


Abstract
The relentless adaptability of pathogen populations is a major obstacle to effective disease control measures. Increasing evidence suggests that gene transcriptional polymorphisms are a strategy deployed by pathogens to evade host immunity. However, the underlying mechanisms of transcriptional plasticity remain largely elusive. Here we found that the soybean root rot pathogen Phytophthora sojae evades the soybean Resistance gene Rps1b through transcriptional polymorphisms in the effector gene Avr1b that occur in the absence of any sequence variation. Elevated levels of histone H3 Lysine27 tri-methylation (H3K27me3) were observed at the Avr1b locus in a naturally occurring Avr1b-silenced strain but not in an Avr1b-expressing strain, suggesting a correlation between this epigenetic modification and silencing of Avr1b. To genetically test this hypothesis, we edited the gene, PsSu(z)12, encoding a core subunit of the H3K27me3 methyltransferase complex by using CRISPR/Cas9, and obtained three deletion mutants. H3K27me3 depletion within the Avr1b genomic region correlated with impaired Avr1b gene silencing in these mutants. Importantly, these mutants lost the ability to evade immune recognition by soybeans carrying Rps1b. These data support a model in which pathogen effector transcriptional polymorphisms are associated with changes in chromatin epigenetic marks, highlighting epigenetic variation as a mechanism of pathogen adaptive plasticity.
INTRODUCTION
Filamentous eukaryotic pathogens such as fungi and oomycetes exhibit rapid adaptability to host immunity, fungicides and other environmental changes, threatening global human health and food security. Genomes of some filamentous pathogens with high adaptability display a bipartite architecture with gene-sparse, repeat-rich compartments serving as sites of dynamic variation. In these particular genomic compartments, genes encoding virulence effector proteins are enriched, and display signs of accelerated genome evolution (1–3). In natural populations, effector genes display rich genetic diversity including presence/absence polymorphisms, nucleotide polymorphisms and copy number variations (4–7). These DNA sequence changes are considered to be signatures of pathogen co-evolution with host plants. Although understanding the adaptation of pathogens is critical for rational disease management, we still know little about how pathogens and their effector genes adapt to resistant hosts.
Increasing evidence has suggested that gene expression polymorphisms contribute to microbial responses to environmental changes (8–12). Epigenetic mechanisms can create heritable changes in gene expression that are independent of DNA sequence changes. A variety of small RNA molecules, and chemical modifications of histone tails and DNA bases can serve as mechanisms of epigenetic regulation (13). For example, RNAi-dependent epimutations mediated antifungal drug resistance in the human pathogen Mucor circinelloides (10). Histone H3 Lys27 tri-methylation (H3K27me3) in Neurospora crassa modulated the response to genotoxic stress (11). Heritable trans-generational gene silencing of the effector gene PsAvr3a, mediated by small RNAs, enabled the soybean root pathogen Phytophthora sojae to evade the resistance of soybeans carrying the Rps3a resistance gene (12). Compared to DNA sequence changes, epigenetic variation is dynamic and reversible upon environmental and physiological changes. However, how filamentous pathogens epigenetically adapt to host plants remains largely unexplored.
The histone modification H3K27me3, in general, marks facultative heterochromatin and represses the expression of genes in spatial and temporal patterns (14). In all eukaryotes studied so far, the polycomb repressive complex 2 (PRC2) generates H3K27me2/3 marks via its methyltransferase subunits EZH2 or KMT6. PRC2 also comprises core components such as Su(z)12 and ESC that act in a regulatory manner (15–18). In several plant-associated fungi, genetic manipulations of PRC2 subunits impaired H3K27me3 homeostasis and broadly impacted fungal development, metabolism and chromosome functions, suggesting the H3K27me3 machinery plays important regulatory roles in these filamentous organisms (19–21).
Phytophthora sojae is a devastating root rot pathogen of soybean, and is one of the most damaging disease problems confronting soybean growers (22). The interaction between P. sojae and soybean is regulated by gene-for-gene interactions. Typical gene-for-gene interactions involve plants that carry a specific disease resistance (R) gene that confers resistance towards a pathogen that expresses a matching avirulence (AVR) effector gene. In such cases, the R gene product can directly or indirectly detect the presence of the AVR effector protein, triggering a robust and effective defense response. Pathogen strains containing variations in the sequence or expression of an AVR gene can evade plant R protein recognition, resulting in a failure of plant immunity and onset of disease (23). Phytophthora sojae strains expressing the Avr1b avirulence allele cannot efficiently infect soybean cultivars carrying the Rps1b resistance gene (24). Field strains of P. sojae that can evade Rps1b-mediated immunity exhibit a variety of Avr1b polymorphisms including nucleotide point mutations, Avr1b deletions, and natural silencing of Avr1b (24,25). These polymorphisms provide an opportunity to study the mechanisms of P. sojae adaption to host resistance. Here, we show that H3K27me3 is required to maintain the naturally silenced state of Avr1b in a P. sojae strain.
MATERIALS AND METHODS
Phytophthora sojae and plant cultivation
Phytophthora sojae strains were routinely cultivated at 25°C in the dark on 10% vegetable (V8) juice agar medium. Non-sporulating hyphae were collected after 3-day cultivation at 25°C in the dark using 10% V8 liquid medium. Soybean cultivars Williams and Harosoy13xy were used for virulence assays. Seedlings for inoculation were grown at 25°C (16 h, light) and 22°C (8 h, dark) for 7–10 days. Etiolated hypocotyls harvested after growth at 25°C (16 h, dark) and 22°C (8 h, dark) for 4 days were also used for P. sojae inoculations to produce infected tissue.
P. sojae virulence assays
Phytophthora sojae virulence assays were performed on light-grown seedlings by the hypocotyl split inoculation method (26) using approximately 10 plants per plastic pot (15 cm in diameter). P. sojae strains were grown for 3–5 days on 10% (v/v) V8 agar plates, then 2 mm × 4 mm segments of infested agar were cut from the growing edge of mycelial colonies and inoculated into the splits of the hypocotyls. Inoculated plants then were kept in the greenhouse maintained at high humidity for 12 h. Plants were then photographed after 3 days. A minimum of three independent replicates of the disease assay were performed for each P. sojae strain tested.
