

Abstract
The chemokine CXCL10 and its receptor CXCR3 have been demonstrated to be implicated in cancer cell proliferation and metastasis. CXCR3 has three splice variants: CXCR3A, CXCR3B and CXCR3-alt. CXCR3A and B serve multiple roles in the growth and invasiveness of a number of cancer types. However, the roles of CXCR3 isoforms in colorectal cancer (CRC) cells remain unclear. In the current study, the effects of CXCL10 and CXCR3 isoforms on proliferation and invasion of CRC cells was examined. Proliferation and invasiveness of the CRC cell line HCT116, which were transfected with CXCR3A or CXCR3B in the presence of CXCL10, were evaluated in vitro using MTT, scratch wound healing and transwell assays. MTT assay indicated that regardless of the presence or absence of CXCL10, the proliferative ability of CXCR3A-transfected HCT116 cells was enhanced compared with blank and mock cells. Scratch wound healing and transwell assays indicated that invasiveness of CXCR3A-transfected cells was greater compared with blank and mock cells. However, HCT116 cells transfected with CXCR3B did not exhibit changes in their proliferative or invasive ability. mRNA expression of MMP9, which is associated with signaling downstream of the CXCL10/CXCR3A pathway, was increased 4-fold in CXCR3A-transfected HCT116 cells compared with control cells. The results of the present study indicated that CXCL10-enhanced proliferation and invasiveness of the CRC cell line HCT116 was likely mediated by CXCR3A, but not by CXCR3B.
Keywords: colorectal cancer, CXCR3 isoform, cellular proliferation, cellular invasiveness, cellular migration
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors and is the fourth leading cause of cancer-related deaths worldwide (1). While the 5-year survival rate of patients with localized CRC is 87–91%, prognosis for patients in distant stages is markedly poor, with 5-year-survival rate of 10–14% (2). Prognosis for patients with advanced stages of CRC has recently been improved by newly-developed molecular-targeted drugs and anticancer agents (3). However, satisfactory outcomes have not yet been achieved in patients with recurrent CRC. Novel therapeutic strategies targeting alternative mechanisms of cancer proliferation and metastasis are urgently needed to overcome advanced stages of CRC. For this, we need to explore the molecular mechanisms driving the invasive front of CRC, which likely plays a central role in the invasiveness of cancer cells in advanced stages of cancer.
In our previous study, we have shown that expression of C-X-C motif chemokine ligand (CXCL)9, 10 and 11, combined with other factors related to epithelial mesenchymal transition (EMT), is elevated in the invasive front of CRC tumor tissues (4). CXCL9-11 participate in numerous biological processes including leukocyte trafficking, immune response, and cellular proliferation. CXCL10 is particularly known to promote the proliferation of several carcinoma cell lines (5–7). Clinical studies have shown an association between expression levels of CXCL10 and survival of patients with colorectal cancer (8–11). CXCL9-11 exert their biological effects via common receptor called C-X-C motif chemokine receptor (CXCR)3. CXCR3 is a member of the G protein-coupled receptor family, and is expressed in monocytes and leukocytes (12). Previous studies have reported that CXCR3 is expressed in CRC (9). CXCR3 is also involved in the proliferation and metastasis of renal, melanoma, and breast cancer cells. Three splice variants of CXCR3 (CXCR3A, CXCR3B, and CXCR3-alt) have been identified in humans (13,14); these splice variants play different roles in various cancer cells. CXCR3A promotes the invasiveness and metastasis of gastric- and renal-cancer cells (15,16), while CXCR3B inhibits the invasiveness and migration of prostate-cancer cells (17). The function of CXCR3-alt remains unknown, and functional diversity of CXCR3A and CXCR3B in CRC is also undetermined.
In this study, we examined the diverse roles of CXCR3A, CXCR3B, and the CXCL10-CXCR3 signaling pathway in the proliferation and invasiveness of CRC cells in vitro.
Materials and methods
Cell culture
Human CRC cell line HCT116 was obtained from American Tissue and Cell Culture. HCT116 cells were cultured in Dulbecco's modified Eagles medium (DMEM; Gibco; Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.) and antibiotic-antimycotic solution (100X; Gibco) at 37°C and 5% CO2.
