resulting in C>A transversions. Pol eta is also able to bypass other lesions, including abasic sites (36), O4-methyl thymines and O6-methyl guanines (37), acetylaminofluorene adducts (38), and cisplatin and oxaliplatin-induced DNA adducts (39).
The mechanisms by which UVA light induces mutations and skin cancer are an open question (40). The role of pol eta in suppressing mutations induced by UV light is incontrovertible since mutations in the POLH gene result in the XP-V phenotype, which is characterized by sensitivity to sunlight and a higher propensity to develop skin cancer (3). Although there has been interest in understanding how mutations contribute to the high rate of UV-induced skin cancer in XP-V patients (1,41), little is known about UVA light mutagenesis in XP-V cells. In the present work, we applied a whole-exome sequencing (WES) approach to identify which mutations are induced by UVA in pol eta-deficient patient cells compared with pol eta-complemented cell data. These results were further compared with a skin cancer mutation database to evaluate the involvement of UVA in skin carcinogenesis.
MATERIALS AND METHODS
Cell lineages and cell culture
The cells used in this work were SV40-transformed human skin fibroblasts from an XP-V patient (XP30RO – XP-V) and the same cell line complemented with the POLH/XPV gene (XP30RO complemented, clone 6 – XP-V comp). These cells were kindly provided by Drs. Anne Stary and Patricia Kannouche (Laboratory of Genetic Instability and Cancer, Institut Gustave Roussy, Villejuif, France) (41). The cells were grown in Dulbecco's modified Eagle's medium-high glucose (DMEM, LGC Biotechnologies, Cotia, SP, Brazil) supplemented with 10% foetal bovine serum (FBS, Cultilab, Campinas, SP, Brazil) and 1% antibiotic/antimycotic solution (0.1 mg/ml penicillin, 0.1 mg/ml streptomycin and 0.25 mg/ml fungizone, Life Technologies, Carlsbad, CA, USA) under a humidified 5% CO2 atmosphere at 37°C.
Obtaining clones for DNA sequencing and UVA irradiation
For the identification of UVA-induced mutations, XP-V and XP-V comp cells were cloned after UVA irradiation. Cells (1–3 × 103) were plated in 100 mm Petri dishes, and 16–18 h later, they were irradiated with 120 kJ/m2 of UVA light in PBS at a dose rate of 0.058 kJ/m2/s. This dose was chosen from previous work, as it results in ∼20% survival of XP-V cells (for complemented cells, ∼40% survival), which is enough cell survival for mutagenesis assays (29). A UVA lamp (Osram Ultramed FDA KY10s 1000 W, Munich, Germany) was employed for irradiation with a 3 mm-thick Schott BG39 filter (Schott Glass, Mainz, Rheinland-Pfalz, Germany) to shut off wavelengths of <320 nm (17). Control cells were maintained in the dark during irradiation under similar conditions. After irradiation, the cells were grown for 10–15 days, and clones were randomly selected and transferred to 96-multiwell plates using cloning discs (3 mm – Sigma Aldrich, St. Louis, MI, USA) with trypsin (LGC Biotechnologies, Cotia, SP, Brazil). As soon as the clones reached confluence, they were transferred to a larger dish. Cells from two 75 cm2 flasks were used for genomic DNA extraction (Supplementary Figure S1) with a Blood and Cell Culture DNA Mini Kit (Qiagen, Hilden, Nordrhein-Westfalen, Germany). The selection of clones immediately after irradiation led us to detect induced mutations only in the first round of DNA replication for each cell.
Whole-exome sequencing
To analyse the mutation profile of clones, exome sequencing was performed after UVA irradiation. Libraries were prepared with 5 μg of genomic DNA using the Sure Select QXT kit (Agilent Technologies, Santa Clara, CA, USA). Sure Select QXT Human All Exon V6 baits were used for the exome capture procedure. Library preparation and the exon capture were carried out following the manufacturer's specifications. Quality control of libraries was performed with two kits: a High Sensitivity DNA kit (Agilent Technologies) and an Illumina library Quantification kit with primer premix and Light Cycler 480 qPCR mix (KAPA Biosystems, Wilmington, MA, USA). Libraries were sequenced in paired-end mode (2 × 125 bp) on the Illumina HiSeq platform (Illumina, San Diego, CA, USA). Similar but entirely independent experiments were performed using the SOLiD 5500xl sequencing platform (Applied Biosystems, Cleveland, Ohio, EUA) in paired-end mode (75 × 35 bp) with the TargetSeq Exome Enrichment Kit (Life Technologies, Carlsbad, CA, USA). The results are shown in detail in the Supplementary data (Supplementary Figures S2 and S3).
Identification of point mutations
Data analyses of Illumina reads were performed according to GATK Best Practices for exome variant calling. Reads were mapped to the UCSC GRCh37/hg19 reference genome from the GATK Resource Bundle using BWA, and duplicates were marked using Picard. Reads were realigned around indels, and sequencing errors and experimental artefacts were corrected by using the BQSR method. Raw variant discovery was performed with HaplotypeCaller using discovery as the genotyping mode and a minimum Phred-scaled Qscore confidence threshold of 30, and variants with Qscore <10 were flagged as low-quality calls. SNPs with a QD <2.0, FS >60 and MQ <40.0 were flagged and removed from subsequent polymorphism filtering steps.
To identify induced point mutations and filter out polymorphisms, unique single nucleotide variants (SNVs) were selected, considering the position and nucleotide change in comparison to all other samples sequenced, despite genotyping assignment. Whenever a variant was detected in two or more independent clones, it was considered a polymorphism and was discarded. Comparisons were performed using VCFtools, and unique SNVs for each sample were annotated with ANNOVAR using RefSeq. Alternatively, an additional filtering step involving only SNVs annotated as ‘exonic’ or ‘splicing’ in each clone sample was applied to restrict the analysis only to regions enriched in exome sequencing. Raw data were deposited under BioProject accession number PRJNA556825. All sequencing alignment statistics are specified in Supplementary Table S1 for Illumina data and in Supplementary Table S2 for SOLiD data.
Mutational signature analysis
Exclusive point mutations annotated by ANNOVAR were subjected to exploratory data analysis using Woland (github.com/tiagoantonio/woland). We investigated general nucleotide change patterns and motif sequences associated with common mutagens previously described in the literature. Motif sequence analyses were focused on CPD (YY) and 8-oxoG (RGR) photolesions (42,43). Sequence motifs were visualized using pLogo tool (44) considering the human exonic sequences as background. Mutational signature analyses were performed using SomaticSignatures (45), an R implementation of the NMF method framework developed previously (11,46) to identify signatures in human cancer using trinucleotide context motifs. Evaluation of known single base substitution (SBS) signatures from COSMIC database version 3 (47) as performed using the deconstructSigs package (48) using all unique SNVs without restriction to enriched regions.
Statistical analyses
A non-parametric permutation test was used to infer group differences under the null hypothesis (the mean value of the experimental group is not greater than or equal to the mean value of the control group). P-values were calculated comparing the mean values of two groups using Monte-Carlo simulated data from a total of 4999 permutations of group values. We consider two-sided P-values <0.01 as highly statistically significant, 0.01 < P-values < 0.025 as highly statistically significant, 0.025 < P-values < 0.05 as statistically significant and P-values > 0.05 as non-significant. Motif sequence conservation was shown using probability (log-odds of the binomial probability). Significant nucleotide enrichment at a specific position corresponds to a log-odd threshold to achieve a P-value of 0.05 after Bonferroni correction (44).