RNA extraction, RNA-seq and qRT-PCR
Total RNA of 3-day-old P. sojae hyphae was isolated using the Omega Total RNA Kit I according to the manufacturer's manual. RNA quantity and quality were measured using a Nanodrop ND-1000 and 1% agarose gel electrophoresis. BGI (Shenzhen, China) provided RNA-seq services for this study; two biological replicates of mycelia of T34 were utilized. cDNA synthesis was performed using the PrimeScript RT reagent Kit (Takara). Corresponding cDNA levels were measured by quantitative PCR and normalized to endogenous Actin levels; the primers used for the qRT-PCR Actin reference were previously described (25) and are listed in Supplementary Table S1. Each qPCR reaction was performed using the ABI PRISM 7500 Fast Real-Time PCR System under the following conditions: 95°C for 30 s, 40 cycles of 95°C for 5 s and 60°C for 34 s to calculate cycle threshold (Ct) values, followed by a dissociation step, 95°C for 15 s, 60°C for 1 min and 95°C for 15 s. Relative transcript levels were calculated using the 2−△△CT method.
ChIP-seq and ChIP-qPCR
Chromatin immunoprecipitation (ChIP) experiments were carried out as previously described (27,28), using 30 μg of chromatin and 3 μg of the corresponding antibody. 1.5 g of 3-day old mycelia were harvested for nuclei preparation. 15 μl micrococcal nuclease (MNase: NEB M0247S, 2000 gels units/μl) were added to digest the nuclei in 2 ml MNB buffer (10% sucrose, 0.05 M Tris–HCl, pH 7.5, 4 mM MgCl2, 1 mM CaCl2) for 10 min at 37°C. 200 μl 0.5 M EDTA were then added to stop digestion. After centrifugation, 40 μl of supernatant was stored as input at -20°C. The remainder was incubated with anti-H3K27me3 antibodies for more than 6 h. After incubation, 30 μl pre-washed protein A Dynabeads (Thermofisher, 10001D) were added to the chromatin-antibody solution and agitated for more than 6 h. After the incubation the beads were washed successively with three 1 ml aliquots of buffer A (50 mM Tris–HCl, pH 7.5, 10 mM EDTA), containing 50 mM NaCl, 100 mM NaCl or 150 mM NaCl, respectively. Then, the DNA was eluted by two washes of 400 μl elution buffer (20 mM Tris–HCl, pH 7.5, 50 mM NaCl, 5 mM EDTA, 1% SDS) at 65°C for 15 min. The DNA was then extracted using phenol–chloroform–isoamyl alcohol (25:24:1) to remove remaining proteins, then precipitated using ethanol overnight at –20 °C, and washed twice with 70% ethanol. Finally, the sample was dried and dissolved in 33 μl ddH2O. As controls, input DNA was recovered by phenol extraction after the nuclease digestion step.
The DNA extracted from the H3K27me3 ChIP samples from P. sojae mycelium was subjected to high-throughput sequencing and qPCR. Two gDNA input replicates from P6497 or T34 mycelia, two H3K27me3 ChIP replicates from P6497 mycelia, and three H3K27me3 ChIP replicates from T34 mycelia were collected for sequencing on the Illumina Hiseq4000 platform by BGI (Shenzhen, China). Primers used for quantitative PCR (qPCR) are listed in Supplementary Table S1. Each qPCR reaction was performed using the ABI PRISM 7500 Fast Real-Time PCR System according to the following protocol: 95°C for 30 s followed by 40 cycles of 95°C for 5 s and 60°C for 34 s, followed by a dissociation step, 95°C for 15 s, 60°C for 1 min and 95 °C for 15 s. Relative accumulation levels were calculated using the 2−△△CT method taking the input DNA control as an internal reference.
Phytophthora sojae transformation and gene editing
Genome editing using CRISPR/Cas9 and transformation in P. sojae were performed as previously described (29). The targeted region of PsSu(z)12 is unique in the genome, and two single guide RNA (sgRNA) target sites (sgRNA207: GCTGCCACGCCGGGTCCAGG, sgRNA768: GCAGAGGATCCCAAGTAAGA) were selected using an online tool (http://grna.ctegd.uga.edu/). For construction of the construct encoding the sgRNAs targeting PsSu(z)12, the full-length sense and antisense guide oligos of sgRNA coding sequence (together with the flanking HH-ribozyme) were synthesized by the Genscript and then annealed to one another. Digestion of pYF2.3G-Ribo-sgRNA with Nhe I and Bsa I allowed the direct insertion of the annealed oligos into the cleaved plasmid. Finally, Polyethylene Glycol (PEG)-mediated protoplast transformations were conducted to create mutations. Transformant screening was performed by PCR and sequencing using primers listed in Supplementary Table S1.
Multiple unsuccessful attempts were made to complement the PsSu(z)12 mutations. We tried to express the intact PsSu(z)12 gene in mutant T34 (561 bp deletion) by using the gene expression vector pTor with the constitutive HAM34 promoter or with the PsSu(z)12 native promoter, but never recovered any transformants with significant expression of full length PsSu(z)12. We also tried to carry out complementation in T34 using sgRNA-mediated homology-directed repair with HA-tagged PsSu(z)12. We obtained three complemented mutants (one homozygote and two heterozygotes), but we could not detect the HA-tagged PsSu(z)12 protein by western blotting in any of these transformants.
Western-blot analysis
Total proteins were extracted from 3-day-old P. sojae mycelia and separated in SDS-PAGE gels. The separated proteins were then transferred to PVDF membranes. The membranes were blocked using 5% non-fat milk in PBST buffer (1 × PBS + 0.1% Tween 20) (PBSTM) for 30 min at room temperature with 60 r.p.m. shaking. The corresponding antibodies for histones H3 (Abcam, no. ab1791) and H3K27me3 (Millipore, no. 07-449) were then added to the PBSTM at a dilution of 1:5000. The membranes were incubated with the antibody at room temperature for 4 hr, then were washed three times (5 min each) with PBST buffer. After washing, the membranes were incubated with a goat-anti-rabbit IRDye 800CW antibody (Odyssey, no. 926-32211; Li-Cor) at a dilution of 1:10 000 in PBSTM at room temperature for 30 min with 60 r.p.m. shaking, then followed by three washes (5 min each) with PBST. The signals were excited and detected at 700 and 800 nm, respectively, using a double color infrared laser imaging system (Odyssey, LI-COR company).