Transfection of CXCR3
For transfection of CXCR3, HCT116 cells were seeded into a 6-well plate at a density of 3×105 cells/well. After 16 h, the cells were transfected with 500 ng CXCR3 (Myc-DDK-tagged) transcript variant A plasmid DNA or CXCR3 (Myc-DDK-tagged) transcript variant B plasmid DNA (both from Origene Technologies) using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) per manufacturer's protocol. After 48 h of transfection, cells were cultured in DME medium supplemented with 600 µg/ml G418 for 1 week. These cells were designated as HCT116-CXCR3A and HCT116-CXCR3B, respectively. Similarly, HCT116 cells were transfected with pCMV6 mammalian vector, designated as mock cells, and used as controls. Non-transfected cells, designated as blank cells, were used as additional controls.
Sequence analysis of inserted genome
After HCT116 cells were transfected with CXCR3A or CXCR3B plasmid DNA, insertion was confirmed by DNA sequencing. DNA was extracted from each cell line via QIAamp DNA Mini Prep Kit (Qiagen) and labeled using Big Dye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Inc.); this was followed by direct sequencing on an ABI Prism 3730×l Analyzer (Thermo Fisher Scientific). All kits were used per instructions of respective manufacturers.
Western blot analysis
To examine the expression of total CXCR3 in transfected cells, we performed western blotting using antibodies that recognize both CXCR3A and 3B. HCT116-CXCR3A, HCT116-CXCR3B, mock, and blank cells were lysed in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Inc.) and then centrifuged at 23,000 g for 15 min at 4°C. Supernatants were collected, and protein concentration in the supernatants was assessed using Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Inc.). Lysates (30 µg total protein per lysate) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were then transferred to 0.2 µm polyvinylidene difluoride (PVDF) membranes (EMD Millipore,). The membranes were blocked in Tris-buffered saline containing Tween-20 (TBST) and 5% skim milk at 25°C for 1 h. Subsequently, the membranes were incubated with rabbit anti-human antibodies against CXCR3 (cat. ab154845, 1:1,500; Abcam) overnight at 4°C, and then with horseradish peroxidase-labeled goat anti-rabbit secondary antibody at 25°C for 1 h. The membranes were then washed thrice (15 min per wash) using TBST. Signals were enhanced using the ECL Plus Western Blotting System (Perkin-Elmer) and detected via LAS-4000 Luminescent Image Analyzer. Anti-β-actin (cat. no. 8H10D10, 1:1,500; Cell Signaling Technology) was used as loading control.
RNA preparation
Total RNA was extracted from each cell line using RNA Mini Prep Kit (Qiagen) according to the manufacturer's instructions. The concentration and purity of extracted RNA were evaluated using NanoDrop (Thermo Fisher Scientific, Inc.). To assess the quality of extracted RNA, RNA integrity number (RIN) was calculated using Agilent 2100 Bioanalyzer (Agilent Technologies).
Reverse transcription (RT)-PCR
RT-PCR was performed to examine the expression of CXCR3A and CXCR3B mRNA using specific primers sets. One microgram purified total RNA was used for cDNA synthesis via Affinity Script QPCR cDNA Synthesis Kit (Agilent Technologies) utilized per manufacturer's instructions. RT-PCR was performed using Takara Ex Taq (Takara Bio, Inc.) using the following primers, designed specifically for amplification of CXCR3A or CXCR3B mRNA: CXCR3A forward, 5′-ccatggtccttgaggtgag-3′ and reverse, 5′-tccatagtcataggaagagctgaa-3′; CXCR3B forward, 5′-ttgaggaagtacggccctg-3′ and reverse, 5′-tgagcagctcctcctataac-3′; GAPDH forward, 5′-agccacatcgctcagacac-3′ and reverse, 5′-gcccaatacgaccaaatcc-3′.
Conditions for PCR were as follows: 95°C for 5 min, and then 25 cycles at 95°C for 10 sec, 58°C for 30 sec, and 72°C for 30 sec. Expression level of GAPDH mRNA was used as internal control.
MTT assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to examine the involvement of CXCL10/CXCR3 signaling in the proliferation of CRC cells. While CXCR3 has three ligands (CXCL9, 10 and 11), CXCL10 was most frequently examined in previous studies. Furthermore, the level of CXCL10 expression is associated with poor prognosis in patients with CRC. Therefore, CXCL10 was used in our present study as a ligand for CXCR3.