RESULTS
In the absence of pol eta, UVA light increases mutagenesis in fibroblast cells
Complemented XP-V (XP-V comp, control cells) and XP-V cells were irradiated with 0 or 120 kJ/m2 of UVA light and then subjected to a cloning approach based on clone expansion (Supplementary Figure S1). Four clones from each cell type and treatment were selected for WES. SNVs were called, and unique SNVs from each sample were selected, independent of the cell type, treatment or SNP calling zygosity. Unique SNVs found in both exons and splicing sites were considered. These criteria were used to exclude shared polymorphisms, thus reducing the overall false-positive rate. Considering a typical human cell that divides by mitosis every 24 h on average, the spontaneous mutation rate per generation in exonic regions in this experiment was estimated to be 2.68 × 10−8 mutations per base per generation/replication (Table 1). In pol eta-deficient cells, there was a significant (P < 0.01) increase in the spontaneous mutation rate by 1.4 times (Figure 1A and Table 1), particularly for transversions (P < 0.01), whereas the difference was not significant (P > 0.05) for transitions (Figure 1B). The analysis also revealed a non-synonymous (NS) to synonymous (S) substitution rate (NS/S) of 1.89 in XP-V comp (Table 1), which is consistent with the normal observed genetic variation of a 1.8-fold excess of non-synonymous mutations previously reported using exomes (49). Interestingly, UVA irradiation did not affect the NS/S ratio in either the XP-V UVA or XP-V comp UVA cells, but this ratio was significantly higher (P < 0.05) in non-irradiated XP-V cells (Table 1), suggesting the induction of non-selected mutations due to the absence of pol eta activity.
Table 1.
Somatic point mutations rates of fibroblast cell lines calculated by whole-exome sequencing
| Cell line | Pol eta | Treatment | Point mutation rate per base per generation in exome (10−8) | NS/S ratio |
|---|---|---|---|---|
| XP-V comp | WT | No UVA | 2.68 ± 0.65 | 1.89 ± 0.21 |
| XP-V comp UVA | WT | 120 kJ/m2 UVA | 7.79 ± 6.34 | 1.74 ± 0.78 |
| XP-V | deficient | No UVA | 3.85 ± 0.42 | 2.5 ± 0.82 |
| XP-V UVA | deficient | 120 kJ/m2 UVA | 17.03 ± 10.51 | 1.76 ± 0.22 |
Estimated point mutation rate per base per generation in exome was calculated using only exonic or splicing site SNVs. Exome length of 60 456 963 bp was calculated using total bait targeted regions provided by enrichment kit. Generation was estimated as one cell division cycle each 24 h during a total of 45 days/generations from treatment to genomic DNA extraction. Data represent mean ± SD. Statistical differences were calculated by non-parametric permutation tests; XP-V comp versus XP-V comp UVA: P = 0.065; XP-V versus XP-V UVA: P = 0.0106; XP-V comp versus XP-V: P = 0.0016.
Figure 1.
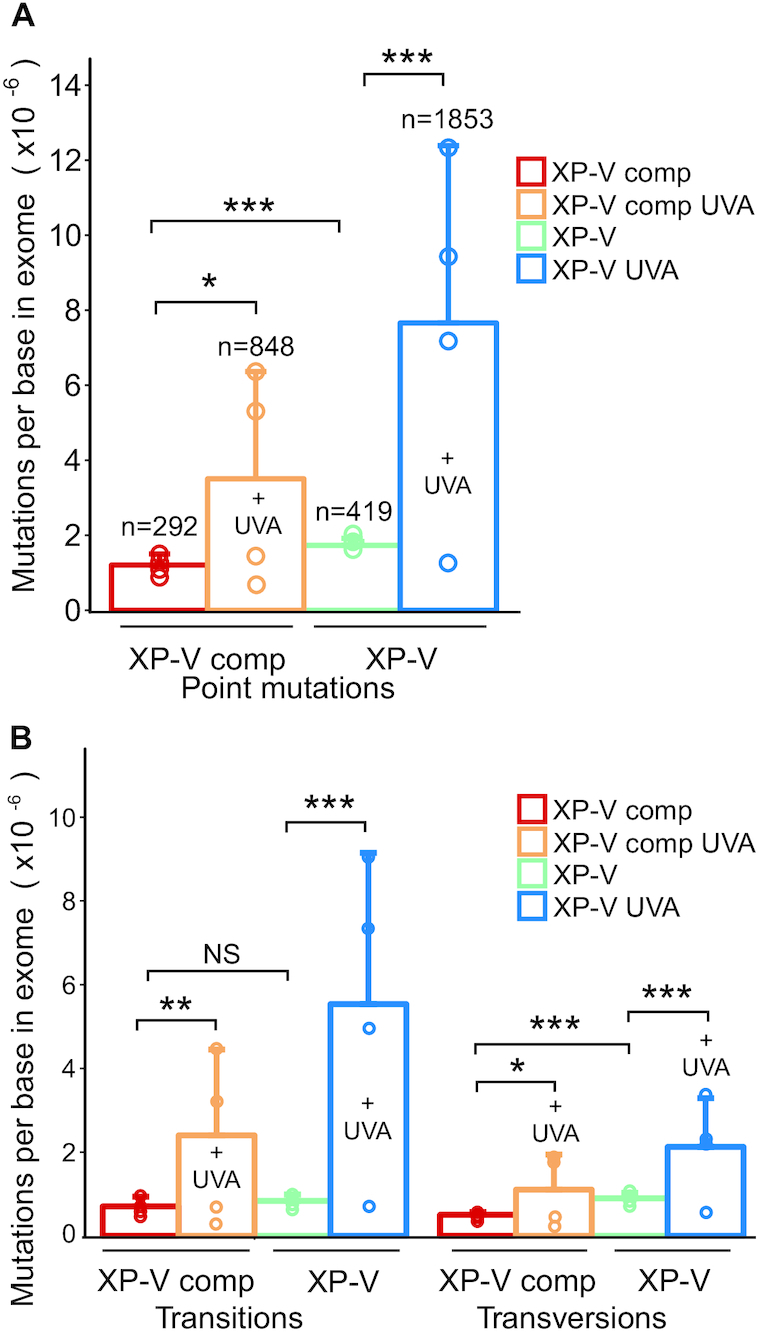
UVA-light and Pol eta absence effects on point mutations detected in exomes. XP-V complemented (XP-V comp), and XP-V cells (XP-V) were irradiated with 120 kJ/m2 of UVA light and then subjected to a 45-day cloning procedure based on colony selection and expansion. Genomic DNA was extracted from four clones of each cell type and treatment. Whole-exome enrichment was performed to construct libraries for sequencing on HiSeq Platform. Read mapping and SNP calling were done using BWA-GATK pipeline. Whenever a variant was detected in two or more independent clones, it was considered a polymorphism – not an induced mutation - and was discarded. (A) Total mutations per base in exome (n = 4 clones per group). (B) Transition and transversion average rates per base in exome. Statistical differences were calculated by non-parametric permutation tests: P < 0.01 (***), 0.01 < P < 0.025 (**), 0.025 < P < 0.05 (*), P > 0.05 (NS). Data represent mean ± SD.
UVA light treatment significantly (P < 0.01) increased the mutation rate to almost three times that in XP-V comp cells compared to non-irradiated clones (Figure 1A). This effect was stronger in XP-V cells, corresponding to a 4.4-fold increase compared to non-irradiated clones (Figure 1A). Both transitions and transversions increased in the UVA treatments in XP-V complemented cells or XP-V cells (Figure 1B). However, transitions were twice as common as transversions after UVA irradiation. Transition levels were also significantly higher in XP-V cells treated with UVA (∼5 times) compared to XP-V cells (∼3 times) (Figure 1B). Similar results obtained in a completely independent set of experiments support the increase in mutagenesis in the absence of pol eta and after UVA exposure (Supplementary Figure S2).
Types of nucleotide changes induced in XP-V cells by UVA light and the absence of pol eta
One of the main goals was to identify the types of mutations and signatures present in UVA-irradiated human cells. The somatic spectra of mutational signatures suggest some different patterns of the trinucleotide context, where C>T and C>A changes are major players (Figure 2, Supplementary Figure S4 for individual clones). Non-irradiated XP-V comp cells display a flat background across changes with a slightly higher contribution of transitions (Figure 2A). This nonspecific pattern may simply correspond to a spontaneous background mutation. UVA irradiation increased the C>T contribution, notably at CCG trinucleotides in the cells (Figure 2B). On the other hand, XP-V cells exhibited a similar spectrum to XP-V comp cells despite more frequent C>A changes, mainly at CCA and TCT trinucleotides, with a small contribution of C>T changes (Figure 2C). Irradiated XP-V cells showed a peculiar spectrum, concentrated in both C>T (mainly) and C>A changes, along with some T>A and T>C changes (Figure 2D). Interestingly, most of these changes seemed to be found at dipyrimidine sites.
