


Abstract
Detection of disease-associated, cell-free nucleic acids in body fluids enables early diagnostics, genotyping and personalized therapy, but is challenged by the low concentrations of clinically significant nucleic acids and their sequence homology with abundant wild-type nucleic acids. We describe a novel approach, dubbed NAVIGATER, for increasing the fractions of Nucleic Acids of clinical interest Via DNA-Guided Argonaute from Thermus thermophilus (TtAgo). TtAgo cleaves specifically guide-complementary DNA and RNA with single nucleotide precision, greatly increasing the fractions of rare alleles and, enhancing the sensitivity of downstream detection methods such as ddPCR, sequencing, and clamped enzymatic amplification. We demonstrated 60-fold enrichment of the cancer biomarker KRAS G12D and ∼100-fold increased sensitivity of Peptide Nucleic Acid (PNA) and Xenonucleic Acid (XNA) clamp PCR, enabling detection of low-frequency (<0.01%) mutant alleles (∼1 copy) in blood samples of pancreatic cancer patients. NAVIGATER surpasses Cas9-based assays (e.g. DASH, Depletion of Abundant Sequences by Hybridization), identifying more mutation-positive samples when combined with XNA-PCR. Moreover, TtAgo does not require targets to contain any specific protospacer-adjacent motifs (PAM); is a multi-turnover enzyme; cleaves ssDNA, dsDNA and RNA targets in a single assay; and operates at elevated temperatures, providing high selectivity and compatibility with polymerases.
INTRODUCTION
In recent years, researchers have identified various enzymes from archaea and bacteria that can be programmed with nucleic acids to cleave complementary strands, among which CRISPR–Cas have attracted considerable attention as genome-editing tools (1–3). In medical diagnostics, CRISPR-associated nucleases have been used to (i) amplify reporter signals during nucleic acid detection (4–8), (ii) detect unamplified targets immobilized on graphene field-effect transistors (9) and (iii) enrich rare alleles to enhance detection limits of oncogenic sequences (10–12). Despite remarkable progress, the applications of CRISPR–Cas are restricted due, among other things, to their reliance on the protospacer-adjacent motif (PAM), which is absent in many sequences of interest (10–12).
Analogous to CRISPR–Cas, Argonaute (Ago) proteins (13) are nucleic acid-guided endonucleases. In contrast to Cas nucleases, Ago nucleases do not require the presence of any specific motifs and are, therefore, more versatile. Under appropriate conditions, Ago from the thermophilic bacterium Thermus thermophilus (TtAgo) (14–17) cleaves with high efficiency both DNA and RNA complementary to its small interfering DNA guides (siDNA), but spares nucleic acids with a single nucleotide mismatch at and around its catalytic site. We formulate a new assay (Figure 1) termed NAVIGATER (Nucleic Acid enrichment Via DNA Guided Argonaute from Thermus thermophilus) to enrich the fraction of rare alleles. We optimize guide length, assay composition, and incubation temperature to enable NAVIGATER to increase rare allele fractions of the cancer markers: KRAS, EGFR, and BRAF mutants in blood samples from cancer patients, greatly improving the sensitivity of downstream mutation detection schemes such as droplet digital PCR (ddPCR) (18), Peptide Nucleic Acid-Mediated PCR (PNA-PCR) (19), PNA-Loop Mediated Isothermal Amplification (LAMP) (20), Xenonucleic Acid clamp PCR (XNA-PCR, Diacarta) and Sanger sequencing. NAVIGATER has the potential to greatly increase the sensitivity and clinical utility of non-invasive tests such as liquid biopsy, especially for monitoring of somatic mutations through cell-free nucleic acids for early diagnostics and for personalized therapy.
Figure 1.

Schematic overview of NAVIGATER. Programmed with a short DNA guide TtAgo specifically cleaves abundant, fully complementary wild type (WT) alleles (both DNA and RNA, blue) while sparing rare alleles (red) with a single nucleotide mutation. This results in the enrichment of the fraction of disease-related rare nucleic acids, enhancing the sensitivity of downstream detection methods.
MATERIALS AND METHODS
TtAgo expression and purification
TtAgo gene, codon-optimized for Escherichia coli BL21 (DE3), was inserted into a pET-His6 MBP TEV cloning vector (Addgene plasmid # 29656) using ligation-independent cloning. The TtAgo protein was expressed in E. coli BL21 (DE3) Rosetta™ 2 (Novagen). Cultures were grown at 37°C in Lysogeny broth medium containing 50 μg ml−1 kanamycin and 34 μg ml−1 chloramphenicol until an OD600 nm of 0.7 was reached. TtAgo expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. During the expression, cells were incubated at 18°C for 16 h with continuous shaking. Cells were harvested by centrifugation and lysed by sonication (Bandelin, Sonopuls. 30% amplitude, 1s on/2s off for 5 min) in lysis buffer containing 20 mM Tris–HCl pH 7.5, 250 mM NaCl, 5mM imidazole, supplemented with EDTA-free protease inhibitor cocktail tablet (Roche). The soluble fraction of the lysate was loaded on a nickel column (HisTrap Hp, GE healthcare). The column was extensively washed with buffer containing 20 mM Tris–HCl pH 7.5, 250 mM NaCl and 30 mM imidazole. Bound proteins were eluted by increasing the concentration of imidazole in the wash buffer to 250 mM. The eluted protein was dialysed at 4°C overnight against 20 mM HEPES pH 7.5, 250 mM KCl, and 1 mM dithiothreitol (DTT) in the presence of 1 mg TEV protease (expressed and purified as previously described (21)) to cleave the His6-MBP tag. Next, the cleaved protein was diluted in 20 mM HEPES pH 7.5 to lower the final salt concentration to 125 mM KCl. The diluted protein was applied to a heparin column (HiTrap Heparin HP, GE Healthcare), washed with 20mM HEPES pH 7.5, 125 mM KCl and eluted with a linear gradient of 0.125–2 M KCl. Next, the eluted protein was loaded onto a size exclusion column (Superdex 200 16/600 column, GE Healthcare) and eluted with 20 mM HEPES pH 7.5, 500 mM KCl and 1 mM DTT (Supplementary Figure S26). Purified TtAgo protein was diluted in a size exclusion buffer to a final concentration of 5 μM. Aliquots were flash frozen in liquid nitrogen and stored at –80°C.
TtAgo-based cleavage assays
5′-Phosphorylated DNA guides and Ultramer® ssDNA and ssRNA targets (100 nt) were synthesized by IDT (Coralville, IA, USA). For ssDNA and ssRNA cleavage experiments, purified TtAgo, DNA guides, and ssDNA or ssRNA targets were mixed with TtAgo and guides at the ratios indicated in the buffers listed in Supplementary Table S1 and incubated at the indicated temperatures. Reactions were terminated by adding 1 μl proteinase K (Qiagen, Cat. No. 19131) solution, followed by 15 min incubation at 56°C. Samples were then mixed with 2× loading buffer (95% (de-ionized) formamide, 5 mM EDTA, 0.025% SDS, 0.025% bromophenol blue and 0.025% xylene cyanol) and heated for 10 min at 95°C before the samples were resolved on 15% denaturing polyacrylamide gels (7 M urea). Gels were stained with SYBR gold Nucleic Acid Gel Stain (Invitrogen) and nucleic acids were visualized using a BioRad Gel Doc XR+ imaging system. For dsDNA cleavage, TtAgo and guides were pre-incubated in LAMP Buffer 3 (Supplementary Table S1) at 75°C for 20 min or on ice for 3 min.