In a preliminary study, we used qPCR to examine the expression of CXCL10 mRNA in HCT116 cells; however, the expression level of CXCL10 was below the detection threshold. Therefore, we cultured the cell lines in the presence of CXCL10 to enhance the activity of CXCR3A/3B. The four types of cells were separately seeded into 24-well plate at a density of 5×104 cells/well. After 16 h, the cells were cultured with or without CXCL10 (100 ng/ml) for 24 h, and MTT was added for final 3 h before measuring. Prior to conducting measurements, the media were removed and isopropanol was added to all the wells. Absorbance was measured using a microplate reader (Hitachi SH-9000Lab; Hitachi) at 570 nm. All experiments were repeated three times.
Scratch wound healing assay
In order to examine CXCL10/CXCR3 signaling at the invasive front of tumor tissue, we performed a scratch wound healing assay as described previously with modifications (16). Each cell line was seeded into a 6-cm dish at a density of 8×105 cells/well. After 36 h, the cell sheets were scratched with a pipette tip (1,000-µl long filter tip; Watson) and then treated with CXCL10 (100 ng/ml) for 24 h. After 0 and 24 h, three different areas of the scratched cell sheets were observed under microscopy. Wounded areas (the areas with no attached cells) were measured using ImageJ software, and rate of migration was calculated as a proportion of the areas at 24/0 h. All experiments were repeated three times.
Transwell assay
The role of CXCL10/CXCR3 signaling in cellular invasiveness was examined using a Transwell assay performed via CytoSelect 24-Well Cell Invasion Assay Kit (Cell Biolabs). A Boyden chamber was used for the assay. Cells from each of the four cell lines were added to the upper chamber and incubated overnight. The medium in the upper chamber was then changed to Opti-minimum essential media, and CXCL10 (100 ng/ml) was added to the medium contained in the lower chamber. After 24 h, cells attached to the lower surface of the membrane were stained with Cell Staining Solution supplied with the CytoSelect 24-Well Cell Invasion Assay Kit. Stained cells were lysed in elution buffer, and each lysate was placed into 96-well plate at 200 µl/well. Absorbance of the lysates was measured using a microplate reader (Hitachi SH-9000Lab) at 560 nm. This assay was repeated six times.
Expression of MMP9 mRNA
CXCR3 potentiates the invasiveness and migration of various cancer cells by upregulating the expression of MMP9, a protein downstream of the ERK1/2 pathway via CXCL10-CXCR3A signaling. In order to confirm the effect of CXCR3A on cancer cell invasiveness and migration, we analyzed the expression of MMP9 in HCT116-CXCR3A cells. Total RNA was extracted from HCT116-CXCR3A cells and controls (blank and mock cells) and used to synthesize cDNA via Affinity Script QPCR cDNA Synthesis Kit. GAPDH was used as the housekeeping gene. Detection was performed using LAS-4000 (GE Healthcare), and band intensity was calculated using ImageQuant TL software (GE Healthcare). These experiments were repeated three times.
Statistical analysis
All data were expressed as mean ± standard deviation of the mean. Kruskal-Wallis test was used to evaluate differences among the groups, and Bonferroni post hoc test was used for multiple comparisons to evaluate the differences between two groups. All statistical analyses were performed using SPSS version 2.0 (IBM, Corp.). P<0.05 was considered statistically significant.
Results
Expression of CXCR3 in transfected HCT116 cells
Using DNA sequencing, we confirmed that the plasmid had been successfully inserted into each cell line (data not shown). CXCR3B Expression of CXCR3A or CXCR3B in transfected cells was then confirmed via western blotting and RT-PCR. Total CXCR3 expression at the protein level was markedly higher in HCT116-CXCR3A and CXCR3B cells than in blank and mock cells (Fig. 1A). RT-PCR was used to examine gene expression of each isoform. Our results show that CXCR3A mRNA was only expressed in HCT116-CXCR3A cells, but not in blank, mock, or HCT116-CXCR3B cells (Fig. 1B). Similarly, CXCR3B mRNA was only expressed in HCT116-CXCR3B cells. These four cell lines were used in subsequent experiments.
Figure 1.
