

Summary
A wide variety of pathogens target chemokine signaling networks in order to disrupt host immune surveillance and defense. Herein, we report a structural and mutational analysis of rodent herpesvirus Peru encoded R17, a potent chemokine inhibitor that sequesters CC- and C-chemokines with high affinity. R17 consists of a pair of β-sandwich domains linked together by a bridging sheet, which form an acidic binding cleft for the chemokine CCL3 on the opposite face of a basic surface cluster that binds glycosaminoglycans. R17 promiscuously engages chemokines primarily through the same N-loop determinants used for host receptor recognition while residues located in the chemokine 40’s-loop drive kinetically stable complex formation. The core fold adopted by R17 is unexpectedly similar to that of the M3 chemokine decoy receptor encoded by MHV-68, although strikingly neither the location of ligand engagement nor the stoichiometry of binding is conserved suggesting that their functions evolved independently.
In Brief
Lubman and Fremont describe the atomic structure of the herpesvirus-encoded chemokine binding protein R17 alone and in complex with a high-affinity ligand, CCL3. The study offers novel insights into the conserved and unique mechanisms that different pathogens use to undermine host chemokine signaling networks.
Graphical Abstract
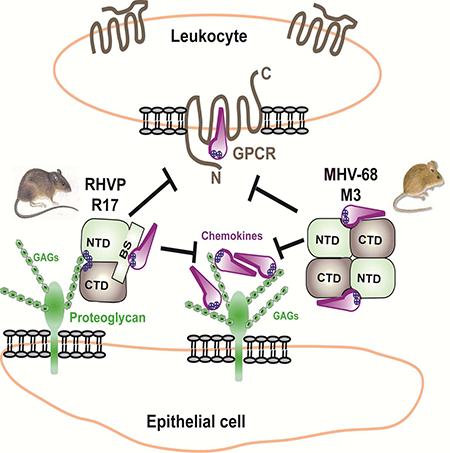
Introduction
Chemokines are a group of small cytokines that orchestrate host defense against microorganisms in vertebrates (Esche et al., 2005; Gerard and Rollins, 2001). Pro-inflammatory chemokines play an essential role in the clearance of a broad array of pathogens through the recruitment of effector leukocytes (Luster, 1998). Chemokines establish gradients through specific interactions with glycosaminoglycans (GAGs), and direct target cell migration and activation by binding to G-protein coupled chemokine receptors (Allen et al., 2007; Handel et al., 2005). Chemokine networks are characterized by ligand-receptor promiscuity, antagonistically acting ligands and non-signaling decoy receptors (Allen et al., 2007; Fernandez and Lolis, 2002; Handel and Lau, 2004). All chemokines adopt a similar fold consisting of an extended N terminus followed by a long flexible loop (N-loop), a three stranded β-sheet and C-terminal α-helix (Fernandez and Lolis, 2002). The structural determinants of chemokine-GPCR recognition have recently been illuminated by studies of CXCR4 in complex with a herpesvirus-encoded chemokine, and CX3CL1 in complex with a herpesvirus-encoded chemokine receptor (Burg et al., 2015; Qin et al., 2015). Receptor activation is thought to occur in several steps, where initial binding of the chemokine N-loop causes conformational changes in the receptor allowing the N-terminal residues of the chemokine to insert between transmembrane helices of the GPCR (Kufareva et al., 2015).
Pathogens undermine host chemokine signaling networks using a number of different strategies. Large DNA viruses, such as herpes- and pox- viruses, encode versions of chemokines, chemokine receptors, and unique soluble chemokine binding proteins capable of sequestering host chemokines with distinct specificity (Alcami, 2003; Alcami and Lira, 2010; Epperson et al., 2012). The first secreted chemokine decoy receptor was discovered in orthopoxviruses, and it is now established that a wide array of chemokine binding proteins are encoded by poxviruses (Patel et al., 1990; Smith et al., 1997). Unique chemokine binding proteins had been identified in all three subfamilies of herpesviruses, with perhaps the best characterized being M3 encoded by mouse gammaherpesvirus 68 (MHV-68) (Heidarieh et al., 2015). Blood sucking ticks and the helminth parasite Schistosoma mansoni have also been shown to produce chemokine-binding proteins (Deruaz et al., 2008; Smith et al., 2005).
We recently discovered a novel chemokine decoy receptor encoded by rodent herpesvirus Peru (RHVP) (Lubman et al., 2014). RHVP is a gammaherpesvirus (rhadinovirus) related MHV-68 (Stevenson and Efstathiou, 2005) and Kaposi’s sarcoma-associated herpesvirus (KSHV) (Lee et al., 2015) that establishes acute and latent infection in laboratory mice with overt pathology evident only in immunocompromised animals (Loh et al., 2011). We demonstrated that R17 binds all human and murine CC and C chemokines tested (mCCL2 and hCCL2, mCCL3 and hCCL3, mCCL4, mCCL5 and hCCL5, mCCL8, mCCL11, mCCL20, mCCL24, mCCL19, mCCL12 and mXCL1) but not any of the CXC or CX3C chemokines (mCXCL8, mCXCL10, mCXCL9, mCXCL2, mCXCL12, mCXCL1, CX3C). Functionally, recombinant R17 potently inhibits CCL3-driven chemotaxis of human peripheral blood mononuclear cells (PBMCs) and CCL2-driven transmigration of human THP-1 monocytes (Lubman et al., 2014). Our initial studies also revealed that in addition to chemokines, R17 interacts with cell surface GAGs in a process dependent upon two BBXB motifs (where B represents a basic residue) (Lubman et al., 2014). Taken together, our results suggest that R17 plays a role in RHVP immune evasion through targeted sabotage of chemokine-mediated immune surveillance.