Bioinformatics analysis
A detailed list of all programs and commands used for RNA-seq, ChIP-seq and small RNA-seq data analyses can be found in the Supplementary Text S1.
For ChIP-seq, reads were mapped to the P. sojae genome sequence v3.0 (http://genome.jgi-psf.org/Physo3/Physo3.home.html) using bowtie2 (30), normalized using deepTools (31) and visualized using IGV (32). Original files of sequencing data from P6497 and pssu(z)12 mutant T34 mycelia (two replicates of H3K27me3 IP and input samples from P6497, three replicates of H3K27me3 IP samples from T34 and two replicates of input samples from T34) were submitted to NCBI with accession No. GSE127206.
For RNA-seq, reads were aligned to the P. sojae P6497 genome assembly v3.0 using Hisat2 (33) and visualized using IGV, the TPM value and raw read count of each gene were calculated using Stringtie (34). DESeq2 (35) was then used to determine 2-fold or 10-fold differentially expressed genes with its default settings for scaling and transforming the data. Three biological replicates of P6497 RNA-seq data were accessed from NCBI (GSE116089), two biological replicates of pssu(z)12 mutant T34 RNA-seq data were submitted to NCBI (GSE127207). The mean values of TPM from the biological replicates were adopted to draw the transcriptome comparison, and the 2-fold and 10-fold differentially expressed genes identified by DESeq2 were marked in Figure 3C with different colors.
Figure 3.

Editing the PsSu(z)12 locus abolished Avr1b gene silencing and changed the virulence phenotype. (A) Avr1b transcript levels in mycelia of pssu(z)12 mutants measured by RT-qPCR. Transcript levels were normalized with Actin as the internal standard and presented as means ± SEM of three biological replicates. Asterisks represent significant differences (P < 0.01, compared with P6497, Student's t test). sc10 served as the positive (expressed) control, Ctrl1 and Ctrl2 are negative (silenced) controls described in Figure 2C. (B) RT-PCR experiment as (A). Three biological replicates were conducted with similar results. (C) Comparative transcriptome analysis of wild-type P6497 and pssu(z)12Δ mycelia. The x-axis (wild-type P6497) and y-axis (pssu(z)12Δ T34) show log2(TPM+1) of each gene (shown as a dot) based on the mean value of biological replicates, with three replicates of P6497 and two replicates of T34. Black dots represent genes with transcript level differences of >2×-fold change identified by DESeq2 (|log2FoldChange(T34/P6497)| > 1, adjusted P value < 0.01), red dots represent genes with more than ten-fold elevation in T34 compared to P6497 with an adjusted P value < 0.01, while blue dots represent genes with more than ten-fold reduction with adjusted P value < 0.01. Grey dots represent genes lacking differential transcript levels. Avr1b is shown in orange and indicated by an orange arrow. (D) H3K27me3 ChIP-seq and RNA-seq data in the Avr1b region from P6497 and pssu(z)12. Samples were harvested from mycelia, symbols as in Figure 1D with P6497 data in blue and pssu(z)12 data in red. Two and three biological replicates of ChIP-seq were performed for P6497 and T34, two biological replicates were performed for pssu(z)12 mutant T34 RNA-seq, IGV visualization shows similar results from different replicates. (E) H3K27me3 deposition in P6497, pssu(z)12 and controls measured by ChIP-qPCR. Values represent the mean fold changes (±SEM) of the transformants relative to tubulin, which was set as 1. Experiments were repeated twice with similar results.
For Gene Ontology analysis, P. sojae gene GO terms were updated by using the program BLAST2GO (36) (Supplementary Table S2). Both 10-fold upregulated and downregulated genes (Supplementary Table S3) were utilized for GO functional classification using Web Gene Ontology Annotation Plotting (WEGO) tool, Version 2.0 (37), the differentially enriched GO terms versus genome-wide P. sojae gene GO terms were then visualized in Supplementary Figure S6, P-values are reported as –log10P-value.
For small RNA-seq, three biological replicates from P6497 and ACR10 were downloaded from NCBI Bioproject PRJNA300858. Sequencing reads were parsed to remove adaptors and collapsed to a unique set with read counts; any reads shorter than 18 nt were discarded. Clean reads were mapped to P. sojae v3.0 using Bowtie (38) with no mismatch (-v 0). Finally, sRNA-seq data was normalized and visualized using IGV, reads on loci of interest were counted using ShortStack (39) and calculated in reads per million mapped (rpmm).
The sc10 genome was assembled de novo into 145 contigs, with an average genome coverage of 80.19×, using the Hierarchical Genome Assembly Process version 3 (HGAP3) pipeline using SMRT Link 3.1.1 (40).
RESULTS
Avr1b gene expression differs between two P. sojae strains in the absence of sequence variation
To examine the mechanisms underlying the lack of Avr1b transcripts in field isolates, we selected two P. sojae strains from our collection, P6497 and sc10, that exhibit virulent and avirulent phenotypes on Rps1b-carrying soybean plants (Harosoy13xy) respectively (Figure 1A). PCR amplification of the Avr1b coding region followed by sequencing revealed no sequence differences between P6497 and sc10 (Supplementary Figure S1A). To re-examine Avr1b transcript levels in the two strains under different conditions, a reverse transcription polymerase chain reaction (RT-PCR) assay was used. Stages examined included in vitro growth of mycelium on media and 3-day post-inoculation on etiolated soybean or hypocotyls (Figure 1B). Avr1b transcripts were detected in sc10 but absent in P6497 in all three tissues examined. To further investigate polymorphisms in the Avr1b region as well as the genetic background between P6497 and sc10, we performed Pacific Biosciences long read sequencing of the sc10 genome and compared the de novo genome assembly (original sequencing data were submitted to NCBI with accession no. PRJNA524096) to the P6497 genome sequence v3.0 (NCBI Reference Sequence: NW_009258120.1). The result showed that a large region around Avr1b (up to 50 kb upstream and 31 kb downstream) was completely identical between the two strains (Supplementary Figure S1B). The Avr1b sequence and transcriptional status, together with the corresponding phenotypes of the two strains are summarized in Figure 1C. Based on the identical DNA sequences but differing transcript levels, we hypothesized that epigenetic variations may drive the differences in Avr1b transcript levels.
Figure 1.