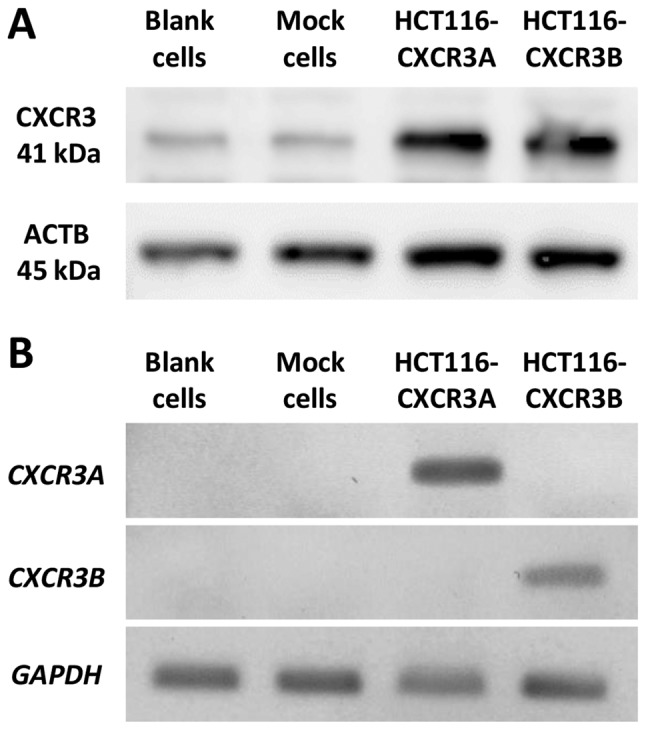
Expression of CXCR3 in transfected HCT116. (A) Western blot analysis of total CXCR3. ACTB was used as an internal control. (B) Reverse-transcription PCR analysis of CXCR3A and 3B mRNA. GAPDH mRNA was used as an internal control.
Proliferation of CXCR3-transfected cells
We first used an MTT assay to examine whether overexpression of CXCR3A or CXCR3B can promote proliferation of human colorectal cancer cells. After 24 h without CXCL10 treatment, HCT116-CXCR3A cell proliferation was moderately enhanced by approximately 1.1-fold compared to the proliferation of blank and mock cells, whereas HCT116-CXCR3B cell proliferation was reduced by approximately 20% compared to that in blank and mock control cells (Fig. 2A). The proliferation of HCT116-CXCR3A cells cultured in media containing CXCL10 for 24 h was enhanced by approximately 1.5-fold compared with that of blank cells and mock cells, and the difference from mock cells narrowly missed the set threshold for statistical significance (P=0.076). Conversely, the proliferation of HCT116-CXCR3B cells was similar to that of blank and mock cells (Fig. 2B).
Figure 2.

Proliferative properties of HCT116-CXCR3A and 3B analyzed using MTT assay. (A) Without treatment with CXCL10, the proliferation of HCT116-CXCR3A cells was moderately enhanced compared with control cells. (B) In the presence of CXCL10, proliferation of HCT116-CXCR3A cells was significantly greater compared with blank and mock cells at 24 h. P=0.5 and P=0.076 compared with blank and mock cells, respectively.
Scratch wound healing assay
A scratch wound healing assay is used to assess cellular invasiveness and proliferative ability by assessing the state of the scratched areas 24 h after induction of the wound. Scratched areas in the blank and mock cell sheets, assessed at 24 h after scratching, were reduced to 88 and 80%, respectively, compared with those assessed at 0 h. The scratched area of the HCT116-CXCR3A cell sheet, assessed at 24 h, was reduced to 40% of that assessed at 0 h; this reduction in the wounded area was significantly greater than those shown by blank and mock cell sheets, and the difference from blank cells was statistically significant (P=0.013). The scratched area on the HCT116-CXCR3B cell sheet was similar to that of blank and mock cells (Fig. 3).
Figure 3.

Scratch wound healing assay examining HCT116-CXCR3A and HCT116-CXCR3B cells: (A) Upper: Scratched cell sheet at 0 h; lower: Cells treated with CXCL10 for 24 h. (B) The percentage of scratched area at 24 h compared with 0 h. The scratched area was reduced, showing enhanced invasiveness of HCT116-CXCR3A cells compared with control cells. *P=0.013 compared with blank cells.
Transwell assay
In the Transwell assay, the number of cells attached to the lower surface of the membrane reflects the invasiveness of the cells. The number of invasive HCT116-CXCR3A cells was increased approximately 2-fold compared with those of blank and mock cells, and the difference from blank cells was statistically significant (P=0.005) (Fig. 4). In contrast, the number of invasive HCT116-CXCR3B cells was similar to those of blank and mock cells.