To gain insight into the mechanism by which R17 sequesters chemokines, we determined crystal structures alone and bound to CCL3. R17 adopts a two-lobed structure that engages the N-loop region of CCL3 important for recognition by its cognate receptor – a “hotspot” commonly targeted by other pathogen derived chemokine decoy receptors. A unique element of the R17-CCL3 interaction, however, is the engagement of the 40’s-loop BBXB motif that serves an important role in both receptor and GAG binding for a number of pro-inflammatory CC-chemokines. Gain of function mutational analysis was used to establish that R17 selectively engages this GAG binding determinant of chemokines to form kinetically long-lived complexes. The structure of R17 also revealed an unexpected similarity to the M3 chemokine decoy receptor encoded by MHV-68, although the chemokine binding locations are completely distinct (Alexander et al., 2002).
Results
Structure determination of RHVP R17
To enable structural studies, recombinant R17 protein was purified from 293F cells cultured in kifunensine, an inhibitor of class I α-mannosidase (Elbein et al., 1991). Before crystallization, R17 was treated with Endo H to trim carbohydrate (see Materials and Methods). The structure of unligated R17 was determined by iodide SAD with sites located by SHELXD (Sheldrick, 2008), phases estimated using MLPHARE (Dodson et al., 1997), and density modification using PARROT (Cowtan, 2010)(Table 1). The initial model of R17 was built using ARP/wARP (Murshudov et al., 1997) and the final model was produced after numerous rounds of manual building using Coot (Emsley and Cowtan, 2004) and refinement in Phenix (Adams et al., 2011). The model spans residues 14–400 of the mature protein along with GlcNAc linkages to Asn103 and Asn205 along with 355 water molecules (Table 1, Figure.1A).
Table 1.
Crystallographic data collection and refinement statistics.
| Data collection | R17_native | R17_KI_derivative 1 | R17_KI_derivative 2 | R17 (KI_ merged) | R17-CCL3 complex |
|---|---|---|---|---|---|
| Source | APS_23ID | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 | |
| Wavelength (Å) | 1.0 | 1.77 | 1.77 | 1.0 | |
| Resolution (Å) | 50–1.9(1.93–1.90) | 50–2.64(2.69–2.64) | 50–2.90(2.94–2.90) | 90–2.64 | 50–3.0(3.06–3.00) |
| # Observations | 281521 | 685862 | 641455 | 45751 | 826635 |
| Unique reflections | 45516 | 17071 | 13291 | 16547 | 26825 |
| Redundancy | 3(2.8) | 13.8(9.7) | 13.8(12.0) | 2.8(1.0) | 4.7(2.3) |
| I/σI | 20.8 (1.83) | 39.04 (3.05) | 26.9 (2.4) | 17.8 (2.7) | 7.4 (1.0) |
| Rmerge* | 5.2(65.4) | 6.8(83.1) | 11.7(100) | 2.1(100) | 11.2(100) |
| Completeness | 93.8(61.2) | 99.8(97.6) | 100(100) | 970(56.8) | 100(64.8) |
| Space group | P212121 | P212121 | P212121 | P212121 | I222 |
| Cell dimensions (Å) | 69.5, 75.8, 106.9 | 69.5, 75.3, 106.1 | 69.5, 76.0, 107.6 | 98.4, 109.4, 210.9 | |
| Phasing Statistics | |||||
| # iodides (SHELXD) | 14 | ||||
| FOM (centric) | 0.22 | ||||
| FOM (acentric) | 0.27 | ||||
| Phasing resolution | 46–3.10 | ||||
| Rcullis | 0.79 | ||||
| Refinement Statistics | |||||
| Resolution (Å) | 50–1.9 | 50–3.0 | |||
| (outer shell) (Å) | (1.94–1.89) | (3.08–3.0) | |||
| No. of reflections/No. in Rfree | 39897/2024 | 24120/1817 | |||
| Rcryst% | 18.44(22.3) | 21.52(32.9) | |||
| Rfree% | 22.08(34.2) | 27.40(45.5) | |||
| Rmsd bond lengths | 0.005 | 0.003 | |||
| Rmsd bond angles | 0.992 | 0.462 | |||
| Ramachandran favored (%) | 95.6 | 94.2 | |||
| Ramachandran outliers (%) | 0.0 | 0.0 | |||
| Average B-factor | 21.65 | 66.92 | |||
| PDBID | 4ZKQ | 4ZLT | |||
Numbers in parentheses refer to the highest resolution shell.
Rmerge = ∑jI – <I>j/∑<I>, where I is the intensity of each individual reflection
Figure 1: Crystal structure of RHVP R17 alone and in complex with murine CCL3.

(A) Ribbon diagram of apo R17. The N terminal domain (NTD), bridging sheet (BS) and C-terminal domain (CTD) are colored based on secondary structure: β-strands are depicted in green, α helices in cyan and connecting loops in brown. β-strands of the NTD are labeled 1–10, BS is labeled B1-B4 and β-strands of the CTD are labeled A-I. During purification, R17 was treated with EndoH to remove complex carbohydrates. Of the three predicted N-linked glycosylation sites, electron density was visible for the N-glycan linked to Asn 205. N-acetyl glucosamine (NAG) followed by a mannose ring is shown in stick representation. Disulfide bonds are shown in stick and colored yellow. (B) Crystal structure of the R17GAG2 in complex with murine CCL3 (D26A) at 3.0Å resolution. R17 is colored as in (A) while the chemokine is colored magenta and labeled according to accepted chemokine convention. Two NAGs linked to Asn 103 and Asn 205 are in ball and stick representation. (C) Displayed in white cartoon are superimposed free and ligated R17 structures. Conformational changes in the loops around chemokine binding cleft are colored green (free R17) and magenta (chemokine bound R17). Two GAG binding sites on R17 are located on the opposite surface from chemokine binding and are circled with blue dashed lines. (D) Electrostatic complementarity between R17 and CCL3. The molecular surface is colored as calculated by APBS ( <−1kT in red, 0kt in white and >+1kT in blue). See also Figure S1.