CRISPR/Cas9-based dsDNA cleavage
S.p. Cas9 Nuclease V3 (Cas9) and CRISPR–Cas9 sgRNA (sgRNA) were purchased from IDT (Coralville, IA). To create 10 μM ribonucleoprotein (RNP) complex which contains both sgRNA and Cas9 in equimolar amounts, 10 μM Cas9 and 10 μM sgRNA were incubated in working buffer (30 mM HEPES, 150 mM KCl, pH 7.5) at room temperature for 10 min. For dsDNA cleavage experiments, RNP complex and dsDNA were mixed in 10:1 ratio (2.5 μM RNP, 0.25 μM dsDNA) in Nuclease Reaction Buffer (20 mM HEPES, 100 mM NaCl, 15 mM MgCl2, 0.1 mM EDTA, pH6.5) to get 10 μl total volume. The mixture was incubated at 37°C for 1 h. 1 μl RNase A (Thermo Scientific™, Cat. No. EN0531) was added and incubated at room temperature for 10 min to digest the sgRNA. Then, Cas9 was digested by adding 1 μl proteinase K (Qiagen, Cat. No. 19131) solution, followed by 15 min incubation at 56°C. Samples were resolved on 15% denaturing polyacrylamide gels with the same procedure as described above.
Samples
Patient cell-free DNA (cfDNA) samples
All pancreatic patient blood samples were obtained from patients either with metastatic pancreatic cancer (Supplementary Table S2) or at start of therapy (Supplementary Table S4) who had provided informed consent under the IRB-approved protocol (UPCC 02215, IRB# 822028). cfDNA was extracted with QIAamp® Circulating Nucleic Acid kit (Qiagen, Valencia, CA, USA). Subsequently, the extracted cfDNA was qualified and quantified with multiplex ddPCR (Raindance).
RNA samples
Total RNA was extracted with RNeasy® mini kit (Qiagen, Valencia, CA, USA) per manufacturer's protocol from Human cancer cell lines U87-MG (WT KRAS mRNA) and ASPC1 (KRAS G12D mRNA).
cfDNA pre-amplification
cfDNA pre-amplification was carried out in 50 μl reaction volumes using 20 ng of cfDNA, 1× Q5 Hot Start High-Fidelity Master Mix (22) (New England Biolabs, Ipswich, MA, USA), and 100 nM each of forward and reverse KRAS ddPCR primers (Supplementary Table S3). Reaction mixes without DNA were included as no-template (negative) controls (NTCs). Nucleic acids were preamplified with a BioRad Thermal Cycler (BioRad, Model CFD3240) with a temperature profile of 98°C for 3 min, followed by 30 cycles of amplification (98°C for 10 s, 63°C for 3 min and 72°C for 30 s), and a final 72°C extension for 2 min.
mRNA pre-amplification
mRNA pre-amplification was performed in 50 μl reactions using 30 ng of total RNA, 1× Q5 Hot Start High-Fidelity Master Mix (New England Biolabs, Ipswich, MA, USA), 100 nM each of forward and reverse KRAS RT-PCR primers (Supplementary Table S3), and 1 μl reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The reaction mix was incubated at 55°C for 30 min and 98°C for 3 min, followed by 30 cycles of amplification (93°C for 15 s, 62°C for 30 s and 72°C for 30 s), and a final 72°C extension for 4 min.
Mutant allele enrichment (NAVIGATER)
The same setup as for synthetic dsDNA cleavage was used for cfDNA and mutant mRNA enrichment. TtAgo, S-guide, and AS-guide were mixed in 1:10:10 ratio (1.25 μM TtAgo, 12.5 μM S-guide, 12.5 μM AS-guide) in Buffer 3 and pre-incubated at 75°C for 20 min or on ice for 3 min. 2 μl nucleic acids were added to 8 μl TtAgo-guides complexes and incubated at 83°C for 1 h. When using preamplified nucleic acids, the enriched products were diluted 104-fold before downstream mutation analysis or second-round enrichment. TtAgo activity was terminated with the addition of 1 μl proteinase K solution, followed by 15 min incubation at 56°C. Alternatively, TtAgo can be deactivated by incubating the assay at 95°C for over 20 min (Supplementary Figure S27).
NAVIGATER combined with downstream mutation detection methods
Droplet digital PCR (ddPCR)
ddPCR was carried out with the RainDrop Digital PCR system (RainDance Technologies, Inc.) to verify mutation abundance before and after TtAgo enrichment. 2 μl of the 104-fold diluted, TtAgo-treated sample was added to each 30 μl dPCR. Each dPCR contained 1× TaqMan Genotyping Master Mix (Life Technologies), 400 nM KRAS ddPCR primers, 100 nM KRAS wild type probe, 100 nM KRAS mutant probe (Supplementary Table S3) and 1× droplet stabilizer (RainDance Technologies, Inc.). Emulsions of each reaction were prepared on the RainDrop Source instrument (RainDance Technologies, Inc.) to produce 2–7 million, 5-pl-volume droplets per 25 μl reaction volume. Thereafter, the emulsions were placed in a thermal cycler to amplify the target and generate signal. The temperature profile for amplification consisted of an activation step at 95°C for 10 min, followed by 45 cycles of amplification [95°C for 15 s and 60°C for 45 s]. Reaction products were kept at 4°C before placing them on the RainDrop Sense instrument (RainDance Technologies, Inc.) for signal detection. RainDrop Analyst (RainDance Technologies, Inc.) was used to determine positive signals for each allele type. Gates were applied to regions of clustered droplets to define positive hits for each allele, according to manufacturer's instructions.
PNA-PCR
PNA-PCR was performed in 20 μl reaction volumes, each containing 4.5 μl of 104-fold diluted TtAgo-treated products, 1× Q5 Hot Start High-Fidelity Master Mix (New England Biolabs, Ipswich, MA, USA), 0.5 μl of EvaGreen fluorescent dye (Biotium, Hayward, CA, USA), 500 nM KRAS PNA clamp (Supplementary Table S3), and 100 nM each of forward and reverse KRAS ddPCR primers. Reactions (quantitative PCR) were amplified with a BioRad Thermal Cycler (BioRad, Model CFD3240) with a temperature profile of 98°C for 3 min, followed by 40 cycles of amplification (98°C for 10 s, 63°C for 3 min and 72°C for 30 s).
Sanger sequencing
RNA extracted from cell lines were pre-amplified with KRAS RT-PCR primers as described above and treated by our TtAgo enrichment system. 2 μl of the 104-fold diluted, TtAgo-treated sample was then amplified by KRAS PCR protocol (same as for mRNA pre-amplification but without the reverse transcription step) for 30 cycles. PCR products were inspected for quality and yield by running 5 μl in 2.2% agarose Lonza FlashGel DNA Cassette and processed for Sanger sequencing at the Penn Genomic Analysis Core.
Point-of-care (POC) mutation detection
PNA-LAMP (SMAP-2) was prepared in 20 μl reaction volumes according to the previously described protocol with minor modifications (20). The reaction mix contained 2 μl of the 104-fold diluted TtAgo-treated products (same as used for Sanger sequencing), 1× LAMP buffer 3 (Eiken LAMP buffer), 1 μl Bst DNA polymerase (from Eiken DNA LAMP kit), 2.5 μl of BART reporter (Lot: 1434201; ERBA Molecular, UK) (23), KRAS PNA clamp and LAMP primers (sequences and concentrations listed in Supplementary Table S3). The prepared mixtures were injected into reaction chambers of our custom made, multifunctional chip (24,25). The inlet and outlet ports were then sealed with transparent tape (3M, Scotch brand cellophane tape, St Paul, MN, USA) and the chip was placed in our portable Smart-Connected Cup and processed according to previously described protocol (23).
Comparison of TtAgo and CRISPR/Cas9 -based multiplexed enrichment
Multiplexed pre-amplification
Triplex PCR were carried out with primers from mutation detection kit (DiaCarta, Inc.). The 10 μl reaction mixture contains 60 ng of cfDNA (reference standard that includes various mutant alleles, Horizon Discovery, HD780), 1× PCR Master Mix, 1 μl of mixed PCR primers (1:1:1) for targets of interest. Nucleic acids were pre-amplified with a BioRad Thermal Cycler (BioRad, Model CFX96) with the temperature profile of 95°C for 5 min, followed by 35 cycles of amplification (95°C for 20 s, 70°C for 40 s, 60°C for 30 s and 72°C for 30 s) and a final 72°C extension for 2 min.