Figure 4.
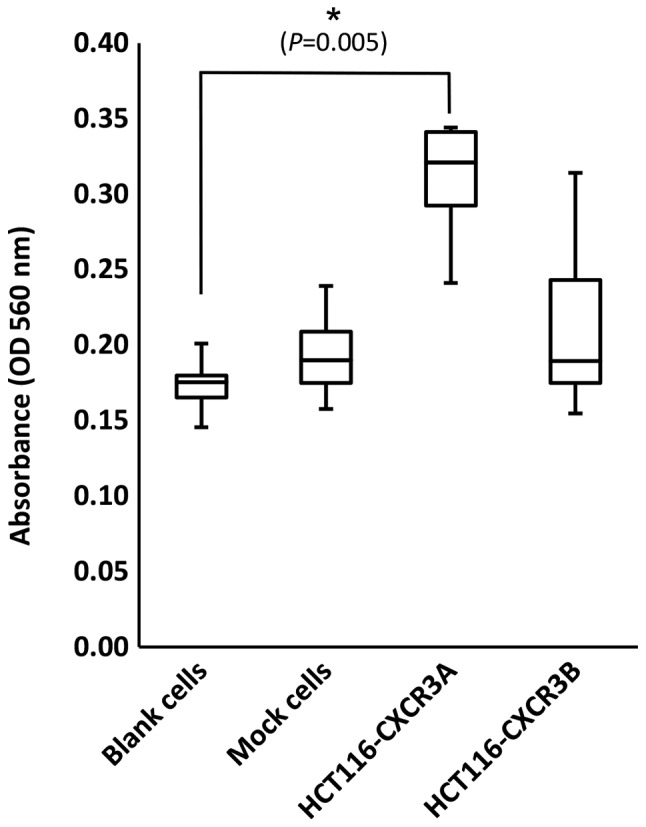
Transwell assay examining HCT116-CXCR3A and HCT116-CXCR3B cells. Invasiveness of HCT116-CXCR3A cells was enhanced in the presence of CXCL10 compared with blank and mock cells. *P=0.005 compared with blank cells.
Expression of MMP9
To examine the status of CXCR3A downstream molecules, we investigated mRNA expression of MMP9, which was reported to be involved in ERK1/2 signaling in human CRC cell lines. MMP9 mRNA expression in HCT116-CXCR3A cells was 4-fold higher than that in blank or mock cells, and the difference from mock cells was statistically significant (P=0.019) (Fig. 5).
Figure 5.
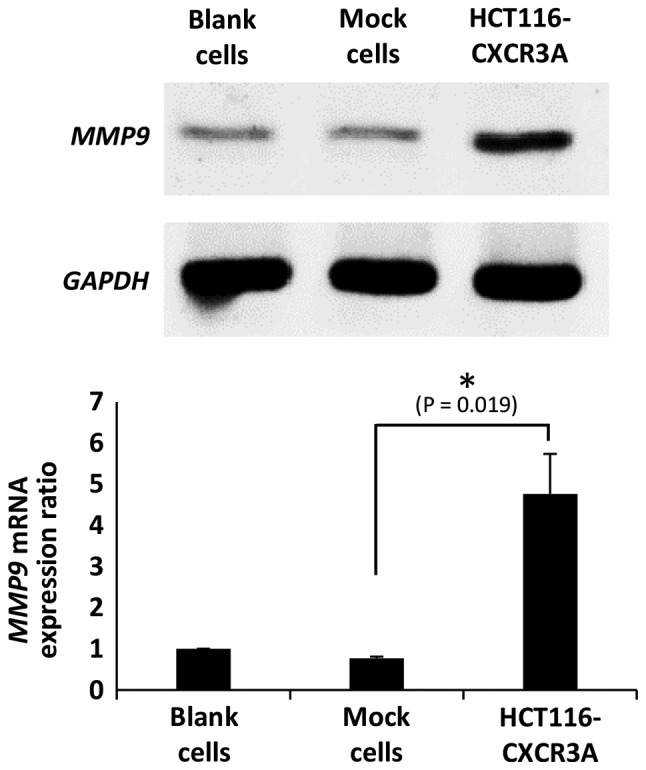
Expression of MMP9 mRNA in HCT116-CXCR3A cells. HCT116 cells transfected with CXCR3A showed significantly enhanced expression of MMP9 mRNA compared with control cells. *P=0.019 compared with mock cells.