R17 adopts a two-lobed structure with an N-terminal domain (NTD) positioned perpendicular to a C-terminal domain (CTD) linked together by a bridging sheet (BS). The terminal domains consist of β−sandwich folds decorated by loops and helical segments while the BS is composed of 4 strands packed with the NTD (residues 190–216 and 233–266), and 2 strands inserted into the CTD (residues 218–232) (Figure 1A). The NTD spans residues 14–187 and is composed of a 7 and 3-stranded sheet (Figure.1A). Three disulfide bonds occur in the NTD; one pins the end of helix h1 to the end of strand s10, one bridges the turn at the start of s4 and another links the end of s4 to the start of s9. The CTD spans residues 285–400 and adopts an approximately I-type immunoglobulin fold composed of 9 β-strands and a disulfide linking the C’-strand with the beginning of the D-strand. A long flexible linker connects the BS and CTD, specifically residues 265 – 288, of which residues 267–270 refined with high B-factors. One disulfide is found within the BS B1-B2 loop while another joins the end of the flexible BS linker to the A” strand inserted into the CTD.
Structural relatives of R17
We looked for proteins of related structure to R17 using the DALI server (Holm and Sander, 1995), and remarkably found that the two closest relatives are both from gammaherpesviruses: M3 encoded by MHV-68 (Z-score=14.1) and GP350 encoded by Epstein Barr virus (EBV) (Z-score=8.2). The structures of R17 and M3 aligned with an rmsd of 4.2 Å over 262 residues including five disulfide bonds despite displaying only 8% sequence identity (Figure.2A & B). While similarities with both terminal domains are readily apparent, the NTDs of R17 and M3 align best (rmsd=3.3 Å for aligned 150 residues with 11% sequence identity). The R17 bridging sheet architecture between the NTD and CTD is absent in M3, which instead has a series of large helical loops that decorate the CTD.
Figure 2: Structural comparison of R17 with M3.

(A) Comparison of CCL3 (magenta) bound R17 with CCL2 (yellow) bound M3 where shared core secondary structure elements are depicted in green for R17 and dark blue for M3. Divergent structural elements are depicted in light gray in both R17 and M3. (B) Structure-based sequence alignment of R17 with M3. Secondary structure elements of R17 are on top while secondary structure elements of M3 are on the bottom. Both are colored as in Figure 1A. Structurally similar residues are colored grey, while identical residues including conserved three cysteines are in black. Yellow circles denote M3 chemokine binding interface residues, while down-pointing magenta triangles denote R17 chemokine binding interface residues. Residues buried in the M3 dimer are boxed. BBXB motifs on R17 are boxed in cyan. See also Figure S2.
The core of the R17 CTD domain also displays structural similarity to the second Ig domain of the viral surface glycoprotein GP350 encoded by EBV (rmsd of 2.8Å for 109 aligned residues, with 9% sequence identity). EBV infects B cells through binding of GP350 to complement receptor 2 (CR2) (Nemerow et al., 1987) using residues from the N-terminal Ig domain and the linker connecting to the second (Szakonyi et al., 2006). We tested the ability of biotinylated R17 to bind CR2 positive B cells in wild type versus CR2 knock-out mice (Molina et al., 1996; Wu et al., 2000). No differences in cell staining were noted (data not shown).
Mutational analysis based on M3 chemokine binding determinants
We attempted to define the chemokine-binding site on R17 based upon the structural similarity to M3 (Figure S1A.) (Alexander et al., 2002). As opposed to monomeric R17, M3 is an anti-parallel homodimer with deep chemokine-binding clefts formed between the NTD and CTD of the opposing monomers. We reasoned that the loop connecting strands 2b to 3 of R17 is structurally equivalent to the chemokine binding loop s2b-3 from the NTD of M3. By the same token, the large loop connecting strands B3 to B4 in the bridging sheet of R17 could mimic the chemokine-binding region in the CTD of M3 (Figure S1A, dotted circles). To experimentally address our hypothesis, we constructed two R17 variants: R1765AAAA68 has residues 65LEKE68 of the s2b-3 loop mutated to Ala and R17Δ248−254 has residues 248 through 254 of the B3-B4 loop deleted. To our surprise, neither of the R17 variants had altered binding to two chemokines we tested: CCL2 and CCL3 (Lubman et al., 2014) (Figure S1.B) indicating that ligand engagement is most likely localized elsewhere on the protein.
Crystal structure of the R17-CCL3 complex
To address where chemokines bind R17 we initiated co-crystallization experiments with CCL3, a chemokine we previously had shown binds the decoy receptor with an exceptionally long kinetic half-life leading to the potent inhibition of PBMC transmigration (Lubman et al., 2014). Diffraction quality crystals were obtained using a CCL3 mutant (D26A) reported to reduce aggregation(Czaplewski et al., 1999) and a R17GAG2 variant that could no longer interact with cell surfaces due to the mutation of residues 333KGRRK337 to 333DGEED337. The structure of the complex was solved by molecular replacement with a final atomic model refined to 3.0 Å resolution (Table 1, Figure 1B). Each asymmetric unit contained two R17GAG2-CCL3 complexes, with two GlcNAc linkages to Asn103 and Asn205 of R17 in chain A and a single GlcNAc linkage to Asn205 built for chain B. Using multi-angle static light scattering (MALS), we determined that R17 binds CCL3 with 1:1 stoichiometry suggesting that additional lattice interactions observed in the crystal structure are not functionally relevant (Figure S2).