Multiplexed enrichment
For triplex NAVIGATER, guides (1:1:1) for targets of interest were mixed with TtAgo in 10:1 ratio (12.5 μM S-guides, 12.5 μM AS-guides, 1.25 μM TtAgo) in Buffer 3 and pre-incubated on ice for 3 min. Samples consisted of 1 μl pre-amplified triplex PCR products mixed with pre-incubated TtAgo-guide complexes. The reaction mixes were incubated at 83°C for 1 h. The triplex Cas9-DASH (Depletion of Abundant Sequences by Hybridization) (10) used the pre-amplified triplex PCR products as the sample and three sgRNAs in 1:1:1 ratio (10 μM sgRNA in total). The products with and without treatment were resolved on 15% denaturing polyacrylamide gels (7 M urea).
XNA-PCR
NAVIGATER products were tested by the mutation detection method XNA-PCR (DiaCarta, Inc.). XNA-PCR was carried out for individual mutants in 10 μl reaction volumes, containing 3 μl of the 107-fold diluted NAVIGATER products, 1× PCR Master Mix, 1 μl of PCR primer/probe mix, and 1 μl of XNA clamp. Reactions (quantitative PCR) were amplified with a BioRad Thermal Cycler (BioRad, Model CFX96) with a temperature profile of 95°C for 5 min, followed by 45 cycles of amplification (95°C for 20 s, 70°C for 40 s, 60°C for 30 s and 72°C for 30 s).
RESULTS
Betaine, Mg2+ and dNTPs enhance TtAgo's endonucleolytic activity
We first examined TtAgo activity in various buffers (Supplemental Section 1) and selected a buffer that provided the best performance (Buffer 3, Supplemental Table 1). Significantly, dNTPs additives greatly enhance cleavage efficiency of RNA (from 20% to 100%) while betaine and high concentrations of Mg2+ (≥8 mM) enable high cleavage efficiency (>90%) of DNA at temperatures ≥80°C without adversely impacting selectivity. The same buffer is suitable for cleaving both DNA and RNA.
Single guide-target-mismatches curtail target cleavage
The conformation and, therefore, cleavage efficiency of Ago proteins depend sensitively on the position and type of single and dinucleotide mismatches between a guide oligo and target strand (15,26–28). Since we require efficient cleavage of wild type (WT) alleles while sparing mutant alleles with a single nucleotide mismatch, we examined the effects of guide nucleotide mismatch position (MP, counted from the siDNA’s 5′ end) on cleavage efficiency of WT KRAS, BRAF, EGFR and their mutants—both DNA and RNA (Figure 2 and Supplementary Section 2). Figure 2A depicts the various siDNAs tested with KRAS G12D. TtAgo-mediated cleavage of KRAS G12D strand is nearly completely curtailed when siDNA has a nucleotide mismatch at MP7 or MP11-13 (Figure 2B). Guides designed for enriching KRAS G12D also enrich the fractions of other KRAS G12 mutants.
Figure 2.

NAVIGATER discrimination efficiency (DE) depends sensitively on the position of the mismatch (MP) between siDNA and off-target allele. (A) KRAS – sense (S) guide and S target (WT) / off-target (mutant) sequences. Column t1 identifies the target's nucleotide at the opposing position of the 5′-terminal nucleotide of the guide. (B) Cleavage efficiencies of WT KRAS DNA (S) and KRAS G12D DNA (S) as functions of MP. Error bars show 1 standard deviation, N = 3. (C) Stacked DE for KRAS G12D, EGFR L858R, EGFR T790M and BRAF V600E DNA as functions of MP. AS = Antisense. (D) Cleavage efficiencies of WT and off-target as functions of target nucleotide at position 1. Experiments were carried out with short guides (15/16 nt) in Buffer 3 at 80°C. TtAgo: guide: target = 5:1:1 (1.25 μM:0.25 μM:0.25 μM). The notation MP-X indicates the position (X) of the mismatch (MP) between guide oligo and allele counted from the 5′ end of the guide.
To characterize siDNA performance, we define the discrimination efficiency (DE) as the difference in cleavage efficiency of the WT and mutant strand. Figure 2C depicts DE as a function of MP for KRAS, EGFR and BRAF DNA and their mutants. Mismatches MP7 and MP9–MP13, located around the cleavage site (g10/g11), yielded the best discrimination in most cases (Figure 2C and Supplementary Figure S10e). The optimal MP depends, however, on the allele's specific sequence. Cleavage of RNA was less tolerant to mismatches than cleavage of DNA. Single mismatches anywhere between MP4–MP11 near completely prevented RNA cleavage (Supplementary Figures S6e and S8).
Despite being loaded with guides that were fully complementarity to the target, WT sequences were cleaved by TtAgo at variable efficiencies (Figure 2B and Supplementary Section 2). This suggests that besides the different buffer components, the activity of TtAgo also depends on the sequences of the guide and target, which might affect the conformation of the ternary TtAgo–siDNA–DNA and TtAgo–siDNA–RNA complexes (29).
TtAgo encompasses two distinct binding pockets in the MID and PIWI domains to, respectively, accommodate the guide's 5′-terminal nucleotide (g1) and its opposing target nucleotide (t1) (Figure 2A) (29). Although a t1 guanosine (t1G) binds preferentially into the pocket of the PIWI-domain (30), this was not reflected in the TtAgo's cleavage efficiencies of fully complementary targets. That is the cleavage efficiencies of targets (WT) did not significantly depend on the nucleotide type at target position 1 (Supplementary Note 1) (30). In contrast, there is a significant increase in the cleavage of mutant alleles when target position 1 is occupied with guanosine (Figure 2D) even in the presence of a mismatch between guide and target (Figure 2B and D), resulting in a poor discrimination efficiency. Therefore, when designing guides for NAVIGATER, t1G should be avoided.
TtAgo cleaves most specifically with short guides (15/16 nt)
Heterologously-expressed TtAgo is typically purified with siDNAs ranging in length from 13 to 25 nt (17), whereas in vitro, TtAgo operates with siDNAs ranging in length from 7 to 36 nt (15). Since little is known on the effect of guide length on TtAgo discrimination efficiency, we examine this effect in our in vitro assay. TtAgo efficiently cleaves WT KRAS (sense, S) with complementary guides, ranging in length from 16 to 21 nt at both 70°C and 75°C (Figure 3A and Supplementary Figures S11 and S12). Guides 17–21 nt in length with a single nucleotide mismatch at MP12 cleave mutant DNA strands at 75°C, but not at 70°C (Figure 3A). We compared cleavage efficiencies of 16 nt and 19 nt long guides as functions of temperature (Figure 3B and C). The 19 nt long guide cleaves DNA mutant strands at temperatures >75°C, but not at temperatures below 70°C (Figure 3B). Cleavage of the mutant allele at ≥75°C is, however, completely suppressed with the 16 nt guide (Figure 3A, bottom and Figure 3C). We observe a similar behavior when a 15 nt guide with a single mismatch at MP13 targets the antisense (AS) KRAS strand (Supplementary Figure S11a–ii). We hypothesize that short guides form a less stable complex with off-targets than longer guides, preventing undesired cleavage.
Figure 3.

TtAgo cleaves more specifically with short guides. (A) Cleavage efficiencies as functions of guide length at 70°C (top) and 75°C (bottom). TtAgo in complex with the MP12 guide was used to target either KRAS (S) WT or G12D. (B) Cleavage efficiencies of (top) DNA (S) and (bottom) RNA (S) as functions of temperature with 19 nt long guide (KRAS-S (19 nt)-MP12). (C) Cleavage efficiencies of DNA (S) and RNA (S) as functions of temperature with 16 nt long guide (KRAS-S (16 nt)-MP12). Experiments were carried out in Buffer 3 with TtAgo: guide: target ratio 5:1:1. Incubation time 1 h. N = 3.