Figure 2.

Somatic spectrum of mutations grouped by cell line and treatment by UVA. Somatic spectrum of all point mutations and its trinucleotide context extracted in each cell line group: (A) XP-V comp, (B) XP-V comp UVA, (C) XP-V and (D) XP-V UVA. Trinucleotide contribution was calculated by its frequency. The two most frequent trinucleotides were highlighted in XP-V comp UVA and XP-V groups as trinucleotides mutations in C>A, C>T, T>A and T>C within pyrimidine dimers (Pyr dimers) in the XP-V UVA group.
The results indicate that UVA irradiation mainly induces C>T mutations but also C>A mutations in XP-V cells. Moreover, a significant increase in C>A mutations was detected in non-irradiated XP-V cells.
The absence of pol eta increases spontaneous C>A mutations potentially induced by oxidation, also increased by UVA irradiation
The higher levels of point mutations (mainly C>A transversions) found in non-irradiated XP-V cells (Figure 1B) compared to control XP-V comp cells are curious, and these types of changes were therefore further investigated (Figure 3). These changes are often associated directly with 8-oxoG lesions due to the oxidization of guanines. In fact, the absence of pol eta significantly increased (P < 0.01) C>A changes compared to those in complemented cells (Figure 3A). UVA irradiation did not increase C>A changes (P > 0.05) in XP-V comp cells, but it significantly increased (P < 0.01) this type of change when pol eta was mutated (Figure 3A). Sequence context is considered to be very important for the efficient removal of 8-oxoG lesions (50). The Escherichia coli protein MutM removes 8-oxoG more efficiently in pyrimidine-rich than purine-rich sequences (51). Similarly, the context sequence is expected to affect the efficiency of 8-oxoG removal by OGG1 (50). Thus, C>A transversions present within the RGR motif were evaluated, and the data were similar to what was observed for all C>A changes (Figure 3B), suggesting that both the absence of pol eta and UVA increased mutations potentially caused by 8-oxoG. Additionally, less than ∼28% of C>A changes were located at RGR motifs in XP-V comp cells—whether irradiated by UVA or not—while more than ∼35% and ∼45% were located at RGR motifs in XP-V non-irradiated and irradiated cells, respectively (Figure 3B).
Figure 3.

Distribution of C>A mutations across cell groups in the presence and absence of Pol eta and UVA irradiation. (A) Comparison of C>A mutations (C:G > A:T) in the four groups using mutations per base in exome. (B) Selection of all C>A mutations within RGR sequence motif beyond strand bias. (C–F) Context sequence logo of all C>A mutations adjusted by strand to mutations at G on all clones grouped by the cell line. Sequences were aligned and visualized with pLogo tool using human exome as background. Sequence conservation was shown using probability (log-odds of the binomial probability). A highlight (*) indicates a significant nucleotide enrichment at a specific position correspondent to a log-odd threshold to achieve a P-value of 0.05 after Bonferroni correction. A/T nucleotides were colored in orange and G/C nucleotides in blue. Statistical differences were calculated by non-parametric permutation tests: P < 0.01 (***), 0.01 < P < 0.025 (**), 0.025 < P < 0.05 (*), P > 0.05 (NS). R = IUPAC code for A or G nucleotides.
The sequence context was also checked considering strand orientation based on G>T changes (Figure 3C–F). Non-irradiated XP-V comp cells did not exhibit any apparent preferred sequence neighbouring mutated G nucleotides (Figure 3C). However, in XP-V cells, we found a significant T enrichment bias 5′ of the mutated G (Figure 3D). UVA irradiation did not result in any specific change in sequence context bias in XP-V complemented cells (Figure 3E). However, when pol eta-deficient cells were irradiated with UVA, an intriguing, more complex, sequence-oriented bias was observed: G>T transversions were mainly flanked by G or A in the 3′ direction from the mutated base, which was specific to UVA irradiation (Figure 3F). Moreover, G bases at G>T mutations in RGR motifs were less frequent in the transcribed strand of genes in XP-V cells (irradiated or not), suggesting an involvement of TCR activity in the removal of potential lesions that were not removed by OGG1 (Supplementary Table S3). Thus, we hypothesize that translesion synthesis in the absence of pol eta may increase the mutagenic potential of unexcised 8-oxoG lesions, leading to G>T (C>A) mutations. The possibility of other UVA-induced or spontaneously generated oxidized bases participating in mutagenesis cannot be discarded.
UVA irradiation increases C>T transitions at dipyrimidine sites mainly in the absence of pol eta
CPDs are the most important UV light-induced DNA lesions that preferentially lead to C>T transitions. In non-irradiated cells, the absence of pol eta did not lead to a significant increase in C>T transitions compared to the XP-V comp control (Figure 4A). However, UVA irradiation significantly increased (0.01 < P < 0.05) the frequency of C>T changes in XP-V comp cells, and this increase was even higher in XP-V cells (2.3 times) (Figure 4A). In addition, there was a significant increase in mutations within dipyrimidine motifs (YY) after UVA irradiation in both XP-V comp (P <0.05) and XP-V cells (P <0.01) (Figure 4B). The data analyses also showed that nearly all of the C>T changes (97.3%) in irradiated XP-V cells were found in YY motifs, whereas this percentage was only 66.6% (Figure 4B) in XP-V complemented cells. A significant 7.1-fold increase (P < 0.05) in tandem double mutations (CC>TT) was also observed in UVA-irradiated XP-V cells, not observed in control cells (Figure 4C). No clear sequence context bias was observed in the XP-V comp cell line for C>T transitions (Figure 4D), while UVA-irradiated XP-V complemented cells were biased to a CCG context sequence (Figure 4E). However, a significant sequence bias (C and T) was observed 5′ of C>T changes both in XP-V and UVA-irradiated XP-V cells (Figure 4F and G). This finding confirms that, in XP-V cells, C>T mutations rely on potential pyrimidine dimer sites and preferentially occur at the 3′ base of the dimer. Moreover, when C>T mutations at dipyrimidine sites were analysed for strand bias, these mutations were found to occur less frequently in the transcribed strand of genes in XP-V cells, which is subject to TCR activity for damage removal (Supplementary Table S1). Taken together, these results indicate that other translesion DNA polymerases are more mutagenic than pol eta when replicating potential UVA-induced CPD lesions.
Figure 4.