The primary structural element used by R17 to create a chemokine-binding platform is the flexible linker that connects the bridging sheet of R17 with the CTD and forms a hydrophobic cavity between the two β-sandwiches. While this region is not well ordered in the crystal structure of the unligated R17, it becomes partially ordered upon ligand binding (Figure 1C). There are 31 residues from CCL3 and 46 residues from R17 at the R17/CCL3 interface, leading to 2700 Å2 of buried solvent accessible surface area (1385 Å2 buried for CCL3 and 1298 Å2 buried for R17). The shape complementarity at the R17-CCL3 interface is calculated to be Sc=0.70 (Lawrence and Colman, 1993). In addition to the linker that connects the two domains, CCL3 is “clamped” through multiple interactions with both the BS and CTD. A primary structural element of the BS used to bind chemokines is the B1-B2 loop. A notable hydrophobic pocket is formed by R17 residues Val 195, Leu198, Leu239 and Leu264 that serves to sequester CCL3 Phe13, a critical residue for GPCR binding (Laurence et al., 2000). The hydrogen bonds observed between the main chain carbonyl oxygens of Glu199 and Thr200 in R17 with Ser35 of CCL3 serve as yet another anchor to the BS of R17. Another pocket buries Arg45 and Asn46 of the CCL3 40’s-loop BBXB motif, formed mainly by R17 residues Tyr272, Tyr275, Trp313, Phe378 and. Tyr395. Within this acidic pocket a prominent salt bridge is formed between Glu393 of R17 and Arg45 of CCL3. Arg45 is the first B (basic residue) of the BBXB GAG binding motif on CCL3 and was shown to be critical for the ability of CCL3 to bind heparin sulfate and the CCR5 receptor (Kim et al., 2001; Koopmann et al., 1999; Teng et al., 2008).
Comparison of apo versus chemokine bound R17 points to several conformational variations associated with ligand binding (Figure 1C). Significant conformational differences are observed in the linker region connecting the bridging sheet and CTD that makes numerous chemokine contacts. Large conformational differences are also observed in the B1-B2 loop, B3-B4 loop, and CC’ loop of the CTD, each of which flank the engaged chemokine (Figure 1C). The fact that R17 uses structurally labile elements to engage chemokines suggests that structural plasticity may be associated with its broad ligand binding specificity.
Mutational analysis of the R17 chemokine recognition site
To experimentally assess our crystallographic observations, we mutagenized the linker region of R17 and selectively removed the negative charge from 266DSGSE270 to 266NAGAQ270. The resulting R17 variant could no longer bind to CCL2 (concentration range tested up to 150nM) and bound to CCL3 with more than a 100 fold (t 1/2=11s) faster kinetic off-rate compared to wild type R17-CCL3 interactions (Figure 3). Thus, the deleterious effects of the linker mutations are more pronounced for R17-CCL2 interactions. This mutational analysis of R17 establishes that our structurally defined recognition site for CCL3 is shared by CCL2 and likely the additional CC and C chemokines it binds.
Figure 3. Experimental assessment of crystallographic observations.

(A) Structure of the R17-CCL3 complex where chemokine is in electrostatic surface representation. Inset shows a part of the linker region connecting BS and CTD domains. Residues 266DSGSE270 were mutated 266NAGAQ270. (B) SPR analysis of mCCL2 and mCCL3 binding to the R17 266NAGAQ270 mutant immobilized to a CM5 chip. Shown are response curves for a typical chemokine titration. The experimental curves were globally fit using a 1:1 mass transport model to determine the kinetic KD and half-life (t1/2) presented above each sensogram. Values for KD are means of three independent experiments where KD=kd/ka and t1/2=0.693/kd.
R17 binds chemokines and cell surface GAGs at two distinct sites
We previously reported that R17 contains two BBXB motifs located at distal ends of its linear sequence that allow it to interact with cell surfaces (Lubman et al., 2014). We hypothesized that cell surface binding will permit R17 to sequester chemokines locally, perhaps at the site of infection. Charge reversal of either one of these motifs abrogated the ability of R17 to bind to the surface of CHO cells but did not compromise its ability to interact with chemokines (Lubman et al., 2014). The crystal structure of R17 supports our initial observations and provides insight as to how GAG binding by R17 is accomplished. Despite being far apart in the linear sequence, the two BBXB motifs found on R17 are in physical proximity of one another, coming together to create a large positively charged surface patch at the junction of the NTD and CTD (Figure 1C). These GAG binding determinants are located over 40 Å away from the chemokine-binding site on the opposite face of R17 (Figure 1D). Interestingly, no basic clusters are located on the surface of M3 (Figure 2B). Mechanistically, these findings are in agreement that R17, but not M3, can bind cell surfaces while simultaneously interacting with chemokines (Lubman et al., 2014).
Kinetically stable R17 interactions are imparted by chemokine 40’s-loop residues
Kinetic analysis of R17 binding to different chemokines identified two types of R17-chemokine interactions. Kinetically stable complexes were formed with CCL3, CCL4, CCL5, CCL24 and XCL1 (t1/2 >1000s), while significantly faster off rates were observed for the binding of CCL2, CCL8, CCL9 and CCL20 (Lubman et al., 2014). In order to address the structural basis for these distinct kinetic off rates, we undertook a comparative analysis of R17 binding chemokines in the context of our R17-CCL3 structure (Figure 4A). The alignment of characterized chemokines suggested to us that kinetic stability of the R17-CCL3 complex might be regulated by basic residues in the chemokine 40’s-loop. If true, replacement of structurally equivalent residues in CCL2 with residues found in CCL3’s 40’s-loop could extend the kinetic half-life of the R17-CCL2 complex. To test our hypothesis, we created a CCL2L46RK47N variant (Figure 4B) and evaluated its binding to an R17 coated CM5 chip (Figure 4C, bottom right). The SPR binding profile of the CCL2L46RK47N double mutant resembled that of CCL3, characterized by an apparently slow kinetic on-rate and a half-life exceeding 15 minutes (Lubman et al., 2014). In order to further dissect the contribution of individual 40’s-loop residues to complex stability, we created two additional CCL2 variants: CCL2L46R and CCL2K47N. We found that the CCL2L46R variant has a two-fold longer half life compared to the wild type CCL2-R17 interaction (Figure 4C, top right), while the CCL2K47N mutant forms a less stable complex with t1/2 of only 5.5s (Figure 4C, bottom left). Thus, the single site mutations only partially explain the binding profile of the CCL2L46RK47N variant, which could be stabilized by energetic coupling at the 40’s-loop binding interface (Lubman and Waksman, 2002).