In contrast to mutant DNA, increases in assay temperature and guide length do not increase undesired cleavage of mutant RNA (Figure 3A and B), likely due to differences in the effects of ssDNA and ssRNA on TtAgo conformation. In summary, when operating with short guides (15/16 nt), TtAgo efficiently cleaves both WT RNA and DNA targets in the same buffer while avoiding cleavage of mutant alleles at temperatures ranging from 66 to 86°C (Figure 3C).
TtAgo efficiently cleaves targeted dsDNA at temperatures above the dsDNA’s melting temperature
Guide-free (apo-) TtAgo can degrade dsDNA via a ‘chopping’ mechanism, autonomously generating functional siDNA (30). This is, however, a slow process that takes place only when the target DNA is rich in AT (<17% GC) (17), suggesting that TtAgo lacks helicase activity and depends on dsDNA thermal breathing to enable chopping (13,17,31). In our assays TtAgo is incubated with an excess of siDNAs to suppress chopping of dsDNA. TtAgo's activity at high temperatures provides NAVIGATER with a clear advantage since dsDNA unwinds as the incubation temperature increases.
We determined the optimal temperature at which TtAgo, saturated with guides, efficiently cleaves dsKRAS WT while sparing mutant alleles. The estimated melting temperature of 100 bp dsKRAS (S strand sequence listed in Figure 2A) in Buffer 3 is 79.7°C (IDT-OligoAnalyzer. https://www.idtdna.com/calc/analyzer). Consistent with this estimate, very little cleavage of dsDNA takes place at temperatures below 80°C, but TtAgo cleaves dsDNA efficiently at temperatures above 80°C (Figure 4A-i). Cleavage efficiency increases as the incubation time increases and nearly saturates after about one hour (Figure 4B). TtAgo efficiently cleaves high GC-content (∼70%) dsDNA at 83°C (Supplementary Figure S13), presumably due to the high betaine concentration of our buffer (32).
Figure 4.

Excess guide concentration provides high cleavage discrimination efficiency. (A) The cleavage efficiencies of KRAS WT and KRAS G12D dsDNA as functions of temperature: (i) TtAgo/guide ratio: 1:1 (1.25 μM:1.25 μM), 40 min and 120 min incubation times; (ii) TtAgo/guide ratio: 5:1 (1.25 μM:0.25 μM), 40 min and 120 min incubation times. (B) Electropherograms of NAVIGATER’s products as a function of incubation time (incubation temperature: 83°C): (i) TtAgo/guide ratio 1:1 and (ii) TtAgo/guide ratio 1:10 (1.25 μM:12.5 μM). (C) Cleavage efficiency of RNA (250 nM), dsDNA (250 nM) and a mixture of RNA (125 nM) and dsDNA (125 nM) (RNA/dsDNA) (1:1) at 83°C for 60 min (i) and corresponding electropherograms (ii). The differences in fluorescence intensity and electrophoretic migration between RNA and dsDNA are, respectively, caused by the different binding affinity of SYBR Gold dye to RNA and dsDNA (49) and the different electrophoretic mobility of DNA and RNA (50). All experiments were carried out in Buffer 3 with KRAS-S (16 nt)-MP12 and KRAS-AS (15 nt)-MP13 guides. N = 3.
Guide saturation is necessary to avoid off-target cleavage
In the presence of guide concentrations smaller than the TtAgo concentration, e.g. a TtAgo/guides ratio of 5:1, non-specific, undesired cleavage of ds mutant alleles takes place (Figure 4A-ii). This off-target cleavage increases as the incubation time increases (Figure 4A-ii). Based on data for dissociation rate constants of other prokaryotic Ago homologs (33), we hypothesize that TtAgo binds siDNA with high affinity (17). Thus, excess siDNA prevents cleavage products from acting as guides (Figure 4B and Supplementary Figure S14). Indeed, at a TtAgo/guide ratio of 1:10, TtAgo efficiently cleaves WT KRAS, BRAF and EGFR dsDNA while sparing the mutant dsDNAs harboring point mutations: KRAS G12D (Figure 4B-ii), KRAS G12V (Supplementary Figure S15), BRAF V600E, EGFR L858R and EGFR T790M (Supplementary Figure S16), and deletion mutations in EGFR exon 19 (Supplementary Figure S17). For optimal discrimination between ds WT and mutant alleles, it is desirable to operate with excess siDNA and pre-incubate TtAgo with guides to form TtAgo–siDNA complexes (Supplementary Figure S15).
Since clinical samples include both DNA and RNA, an important advantage of NAVIGATER is its ability to concurrently enrich the fractions of both mutant DNA and mutant RNA (Figure 4C).
TtAgo-mediated cleavage outperforms CRISPR/Cas9 cleavage
The recently developed Cas9-DASH (10) and CUT-PCR (CRISPR-mediated Ultra-sensitive detection of Target DNA-PCR) (11) take advantage of the low tolerance of CRISPR/Cas proteins to mismatches at the PAM recognition site to discriminate mutant from WT alleles. We examine the discrimination efficiency of CRISPR/Cas9 with a previously reported guide RNA (10–12) (Supplementary Figure S18a). CRISPR/Cas9 nonspecifically cleaves both ds WT and mutant alleles harboring KRAS G12D and G12V mutations (Supplementary Figure S18b and c). Consistent with previous reports (11,34,35), we suspect that these non-specific cleavages are caused by non-canonical PAM recognition. CRISPR/Cas9 also failed to differentiate between ds WT and mutant alleles harboring EGFR L858R mutation (Supplementary Figure S18b and c), presumably because of the presence of a PAM site in EGFR L858R allele and a non-canonical PAM in the WT allele (Supplementary Figure S18a), which complicates the design of a guide to specifically cleave the WT while sparing the mutant. In contrast, CRISPR/Cas9 specifically cleaved ds WT EGFR while sparing the ds mutant allele harboring the deletion mutation E746-A750 del [1]. In summary, CRISPR/Cas9 shows a lower discrimination efficiency compared to our TtAgo system.
NAVIGATER increases the sensitivity of downstream rare allele detection
In recent years, there has been a rapidly increasing interest in applying liquid biopsy to detect cell-free circulating nucleic acids associated with somatic mutations for, among other things, cancer diagnostics, tumor genotyping, and monitoring susceptibility to targeted therapies. Liquid biopsy is attractive since it is minimally invasive and relatively inexpensive. Detection of mutant alleles is, however, challenging due to their very low concentrations in liquid biopsy samples among the background of highly abundant WT alleles that differ from mutant alleles by as little as a single nucleotide. To improve the sensitivity and specificity of detecting rare alleles that contain valuable diagnostic and therapeutic clues, it is necessary to remove and/or suppress the amplification of WT alleles (36–38). NAVIGATER meets this challenge by selectively and controllably degrading WT alleles in the sample, thereby increasing the fraction of mutant alleles. We demonstrate here that NAVIGATER increases sensitivity of downstream mutation detection methods such as gel electrophoresis, ddPCR (18), PNA-PCR (19), PNA-LAMP (20), Sanger sequencing, and XNA-PCR; and enables multiplexed enrichment. To demonstrate NAVIGATER’s potential clinical utility, we enriched blood samples from pancreatic cancer patients (Supplementary Table S2) whose tumor genotype was previously analyzed with next generation sequencing (NGS) and blood samples with RainDance ddPCR. Due to limited sample volume, these samples were pre-amplified with high fidelity PCR to increase WT and mutant KRAS total content before NAVIGATER enrichment.