Discussion
In our previous study, we demonstrated that mRNA expression of CXCL9, 10 and 11 was upregulated at the invasive front of CRC tissue (4), suggesting that these chemokines play crucial roles in tumor development during CRC. Receptors, expressed by cancer cells, can determine how these cells are affected by chemokines. The role of one such receptor, CXCR3, has not been fully investigated in CRC development, particularly with respect to differential properties of CXCR3 subtypes. Therefore, in this study, we examined the function of two CXCR3 variants (CXCR3A and CXCR3B) in CRC cells.
CXCR3 is involved in the proliferation and metastasis of renal, melanoma, and breast cancer cells. CXCR3 expression, which is upregulated in cancer tissues and metastatic carcinoma compared with that in normal tissues, is correlated with poor prognosis in patients with renal, melanoma, gastric, and breast cancer (18–21). Molecular analysis has shown that CXCR3 promotes tumor progression and metastasis by regulating proliferative, migratory, and invasive abilities in these cancer cells (6,16,20,22). Ma et al has shown that in a murine model of metastatic breast cancer, small-molecular-weight antagonists or siRNAs inhibit metastatic spread into lungs; this anti-metastatic effect, however, is compromised by depletion of natural killer cells in these mice (21,23).
CXCR3A and CXCR3B play different roles in the growth and invasiveness of several types of cancer cells. Wu et al has shown that overexpression of CXCR3A promotes proliferation and invasiveness, while overexpression of CXCR3B ameliorates proliferation and invasiveness, of prostate cancer cells (17). Shen and Cao used siRNA interference to show that proliferative and invasive abilities of prostate cancer cells are reduced by suppressing the expression of CXCR3A and enhanced by suppressing the expression of CXCR3B (24).
In our present study, we have shown that overexpression of CXCR3A promoted the proliferation of HCT116 colorectal cancer cells. In particular, the proliferation of HCT116 cells exposed to CXCL10 was enhanced compared with that of untreated control HCT116-CXCR3A cells. This indicates that CXCL10-CXCR3A signaling plays an important role in the proliferative ability of CRC cells. Conversely, overexpression of CXCR3B had no effect on the proliferation of HCT116 cells. These results suggest that CXCR3A, but not CXCR3B, was responsible for CXCL10-mediated enhancement of proliferation in CRC cells. Scratch wound healing assays showed that recovery of the wounded area was promoted in HCT116-CXCR3A cells compared with that in non-transfected cells. Transwell assays showed that the number of invasive HCT116-CXCR3A cells was increased compared to those of blank cells. These results indicate that overexpression of CXCR3A contributed to the proliferation and invasiveness of HCT116 cells. These results also agree with those obtained in previous studies analyzing carcinomas, suggesting that CXCR3A, in conjunction with CXCL10, plays crucial roles in the proliferation and invasiveness of colorectal cancer cells. Conversely, previous studies indicate that CXCR3B overexpression did not affect cellular invasiveness. Whereas these previous studies indicate that CXCR3B inhibits the migration of cancer cells, we did not observe this effect of CXCR3B on CRC cells in our present study. This discrepancy may stem from differences in the tumor microenvironment of different types of cancer.
Previous studies have shown that CXCR3A induces cancer cell proliferation and invasiveness by activating the ERK1/2 and PI3K pathway via Gαi or Gαq (15,25,26). In addition, CXCR3A promotes cancer cell invasiveness and migration by activating Gαq. CXCR3B activates Gαs by promoting enhanced cAMP concentrations; this cascade inhibits the migratory ability of cancer cells (17). Shen and Cao reported that MMP activity is involved in CXCR3A signaling downstream of PI3K in prostate cancer (24). Zipin-Roitman et al reported that CXCL10 promotes the invasiveness of human colorectal cancer cells by upregulating MMP9 expression (27). In our present study, we have shown that CRC cells transfected with CXCR3A showed enhanced expression of MMP9. This finding agrees with results obtained in previous studies, showing that MMPs play crucial roles in promoting the invasiveness and proliferation of cancer cells via CXCR3A.