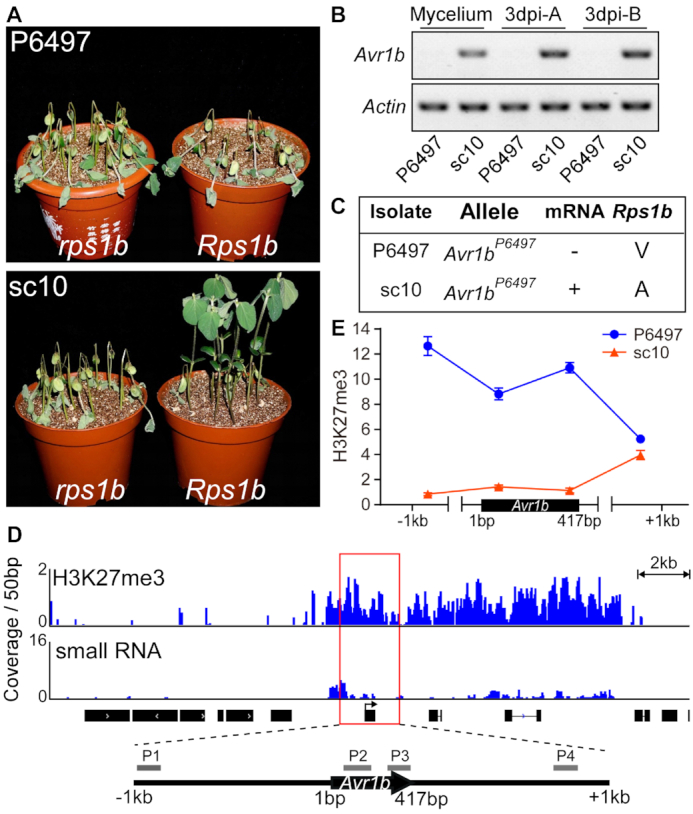
H3K27me3 epigenetic status at the Avr1b locus differs between silenced and non-silenced isolates. (A) Virulence assays of P. sojae strains P6497 and sc10. Shown are disease outcomes 3 days after hypocotyl inoculation of soybean cultivars Williams (rps1b) and Harosoy13xy (Rps1b). Three biological replicates were performed with similar results. (B) RT-PCR experiments performed with P6497 and sc10 cDNA samples from mycelia or infected plants. Primers specific for the Avr1b gene were used for RT-PCR. Actin was amplified as the positive control. A and B stand for etiolated seedling infection and hypocotyl infection, respectively. Three biological replicates were performed with similar results. (C) A summary of Avr1b alleles, transcript presence, and specific virulence of P6497 and sc10. ‘–’ indicates silenced, ‘+’ indicates expressed. V and A indicate successful or failed infection of Rps1b plants, respectively. (D) Abundant H3K27me3 methylation but few sRNAs around the Avr1b locus in P6497. H3K27me3 ChIP-seq data and small RNA-seq data from P6497 mycelium are shown in the IGV browser. The coverage of ChIP-seq data was normalized by input using RPKM, the coverage of small RNA-seq reads was normalized using CPM. Profiles of H3K27me3 and sRNA signals (25 kb around Avr1b) are shown in blue. Black boxes with arrows represent the open reading frames of predicted genes. Four pairs of primers (P1, P2, P3, P4) were designed to quantify H3K27me3 deposition around Avr1b. (E) ChIP-qPCR measurements of H3K27me3 deposition at Avr1b in P6497 and sc10. H3K27me3 enrichment signals were normalized to the tubulin gene. The values are the means ± SEM of three biological replicates.
Avr1b gene silencing is correlated with H3K27me3 hypermethylation in the virulent strain
We examined the epigenetic landscape of Avr1b in P6497 including H3K27me3 and sRNA levels to explore which epigenetic features might be associated with Avr1b gene silencing. We downloaded the in-depth sRNA sequencing data of strains P6497 and ACR10 from NCBI (41), then examined the occurrence of sRNAs in the Avr1b region. The results revealed little occurrence of sRNAs in the region, other than a short region 1 kb upstream of Avr1b (Figure 1D). As a control, we examined the levels of sRNAs at the PsAvr3a locus, observing that abundant sRNAs accumulated at the silenced PsAvr3a locus of ACR10 but not P6497, consistent with the Avr3a phenotypes of the two strains (Supplementary Figure S2). Next, we performed ChIP-seq experiments with chromatin from P6497 using H3K27me3 antibodies. The results illustrated a high accumulation of H3K27me3 marks at the Avr1b locus (Figure 1D). To validate the ChIP-seq results, we compared H3K27me3 accumulation at Avr1b between P6497 and sc10 by using ChIP-qPCR. Four pairs of primers located in a 2.4-kb genomic region in the vicinity of Avr1b were used (Figure 1D). We observed elevated H3K27me3 levels across the Avr1b region in the non-expressing strain, P6497, but little methylation in the Avr1b-expressing strain, sc10 (Figure 1E), supporting our hypothesis that H3K27me3 plays a role in maintaining Avr1b silencing.
Editing the H3K27me3 writer complex gene, Su(z)12, in the Avr1b-silenced strain
To test the hypothesis that H3K27me3 is involved in maintaining Avr1b silencing, we set out to generate mutants of subunits of the major H3K27me3 writer complex – Polycomb Repressive Complex 2 (PRC2). We searched the P. sojae genome (v3.0) for the orthologs of D. melanogaster PRC2 subunits E(z), ESC, Su(z)12 and Nurf55 (Figure 2A), by bidirectional blastp searches with an E-value cut-off of 10−20 (42,43). Genes encoding these subunits were conserved in P. sojae, a total of seven E(z), one ESC, two Nurf55 and one Su(z)12 homologs were identified and verified by constructing phylogenetic trees (Supplementary Figure S4). As there are seven orthologs of PRC2 catalytic active subunit E(z), it was not feasible to disrupt so many genes simultaneously. Ps326596 (PsSu(z)12, protein ID Ps324933) appeared to be the only unique ortholog of Su(z)12 in P. sojae. It encoded a protein of 620 amino acids, with 55.97% similarity compared to D. melanogaster Su(z)12, and contained a typical VEFS-Box which is conserved in polycomb proteins. We set out to knock out this gene by using CRISPR/Cas9 technology recently developed in P. sojae (29).
Figure 2.