Gel electrophoresis (Supplementary Figure S19)
We subjected NAVIGATER products to gel eIectrophoresis. In the absence of enrichment (control), the bands at 80 bp (KRAS) on the electropherogram are dark. After 40 min of TtAgo enrichment, these bands faded, indicating a reduction of KRAS WT alleles. After 2 h enrichment, all the bands at 80 bp, except that of patient P6, have essentially disappeared, suggesting that most WT alleles have been cleaved. The presence of an 80 bp band in the P6 lane is attributed to the relatively high (20%) mutant allele frequency (MAF) that is not susceptible to cleavage. We also PCR amplified the products of a 2-hour NAVIGATER treatment, and subjected the amplicons to a second NAVIGATER (2 h). The columns P3, P4 and P6 feature darker bands than P1, P2 and P5, indicating the presence of mutant alleles in samples P3, P4 and P6 (Supplementary Figure S19) and demonstrating that NAVIGATER renders observable otherwise undetectable mutant alleles.
Droplet Digital PCR (ddPCR) (Supplementary Figure S20)
To quantify our enrichment assay products, we subjected them to ddPCR. The detection limit of ddPCR is controlled by the number of amplifiable nucleic acids in the sample, which must be a small fraction of the number of ddPCR droplets. The large number of WT alleles in the sample limits the number of pre-ddPCR amplification cycles that can be carried out to increase rare alleles’ concentration. Since NAVIGATER drastically reduces the number of WT alleles in the sample, it enables one to increase the number of pre-amplification cycles, increasing the number of mutant allele copies, and thus, the ddPCR sensitivity. When operating with a mixture of WT and mutant allele, NAVIGATER products include residual uncleaved WT (NWT), mutant (NM), and WT-mutant hybrids (NH) alleles. Hybrid alleles form during re-hybridization of an ss WT with an ss mutant allele. The MAF is fM= (NM+ 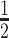 NH)/(NWT + NM+ NH).
NH)/(NWT + NM+ NH).
We carried out ddPCR of un-enriched (control), once-enriched, and twice-enriched samples (Supplementary Figure S20b), increasing MAF significantly (Figure 5A). For example, the MAF increased from 0.5% in the un-enriched P5 (G12D) sample to ∼30% in the twice-enriched sample. This represents an ∼60-fold increase in the fraction of droplets containing the mutant allele (Figure 5B), similar to previously reported methods (10,39,40). The same assay also enriched G12R, increasing MAF from 3% to ∼66% in sample P3 and G12V, increasing MAF from 5% to ∼68% in sample P4 (Figure 5B).
Figure 5.

NAVIGATER enhances the sensitivity of downstream detection methods. (A, B) ddPCR of samples from pancreatic cancer patients containing KRAS mutants (Supplementary Table S2): (A) Fraction of droplets reporting mutant alleles in the absence of enrichment (control), once enriched, and twice enriched. (B) Increase in mutant allele frequency after NAVIGATER enrichment. (C) PNA-PCR’s amplification curves of pancreatic cancer patients’ samples before (i) and after (ii) NAVIGATER, and amplification threshold time as a function of MAF (iii). (D) PNA-LAMP of spiked RNA samples before and after NAVIGATER carried out with a minimally-instrumented, electricity-free Smart-Connected Cup (SCC) (23). (E) Sanger sequencing before and after NAVIGATER of samples spiked with RNA extracted from cell lines. All the controls were pre-processed in the absence of TtAgo. N = 3
PNA-PCR
PNA-PCR engages a sequence-specific PNA blocker that binds to WT alleles, suppressing WT amplification and providing a limit of detection of ∼1% MAF (19). To demonstrate NAVIGATER’s utility, we compared the performance of PNA-PCR when processing pancreatic cancer patient samples (Supplementary Table S2) before and after NAVIGATER (Figure 5C). Before enrichment, PNA-PCR real-time amplification curves in the order of appearance are P6, P4 and P3, as expected. Samples P1 (MAF = 0%), P2 (MAF = 0%), and P5 (MAF = 0.5%) nearly overlap, consistent with a detection limit of ∼1% (19). Enrichment (Figure 5C-ii) significantly increases the threshold times of samples P1 and P2 and reveals the presence of mutant alleles in sample P5. PNA-PCR combined with NAVIGATER provides the linear relationship T1/2 = 22.9 – 5 × log(MAF) between threshold time (the time it takes the amplification curve to reach half its saturation value) and MAF (Figure 5C-iii), allowing one to estimate mutant allele concentration. The data suggests that NAVIGATER can improve PCR-PNA limit of detection to <0.1%.
PNA-LAMP
Genotyping with PNA blocking oligos can be combined with the isothermal amplification method LAMP (20). To demonstrate the feasibility of genotyping at the point of care and in resource-poor settings, we use a minimally-instrumented, electricity-free Smart-Connected Cup (SCC) (23) with smartphone and bioluminescent dye-based detection to incubate PNA-LAMP and detect reaction products. To demonstrate that we can also detect RNA alleles, we used samples comprised of mixtures of WT KRAS mRNA and KRAS-G12D mRNA extracted from the human cancer cell lines U87-MG and ASPC1. In the absence of pre-enrichment, SSC is unable to detect the presence of 0.1% KRAS-G12D mRNA whereas with pre-enrichment 0.1% KRAS-G12D mRNA is readily detectable (Figure 5D).
Sanger sequencing
In the absence of enrichment, Sanger sequencers detect >5% MAF (41). The Sanger sequencer failed to detect the presence of MAF ∼3% and 0.5% KRAS-G12D mRNA in our un-enriched samples, but readily detected these mutant alleles following NAVIGATER enrichment (Figure 5E).
Multiplexed enrichments
We carried out triplex NAVIGATER and compared it with triplex Cas9-based DASH (Cas9-DASH) (10). Our experiment demonstrates that NAVIGATER can operate as a multiplexed assay, enriching multiple mutant allele fractions in a single sample; it is more specific than CRISPR/Cas9’s PAM site recognition-based enrichment; and it can be combined with clamped assay XNA-PCR that is similar to PNA-PCR to significantly improve XNA-PCR sensitivity (Supplementary Section 7). NAVIGATER enriches three different KRAS G12 mutants with a single pair of guides (Figure 5A). When mutations are proximate such as in the KRAS region, one can enrich for different mutant allele fractions with a single guide, otherwise multiple guides with different sequences in multiple reactions are needed.
NAVIGATER applied directly to cfDNA
TtAgo cleaves cfDNA (Horizon) with high efficiency at its optimal operating temperature (Supplement Figure 23a) and genomic DNA with reduced efficiency (Supplement Figure 23b). In appropriately prepared clinical samples, the vast majority of cfDNA is fragmented with average length of ∼160 bp (42,43), which our assay handles efficiently. Furthermore, our assay also targets RNA that is likely present at much greater abundance than DNA fragments. Thus, for enrichment of cell-free mutant alleles, pre-amplification is not needed for the operation of our assay.
NAVIGATER enables detection of low-frequency (<0.2%) mutant alleles in blood samples of pancreatic cancer patients
Blood samples were collected from 18 pancreatic cancer patients and 4 healthy control donors, processed to plasma, and banked at –80°C. All patients had clinical NGS performed on a routinely collected tissue sample, 14 tested positive and 4 tested negative for KRAS G12D, V and R mutations (Supplementary Table S4). Below, we compare our blood test results with the tissue data (gold standard). Pre-amplification followed by KRAS G12D/V/R mutation detection by RainDance ddPCR was performed on cfDNA extracted from 0.75 ml of patient plasma. An equivalent (blinded) cfDNA sample was pre-amplified for the KRAS G12 region (DiaCarta, Inc.). Equal volumes of the resultant amplicons were subjected in duplicate to (i) XNA-PCR without pre-enrichment, (ii) Cas9-DASH followed by XNA-PCR and (iii) single NAVIGATER (once-NAVIGATER) followed by XNA-PCR. These results were compared to tissue NGS prior to subsequent testing with two NAVIGATER enrichment steps. The XNA-PCR test was deemed positive when the threshold cycle (Ct) was less than a cutoff (CtC)—the average Ct minus 3SD (95% confidence level) of XNA-PCR of standard (Horizon) WT KRAS allele samples (in the absence of mutant alleles) at DNA concentrations greater than in our clinical samples (Supplementary Table S5).