Our present study has several limitations. In this study, we used only CXCL10 as a ligand of CXCR3 for analysis, and did not examine the functions of CXCR3 using CXCL9 and CXCL11. These ligands may act differently on CXCR3 than did CXCL10. Additionally, we used only the HCT116 line of CRC cells for functional analysis. Therefore, our results may have been affected by the individual characteristics of this cell line. Future studies will need to determine the functions of CXCR3 using other ligands and different CRC lines. Furthermore, among the molecules involved in the CXCL10-CXCR signaling pathway in CXCR3A-transfected cells, we only examined the expression of MMP9 mRNA. In our future studies, we will examine the expression of other proteins, such as Gαi and ERK1/2 involved in CXCR3-mediated proliferation and invasiveness of CRC cells. Phosphorylation of ERK1/2 should be also investigated to clarify the effect of ERK1/2-MMP9 signals. In addition, it would be interesting to examine the roles of CXCR3 proteins by increasing or decreasing their expression. Actually, we attempted to perform an siRNA-inhibition experiment with the transfected cells; however, the results we obtained were highly variable, probably due to insufficient effect of the synthesized siRNAs. Future studies should include such experiments with a modified experimental design.
In conclusion, our results indicate that CXCL10-induced proliferation and invasiveness of colorectal cancer cells was likely mediated by CXCR3A, but not by CXCR3B. Further analysis of proteins will provide additional insights into the molecular mechanisms involved in CRC development via CXCR3 and CXCL pathways.
Acknowledgements
The authors would like to thank Dr Kanae Karita from the Department of Public Health, Kyorin University School of Medicine, for her assistance in statistical analysis.
Glossary
Abbreviations
- CRC
colorectal cancer CXC
- C-X-C motif chemokine ligand CXCR
C-X-C motif chemokine receptor
- siRNA
small interfering RNA
- MMP
matrix metalloproteinase
Funding
The present work was supported by the Japan Society for the Promotion of Science (grant no. 21791305).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
EN, TW and MS made substantial contributions to conception and design. EN, TK and TM made substantial contributions to acquisition of data. EN, TK, HO and KO made substantial contributions to analysis and interpretation of data. In addition, all authors have been involved in drafting the manuscript or revising it critically for intellectual content, given final approval of version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–1502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Kopetz S, Chang GJ, Overman MJ, Eng C, Sargent DJ, Larson DW, Grothey A, Vauthey JN, Nagorney DM, McWilliams RR. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–3683. doi: 10.1200/JCO.2008.20.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi T, Masaki T, Nozaki E, Sugiyama M, Nagashima F, Furuse J, Onishi H, Watanabe T, Ohkura Y. Microarray analysis of gene expression at the tumor front of colon cancer. Anticancer Res. 2015;35:6577–6581. [PubMed] [Google Scholar]
- 5.Datta D, Flaxenburg JA, Laxmanan S, Geehan C, Grimm M, Waaga-Gasser AM, Briscoe DM, Pal S. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: Relevance for the development of human breast cancer. Cancer Res. 2006;66:9509–9518. doi: 10.1158/0008-5472.CAN-05-4345. [DOI] [PubMed] [Google Scholar]
- 6.Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–1549. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodnar RJ, Yates CC, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ Res. 2006;98:617–625. doi: 10.1161/01.RES.0000209968.66606.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wightman SC, Uppal A, Pitroda SP, Ganai S, Burnette B, Stack M, Oshima G, Khan S, Huang X, Posner MC, et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br J Cancer. 2015;113:327–335. doi: 10.1038/bjc.2015.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai M, Chen X, Ba YI. CXCL10/CXCR3 overexpression as a biomarker of poor prognosis in patients with stage II colorectal cancer. Mol Clin Oncol. 2016;4:23–30. doi: 10.3892/mco.2015.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toiyama Y, Fujikawa H, Kawamura M, Matsushita K, Saigusa S, Tanaka K, Inoue Y, Uchida K, Mohri Y, Kusunoki M. Evaluation of CXCL10 as a novel serum marker for predicting liver metastasis and prognosis in colorectal cancer. Int J Oncol. 2012;40:560–566. doi: 10.3892/ijo.2011.1247. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Z, Xu Y, Cai S. CXCL10 expression and prognostic significance in stage II and III colorectal cancer. Mol Biol Rep. 2010;37:3029–3036. doi: 10.1007/s11033-009-9873-z. [DOI] [PubMed] [Google Scholar]