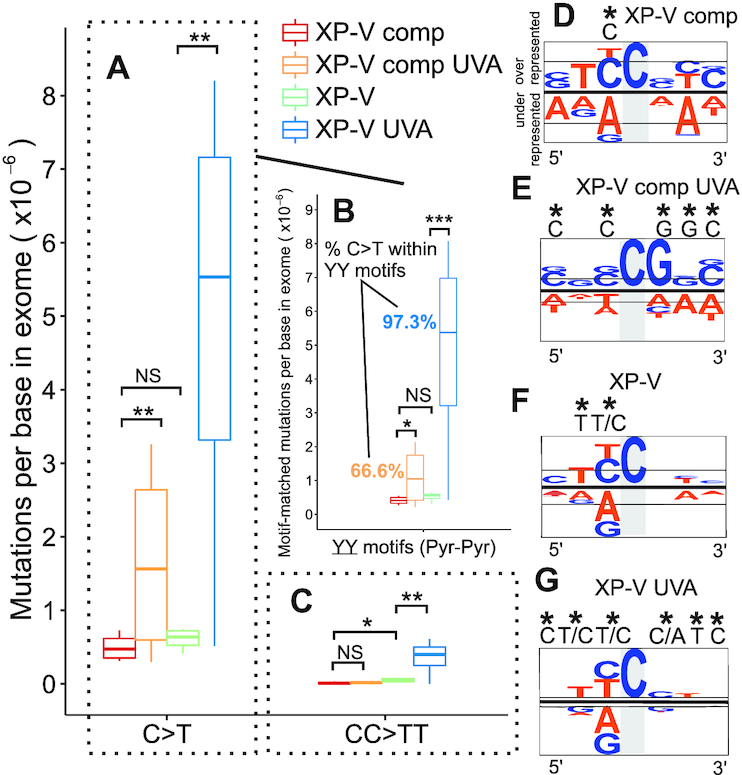
Evaluation of C>T point mutations at pyrimidine dimers in XP-V cells and UVA treatment. (A) Comparison of C>T mutations (C:G > T:A) in the four groups using mutations per base in exome. (B) Selection of all C>T mutations within YY sequence (pyrimidine dimer) motif beyond strand bias. (C) Comparison of double in tandem C>T mutations (CC>TT) in the four groups using double mutations per base in exome. (D–G) Context sequence logo of all C>T mutations adjusted by strand to mutations at C on all clones grouped by the cell line. Sequences were aligned, and visualized with pLogo tool using human exome as background. Sequence conservation was shown using probability (log-odds of the binomial probability). A highlight (*) indicates a significant nucleotide enrichment at a specific position correspondent to a log-odd threshold to achieve a p = value of 0.05 after Bonferroni correction. A/T nucleotides were colored in orange and G/C nucleotides in blue. Statistical differences were calculated by non-parametric permutation tests: P < 0.01 (***), 0.01 < P < 0.025 (**), 0.025 < P < 0.05 (*), P > 0.05 (NS). Y = IUPAC code for T or C nucleotides.
The absence of pol eta also results in more mutagenic events at T:A base pairs within dipyrimidine sites
DNA translesion synthesis by pol eta is considered to be error prone, and it is well accepted that pol eta follows the ‘A-rule’, indicating that the insertion of an A nucleotide is more frequent when performing translesion synthesis at sites of DNA damage. If this is correct, then pol eta should mainly protect against mutations at T:A base pairs, which were further investigated (Figure 5). In fact, mutations at T:A sites were less frequent (all changes with <1.5 × 10−6 mutations per base) than at C:G base pairs under all examined conditions (Supplementary Table S4). After UVA irradiation, mutations at T:A base pairs increased ∼3-fold in both XP-V and XP-V comp cells (Figure 5A and Supplementary Table S4). The potential effects of a prospective ‘A-rule’ for pol eta were investigated in more detail at dipyrimidines. Mutations at T:A base pairs increased at YY sites after UVA irradiation in both XP-V comp and XP-V cells (Figure 5B), but nearly 90% of all T:A base pair mutations were located at dipyrimidine sites in XP-V UVA clones, compared to only 67% when pol eta was present (UVA-irradiated) XP-V comp cells (Figure 5B).
Figure 5.

Distribution of mutations at T:A after UVA irradiation in human cells. (A) T>A, T>C, and T>G (T:A) mutations per base in exome in all cell clone groups. (B) Mutations per base in exome of each type of thymine (T:A) changes at dipyrimidine motifs (YY). (C) T>A, T>C, and T>G mutations per base in exome in all cell clone groups. (D) T>A, T>C and T>G mutations per base in exome in all cell clone groups within YY motifs. Statistical differences were calculated by non-parametric permutation tests: P < 0.01 (***), 0.01 < P < 0.025 (**), 0.025 < P < 0.05 (*), P > 0.05 (NS). Y = IUPAC code for T or C nucleotides.
All specific nucleotide mutations at T:A base pairs (T>A, T>C, and T>G) increased significantly (P <0.01) in UVA-irradiated XP-V cells, while in XP-V comp cells, UVA irradiation led to significant increases (P <0.05) in T>C and T>G mutations, but not T>A mutation (Figure 5C). Similar results were observed in the analysis of these mutations at YY motifs (Figure 5D). Interestingly, T>A mutations that occur specifically in UVA-irradiated XP-V cells are not reported to be related to CPDs, and may occur due to a different type of UVA-induced lesion. These observations and the high frequency of mutations at C-containing dimers compared to T-containing dimers in XP-V cells indicated that the ‘A rule’ is not the main pathway whereby pol eta protects human cells by preventing UVA light-induced mutations.
A single UVA irradiation of XP-V cells induces a mutational signature related to cutaneous skin melanoma
Profiling of mutational signatures hidden within SNVs has been considered a successful method for understanding the mutagenic mechanisms and complex processes related to carcinogenesis. To evaluate the similarities between trinucleotide changes among all samples sequenced, we used a clustering approach to compute Euclidean distances between them (Figure 6A). Two main groups were clustered: one formed mainly by XP-V comp clones and the other formed by XP-V clones (Figure 6A), suggesting a potential signature of the absence of pol eta. UVA-irradiated XP-V comp and XP-V clones clustered into separated subgroups, suggesting distinct but singular UVA mutation effects in the absence and presence of pol eta. Similar results were obtained from in another set of experiments, with MRC5 and XP-V cell lines (Supplementary Figure S3). Our hypothesis was a model involving three signatures: two for UVA irradiation (in the presence and absence of pol eta) and one mutational signature of the absence of pol eta without irradiation.
Figure 6.

Linking mutational signatures to UVA light-induces mutagenesis: Pol eta and cancer. (A) Hierarchical clustering trinucleotide mutational spectrum of cell-line clones using Euclidean distances. XP-V comp, XP-V comp UVA, XP-V and XP-V UVA clones were highlighted in red, orange, green, and blue respectively. (B) De novo mutational signatures SA, SB, and SC extracted using NMF-method implemented in SomaticSignatures package based on a method developed by Alexandrov et al. (11). (C) Contribution of each extracted signature (SA, SB, SC) occurrences on the 16 samples of each cell-line group. (D) Contribution of known mutational signatures from COSMIC database version 3 on XP-V comp, XP-V comp UVA, XP-V and XP-V UVA cell-line groups using deconstructSigs package. The 14 COSMIC signatures detected were grouped into seven classes according to its proposed aetiology and/or mutational features.
The non-negative matrix factorization (NMF) is one of the most used methods for extracting mutational signatures using SNVs and their trinucleotide sequence context (46). To investigate whether we could identify mutational signatures from individual clones as suggested by hierarchical clustering, we used an NMF approach to decompose mutation data to fit the model based on three de novo signatures (Figure 6B). Signature SA consisted of C>T changes at ACG, CCG and GCG motifs, whereas SB consisted of nonspecific C>T changes with a higher contribution of C>A changes at CCG, GCT and TCT motifs. Signature SC was characterized by C>T at pyrimidine dimers, and C>A changes were also enriched at some dipyrimidine sites (CCA, TCA, TCG and TCT). XP-V comp clones presented a mixture of all three signatures, sometimes with a substantial contribution of SB (Figure 6C). Irradiation by UVA strongly increased the contribution of SA in XP-V comp clones (Figure 6C). XP-V clones presented a mixture of all signatures with a slight prevalence of SC, which was the signature of almost all UVA-irradiated XP-V clones (Figure 6C). We propose that SC is associated with higher error-prone activity of other polymerases with higher mutagenic activities at CPDs at pyrimidine dimers and 8-oxoG lesions. Signature A could be related to UVA mutations in the presence of pol eta and is characterized only as increased C>T changes (not specifically at dipyrimidines) (Figure 6C). We also hypothesize that Signature B is a spontaneous mutagenic signature found mainly non-UVA-irradiated clones (Figure 6C).