Generating PsSu(z)12 mutants in P. sojae. (A) P. sojae contains conserved components of PRC2 complexes, including Su(z)12, which regulate H3K27 methylation in eukaryotes. (B) PsSu(z)12 locus edited using CRISPR/Cas9. All three recovered mutants were homozygotes. T34 and T21, with identical editing events, were recovered from two independent transformation attempts. (C) Western blot analysis of global H3K27me3 levels in pssu(z)12 mutants. Ctrl1 and Ctrl2 are regenerated lines from the CRISPR transformation experiment that lacked editing of the PsSu(z)12 locus. (D) Statistics of relative H3K27me3 levels in (C). Five biological replicates were carried out and represented with quartiles, asterisks indicate significance compared with P6497 (P < 0.01, Student's t test).
Although extensive attempts were made, we never succeeded in isolating homozygous full-length deletion mutants of PsSu(z)12 through the sgRNA-mediated homology-directed repair (HDR) gene replacement approach. This result suggested that complete PsSu(z)12 removal may be lethal in P6497. However, we did isolate three independent homozygous mutants (T34, T21, T12) comprising two types of in-frame deletion events, having 561 bp or 12 bp deletions in the PsSu(z)12 coding sequence, respectively. (Figure 2B, Supplementary Figure S3A). RNA-seq analysis of one of the mutants, T34, revealed that PsSu(z)12 transcript levels in the mutant were similar to those in P6497, but lacked reads matching the 561 bp deleted region (Supplementary Figure S5). To characterize the mutants further, we employed an additional two control strains, Ctrl1 and Ctrl2 in which the PsSu(z)12 locus was not altered, that were recovered from the same transformation experiment as the three mutants. Firstly, we performed a mycelial growth assay on regular cleared V8 media and did not observe an in vitro growth defect (Supplementary Figure S3B, C). Then we performed western blots to examine the global H3K27me3 levels in each line. The results showed that all three mutants exhibited a significant reduction in global H3K27me3 levels, 58%, 57% and 43%, respectively in comparison to P6497 and the control strains (Figure 2C, D). However, the H3K27me3 levels in Ctrl1 and Ctrl2 were not significantly different than in P6497. These results indicated that we had partially reduced H3K27me3 methylation in the three independent PsSu(z)12 mutants.
Despite several attempts (see Materials and Methods) we unable to complement the PsSu(z)12 mutants with the wild-type gene, due to poor expression of the introduced gene. Further work will be needed to resolve this issue.
Reduced H3K27me3 in PsSu(z)12 mutants is associated with release of Avr1b gene silencing and failure to infect Rps1b soybean cultivars
We measured Avr1b transcript levels in the three PsSu(z)12 mutants. Both reverse transcription quantitative real-time PCR (qRT-PCR) analysis and RT-PCR results revealed that Avr1b silencing was released in all three mutants, with Avr1b transcript levels elevated >100-fold in all three mutants. (Figure 3A, B). We also used RNA-seq analysis of T34 to examine the global effect of the 561 bp deletion in T34. We found a significant increase in the transcript levels of 1434 genes and a decreased level for 473 genes in the PsSu(z)12 mutant T34 compared to wild type, based on a minimum 2X Fold Change and adjusted P value <0.01. Among these genes with >2-fold differential transcript levels, a total of 687 genes including Avr1b exhibited transcript levels that were elevated more than ten-fold in T34 compared to P6497 (FoldChange(T34/P6497) > 10, adjusted P value < 0.01). In contrast, 99 genes were found with significantly lower transcript levels of more than ten-fold in T34 compared to P6497 (FoldChange(T34/P6497) < 0.1, adjusted P value < 0.01) (Figure 3C). Then we conducted a Gene Ontology (GO) category analysis based on these 786 genes with ten-fold differential transcript levels. Associated with H3K27me3 defects in the pssu(z)12 mutants, we found significant enrichment of genes annotated for nucleotide binding (P = 1.0 × 10−10), protein binding (P = 1.0 × 10−10), methyltransferase activity (P = 1.8 × 10−3), DNA binding (P = 2.3 × 10−2) and other molecular function terms (Supplementary Figure S6). Interestingly, we found significantly enriched genes associated to drug binding (P = 1.0 × 10−10), indicating a possibility of new findings on rational disease management. Next, we examined H3K27me3 levels across the 25 kb region spanning Avr1b by conducting ChIP-seq analysis. The ChIP-seq data confirmed that the Avr1b region is hypomethylated in T34, especially in the immediate region of Avr1b. In the same region, the RNA-seq data confirmed elevated Avr1b transcript signals in T34 compared to P6497, whereas neighboring genes showed little difference between T34 and P6497. Ps337246, a gene located 3.5 kb downstream of Avr1b, maintained high H3K27me3 methylation levels in both P6497 and T34 and remained silenced in both strains, providing an excellent control for Avr1b. Similarly, Ps337247 was another downstream gene that remained highly methylated and silenced in both P6497 and T34. Furthermore, three genes (Ps512252, Ps513314, Ps249437) upstream of Avr1b displayed negligible methylation and remained fully expressed in both strains (Figure 3D). To confirm the reduction of H3K27me3 modification in the Avr1b region, we performed ChIP-qPCR assay at four locations across the Avr1b region. The data demonstrated that levels of H3K27me3 across the Avr1b locus were reduced 3–4-fold in all three PsSu(z)12 mutants in comparison to P6497 (Figure 3E).
To further investigate the impact of reducing H3K27me3 levels at the Avr1b locus, we assessed the virulence of the mutants on Rps1b plants compared to non-Rps1b plants. All three mutants produced typical infection levels on a susceptible soybean cultivar (Williams). However, the mutants were avirulent on Rps1b soybean plants (Harosoy13xy). In contrast, the parent strain P6497 and the two control lines remained virulent on both Williams and Harosoy13xy soybeans (Figure 4A). This experiment demonstrated that the release of Avr1b gene silencing through mutation of PsSu(z)12 led the mutant to being recognized by Rps1b plants.
Figure 4.

H3K27me3 deposition at Avr1b matches virulence phenotypes on Rps1b plants. (A) pssu(z)12 mutants are avirulent on Rps1b soybean plants. Soybean seedlings of cultivars Williams (rps1b) and Harosoy13xy (Rps1b) were inoculated with P. sojae mycelia agar plugs. Phenotypes were photographed 3 days after inoculation. Three biological replicates were performed with similar results. (B) Model of Avr1b silencing maintained by elevated H3K27me3 methylation. Disruption of H3K27me3 methylation machinery changes virulence against Rps1b resistant plants.