XNA-PCR without pre-enrichment identified 1/14 samples as positive and 1/14 as inconclusive (only one of the duplicates tested positive). Cas9-DASH XNA-PCR identified 3/14 as positive; the Ct values of other samples were clustered together (Supplementary Figure S24b) complicating discrimination between positives and negatives. Once-NAVIGATER followed by XNA-PCR identified 6/14 samples as positive; twice-NAVIGATER followed by XNA-PCR identified 9/14 as positive (Figure 6B, C, E). ddPCR identified 8/14 as positive (Figure 6B and C). The G12D/V/R tissue negative samples and healthy controls are all negative in these tests. The sensitivity, specificity, positive predictive value, negative predictive value, and concordance with tissue NGS genotyping for KRAS G12 mutations were, respectively, 64%, 100%, 100%, 62%, 77% for twice-NAVIGATER followed by XNA-PCR, and 57%, 100%, 100%, 57%, 73% for ddPCR (Figure 6B). The receiver operating characteristic (ROC, Figure 6D) curve for twice-NAVIGATER followed by XNA-PCR has an area under the ROC curve (AUC) of 0.88 (95% CI: 0.67–0.98), indicating that with an optimal CtC, our assay has 79% sensitivity and 100% specificity.
Figure 6.

Detection of low-frequency (<0.2%) KRAS G12 mutations in cfDNA samples from healthy donors [4] and pancreatic cancer patients [18] that are testing positive [14] and negative [4] to KRAS G12 mutations in tissue biopsy. (A) Schematic of patient sample workflow. cfDNA corresponding to equivalent volumes of plasma were assayed by RainDance ddPCR after pre-amplification (9 cycles) and by XNA-PCR without enrichment, cas9-DASH XNA-PCR, and NAVIGATER XNA-PCR after pre-amplification (35 cycles). (B) Performance of twice-NAVIGATER XNA-PCR and ddPCR compared to tissue NGS genotyping (‘gold standard’). M+ = mutation positive. M– = mutation negative. (C) Summary of test results. Gray boxes indicate that mutation was not detected either in tissue or in blood (negative control); red boxes indicate that mutation was detected in tissue but not by the listed method; green boxes indicate that mutation was detected both in tissue and the listed method; and yellow boxes indicate that mutation was detected in tissue and one of the duplicate test by the listed method. ‘XNA-PCR only’ are the results of XNA-PCR of non-enriched patient samples, ‘cas9-DASH’, ‘once-NAVIGATER’, and ‘twice-NAVIGATER’ are, respectively, the results of XNA-PCR of the enriched products of cas9-DASH, one round of NAVIGATER, and two rounds of NAVIGATER. N = negative. P = positive. ND = not detected. NA = not available because healthy donors didn’t get tissue NGS testing. (D) Receiver operator characteristic (ROC) curve of twice-NAVIGATER XNA-PCR versus tissue NGS genotyping. (E) Summary of results of NAVIGATER XNA-PCR compared with RainDance ddPCR.
NAVIGATER XNA-PCR identified three positives that were undetectable by ddPCR, while ddPCR identified two positives that were undetectable by NAVIGATER XNA-PCR (Figure 6E). All positive twice-NAVIGATER-enriched samples were subjected to Sanger sequencing to verify that the KRAS G12 variants were identical to those detected in tissue biopsy (Supplementary Figure S25). Since a few samples had single copies of mutant allele (based on ddPCR, Supplementary Table S4), we attribute the discordance between NAVIGATER XNA-PCR and ddPCR to sampling bias, resulting in some of the samples having no targets. In summary, NAVIGATER XNA-PCR (AUC = 0.85, 95% CI: 0.64–0.96 for once-NAVIGATER, and 0.88 for twice-NAVIGATER) is effective at detecting the presence of rare mutant alleles and outperforms XNA-PCR in the absence of pre-enrichment (AUC = 0.65, 95% CI: 0.42–0.84) and Cas9-DASH XNA-PCR (AUC = 0.83, 95% CI: 0.62–0.96) (Supplementary Figure S24), consistent with our earlier observations that Cas9 is less specific than TtAgo in sparing mutant alleles.
DISCUSSION
Liquid biopsy is a simple, minimally invasive, rapidly developing diagnostic method to analyze cell-free nucleic acid fragments in body fluids and obtain critical information on patient health and disease status. Currently, Liquid biopsy helps personalize and monitor treatment for patients with advanced cancer, but the sensitivity of available tests is insufficient for patients with early stage disease (38) and for cancer screening. Detection of alleles that contain critical clinical information is challenging since they are present at very low concentrations among abundant background of nucleic acids that differ from alleles of interest by as little as a single nucleotide.
Here, we report on a novel enrichment method (NAVIGATER) for rare alleles that uses the endonuclease TtAgo. TtAgo is programmed with short ssDNA guides to specifically cleave guide-complementary alleles, both DNA and RNA, and stringently discriminate against off-targets with single nucleotide precision. Sequence mismatches between guide and off-targets reduce hybridization affinity and cleavage activity by sterically hindering formation of a cleavage-compatible state (15,16). TtAgo's activity and discrimination efficiency depend sensitively on the (i) position of the mismatched pair along the guide, (ii) buffer composition, (iii) guide length, (iv) Ago/guide ratio, (v) incubation temperature and time, (vi) target sequence, and (vii) type of nucleotide at target position 1 (t1). TtAgo appears to discriminate best between target and off-target in the presence of a mismatch at or around the cleavage site located between guide nucleotides 10 and 11 and in the absence of t1G. Optimally, the buffer should contain ≥8 mM [Mg2+], ≥0.8 M betaine and 1.4 mM dNTPs. Optimal ssDNA guides should be 15–16 nt in length and at concentrations exceeding TtAgo's concentration. The incubation temperature should exceed the target dsDNA melting temperature. NAVIGATER is amenable to multiplexing and can concurrently enrich multiple mutant alleles in a single sample.
NAVIGATER successfully enriches the fraction of cancer biomarkers such as KRAS, BRAF and EGFR mutants in various samples. NAVIGATER increased KRAS G12D fraction from 0.5% to 30% (60-fold) in a blood sample from a pancreatic cancer patient. The presence of 0.5% KRAS G12D could not be detected with Sanger sequencing or PNA-PCR. However, after NAVIGATER processing, both the Sanger sequencer and PNA-PCR readily identified the presence of KRAS G12D. Additionally, NAVIGATER combined with PNA-LAMP detects low fraction (0.1%) mutant RNA alleles, enabling genotyping at the point of care and in resource-poor settings. NAVIGATER improves the detection limit of XNA-PCR by >10-fold, enabling detection of rare alleles with frequencies as low as 0.01%. NAVIGATER combined with XNA-PCR exhibits higher sensitivity for detecting KRAS mutants in Pancreatic cancer patients than XNA-PCR alone and Cas9-DASH XNA-PCR.