Mutational signatures also provide an interesting approach for characterizing and inferring the processes of carcinogenesis and improving cancer treatment and diagnosis. UV light is the main mutagenic driver of skin melanoma, and patients with mutations in the pol eta gene exhibit an increased incidence of skin tumours. It is proposed that this higher incidence is due to other error-prone polymerases that replace the role of pol eta and bypass UV light-induced lesions, such as CPDs. Thus, we analysed the contribution of known COSMIC single base substitution (SBS) signatures database v3 in our data (Figure 6D). XP-V comp cells exhibited a mixture of three different signatures (SBS5, SBS10b and SBS21), which could be associated with a broad spontaneous mutational profile. UVA irradiation of these cells resulted in high contribution of clock-like signatures SBS1 and SBS5, which are found in many types of cancer. SBS1 is a signature related to an increase in C>T changes probably due to spontaneous deamination of 5-methylcytosine. Non-irradiated XP-V cells displayed the signature SBS1, similar to that in XP-V comp cells, but with a contribution of signatures SBS18 and SBS36. These signatures are associated with C>A mutations, possibly due to damage by reactive oxygen species.
Nevertheless, UVA irradiation of XP-V cells resulted in key contribution (>49%) of signatures SBS7a, SBS7b and SBS7c, which are the top signatures in those cells. Signatures SBS7 are characteristic signatures found predominantly in skin cancers and are associated with C>T mutations at dipyrimidines (Figure 6D). These signatures are very similar to de novo Signature C extracted from irradiated XP-V clones, despite the contribution of C>A changes at dipyrimidines (Figure 6B), which we proposed is due to the increase in mutagenic events at CPDs caused by UVA and 8-oxoG lesions in a context where pol eta is not present. In addition, there is also a contribution of ∼9% of signature SBS18, which is related to damage by reactive oxygen species. These data reveal the participation of UVA light in skin carcinogenesis, especially in XP-V cells, involving pyrimidine dimers as the main mutation causes.
DISCUSSION
Here, a successful experimental cloning strategy was developed to identify mutations induced after UVA irradiation, potentially associated with carcinogenesis in pol eta-deficient patients, using WES. This strategy allowed a more complete evaluation of induced point mutations compared to experiments targeting specific genes. Moreover, the fact that mutations are observed directly within the chromosomal DNA may better reflect what occurs in skin cells’ genome after sunlight exposure. Another important aspect of this approach is that most mutations detected are mainly induced during the first round of DNA synthesis after treatment, immediately after cloning. The reason for this is that if mutations were induced later, when the clones exhibit two or more cells, they would be diluted and therefore not considered a mutation. The spontaneous mutation rate estimated in the exomes of XP-V comp cells (Table 1 and Figure 1) is on the same order of magnitude as that in germline and one order of magnitude lower than somatic mutation rates calculated using whole-genome sequencing (52). In fact, fewer mutations in exons are expected due to purifying selection simply via cell growth, but recent studies demonstrate that that this situation can also be caused by higher mismatch-repair activity in exonic regions (53). However, mutational background rate calculation could be affected by heterogeneous clones with different somatic mutations, since parental cell lines were not sequenced. A recent study (54), using a high-controlled strategy based on sequencing of subclones and parental clones, estimated a background mutation rate of ∼1.11 × 10−8 mutations per base per generation. Therefore, the estimated spontaneous mutation rate in this work (2.68 × 10−8 mutations per base per generation) is comparable to other measurements, indicating reasonable SNP calling, mutation-selection, and polymorphism exclusion criteria.
XP-V patients exhibit an increased frequency of skin cancer due to the absence of pol eta, which confers a hypermutation phenotype in sunlight-exposed skin areas (3,6). Previous work using a UVC-irradiated shuttle vector transfected in XP-V cells and complemented cells showed a significant increase in mutagenesis in a reporter gene in pol eta-deficient cells (41). More recently, the endogenous HPRT gene was used to detect increased UVB mutagenesis in XP-V cells in comparison to healthy cells (1). The results reported here are generally in agreement with those reported in association with UVC and UVB, with a clear increase in mutations detected in pol eta-deficient cells after UVA irradiation but with many particularities. Curiously, however, there was an unexpected significant increase in mutagenesis in non-irradiated XP-V cells compared to complemented cells, which was related to C>A transversions.
Previous work has shown that pol eta correctly bypasses 8-oxoG in vitro, and an absence of Rad30 (the homologue of pol eta) in yeast leads to increased C>A mutagenesis, consistent with its role in suppressing mutations due to oxidatively generated DNA damage (55). In this work, we observed that pol eta complementation decreased C>A mutations by 3-fold compared to that in pol eta-deficient cells, even in the absence of UVA irradiation (Figure 3). These C>A transversions were mainly observed in sequence motifs (RGR) related to 8-oxoG lesions that were not efficiently removed by OGG1. Thus, these data are consistent with the participation of pol eta to correctly bypass oxidized bases (24,35,55), suppressing mutagenesis due to oxidized bases induced endogenously, including 8-oxoG bases (3), which are by products of normal cell metabolism. These data have important consequences for individuals with XP-V, as they imply an increase in mutagenesis even in the absence of sunlight exposure, which could be responsible for internal tumours.
The role of UVA light in mutagenesis has been related to its ability to generate DNA damage directly by light absorption (2,17,56) as well as due to oxidative stress through type I and II photosensitization reactions (14). The main mutations induced after UVC light exposure are C>T and CC>TT transitions (3,40,41). These changes are considered a UV signature when they are located within potential dipyrimidine (YY) sites (57,58) and because they are rarely observed due to non-UV carcinogens (59,60). However, the participation of UVA and UVB light in inducing these types of mutations is still a matter of debate (2). Pointed efforts to map and understand mutagenesis induced by UVA and UVB have been made. The sequencing of the p53 gene from human keratinocyte cells exposed to UVA light showed only G>T transversions, although in a limited number of mutations analysis (61). However, the mutations observed in the UVA-irradiated skin of lacZ transgenic mice are primarily C>T transitions at dipyrimidine sites (23).
Our data clearly showed that UVA irradiation, similar to UVC and UVB irradiation, induces increased mutagenesis in pol eta-deficient cells compared to non-irradiated cells (Figure 1 and Table 1). This increase in UVA mutagenesis is also observed, although to a lower extent, in cells expressing pol eta. While almost all types of point mutations showed a significant increase in both cell lines upon UVA irradiation, C>T mutations were especially increased in XP-V cells (Figures 2 and 4). CC>TT tandem transitions were also significantly increased by UVA irradiation in the absence of pol eta (Figure 4). These results confirm the high relevance of pyrimidine dimers in mutagenesis induced by UVA light in human cells, as observed by others in different models (62,63). Additionally, our results support the role of pol eta in bypassing UVA light-induced pyrimidine dimers and participating in the reduction of C>T mutations, as previously reported in association with UVC and UVB (2,41,64).
However, predicting what lesion led to a particular mutation is not trivial. For example, C>T mutations can be generated either at dipyrimidine sites or by pyrimidine oxidation (65). In fact, in vitro data indicated that 5-hydroxypyrimidine lesions may induce C>T and C>G mutations (66). Similarly, uracil glycol (a product of C oxidation) may mispair with A, inducing C>T transitions (67). Moreover, works with shuttle vectors replicating in mammalian cells revealed that 2-hydroxy-adenine may induce A>G (T>C) (68) and the open ring-opened formamido-deoxyguanosine (Fapy.dG) also induces G>T (C>A) mutations (69). Therefore, the contribution of oxidative stress in the UVA-induced mutagenesis cannot be ruled out. To minimize this bias, we analysed C>T and C>A changes within motif sequences described for CPDs and 8-oxoG (42,70). The occurrence of C>A mutations was significant at 8-oxoG motif sites in the absence of pol eta and when XP-V cells were irradiated with UVA (Figure 3). In fact, the ratio on the yields of CPD and 8-oxoG is around 5 for UVA light, while it is 2 to 3 orders of magnitude larger for UVB (22,56,57), which may explain the fact oxidation may contribute to mutagenesis in UVA irradiated cells. Moreover, we recently demonstrated that oxidative stress strongly affects particularly pol eta-deficient cells, after UVA exposure, with induction of oxidized bases in the genome (29). This increased oxidative stress can also explain the UVA-induced C>A mutations observed in the experiments described here (Figures 2 and 3). However, CPD motifs (dipyrimidines) were mostly present under UVA mutagenesis in XP-V clones: 97.3% of all C>T changes induced by UVA in XP-V samples were induced within CPD motifs, whereas in complemented cells, these motifs were less represented (66.6%) (Figure 3). These results showed that UVA predominantly induced mutations at dipyrimidine sites in pol eta-deficient cells, in agreement with previous data demonstrating that UVA could induce mutations at these sites in hamster and human skin cells (57,71). This pattern of mutation is also observed in human skin tumours (72,73).