DISCUSSION
While the contributions of DNA sequence polymorphisms to pathogenic plasticity have been well documented in oomycetes (44,45), the basis of gene expression polymorphisms in pathogenic adaptation remain poorly explored (8). Previous studies of gene silencing in Phytophthora species have mainly focused on the roles of sRNAs. Internuclear spread of RNAi-mediated gene silencing of a PAMP gene, INF1, was observed in Phytophthora infestans by van West et al. (46). More recently, heritable transgenerational gene silencing of effector gene PsAvr3a in P. sojae was found to be mediated by 24 nt sRNAs (13). Mechanistic studies have revealed that RNAi-triggered silencing of the INF1 elicitin gene in P. infestans could be released by chemical inhibitors of DNA methylation and histone deacetylation (47). Similarly, RNAi-mediated silencing of the P. infestans sporulation gene cdc14 could be relieved not only by silencing of Dicer-like and Argonaute genes, but also by silencing of histone deacetylase genes (48). No cytosine methylation nor the required enzymes can be detected in Phytophthora. The methylome of adenine methylation 6mA in Phytophthora was recently profiled by our group. However, whether m6A plays a role in regulating gene expression remains to be examined (49). All these data led us to consider whether histone modifications could be players in gene silencing in addition to the sRNA-associated silencing machinery in Phytophthora.
Here, we have examined the epigenetic mechanisms underlying the silencing of Avr1b in in P. sojae strain P6497. The DNA sequences surrounding Avr1b were identical to a strain (sc10) expressing Avr1b, for a least 50 kb upstream and 31 kb downstream. There was little accumulation of sRNAs matching Avr1b in P6497, although there was some accumulation in a region 1kb away upstream of Avr1b. In previous reports, Phytophthora sRNA deposition in coding sequences was well researched (50–52). However, no clear association between promoter regions 1 kb away with gene silencing was reported. Whether sRNAs within promoter regions impact Phytophthora gene expression at the transcriptional level remains to be explored. On the other hand, the repressive histone mark H3K27me3 was abundant in the region of Avr1b in P6497 compared to sc10. Deletion mutations introduced into the key PRC2 component gene, PsSu(z)12, reduced H3K27me3 levels more than 3-fold in the vicinity of Avr1b and concomitantly resulted in the release of silencing of Avr1b. These results suggest that local H3K27me3 methylation is essential for the maintenance of Avr1b silencing (Figure 4B), whereas sRNAs are not required. Our analyses found relatively low levels of sRNA accumulation at the Avr1b locus in mycelia of P6497 (4.7 RPMM). On the other hand, Wang et al. (2019) found somewhat higher levels of Avr1b sRNAs (34.5 RPMM) in a pool of RNA from P6497 oospores, mycelia, sporangia, cysts, and germinated cysts, including several anti-sense sRNAs. They speculated that those sRNAs may be responsible for silencing of Avr1b. However, their study did not include a strain like sc10 that expressed Avr1b but had no sequence differences in the neighborhood of Avr1b. Therefore, it is difficult to assess if those higher levels of sRNAs are tissue-specific or are related to Avr1b silencing. Cui et al. (25) reported that multiple strains of P. sojae have independently acquired silencing of Avr1b, indicating that silencing is a common basis for Avr1b phenotypic variation. Mechanistically, it is plausible that some of those strains may have acquired sRNA-mediated silencing while others may have acquired H3K27me3-mediated silencing. It is also plausible that in some strains, silencing was initiated via small RNAs then maintained via H3K27me3 methylation.
Shan et al. (24) showed that when P6497 was crossed with an Avr1b-expressing strain (P7064 or P7076) the Avr1b allele derived from P6497 was re-activated in the heterozygous F1 progeny, but that silencing was re-established in F2 progeny that were homozygous for the P6497-derived allele. Those observations suggested that a recessive allele in P6497 was responsible for the silencing of Avr1b, and that the locus responsible for silencing (called Avr1b-2 by Shan et al. (24)) lay within a 125 kb region encompassing Avr1b itself. The findings of our current paper do not reveal whether the PRC2 complex is directly involved in the re-establishment of Avr1b silencing, or whether the role of the PRC2 complex is to maintain a silenced state that is established via a separate mechanism. The signal for re-establishment of silencing also remains unclear given the 81 kb of sequences encompassing Avr1b that are identical between P6497 and sc10. Interestingly, the H3K27me3 levels within an 800 bp region, located 8.3 kb downstream of Avr1b, were reduced in T34 and the corresponding transcripts were elevated in RNA-seq data. However, no ORFs, domains, repeats, motifs or features could be predicted with confidence. Why do H3K27me3 marks specifically accumulate at the Avr1b locus in silent strain P6497? In Drosophila, Polycomb response elements (PRE) have been identified as cis sites for Polycomb recruitment and provide sequence-specific ‘memory’ modules to maintain silent and active gene expression states (53). In contrast, in mammals, PRC2 and the H3K27me3 mark localize mainly to transcriptionally inactive regions rich in CpG dinucleotides (54,55). However, there have been no reports that such elements exist in Phytophthora. If a cis element is critical for H3K27me3 deposition, then that element might be the target of whatever trans-acting mechanism is controlled by the Avr1b-2 locus. In Arabidopsis, Polycomb-mediated silencing of FLOWERING LOCUS C (FLC) is initiated by the binding of the transcriptional repressor protein VAL1 to a DNA sequence in intron 1 of FLC. VAL1 recruits the histone deacetylase SAP18 together with other subunits of the apoptosis- and splicing-associated protein (ASAP) complex. Recruitment of the ASAP complex and histone deacetylation are thought to allow nucleation of PRC to FLC (56). Further work will be required to determine if a similar process controls the silencing of Avr1b.
The Avr1b gene in P. sojae strain P6497 exists in a state of natural silencing, allowing P6497 to evade defense surveillance mediated by the soybean Rps1b disease resistance gene (22; this study). Here, we propose a model in which P. sojae Avr1b effector gene silencing is maintained in P6497 by H3K27me3 methylation. In this model, the state of H3K27me3 at a specific genomic locus is a key regulator of effector gene expression. This example of natural epigenetic gene silencing is novel for oomycetes, and highlights how histone modifications can determine pathogen virulence type. This strategy could enable other pathogens to adapt to resistant host plants (Figure 4B). How broadly similar mechanisms regulate pathogen gene expression and contribute to pathogen adaptation to changes in the biotic and abiotic environment requires further investigation. Technologies to probe epigenetic changes at specific loci could form a component of accurate disease diagnoses in the future. Furthermore, our findings suggest that interfering with pathogen epigenetic processes could be a potential strategy for protecting plants from infection.