NAVIGATER differs from previously reported rare allele enrichment methods (10–12,39,40,44–46) in several important ways (Table 1). First, NAVIGATER is versatile. In contrast to CRISPR–Cas9 (10–12) and restriction enzymes (44), TtAgo does not require PAM motif or a specific recognition site. A siDNA can be designed to direct TtAgo to cleave any desired target. Second, TtAgo is a multi-turnover enzyme (17): a single TtAgo-guide complex can cleave multiple targets. In contrast, CRISPR–Cas9 is a single turnover nuclease (47). Third, whereas CRISPR–Cas9 exclusively cleaves DNA, TtAgo cleaves both DNA and RNA targets with single nucleotide precision. In fact, NAVIGATER can enrich for both rare DNA alleles and their associated exosomal RNAs (48) in the same assay, further increasing sensitivity. Fourth, TtAgo is robust as it operates over a broad temperature range (66–86°C) and unlike PCR-based enrichment methods, such as COLD-PCR (45) and blocker-PCR (40,46), does not require tight temperature control. Moreover, as we have shown, NAVIGATER can complement PCR-based enrichment methods such as XNA and PNA PCR. Fifth, TtAgo uses DNA guides rather than RNA guides, which reduce cost and increase assay stability. Sixth, TtAgo is more specific than thermostable duplex-specific nuclease (DSN) (39). Since DSN non-specifically cleaves all dsDNA, DSN-based assays require tight controls of probe concentration and temperature to avoid non-specific hybridization and cleavage of the rare nucleic acids of interest. Most importantly, NAVIGATER is compatible with many downstream genotyping analysis methods such as ddPCR, PNA-PCR, XNA-PCR, and sequencing. Last, but not least, NAVIGATER can operate with isothermal amplification methods such as LAMP, enabling integration of enrichment with genotyping for use in resource poor settings.
Table 1.
Comparison of rare allele enrichment methods
| dCas9-based method (12) | Cas9-DASH (10) and Cut-PCR (11) | NAVIGATER | NaME-PrO (36) | COLD-PCR (40) | Blocker (PNA or DNA)-PCR (37,41) | |
|---|---|---|---|---|---|---|
| Enzyme | deactivated CRISPR Cas9 (high cost) | CRISPR Cas proteins (low cost) | TtAgo (low cost) | DSN (high cost) | N/A* | N/A |
| Guide/blocker | ∼120 nt sgRNA (high cost) | ∼120 nt sgRNA (high cost) | 15/16 nt DNA (low cost) | 20–25 nt DNA (low cost) | N/A | PNA (high cost) DNA (low cost) |
|
PAM site
requirement |
Yes | Yes | No | No | No | No |
| Multi-turnover enzyme | No | No | Yes | Yes | N/A | N/A |
| Target | DNA | DNA | DNA & RNA | DNA | DNA | DNA |
| Incubation time | ∼7 h | 2 h | <1 h | 22 min | ∼2 h | ∼2 h |
| Tight temperature control | No (37°C) | No (37°C) | No (66–86°C) | Yes** (98°C, 67°C) | Yes (thermal cycling) | Yes (thermal cycling) |
| Fraction detection limit in clinical samples | 0.1% with allele-specific qPCR | 0.01% with targeted deep sequencing | 0.01% with XNA-PCR | 0.01% with HRM/Sanger sequencing | 0.1%-0.5% with MALDI-TOF | 0.1%-1% |
| Enrichment level | Medium | High | High | High | High | High |
| Specificity | Medium | Medium-high | High | Medium | Medium | Medium |
| Multiplexing capability | Yes | Yes | Yes | Yes | Yes | Yes |
| Requirement for pre-amplification | No | No | No | No | No | No |
| Enrichment of cfDNA mutant fragments | No limitation | No limitation | No limitation | No limitation | No limitation | No limitation |
| Enrichment of mutant genomic DNA | No limitation | No limitation | Low efficiency | No limitation | No limitation | No limitation |
*N/A – not applicable.
**After incubation at 98°C for 2 min, additional operations are required such as incubation at 67°C and then opening the tube to add DSN.
Our study indicates that TtAgo-based enrichment has the potential to significantly increase the clinical utility of liquid biopsy and enable, in combination with downstream detection methods (including NGS), detect low concentrations of mutant alleles indicative of various types of cancer. NAVIGATER could be leveraged to help (i) detect other rare alleles related to such as genetic disorders in fetal nucleic acids and drug resistant pathogens; (ii) recognize the minor populations of interest in biological studies; and (iii) prepare libraries for NGS.
HUMAN SUBJECTS
This study was approved by Penn Institutional Review Board (IRB PROTOCOL #: 822028).
DATA AVAILABILITY
The authors declare that all data supporting the findings in this study are available within the paper and its Supplementary Information files.
Supplementary Material
ACKNOWLEDGEMENTS
Dr Robert M. Greenberg helped with PAGE electrophoresis. Dr Jennifer E. Phillips-Cremins provided us with access to gel imager. Dr Changchun Liu provided helpful comments early in this project. Stephanie Yee and Taylor Black assisted with ddPCR. Yaguang Fan assisted with statistical analysis.
Author contributions: J.S. and H.H.B. conceived the project and designed the experiments. J.W.H. expressed and purified TtAgo protein. J.S., J.W.H., J.P. and L.T.A. carried out the experiments. M.G.M. and J.C. assisted, respectively, with PNA-PCR and XNA-PCR. N.B. and J.E.T. extracted cfDNA from patients’ blood and quantified KRAS G12 mutations with ddPCR. J.E.T. extracted RNA from cell lines. N.B. and J.E.T. assisted with ddPCR experiments. M.S. and J.M. cultured cell lines. E.C. supervised N.B., J.E.T., M.S. and J.M. and advised on patient samples and ddPCR experiments. J.S., J.W.H., M.G.M., J.v.d.O. and H.H.B. analyzed the data and wrote the manuscript. All authors read and commented on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [NCI 1R21CA227056-01, FIC 1R21TW011496-01A1, FIC 1K01TW011190-01A1 to the University of Pennsylvania]; Netherlands Organization of Scientific Research [NWO-ECHO 711013002, NWO-TOP 714015001 to J.v.d.O.]. Funding for open access charge: NIH [NCI 1R21CA227056-01].
Conflict of interest statement. University of Pennsylvania and Wageningen University have applied for a patent on NAVIGATER with J.S., J.W.H., M.G.M., J.v.d.O. and H.H.B. listed as co-inventors.