One interesting feature of pol eta is its remarkable capacity to bypass TT dimers by inserting two adenines opposite TT (28), which is known as the ‘A-rule’. Thus, it is believed that in healthy cells, pol eta avoids UV-induced mutagenesis mainly in T-containing photoproducts (41). In control cells, the data showed that only ∼67% of mutations at T:A base pairs were located at YY sites, which increased to >90% in XP-V cells (Figure 5). However, most of the differences between the cells took the form of T>A mutations, which are probably not related to pyrimidine dimers. Interestingly, UVA induces CPDs predominantly at thymine-containing dipyrimidine sites (16,74), but as most of the mutations induced by UVA do not involve Ts, the mutagenicity of these dimers in human cells must be low, independent of pol eta activity. Consistent with our data, UVA induces more mutagenesis at CT and CC sites than at TT sites in skin cells (71). Therefore, the small contribution of mutations at T-containing dimers indicates that, human DNA polymerases can in fact insert an A opposite these lesions, but the participation of pol eta in this process is lower than previously proposed (37).
The mutation data were also analysed by examining the mutational pattern within the sequence context. Remarkably, UVA light induced C>T mutations in different trinucleotide context sequences in the presence or absence of pol eta (Figure 6). Non-irradiated XP-V comp clones did not show any clear mutational pattern. However, UVA-irradiated XP-V comp cells exhibited C>T changes mainly in CCG sequences but also at sites outside of potential pyrimidine dimers (GCG and ACG) (signature SB). Due to the proximity to guanine, the presence of C>T sites outside of dimers indicate that the mutations may have been caused by lesions other than pyrimidine dimers, such as those resulting from oxidative stress processes. These data differ from the sequence contexts associated with UVB (5′TCG3′) (64), but these experiments were obtained in a mice model, which may differ from human cells. UVA-irradiated XP-V cells presented C>T mutations predominantly at YC sites (i.e. precisely at dipyrimidine sites) and mostly at the first base to be replicated. Recent work with in vitro polymerization by TLS polymerases of UVC and UVB irradiated DNA templates indicated that pol eta is responsible to insert A opposite C-containing dimers, increasing the frequency of C>T mutations, supporting a mutagenic role for this polymerase, and following the idea that pol eta is responsible for the ‘A-rule’ (75,76). This is not consistent with our results where the increase in UVA-induced C>T mutations corroborates the error-prone bypass performed by the TLS proteins in the absence of pol eta at pyrimidine dimers (31,41,64,77,78). The lack of mutations involving T-containing dimers also contradicts the idea that pol eta has such activity within the cells, contrary to what was obtained in the in vitro system. The simplest explanation for the results shown here is that pol eta is prone to replicate C-containing dimers by, correctly, inserting G, thereby controlling UV-induced mutagenesis, thus following a ‘G-rule’. Another possible explanation for C>T mutations is that C-containing pyrimidine dimers are highly unstable and can be deaminated to uracil or, if methylated (5-methylC), to thymine (79), which would replicate through the insertion of an A opposite dimers. This interesting hypothesis would imply that pyrimidine dimers would have to be first monomerized before encountered by a replicative polymerase, as suggested by in vitro experiments (80). Alternatively, pyrimidine dimers containing a U (deaminated C) should exhibit instructional coding capacity during replication by pol eta. In fact, some groups have proposed that TLS polymerases (including pol eta homologs) may read the bases correctly when replicating dipyrimidine photoproducts (33,81). Thus, if these lesions are instructional, deamination of C (which occurs at high frequency within pyrimidine dimers) to U would be one of the main causes for C>T mutations. In the absence of pol eta, pyrimidine dimers would take longer to be replicated increasing the possibility of C deamination and the eventual lesion bypass would result in increased mutagenesis, as observed in UVA-irradiated XP-V cells.
Finally, we measured the contribution of known published mutational signatures from the COSMIC database (Figure 6). Compared with other types of cancer, skin cancer exhibited more C>T at dipyrimidine changes than C>A changes. Here, we described the participation of UVA in the carcinogenesis process in comparison to skin cancer related signatures SBS7s, which presented high similarity to Signature SC obtained in XP-V cells irradiated by UVA (Figure 6). We also shine a light into aetiology of SBS7a-b signatures, which can be more related to CPD lesions than 6–4 photoproducts, and the involvement of reactive oxygen species in SBS18 and SBS36 signatures (Figure 6D). Through these analyses, we provided consistent evidence of the contribution of UVA-induced mutagenesis to the development of skin cancer in XP-V patients, which was greater than is usually assumed, highlighting the participation of CPD lesions in this process.
CONCLUSIONS
This work provides a detailed evaluation of the UVA-induced increase in point mutations in human cells, especially in cells from XP-V patients, which may help us to understand the increased frequency of skin tumour formation in these patients. The absence of pol eta contributed to higher accumulation of mutations, mainly due to C-containing pyrimidine dimers (C>T or CC>TT). The results also demonstrate the role of pol eta in correctly bypassing oxidatively generated lesions, suppressing C>A mutations, even in the absence of irradiation, which could be related to internal tumours in XP-V patients. Additionally, we observed low levels of mutations at T-containing dimers, due to A insertions opposite these lesions, as suggested by the ‘A-rule’, but these mutations seem to be highly independent of pol eta. On the other hand, pol eta seems to be more important in the insertion of a G (this would be the ‘G-rule’) at C-containing dimers, preventing mutations. The examination of mutational signatures revealed that a single UVA irradiation of XP-V cells, corresponding to 30 min of sunlight exposure, was enough to allow the detection of mutational signatures related to cutaneous skin melanoma in the human population. Thus, this work provides data for better understanding the skin cancer initiation process related to DNA damage in XP-V patients as well as healthy individuals.
DATA AVAILABILITY
Woland is a tool to analyze point mutation patterns using resequencing data available in the GitHub Repository (github.com/tiagoantonio/woland).
Sequencing data have been deposited with the Sequence Read Archive (SRA) under SRA accession number PRJNA556825.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Core Facility for Scientific Research - USP (CEFAP-USP/GENIAL) and Multi-user genomic Section of the Human Genome & Stem Cell Research Center (HUG-CELL) for providing access to NGS platforms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP, São Paulo, Brazil) [2014/15982-6, 2012/16929-6, 2013/08028-1, 2018/06619-6]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, DF, Brazil) [308868/2018-8], including scholarships [150049/2018-8 to N.C.M., 141411/2017 to T.A.S.]; Coordenação de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES, Brasília, DF, Brazil, financial code 001). Funding for open access charge: Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP).
Conflict of interest statement. None declared.