Figure 4. Analysis of chemokine binding kinetics.

(A) Structure of the R17-CCL3 complex where R17 is in electrostatic surface representation. The inset shows how Arg45 and Gln46 of CCL3 are buried into a surface pocket of mixed acidic and hydrophobic character. A prominent salt bridge is observed between Arg45 of CCL3 and R17 Glu393, while the side chain of Asn46 makes a hydrogen bond with the hydroxyl of R17 Tyr323. (B) Structure based sequence alignment of CC chemokines known to interact with R17 ordered according to the kinetic stability (as measured by t1/2) of the complex they form. Conserved cysteines are colored red; residues of the BBXB motif in the 40’s-loop are boxed; residues of CCL2 mutated to structurally equivalent residues in CCL3 are colored cyan. Down-pointing magenta triangles denote CCL3 side chains beyond Cα that make direct contact (<4 Å) with R17 and are conserved in both R17-CCL3 complexes in the asymmetric unit of the crystal. Additional residues that lose any solvent accessible surface area in either complex are marked with open black triangles. Note, the recombinant CCL3 protein used for co-crystallization has D26A mutation.(C) SPR curves showing the effect of chemokine mutations on binding to R17GAG2 coupled CM5 chip: wild type mCCL2 (left), mCCL2L46R (right), mCCL2 K47N (bottom left) and mCCL2L46R K47N mutant (bottom right). The experimental curves were globally fit using 1:1 mass transport model to determine the kinetic KD and half-life (t1/2) presented above each sensogram. Values for KD are means of three independent experiments where KD=kd/ka and t1/2=0.693/kd.
R17 can inhibit chemokine-GAG interactions
We next sought to address the question of whether CCL2, whose GAG binding site is localized outside of the BBXB motif in the 40’s-loop, loses its ability to interact with GAGs when bound to R17. The addition of wild-type R17 results in a dramatic increase in CCL2 staining of Chinese hamster ovary (CHO) cells due to the decoy receptor’s ability to bind cell-surface GAGs and chemokines simultaneously (Figure 5A)(Lubman et al., 2014). We therefore mutated the two GAG binding sites on R17 and examined whether our R17GAG1 and R17GAG2 variants (Lubman et al., 2014) were capable of disrupting the binding of biotinylated CCL2 to CHO cells (Figure 5). The addition of either GAG-binding null R17 variant resulted in significantly decreased CCL2 staining. To further examine this issue we designed an SPR-based competition experiment where a fixed concentration of CCL2 was complexed with varying amounts of heparin sulfate and flown over immobilized R17GAG1 or R17GAG2. We found that addition of heparin sulfate to 100 nM of CCL2 blocked R17GAG1 and R17GAG2 interactions in a concentration-dependent manner, with 50% of binding disrupted using 50-fold excess of heparin sulfate. Together these experiments indicate that R17 is capable of disrupting direct chemokine-GAG interactions for chemokines like CCL2 that employ determinants outside the 40’s-loop.
Figure 5. R17 inhibits CCL2 interaction with cell surface GAGs.

(A) FACS analysis monitoring the effect of wild type R17, R17GAG1 and R17GAG2 on the interaction of CCL2 with cell surface GAGs as measured by changes in MFI (mean florescence intensity). 50nM of biotinylated CCL2 was added to CHOK1 cells in the presence or absence of 100nM R17GAG1 (orange line) 100nM R17GAG2 (red line) or 100nM wild type R17 (blue line). Cell surface bound CCL2 was detected with Streptoavidin-PE using FACSCalibur (BD Biosciences) and data analyzed with FlowJo. Representative histogram plot shows inhibition of CCL2-GAG interactions by R17GAG1 and R17GAG2. (B) R17 competes with soluble heparin sulfate for chemokine binding. R17GAG1 and R17GAG2 was immobilized to a CM5 chip and mCCL2 was injected at a concentration of 100 nM alone or in combination with the indicated increasing concentrations of heparin sulfate (0, 50nM, 500nM, 1μM, 5μM and 10μM). The error bars represent the standard error of three independent experiments.
Discussion
Parallel evolution of the R17 and M3 chemokine decoy receptors
With less than 8% sequence identity, the crystal structure of the unligated R17 revealed unexpected structural similarity to MHV-68 encoded M3 (Figure 2A). Thus we proceeded to use the crystal structures of M3 bound to CCL2 and XL1 (Alexander et al., 2002; Alexander-Brett and Fremont, 2007) to assess the chemokine-binding site of R17. Two R17 variants were designed and tested based on structurally equivalent chemokine-binding regions in M3, neither of which exhibited perturbed binding to CCL2 or CCL3 (Figure S1B). We therefore determined the crystal structure of R17 bound to CCL3, which revealed that the spatial location of chemokine binding on R17 is completely distinct from that of M3. In contrast to M3 where two chemokine-binding clefts are formed at the distal ends of an anti-parallel homodimer, R17 engages chemokines as a monomer primarily using a bridging sheet that is completely absent from M3 and likely the related M1 and M4 proteins encoded by MHV-68 as well (Alexander et al., 2002; Clambey et al., 2000; Evans et al., 2006; O’Flaherty et al., 2014). Interestingly, the R17 chemokine binding cleft is located in a structurally analogous position as the M3 dimer interface (Figure 2). Thus, despite a shared structural scaffold, the capacity of these two herpesvirus proteins to disrupt chemokine function appears to have arisen independently. Nevertheless, the chemistry of each chemokine-binding niche is similar, with primarily hydrophobic residues packing against the chemokine N-loop and acidic residues poised to engage basic chemokine regions.