- 12.Groom JR, Luster AD. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billottet C, Quemener C, Bikfalvi A. CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim Biophys Acta. 2013;1836:287–295. doi: 10.1016/j.bbcan.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol. 2004;173:6234–6240. doi: 10.4049/jimmunol.173.10.6234. [DOI] [PubMed] [Google Scholar]
- 15.Yang C, Zheng W, Du W. CXCR3A contributes to the invasion and metastasis of gastric cancer cells. Oncol Rep. 2016;36:1686–1692. doi: 10.3892/or.2016.4953. [DOI] [PubMed] [Google Scholar]
- 16.Utsumi T, Suyama T, Imamura Y, Fuse M, Sakamoto S, Nihei N, Ueda T, Suzuki H, Seki N, Ichikawa T. The association of CXCR3 and renal cell carcinoma metastasis. J Urol. 2014;192:567–574. doi: 10.1016/j.juro.2014.01.100. [DOI] [PubMed] [Google Scholar]
- 17.Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol Cancer. 2012;11:3. doi: 10.3390/cancers11010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jöhrer K, Zelle-Rieser C, Perathoner A, Moser P, Hager M, Ramoner R, Gander H, Höltl L, Bartsch G, Greil R, Thurnher M. Up-regulation of functional chemokine receptor CCR3 in human renal cell carcinoma. Clin Cancer Res. 2005;11:2459–2465. doi: 10.1158/1078-0432.CCR-04-0405. [DOI] [PubMed] [Google Scholar]
- 19.Jacquelot N, Enot DP, Flament C, Vimond V, Blattner C, Pitt JM, Yamazaki T, Roberti MP, Daillère R, Vétizou M, et al. Chemokine receptor patterns in lymphocytes mirror metastatic spreading in melanoma. J Clin Invest. 2016;126:921–97. doi: 10.1172/JCI80071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Wu J, Wang T, Zhang X, Liu D. CXCL10/CXCR3 axis promotes the invasion of gastric cancer via PI3K/AKT pathway-dependent MMPs production. Biomed Pharmacother. 2016;82:479–488. doi: 10.1016/j.biopha.2016.04.069. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Norsworthy K, Kundu N, Rodgers WH, Gimotty PA, Goloubeva O, Lipsky M, Li Y, Holt D, Fulton A. CXCR3 expression is associated with poor survival in breast cancer and promotes metastasis in a murine model. Mol Cancer Ther. 2009;8:490–498. doi: 10.1158/1535-7163.MCT-08-0485. [DOI] [PubMed] [Google Scholar]
- 22.Kawada K, Sonoshita M, Sakashita H, Takabayashi A, Yamaoka Y, Manabe T, Inaba K, Minato N, Oshima M, Taketo MM. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. 2004;64:4010–4017. doi: 10.1158/0008-5472.CAN-03-1757. [DOI] [PubMed] [Google Scholar]
- 23.Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O, Johnson MG, Medina JC, Collins TL, Fulton AM. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006;66:7701–7707. doi: 10.1158/0008-5472.CAN-06-0709. [DOI] [PubMed] [Google Scholar]
- 24.Shen D, Cao X. Potential role of CXCR3 in proliferation and invasion of prostate cancer cells. Int J Clin Exp Pathol. 2015;8:8091–8098. [PMC free article] [PubMed] [Google Scholar]
- 25.Aksoy MO, Yang Y, Ji R, Reddy PJ, Shahabuddin S, Litvin J, Rogers TJ, Kelsen SG. CXCR3 surface expression in human airway epithelial cells: Cell cycle dependence and effect on cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L909–L918. doi: 10.1152/ajplung.00430.2005. [DOI] [PubMed] [Google Scholar]
- 26.Ji R, Lee CM, Gonzales LW, Yang Y, Aksoy MO, Wang P, Brailoiu E, Dun N, Hurford MT, Kelsen SG. Human type II pneumocyte chemotactic responses to CXCR3 activation are mediated by splice variant A. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1187–L1196. doi: 10.1152/ajplung.00388.2007. [DOI] [PubMed] [Google Scholar]
- 27.Zipin-Roitman A, Meshel T, Sagi-Assif O, Shalmon B, Avivi C, Pfeffer RM, Witz IP, Ben-Baruch A. CXCL10 promotes invasion-related properties in human colorectal carcinoma cells. Cancer Res. 2007;67:3396–3405. doi: 10.1158/0008-5472.CAN-06-3087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.