REFERENCES
- 1. Herman K.N., Toffton S., McCulloch S.D.. Detrimental effects of UV-B radiation in a xeroderma pigmentosum-variant cell line. Environ. Mol. Mutagen. Mutagen. 2014; 55:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kappes U.P., Luo D., Potter M., Schulmeister K., Runger T.M.. Short-and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J. Invest. Dermatol. 2006; 126:667–675. [DOI] [PubMed] [Google Scholar]
- 3. Wang Y.C., Maher V.M., Mitchell D.L., McCormick J.J.. Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal, error-prone replication on a template containing photoproducts. Mol. Cell Biol. 1993; 13:4276–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kappes U.P., Runger T.M.. No major role for 7,8-dihydro-8-oxoguanine in ultraviolet light-induced mutagenesis. Radiat. Res. 2005; 164:440–445. [DOI] [PubMed] [Google Scholar]
- 5. D’Errico M., Calcagnile A., Canzona F., Didona B., Posteraro P., Cavalieri R., Corona R., Vorechovsky I., Nardo T., Stefanini M. et al.. UV mutation signature in tumor suppressor genes involved in skin carcinogenesis in xeroderma pigmentosum patients. Oncogene. 2000; 19:463–467. [DOI] [PubMed] [Google Scholar]
- 6. Kannouche P., Stary A.. Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie. 2003; 85:1123–1132. [DOI] [PubMed] [Google Scholar]
- 7. Choi J.H., Pfeifer G.P.. The role of DNA polymerase eta in UV mutational spectra. DNA Repair (Amst). 2005; 4:211–220. [DOI] [PubMed] [Google Scholar]
- 8. Alexandrov L.B., Stratton M.R.. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014; 24:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D., Shendure J.. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011; 12:745–755. [DOI] [PubMed] [Google Scholar]
- 10. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al.. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R.. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013; 3:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poon S., McPherson J.R., Tan P., Teh B., Rozen S.G.. Mutation signatures of carcinogen exposure: genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014; 6:24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schuch A.P., Moreno N.C., Schuch N.J., Menck C.F.M., Garcia C.C.M.. Sunlight damage to cellular DNA: focus on oxidatively generated lesions. Free Radic. Biol. Med. 2017; 107:110–124. [DOI] [PubMed] [Google Scholar]
- 14. Sage E., Girard P.M., Francesconi S.. Unravelling UVA-induced mutagenesis. Photochem. Photobiol. Sci. 2012; 11:74–80. [DOI] [PubMed] [Google Scholar]
- 15. Cadet J., Sage E., Douki T.. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. 2005; 571:3–17. [DOI] [PubMed] [Google Scholar]
- 16. Rochette P.J., Therrien J.P., Drouin R., Perdiz D., Bastien N., Drobetsky E.A., Sage E.. UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells. Nucleic Acids Res. 2003; 31:2786–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schuch A.P., Galhardo R.S., Schuch N.J., Menck C.F.M.. Development of a DNA-dosimeter system for monitoring the effects of solar-ultraviolet radiation. Photochem. Photobiol. Sci. 2009; 8:111–120. [DOI] [PubMed] [Google Scholar]
- 18. Cortat B., Garcia C.C.M., Quinet A., Schuch A.P., Lima-Bessa K.M., Menck C.F.M.. The relative roles of DNA damage induced by UVA irradiation in human cells. Photochem. Photobiol. Sci. 2013; 12:1483–1495. [DOI] [PubMed] [Google Scholar]
- 19. Perdiz D., Gróf P., Mezzina M., Nikaido O., Moustacchi E., Sage E.. Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells: Possible role of dewar photoproducts in solar mutagenesis. J. Biol. Chem. 2000; 275:26732–26742. [DOI] [PubMed] [Google Scholar]
- 20. Courdavault S., Baudouin C., Charveron M., Favier A., Cadet J., Douki T.. Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells. Mutat. Res. 2004; 556:135–142. [DOI] [PubMed] [Google Scholar]
- 21. Brem R., Macpherson P., Guven M., Karran P.. Oxidative stress induced by UVA photoactivation of the tryptophan UVB photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) inhibits nucleotide excision repair in human cells. Sci. Rep. 2017; 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kielbassa C., Roza L., Epe B.. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis. 1997; 18:811–816. [DOI] [PubMed] [Google Scholar]
- 23. Ikehata H., Kudo H., Masuda T., Ono T.. UVA induces C→T transitions at methyl-CpG-associated dipyrimidine sites in mouse skin epidermis more frequently than UVB. Mutagenesis. 2003; 18:511–519. [DOI] [PubMed] [Google Scholar]
- 24. Carlson K.D., Washington M.T.. Mechanism of efficient and accurate nucleotide incorporation opposite 7,8-dihydro-8-oxoguanine by Saccharomyces cerevisiae DNA polymerase eta. Mol. Cell Biol. 2005; 25:2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. David S.S., O’Shea V.L., Kundu S.. Base-excision repair of oxidative DNA damage. Nature. 2007; 447:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friedberg E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer. 2001; 1:22–33. [DOI] [PubMed] [Google Scholar]
- 27. van Steeg H., Kraemer K.H.. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol. Med. Today. 1999; 2:86–84. [DOI] [PubMed] [Google Scholar]
- 28. Masutani C., Araki M., Yamada A., Kusumoto R., Nogimori T., Maekawa T., Iwai S., Hanaoka F.. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999; 18:3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moreno N.C., Garcia C.C.M., Munford V., Rocha C.R.R., Pelegrini A.L., Corradi C., Sarasin A., Menck C.F.M.. The key role of UVA-light induced oxidative stress in human Xeroderma Pigmentosum Variant cells. Free Radic. Biol. Med. 2019; 131:432–442. [DOI] [PubMed] [Google Scholar]
- 30. Moreno N.C., Garcia C.C.M., Rocha C.R.R., Munford V., Menck C.F.M.. ATR/Chk1 pathway is activated by oxidative stress in response to UVA light in human Xeroderma Pigmentosum Variant cells. Photochem. Photobiol. 2018; 95:345–354. [DOI] [PubMed] [Google Scholar]
- 31. Vaisman A., Frank E.G., Iwai S., Ohashi E., Ohmori H., Hanaoka F., Woodgate R.. Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase ι. DNA Repair (Amst). 2003; 2:991–1006. [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Woodgate R., McManus T.P., Mead S., McCormick J.J., Maher V.M.. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase η, DNA polymerase ι causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007; 67:3018–3026. [DOI] [PubMed] [Google Scholar]
- 33. Taylor J.S. New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat. Res. 2002; 510:55–70. [DOI] [PubMed] [Google Scholar]
- 34. Silverstein T.D., Jain R., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. Structural basis for error-free replication of oxidatively damaged DNA by yeast DNA polymerase h. Structure. 2010; 18:1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mcculloch S.D., Kokoska R.J., Garg P., Burgers P.M., Kunkel T.A.. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases delta and eta. Nucleic Acids Res. 2009; 37:2830–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patra A., Zhang Q., Lei L., Su Y., Egli M., Guengerich F.P.. Structural and kinetic analysis of nucleoside triphosphate incorporation opposite an abasic site by human translesion DNA polymerase. J. Biol. Chem. 2015; 290:8028–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Glick E. Mutations in human DNA polymerase eta motif II alter bypass of DNA lesions. EMBO J. 2001; 20:7303–7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masutani C. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000; 19:3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bassett E., Vaisman A., Tropea K.A., McCall C.M., Masutani C., Hanaoka F., Chaney S.G.. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases β and η. DNA Repair (Amst). 2002; 1:1003–1016. [DOI] [PubMed] [Google Scholar]
- 40. Sage E., Lamolet B., Brulay E., Moustacchi E., Chteauneuf A., Drobetsky E.A.. Mutagenic specificity of solar UV light in nucleotide excision repair-deficient rodent cells. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stary A., Kannouche P., Lehmann A.R., Sarasin A.. Role of DNA polymerase η in the UV-mutation spectrum in human cells. J. Biol. Chem. 2003; 278:18767–18775. [DOI] [PubMed] [Google Scholar]
- 42. Ikehata H., Kumagai J., Ono T., Morita A.. Solar-UV-signature mutation prefers TCG to CCG: extrapolative consideration from UVA1-induced mutation spectra in mouse skin. Photochem. Photobiol. Sci. 2013; 12:1319. [DOI] [PubMed] [Google Scholar]