Unifying feature of chemokine recognition
Pathogens often employ a general strategy of molecular mimicry to subvert host defense. To understand how unrelated proteins encoded by distinct pathogens disrupt chemokine signaling, we compared the recently solved crystal structures of vMIP-II/CXCR4 and CX3CL1/US28 (Burg et al., 2015; Qin et al., 2015) with CC-chemokines bound to poxvirus vCCI (Zhang et al., 2006), tick Evasin-1 (Dias et al., 2009), herpesvirus M3 (Alexander et al., 2002; Alexander-Brett and Fremont, 2007) and R17 (Figure 6). This comparative analysis of chemokine signaling and secreted decoy receptors revealed one universal aspect of recognition- the targeting of the invariant disulfide found in all four chemokine classes. Indeed, a similar chemical strategy is used by each chemokine binding protein to engage the disulfide bridge and flanking main-chain. CXCR4, US28, M3, and Evasin-1 all use a Pro residue to contact the disulfide while R17 and vCCI employ Ile. The backbone conformation surrounding the invariant disulfide is also highly conserved in these structures, where the main chain carbonyl of Cys11 serves as hydrogen bond acceptor and the amide of Cys50, Cys51, or Cys52 serves as a hydrogen bond donor. Additionally, all of the receptors employ an extended β-strand to make these contacts, with all but R17 oriented in an anti-parallel configuration. Thus, it appears that many pathogen-encoded chemokine decoys mimic precisely the same structural and chemical environment as GPCRs to engage chemokines. Since the invariant disulfide is present in all four chemokine classes, this unifying aspect of chemokine recognition could potentially be exploited for the design of small molecule inhibitors.
Figure 6. Comparison of the chemokine binding by four pathogen-derived and two host receptors.

Structures of R17 (PDB:4ZLT), M3 (PDB:2NZ1), vCCI (PDB:2FFK), Evasin-1 (PDB:3FPT), CXCR4 (PDB:4RWS) and US28 (PDB:4XT3) in complex with different chemokines were superimposed with all chemokines displayed in the same orientation. Displayed at far left are chemokine bound complexes where pathogen derived decoys and GPCRs are shown in worm diagram while chemokines are shown in surface representation. Depicted in the middle are worm diagrams of the complexes highlighting the chemokine fold and receptor contact regions. Diagrams at right are shown to highlight chemokine surface regions engaged by individual receptor determinants. Chemokine residues making direct contact (<4.0Å) are labeled in magenta. Additional interfacial residues that lose solvent accessible surface area in the complex are colored pink.
Promiscuous versus chaste chemokine engagement
The chemokine signaling network employs approximately 50 chemokines and 20 G-protein coupled receptors (GPCR), with chemokines activating a select few receptors in a class specific fashion. In contrast, many of the characterized pathogen-encoded decoy receptors bind a broad spectrum of different chemokines from multiple chemokine families. For example, M3 is able to bind chemokines from all four classes(van Berkel et al., 2000), R17 interacts with the CC and C family of chemokines and vCCI broadly recognizes CC and some CXC chemokines(Graham et al., 1997). The exceptions include the tick Evasins (Deruaz et al., 2008; Frauenschuh et al., 2007) and HCMV encoded pU21.5 (Wang et al., 2004) that exhibit chemokine selective binding profiles. Structural comparison of four pathogen-encoded chemokine decoys reveals that while they all extensively engage the chemokine N-loop, only Evasin-1 engages the N-term of CCL3 (Figure 7). Consistent with this observation, the N-termini of chemokines are thought to be responsible for specific receptor activation (Clark-Lewis et al., 1995). Indeed, truncations or mutations in the N-termini of chemokines generally lead to a loss in agonist activity, although receptor-binding affinity can be maintained (Pease et al., 1998). Thus, it appears that R17 along with M3 and vCCI achieve promiscuity by making extensive interactions with the invariant disulfide and N-loop regions of chemokines, while the N-term region important for chemokine receptor specificity is ignored. On the other hand, Evasin-1 exclusively interacts with CCL3 and CCL4 perhaps by virtue of extensive N-term engagement (Dias et al., 2009), a trait shared by GPCR chemokine receptors (Burg et al., 2015; Qin et al., 2015)
Figure 7. Conserved recognition of chemokine invariant disulfide.

The invariant disulfide bond (Cys12-Cys51/52) and flanking Cys11 of different chemokines are depicted in ball and stick representation with carbon colored cyan, nitrogen blue, oxygen red and sulfur yellow. Decoy and signaling receptor residues that engage the chemokine invariant disulfide are depicted with carbon green for R17 (PDB:4ZLT), light-blue for M3 (PDB:2NZ1), orange for vCCI (PDB:2FFK), magenta for Evasin-1 (PDB:3FPT), purple for CXCR4 (PDB:4RWS), and pink for US28 (PDB:4XT3). Conserved hydrogen bonds made with chemokine main chain atoms flanking the invariant disulfide are represented by dotted yellow lines.
Roles of GAG binding determinants
R17 broadly binds CC and C-chemokines with nanomolar affinities, but SPR studies indicate that only a subset (CCL3, CCL4, CCL5, CCL24, and XCL1) form kinetically stable complexes with apparent half-lives exceeding an hour (Lubman et al., 2014). While these chemokines serve to recruit a wide array of immune cells during viral infection, what they share in common is a GAG binding BBXB motif in their 40’s-loop. To examine the role of these residues in R17 binding we created a CCL2 variant with the same BBXB motif as found in the 40’s-loop of CCL3 (KRNR). Strikingly, the mutant CCL2 was endowed with an extremely long half-life when bound to R17. Thus, R17 appears to target chemokine 40’s-loop BBXB motifs to drive extremely stable complex formation.