REFERENCES
- 1. Barrangou R., Doudna J.A.. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016; 34:933–941. [DOI] [PubMed] [Google Scholar]
- 2. Komor A.C., Badran A.H., Liu D.R.. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017; 168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu W.Y., Lebbink J.H.G., Kanaar R., Geijsen N., van der Oost J.. Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Chem. Biol. 2018; 14:642–651. [DOI] [PubMed] [Google Scholar]
- 4. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A. et al.. Nucleic acid detection with CRISPR–Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J.S., Ma E.B., Harrington L.B., Da Costa M., Tian X.R., Palefsky J.M., Doudna J.A.. CRISPR–Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018; 360:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P., Cofsky J.C., Kyrpides N.C., Banfield J.F., Doudna J.A.. Programmed DNA destruction by miniature CRISPR–Cas14 enzymes. Science. 2018; 362:839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F.. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018; 360:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hajian R., Balderston S., Tran T., deBoer T., Etienne J., Sandhu M., Wauford N.A., Chung J.-Y., Nokes J., Athaiya M. et al.. Detection of unamplified target genes via CRISPR–Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed. Eng. 2019; 3:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu W., Crawford E.D., O’Donovan B.D., Wilson M.R., Chow E.D., Retallack H., DeRisi J.L.. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016; 17:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S.H., Yu J., Hwang G.H., Kim S., Kim H.S., Ye S., Kim K., Park J., Park D.Y., Cho Y.K. et al.. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene. 2017; 36:6823–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aalipour A., Dudley J.C., Park S.M., Murty S., Chabon J.J., Boyle E.A., Diehn M., Gambhir S.S.. Deactivated CRISPR associated protein 9 for minor-allele enrichment in cell-free DNA. Clin. Chem. 2018; 64:307–316. [DOI] [PubMed] [Google Scholar]
- 13. Hegge J.W., Swarts D.C., van der Oost J.. Prokaryotic Argonaute proteins: novel genome-editing tools. Nat. Rev. Microbiol. 2018; 16:5–11. [DOI] [PubMed] [Google Scholar]
- 14. Swarts D.C., Makarova K., Wang Y.L., Nakanishi K., Ketting R.F., Koonin E.V., Patel D.J., van der Oost J.. The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol. 2014; 21:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y.L., Juranek S., Li H.T., Sheng G., Tuschl T., Patel D.J.. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008; 456:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y.L., Juranek S., Li H.T., Sheng G., Wardle G.S., Tuschl T., Patel D.J.. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature. 2009; 461:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swarts D.C., Jore M.M., Westra E.R., Zhu Y.F., Janssen J.H., Snijders A.P., Wang Y.L., Patel D.J., Berenguer J., Brouns S.J.J. et al.. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014; 507:258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taly V., Pekin D., Benhaim L., Kotsopoulos S.K., Le Corre D., Li X.Y., Atochin I., Link D.R., Griffiths A.D., Pallier K. et al.. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013; 59:1722–1731. [DOI] [PubMed] [Google Scholar]
- 19. Choi J.J., Cho M., Oh M., Kim H., Kil M.S., Park H.. PNA-mediated real-time PCR clamping for detection of EGFR mutations. B. Korean. Chem. Soc. 2010; 31:3525–3529. [Google Scholar]
- 20. Tatsumi K., Mitani Y., Watanabe J., Takakura H., Hoshit K., Kawai Y., Kikuchi T., Kogo Y., Oguchi-Katayama A., Tomaru Y. et al.. Rapid screening assay for KRAS mutations by the modified smart amplification process. J. Mol. Diagn. 2008; 10:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tropea J.E., Cherry S., Waugh D.S.. Doyle SA. High Throughput Protein Expression and Purification: Methods and Protocols. 2009; Totowa: Humana Press; 297–307. [Google Scholar]
- 22. Potapov V., Ong J.L.. Examining sources of error in PCR by single-molecule sequencing. PLoS One. 2017; 12:e0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song J.Z., Pandian V., Mauk M.G., Bau H.H., Cherry S., Tisi L.C., Liu C.C.. Smartphone-based mobile detection platform for molecular diagnostics and spatiotemporal disease mapping. Anal. Chem. 2018; 90:4823–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song J.Z., Liu C.C., Bais S., Mauk M.G., Bau H.H., Greenberg R.M.. Molecular detection of schistosome infections with a disposable microfluidic cassette. PloS Neglect. Trop. D. 2015; 9:e0004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song J.Z., Mauk M.G., Hackett B.A., Cherry S., Bau H.H., Liu C.C.. Instrument-free point-of-care molecular detection of zika virus. Anal. Chem. 2016; 88:7289–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaya E., Doxzen K.W., Knoll K.R., Wilson R.C., Strutt S.C., Kranzusch P.J., Doudna J.A.. A bacterial Argonaute with noncanonical guide RNA specificity. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:4057–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doxzen K.W., Doudna J.A.. DNA recognition by an RNA-guided bacterial Argonaute. PLoS One. 2017; 12:e0177097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheng G., Gogakos T., Wang J.Y., Zhao H.T., Serganov A., Juranek S., Tuschl T., Patel D.J., Wang Y.L.. Structure/cleavage-based insights into helical perturbations at bulge sites within T. thermophilus Argonaute silencing complexes. Nucleic Acids Res. 2017; 45:9149–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheng G., Zhao H.T., Wang J.Y., Rao Y., Tian W.W., Swarts D.C., van der Oost J., Patel D.J., Wang Y.L.. Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Swarts D.C., Szczepaniak M., Sheng G., Chandradoss S.D., Zhu Y.F., Timmers E.M., Zhang Y., Zhao H.T., Lou J.Z., Wang Y.L. et al.. Autonomous generation and loading of DNA guides by bacterial Argonaute. Mol. Cell. 2017; 65:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phelps C., Lee W., Jose D., von Hippel P.H., Marcus A.H.. Single-molecule FRET and linear dichroism studies of DNA breathing and helicase binding at replication fork junctions. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17320–17325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rees W.A., Yager T.D., Korte J., Vonhippel P.H.. Betaine can eliminate the base pair composition dependence of DNA melting. Biochemistry-US. 1993; 32:137–144. [DOI] [PubMed] [Google Scholar]
- 33. Kuzmenko A., Yudin D., Ryazansky S., Kulbachinskiy A., Aravin A.A.. Programmable DNA cleavage by Ago nucleases from mesophilic bacteria Clostridium butyricum and Limnothrix rosea. Nucleic Acids Res. 2019; 47:5822–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Ge X., Yang F., Zhang L., Zheng J., Tan X., Jin Z.B., Qu J., Gu F.. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014; 4:5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamano T., Zetsche B., Ishitani R., Zhang F., Nishimasu H., Nureki O.. Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol. Cell. 2017; 67:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bettegowda C., Sausen M., Leary R.J., Kinde I., Wang Y.X., Agrawal N., Bartlett B.R., Wang H., Luber B., Alani R.M. et al.. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014; 6:224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oxnard G.R., Paweletz C.P., Kuang Y.A., Mach S.L., O’Connell A., Messineo M.M., Luke J.J., Butaney M., Kirschmeier P., Jackman D.M. et al.. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014; 20:1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aggarwal C., Thompson J.C., Black T.A., Katz S.I., Fan R., Yee S.S., Chien A.L., Evans T.L., Bauml J.M., Alley E.W. et al.. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol. 2018; 5:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song C., Liu Y.B., Fontana R., Makrigiorgos A., Mamon H., Kulke M.H., Makrigiorgos G.M.. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 2016; 44:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu L.R., Chen S.X., Wu Y., Patel A.A., Zhang D.Y.. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat. Biomed. Eng. 2017; 1:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsiatis A.C., Norris-Kirby A., Rich R.G., Hafez M.J., Gocke C.D., Eshleman J.R., Murphy K.M.. Comparison of sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations diagnostic and clinical implications. J. Mol. Diagn. 2010; 12:425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Underhill H.R., Kitzman J.O., Hellwig S., Welker N.C., Daza R., Baker D.N., Gligorich K.M., Rostomily R.C., Bronner M.P., Shendure J.. Fragment length of circulating tumor DNA. PLoS Genet. 2016; 12:e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sato A., Nakashima C., Abe T., Kato J., Hirai M., Nakamura T., Komiya K., Kimura S., Sueoka E., Sueoka-Aragane N.. Investigation of appropriate pre-analytical procedure for circulating free DNA from liquid biopsy. Oncotarget. 2018; 9:31904–31914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bielas J.H., Loeb L.A.. Quantification of random genomic mutations. Nat. Methods. 2005; 2:285–290. [DOI] [PubMed] [Google Scholar]
- 45. Li J., Wang L., Mamon H., Kulke M.H., Berbeco R., Makrigiorgos G.M.. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 2008; 14:579–584. [DOI] [PubMed] [Google Scholar]
- 46. Kim H.S., Sung J.S., Yang S.J., Kwon N.J., Jin L., Kim S.T., Park K.H., Shin S.W., Kim H.K., Kang J.H. et al.. Predictive efficacy of low burden EGFR mutation detected by next-generation sequencing on response to EGFR tyrosine kinase inhibitors in non-small-cell lung carcinoma. PLoS One. 2013; 8:e81975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014; 507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krug A.K., Enderle D., Karlovich C., Priewasser T., Bentink S., Spiel A., Brinkmann K., Emenegger J., Grimm D.G., Castellanos-Rizaldos E. et al.. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018; 29:700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tuma R.S., Beaudet M.P., Jin X.K., Jones L.J., Cheung C.Y., Yue S., Singer V.L.. Characterization of SYBR gold nucleic acid gel stain: A dye optimized for use with 300-nm ultraviolet transilluminators. Anal. Biochem. 1999; 268:278–288. [DOI] [PubMed] [Google Scholar]
- 50. Lilley D.M., Bhattacharyya A., McAteer S.. Gel electrophoresis and the structure of RNA molecules. Biotechnol. Genet. Eng. Rev. 1992; 10:379–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings in this study are available within the paper and its Supplementary Information files.