- 43. Rogozin I.B., Babenko V.N., Milanesi L., Pavlov Y.I.. Computational analysis of mutation spectra. Brief. Bioinform. 2003; 4:210–227. [DOI] [PubMed] [Google Scholar]
- 44. Shea J.P.O., Chou M.F., Quader S.A., Ryan J.K., Church G.M., Schwartz D.. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods. 2013; 10:1211–1214. [DOI] [PubMed] [Google Scholar]
- 45. Gehring J.S., Fischer B., Lawrence M., Huber W.. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015; 31:3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al.. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Ng A.W.T., Boot A., Covington K.R., Gordenin D.A., Bergstrom E., Lopez-Bigas N. et al.. The repertoire of mutational signature in human cancer. 2018; bioRxiv doi:03 july 2019, pre-print: not peer-reviewed 10.1101/322859. [DOI]
- 48. Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C.. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Y., Vinckenbosch N., Tian G., Huerta-Sanchez E., Jiang T., Jiang H., Albrechtsen A., Andersen G., Cao H., Korneliussen T. et al.. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010; 42:969–972. [DOI] [PubMed] [Google Scholar]
- 50. Sassa A., Beard W.A., Prasad R., Wilson S.H.. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 2012; 287:36702–36710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hatahet Z., Zhou M., Reha-Krantz L.J., Morrical S.W., Wallace S.S.. In search of a mutational hotspot. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:8556–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Milholland B., Dong X., Zhang L., Hao X., Suh Y., Vijg J.. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 2017; 8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frigola J., Sabarinathan R., Mularoni L., Muinõs F., Gonzalez-Perez A., López-Bigas N.. Reduced mutation rate in exons due to differential mismatch repair. Nat. Genet. 2017; 49:1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zou X., Owusu M., Harris R., Jackson S.P., Loizou J.I., Nik-zainal S.. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 2018; 9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haracska L., Yu S.L., Johnson R.E., Prakash L., Prakash S.. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase η. Nat. Genet. 2000; 25:458–461. [DOI] [PubMed] [Google Scholar]
- 56. Mouret S., Baudouin C., Charveron M., Favier A., Cadet J., Douki T.. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Douki T., Reynaud-Angelin A., Cadet J., Sage E.. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry. 2003; 42:9221–9226. [DOI] [PubMed] [Google Scholar]
- 58. Brash D.E., Seetharam S., Kraemer K.H., Seidman M.M., Bredberg A.. Photoproduct frequency is not the major determinant of uv base substitution hot-spots or cold spots in human-cells. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:3782–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wikonkal N.M., Brash D.E.. Ultraviolet radiation induced signature mutations in photocarcinogenesis. J. Investig. Dermatol. Proc. 1999; 4:6–10. [DOI] [PubMed] [Google Scholar]
- 60. Ziegler A., Leffell D.J., Kunala S., Sharma H.W., Gailani M., Simon J.A., Halperin A.J., Baden H.P., Shapiro P.E., Bale A.E.. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:4216–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Persson A.E., Edström D.W., Backvall H., Lundeberg J., Pontén F., Ros A.M., Williams C.. The mutagenic effect of ultraviolet-A1 on human skin demonstrated by sequencing the p53 gene in single keratinocytes. Photodermatol. Photoimmunol. Photomed. 2002; 18:287–293. [DOI] [PubMed] [Google Scholar]
- 62. Rizzo J.L., Dunn J., Rees A., Runger T.M.. No formation of DNA double-strand breaks and no activation of recombination repair with UVA. J. Invest. Dermatol. 2011; 131:1139–1148. [DOI] [PubMed] [Google Scholar]
- 63. Robert C., Muel B., Benoit A., Dubertret L., Sarasin A., Stary A.. Cell survival and shuttle vector mutagenesis induced by ultraviolet A and ultraviolet B radiation in a human cell line. J. Invest. Dermatol. 1996; 106:721–728. [DOI] [PubMed] [Google Scholar]
- 64. Ikehata H., Chang Y., Yokoi M., Yamamoto M., Hanaoka F.. Remarkable induction of UV-signature mutations at the 3′-cytosine of dipyrimidine sites except at 5′-TCG-3′ in the UVB-exposed skin epidermis of xeroderma pigmentosum variant model mice. DNA Repair (Amst). 2014; 22:112–122. [DOI] [PubMed] [Google Scholar]
- 65. Basu A.K., Loechler E.L., Leadon S.A., Essigmann J.M.. Genetic effects of thymine glycol: site-specific mutagenesis and molecular modeling studies. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:7677–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Purmal A.A., Kow Y.W., Wallace S.S.. Major oxidative products of cytosine, 5-hydroxycytosine and 5-hydroxyuracil, exhibit sequence context-dependent mispairing in vitro. Nucleic Acids Res. 1994; 22:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Purmal A.A., Lampman G.W., Bond J.P., Hatahet Z., Wallace S.S.. Enzymatic processing of uracil glycol, a major oxidative product of DNA cytosine. J. Biol. Chem. 1998; 273:10026–10035. [DOI] [PubMed] [Google Scholar]
- 68. Kamiya H., Kasai H.. Mutations induced by 2-hydroxyadenine on a shuttle vector during leading and lagging strand syntheses in mammalian cells. Biochemistry. 1997; 36:11125–11130. [DOI] [PubMed] [Google Scholar]
- 69. Kalam M.A., Haraguchi K., Chandani S., Loechler E.L., Moriya M., Greenberg M.M., Basu A.K.. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opned formamidopyridines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006; 34:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rogozin I.B., Pavlov Y.I., Goncearenco A., De S., Lada A.G., Poliakov E., Panchenko A.R., Cooper D.N.. Mutational signatures and mutable motifs in cancer genomes. Brief. Bioinform. 2017; 19:1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kappes U.P., Luo D., Potter M., Schulmeister K., Runger T.M.. Short-and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J. Invest. Dermatol. 2006; 126:667–675. [DOI] [PubMed] [Google Scholar]
- 72. Giglia-Mari G., Sarasin A.. TP53 mutations in human skin cancers. Hum. Mutat. 2003; 21:217–228. [DOI] [PubMed] [Google Scholar]
- 73. Wang Y., Digiovanna J.J., Stern J.B., Hornyak T.J., Raffeld M., Khan S.G., Oh K.-S., Hollander M.C., Dennis P., Kraemer K.H.. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Mol. Cell Biol. 2009; 106:6279–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ikehata H. Mechanistic considerations on the wavelength-dependent variations of UVR genotoxicity and mutagenesis in skin: the discrimination of UVA-signature from UV-signature mutation. Photochem. Photobiol. Sci. 2018; 12:1861–1871. [DOI] [PubMed] [Google Scholar]
- 75. Strauss B.S. The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non‐instructional lesions?. Bioessays. 1991; 13:79–84. [DOI] [PubMed] [Google Scholar]
- 76. Strauss B.S. The ‘A’ rule revisited: polymerases as determinants of mutational specificity. DNA Repair (Amst). 2002; 1:125–135. [DOI] [PubMed] [Google Scholar]
- 77. Vaisman A., Takasawa K., Iwai S., Woodgate R.. DNA polymerase ι-dependent translesion replication of uracil containing cyclobutane pyrimidine dimers. DNA Repair (Amst). 2006; 5:210–218. [DOI] [PubMed] [Google Scholar]
- 78. Kanao R., Yokoi M., Ohkumo T., Sakurai Y., Dotsu K., Kura S., Nakatsu Y., Tsuzuki T., Masutani C., Hanaoka F.. UV-induced mutations in epidermal cells of mice defective in DNA polymerase eta and/or iota. DNA Repair (Amst). 2015; 29:139–146. [DOI] [PubMed] [Google Scholar]
- 79. Ikehata H., Ono T.. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011; 52:115–125. [DOI] [PubMed] [Google Scholar]
- 80. Sugiyama T., Chen Y.. Biochemical reconstitution of UV-induced mutational process. Nucleic Acids Res. 2019; 47:6769–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vu B., Cannistraro V.J., Sun L., Taylor J.S.. DNA synthesis past a 5-methylC-containing cis-syn-cyclobutane pyrimidine dimer by yeast pol η is highly nonmutagenic. Biochemistry. 2006; 45:9327–9335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Woland is a tool to analyze point mutation patterns using resequencing data available in the GitHub Repository (github.com/tiagoantonio/woland).
Sequencing data have been deposited with the Sequence Read Archive (SRA) under SRA accession number PRJNA556825.