DATA AVAILABILITY
The Pacific Biosciences long read sequencing dataset obtained in this study is available from the Sequence Read Archive (SRA) under BioProject Number PRJNA524096. The RNA-seq dataset produced during this study were deposited into Gene Expression Omnibus under accession number No. GSE127206, The ChIP-seq dastaset produced during this study were deposited into Gene Expression Omnibus under accession number No. GSE127207.
Supplementary Material
ACKNOWLEDGEMENTS
We appreciate Dr Brent Kronmiller (Oregon State University), Prof. Yufeng Wu and Dr Wenwu Ye (Nanjing Agricultural University) for bioinformatics support. We thank Dr Tingting Gu and Ms. Hairong Xie (Nanjing Agricultural University) for ChIP-seq technical support. We also thank Dr Mark Gijzen (Agriculture and Agri-Food Canada), Prof. Sophien Kamoun (The Sainsbury Laboratory) and Dr Yufeng Fang (Duke University) for helpful discussions. Bioinformatic work station was supported by the Bioinformatics Center, Nanjing Agricultural University.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31721004, 31772144 to S.D. and Y.W.]; Fundamental Research Funds for Central Universities [JCQY201904, KYZ201925]; USDA National Institute for Food and Agriculture [2018-67013-28438 to B.T.]. Funding for open access charge: National Natural Science Foundation of China [31721004, 31772144]; Fundamental Research Funds for Central Universities [JCQY201904,KYZ201925].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tyler B.M., Tripathy S., Zhang X., Dehal P., Jiang R.H., Aerts A., Arredondo F.D., Baxter L., Bensasson D., Beynon J.L. et al.. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006; 313:1261–1266. [DOI] [PubMed] [Google Scholar]
- 2. Dong S., Raffaele S., Kamoun S.. The two-speed genomes of filamentous pathogens: waltz with plants. Curr. Opin. Genet. Dev. 2015; 35:57–65. [DOI] [PubMed] [Google Scholar]
- 3. Rouxel T., Grandaubert J., Hane J.K., Hoede C., van de Wouw A.P., Couloux A., Dominguez V., Anthouard V., Bally P., Bourras S. et al.. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun. 2011; 2:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daverdin G., Rouxel T., Gout L., Aubertot J.N., Fudal I., Meyer M., Parlange F., Carpezat J., Balesdent M.H.. Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathog. 2012; 8:e1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farrer R.A., Henk D.A., Garner T.W., Balloux F., Woodhams D.C., Fisher M.C.. Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet. 2013; 9:e1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L., Ouyang H.B., Fang Z.G., Zhu W., Wu E.J., Luo G.H., Shang L.P., Zhan J.. Evidence for intragenic recombination and selective sweep in an effector gene of Phytophthora infestans. Evol. Appl. 2018; 11:1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qutob D., Tedman-Jones J., Dong S., Kuflu K., Pham H., Wang Y., Dou D., Kale S.D., Arredondo F.D., Tyler B.M. et al.. Copy number variation and transcriptional polymorphisms of Phytophthora sojae RXLR effector genes Avr1a and Avr3a. PLoS One. 2009; 4:e5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gijzen M., Ishmael C., Shrestha S.D.. Epigenetic control of effectors in plant pathogens. Front. Plant Sci. 2014; 5:638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pais M., Yoshida K., Giannakopoulou A., Pel M.A., Cano L.M., Oliva R.F., Witek K., Lindqvist-Kreuze H., Vleeshouwers V., Kamoun S.. Gene expression polymorphism underpins evasion of host immunity in an asexual lineage of the Irish potato famine pathogen. BMC Evol. Biol. 2018; 18:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calo S., Shertz-Wall C., Lee S.C., Bastidas R.J., Nicolas F.E., Granek J.A., Mieczkowski P., Torres-Martinez S., Ruiz-Vazquez R.M., Cardenas M.E. et al.. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature. 2014; 513:555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basenko E.Y., Sasaki T., Ji L., Prybol C.J., Burckhardt R.M., Schmitz R.J., Lewis Z.A.. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6339–E6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qutob D., Chapman B.P., Gijzen M.. Transgenerational gene silencing causes gain of virulence in a plant pathogen. Nat. Commun. 2013; 4:1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kasuga T., Gijzen M.. Epigenetics and the evolution of virulence. Trends Microbiol. 2013; 21:575–582. [DOI] [PubMed] [Google Scholar]
- 14. Freitag M. Histone methylation by SET domain proteins in Fungi. Annu. Rev. Microbiol. 2017; 71:413–439. [DOI] [PubMed] [Google Scholar]
- 15. Schwartz Y.B., Pirrotta V.. Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 2008; 20:266–273. [DOI] [PubMed] [Google Scholar]
- 16. Margueron R., Reinberg D.. The Polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao R., Zhang Y.. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 2004; 15:57–67. [DOI] [PubMed] [Google Scholar]
- 18. Pasini D. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004; 23:4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Connolly L.R., Smith K.M., Freitag M.. The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet. 2013; 9:e1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chujo T., Scott B.. Histone H3K9 and H3K27 methylation regulates fungal alkaloid biosynthesis in a fungal endophyte-plant symbiosis. Mol. Microbiol. 2014; 92:413–434. [DOI] [PubMed] [Google Scholar]
- 21. Schotanus K., Soyer J.L., Connolly L.R., Grandaubert J., Happel P., Smith K.M., Freitag M., Stukenbrock E.H.. Histone modifications rather than the novel regional centromeres of Zymoseptoria tritici distinguish core and accessory chromosomes. Epigenet. Chromatin. 2015; 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wrather J.A., Koenning S.R.. Estimates of disease effects on soybean yields in the United States 2003 to 2005. J. Nematol. 2006; 38:173–180. [PMC free article] [PubMed] [Google Scholar]
- 23. de Wit P.J. Cladosporium fulvum effectors: weapons in the arms race with tomato. Annu. Rev. Phytopathol. 2016; 54:1–23. [DOI] [PubMed] [Google Scholar]