Studies of MHV-68 encoded M3 indicate that it potently disrupts chemokine-GAG interactions, and thereby decoy receptor complexes are likely trafficked away from infected cells (Alexander-Brett and Fremont, 2007). A unique functional element of R17 is the capacity to engage cell surface GAGs using two of its own BBXB motifs positioned distal from the chemokine-binding site. There are no similar basic patches on the surface of M3, and no functional evidence of M3 cell-surface interactions has been reported. R17 is most likely positioned in the local extracellular matrix during infection where it can sequester inflammatory chemokines rendering them inactive. We anticipate that fluid phase chemokines would readily bind R17, while GAG associated chemokines, like CCL2, would likely need to dissociate before R17 engagement. This unique functional attribute of R17 provides a distinct immune evasion strategy that may find therapeutic application in cases where localized disruption of chemokine signaling networks is preferred over systemic disruption, such as allograft rejection (Proudfoot et al., 2015).
Conclusions
A recurrent theme among pathogens is the repurposing of structural scaffoldings to facilitate the evasion of host immune defense. For example, viruses use the MHC fold to engage natural killer (NK) cell receptors in order to protect infected cells from NK cell mediated cytotoxicity(Krmpotic et al., 2005). Still other viruses employ the MHC fold to, for example, prevent NKG2D ligand surface expression or competitively block TNF ligand-receptor interactions (Lodoen et al., 2004; Wang et al., 2012; Zhi et al., 2010). Thus, it is not particularly surprising that RHVP employs a protein of similar structure as MHV-68 M3 to block chemokine signaling networks. What is surprising, however, is that R17 has in parallel developed the capacity to sequester chemokines using determinants completely distinct from those employed by M3.
Experimental Procedures
Mammalian production of RHVP R17 and R17GAG2
The cloning, expression and purification of wild type R17 and its variants has been described in Lubman et al. (Lubman et al., 2014). For crystallization, we employed an alternate version of the published protocol developed to minimize the amount of N-linked carbohydrate (Chang et al., 2007). This involved expression of both R17 and R17GAG2 mutant in medium containing 1mM of glycosylation processing inhibitor kifunensine. The culture medium was collected 10 days after transfection and was purified using Ni-Agarose beads (Qiagen, Valencia, CA). The eluted protein was buffer exchanged into 50mM Hepes pH7.5, 600mM NaCl and incubated at room temperature overnight with Endoglycosidase Hf (3000U of EndoH for 1μg of protein) (New England Biolabs). The digested material passed over an amylose column to remove the EndoHf/maltose-binding protein fusion, followed by size exclusion chromatography (SEC) on a HiLoaD 26/60 Superdex 200pg column (G.E. Healthcare). For purification of the wild type R17, the NaCl concentration was maintained at 600mM throughout purification and crystallization. For the R17GAG2 variant, the NaCl concentration was maintained at 150mM for subsequent co-purification with CCL3 (see below).
E.Coli production of murine CCL3 (D26A)
The gene encoding the mature form of murine CCL3 with optimization for E.coli codon usage was cloned into a pET28A vector (Novogen, EMD Biosciences) using Nhe I and Bam H1 restriction sites. The D26A mutant was generated by site directed mutagenesis (Agilent Biotechnologies). The CCL3 (D26A) mutant was expressed in E.Coli BL21(DE3) cells, and protein production was induced using 1mM IPTG. CCL3 (D26A) partitioned into the inclusion body fraction and was refolded using the arginine oxidative refolding method(Nelson et al., 2014). Briefly, a 400ml volume of arginine refolding buffer (400mM L-Arginine, 100mM Tris pH 8.5, 5mM reduced glutathione, 0.5mM oxidized glutathione, 0.2mM PMSF) was prepared. Into this buffer, 4 injections of 500μl of solubilized inclusion body were made over the course of 2 hours (0, 30min, 60min and 120min). The refolding buffer was then allowed to stir slowly overnight at 4C. The following day, the protein was filtered, concentrated using a YM-10 (10kD cutoff) filter membrane (Millipore) to a volume of 2ml and purified using size exclusion chromatography using a High Load 16/60 Superdex S75 prep grade column (GE Healthcare).
Crystallization and structure determination of R17
R17 was prepared for crystallization by size-exclusion chromatography purification in buffer containing 25mM HEPES pH7.5, 600mM NaCl and 0.01% Na Azide. R17 was then concentrated to 22mg/ml and used to set up crystallization trials by hanging drop vapor diffusion. Crystals of R17 were obtained in 18–25% PEG 550MME and 100mM TrisHCL pH 8.5 in space group P212121.(a=69.561Å,b=75.835Å,c=106.985Å) with 1 molecule in the asymmetric unit. Crystals were soaked for up to 5 min into a solution similar to the precipitant solution, but supplemented with 25–250mM of KI. Diffraction data for several iodide derivatives were collected at ALS beamline 4.2.2 (Lawrence Berkeley Laboratories) at a wavelength of 1.77 Å (iodide edge) at 100K with a CCD detector. 360° of data were collected for all data sets in order to maximize the multiplicity of the data (Cianci et al., 2008; Yogavel et al., 2007; Yogavel et al., 2009) which lowered the error in the measurement of Bijvoet pairs and thereby increased the accuracy of the anomalous signal (Schneider and Sheldrick, 2002). Individual iodide derivative data sets were processed, indexed and scaled using HKL3000 (Minor et al., 2006). Anomalous signal from individual data sets was not sufficient for successful structure determination. However, merging of two iodide derivative data sets with the ratio of anomalous signal defined at 0.2 allowed SHELDX to find 14 iodide sites and subsequent SAD phase calculation lead to interpretable experimental electron density maps. Arp/Warp was used to trace over 85% of the model into experimental density and an initial 2.7Å model was refined to Rworking= 37.16% and Rfree=46.01%. The 2.7Å model was used as a search model for molecular replacement of the 1.9Å native data set collected at Advanced Photon Source (APS) Beamline 23-ID-D. After several rounds of manual model building using Coot, Phenix (Adams et al., 2011) was used to refine the R17 structure to a final Rwork of 18.4% and Rfree of 22.08%. The final R17 model contains mature residues 14–400, 2Asn-GlcNAc linkages and 355 water molecules.