- 24. Shan W., Cao M., Leung D., Tyler B.M.. The Avr1b locus of Phytophthora sojae encodes an elicitor and a regulator required for avirulence on soybean plants carrying resistance gene Rps1b. Mol. Plant. Microbe Interact. 2004; 17:394–403. [DOI] [PubMed] [Google Scholar]
- 25. Cui L., Yin W., Dong S., Wang Y.. Analysis of polymorphism and transcription of the effector gene Avr1b in Phytophthora sojae isolates from China virulent to Rps1b. Mol. Plant Pathol. 2012; 13:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cui L., Yin W., Tang Q., Dong S., Zheng X., Zhang Z., Wang Y.. Distribution, pathotypes, and metalaxyl sensitivity of Phytophthora sojae from Heilongjiang and Fujian Provinces in China. Plant Dis. 2010; 94:881–884. [DOI] [PubMed] [Google Scholar]
- 27. Nagaki K., Talbert P.B., Zhong C.X., Dawe R.K., Henikoff S., Jiang J.. Chromatin immunoprecipitation reveals that the 180-bp satellite repeat is the key functional DNA element of Arabidopsis thaliana centromeres. Genetics. 2003; 163:1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y., Kikuchi S., Yan H., Zhang W., Rosenbaum H., Iniguez A.L., Jiang J.. Euchromatic subdomains in rice centromeres are associated with genes and transcription. Plant Cell. 2011; 23:4054–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fang Y., Cui L., Gu B., Arredondo F., Tyler B.M.. Efficient genome editing in the oomycete Phytophthora sojae using CRISPR/Cas9. Curr. Protoc. Microbiol. 2017; 44:21A.1.1–21A.1.26. [DOI] [PubMed] [Google Scholar]
- 30. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gotz S., Garcia-Gomez J.M., Terol J., Williams T.D., Nagaraj S.H., Nueda M.J., Robles M., Talon M., Dopazo J., Conesa A.. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008; 36:3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ye J., Zhang Y., Cui H., Liu J., Wu Y., Cheng Y., Xu H., Huang X., Li S., Zhou A. et al.. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018; 46:W71–W75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson N.R., Yeoh J.M., Coruh C., Axtell M.J.. Improved placement of multi-mapping small RNAs. G3 (Bethesda, Md.). 2016; 6:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chin C.S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E. et al.. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013; 10:563–569. [DOI] [PubMed] [Google Scholar]
- 41. Shrestha S.D., Chapman P., Zhang Y., Gijzen M.. Strain specific factors control effector gene silencing in Phytophthora sojae. PLoS One. 2016; 11:e0150530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shaver S., Casas-Mollano J.A., Cerny R.L., Cerutti H.. Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics. 2010; 5:301–312. [DOI] [PubMed] [Google Scholar]
- 43. Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G.. Genome regulation by polycomb and trithorax proteins. Cell. 2007; 128:735–745. [DOI] [PubMed] [Google Scholar]
- 44. Jiang R.H., Tyler B.M.. Mechanisms and evolution of virulence in oomycetes. Annu Rev. Phytopathol. 2012; 50:295–318. [DOI] [PubMed] [Google Scholar]
- 45. Tyler B.M., Gijzen M.. The Phytophthora sojae genome sequence: foundation for a revolution. 2014; 133–157.
- 46. van West P., Kamoun S., van ’t Klooster J.W., Govers F.. Internuclear gene silencing in Phytophthora infestans. Mol. Cell. 1999; 3:339–348. [DOI] [PubMed] [Google Scholar]
- 47. van West P., Shepherd S.J., Walker C.A., Li S., Appiah A.A., Grenville-Briggs L.J., Govers F., Gow N.A.. Internuclear gene silencing in Phytophthora infestans is established through chromatin remodelling. Microbiology. 2008; 154:1482–1490. [DOI] [PubMed] [Google Scholar]
- 48. Vetukuri R.R., Avrova A.O., Grenville-Briggs L.J., Van West P., Soderbom F., Savenkov E.I., Whisson S.C., Dixelius C.. Evidence for involvement of Dicer-like, Argonaute and histone deacetylase proteins in gene silencing in Phytophthora infestans. Mol. Plant Pathol. 2011; 12:772–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen H., Shu H., Wang L., Zhang F., Li X., Ochola S.O., Mao F., Ma H., Ye W., Gu T. et al.. Phytophthora methylomes are modulated by 6mA methyltransferases and associated with adaptive genome regions. Genome Biol. 2018; 19:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fahlgren N., Bollmann S.R., Kasschau K.D., Cuperus J.T., Press C.M., Sullivan C.M., Chapman E.J., Hoyer J.S., Gilbert K.B., Grunwald N.J. et al.. Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS One. 2013; 8:e77181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vetukuri R.R., Åsman A.K., Tellgren-Roth C., Jahan S.N., Reimegård J., Fogelqvist J., Savenkov E., Söderbom F., Avrova A.O., Whisson S.C. et al.. Evidence for small RNAs homologous to effector-encoding genes and transposable elements in the oomycete Phytophthora infestans. PLoS One. 2012; 7:e51399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jia J., Lu W., Zhong C., Zhou R., Xu J., Liu W., Gou X., Wang Q., Yin J., Xu C. et al.. The 25–26 nt small RNAs in Phytophthora parasitica are associated with efficient silencing of homologous endogenous genes. Front. Microbiol. 2017; 8:773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steffen P.A., Ringrose L.. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 2014; 15:340–356. [DOI] [PubMed] [Google Scholar]
- 54. Tanay A., O’Donnell A.H., Damelin M., Bestor T.H.. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mendenhall E.M., Koche R.P., Truong T., Zhou V.W., Issac B., Chi A.S., Ku M., Bernstein B.E.. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010; 6:e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Questa J.I., Song J., Geraldo N., An H., Dean C.. Arabidopsis transcriptional repressor VAL1 triggers Polycomb silencing at FLC during vernalization. Science. 2016; 353:485–488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Pacific Biosciences long read sequencing dataset obtained in this study is available from the Sequence Read Archive (SRA) under BioProject Number PRJNA524096. The RNA-seq dataset produced during this study were deposited into Gene Expression Omnibus under accession number No. GSE127206, The ChIP-seq dastaset produced during this study were deposited into Gene Expression Omnibus under accession number No. GSE127207.