Crystallization and structure determination of the R17GAG2/CCL3 complex
The mouse CCL3 variant (D26A) was produced in E.Coli and harbors the mutation D26A. The purification of the R17GAG2 variant is described in the above section. The two proteins were mixed in 1:5 molar ratio of R17 to CCL3. The 1:1 complex was purified using size exclusion chromatography (SEC) on the HiLoaD 26/60 Superdex 200pg column (G.E. Healthcare). Crystals of the R17GAG1/CCL3 complex at 60mg/ml were grown using 22% PEG3350 and 0.4M Mg Nitrate. Crystals of the complex belong to the I222 space group, with two molecules of R17GAG2 and two molecules of CCL3 (D26A) in the asymmetric unit. Native data was collected at the ALS beamline 4.2.2 (Lawrence Berkeley Laboratories) at a wavelength of 1Å at 100K with a CCD detector. The structure of R17 alone and human CCL3 (PDB: 2X69) was used to solve the structure of the complex by molecular replacement using Phaser within Phenix (Adams et al., 2011). The final model has an R-value of 21.52% and Rfree of 27.40%. The refined atomic model of R17GAG2/CCL3(D26A) comprises residues 18–400 Chain A/Chain B of R17 and residues 7–68 Chain D/Chain E of CCL3 along with 2 N-linked glycosylation sites for each of R17 chain. Due to poor electron density, residues 249–254 of Chain A and 247–254 of Chain B were not included in the final model. All of the structural analysis described in the paper was done on AD complex.
Flow Cytometry
To evaluate the effect of R17 and its variants on the ability of CCL2 to interact with cell surfaces, chemokines and a negative control protein (MR1) were non-specifically biotinylated using an EZ-biotin kit (Pierce) with a 2:1 biotin to protein molar ratio, followed by removal of unbound biotin (Thermo Scientific Zebra Desalting Columns). CHOK1 and CHO745 cells were maintained in F-12 media supplemented with 10% FCS and 100X PennStrep. On the day of the experiment, cells were washed once with PBS, detached using 0.2% EDTA and re-suspended in staining buffer containing PBS, 1% BSA and 2mM EDTA. Biotinylated CCL2 was added to cells at a final concentration of 50nM in the presence of absence of R17GAG1 or R17GAG2, incubated for an hour on ice, washed three times and detected with Streptavidin PE (Life Technologies) using a FACSCalibur (BD biosciences).
SPR binding analysis
Surface plasmon resonance (SPR) was used to directly measure the affinity and kinetics of chemokine binding by R17 and its variants and is described in Lubman et al. (Lubman et al., 2014). Briefly R17 was immobilized to on a CM5 chip (GE Healthcare) using standard amine coupling chemistry (BIAcore Amine coupling kit) to a level of 200 to 500 response units (RU) for kinetic binding analysis using a Biacore T-100 biosensor (GE Healthcare). A control flow cell was prepared by coupling a non-chemokine binding protein R7 or neutravidin to the chip at similar level.
Recombinant chemokines
Mutagenesis of mouse CCL2 residues Leu46 to Arg and Lys47 to Asn was performed using Multi-Site Quick Change Mutagenesis Kit (Agilent Technologies) on the background of the wild type mouse CCL2 and verified by DNA sequencing. Murine CCL2, CCL2L46R, CCL2K47N CCL2L46R K47N were expressed in E. Coli, refolded from inclusion bodies, and purified as previously described (Nelson et al., 2014).
Supplementary Material
Highlights.
Crystal structures of RHVP R17 alone and in complex with CCL3 have been determined
R17 is similar to MHV-68 M3 although the location of chemokine binding is distinct
Chemokine residues that stabilize R17 complexes have been mapped by mutagenesis
Pathogen decoys mimic GPCRs in engagement of invariant chemokine determinants
Acknowledgments
The authors would like to thank J. Nix at ALS beamline 4.2.2 (Lawrence Berkeley Laboratories) for help with X-ray data collection and the APS CCP4 School for help with iodide anomalous signal calculation and data collection at macromolecular crystallography beamline 23ID-D. We thank Chris Nelson for discussions and careful reading of the manuscript. This work was supported by the Washington University/Pfizer Biomedical Agreement, the Center for Women’s Infectious Disease Research (cWIDR), and NIAID grants R01 AI019687 and U54 AI057160.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcami A (2003). Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3, 36–50. [DOI] [PubMed] [Google Scholar]
- Alcami A, and Lira SA (2010). Modulation of chemokine activity by viruses. Curr Opin Immunol 22, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JM, Nelson CA, van Berkel V, Lau EK, Studts JM, Brett TJ, Speck SH, Handel TM, Virgin HW, and Fremont DH (2002). Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111, 343–356. [DOI] [PubMed] [Google Scholar]
- Alexander-Brett JM, and Fremont DH (2007). Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J Exp Med 204, 3157–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Crown SE, and Handel TM (2007). Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25, 787–820. [DOI] [PubMed] [Google Scholar]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, et al. (2015). Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VT, Crispin M, Aricescu AR, Harvey DJ, Nettleship JE, Fennelly JA, Yu C, Boles KS, Evans EJ, Stuart DI, et al. (2007). Glycoprotein structural genomics: solving the glycosylation problem. Structure 15, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci M, Helliwell JR, and Suzuki A (2008). The interdependence of wavelength, redundancy and dose in sulfur SAD experiments. Acta Crystallogr D Biol Crystallogr 64, 1196–1209. [DOI] [PubMed] [Google Scholar]
- Clambey ET, Virgin H.W.t., and Speck SH (2000). Disruption of the murine gammaherpesvirus 68 M1 open reading frame leads to enhanced reactivation from latency. J Virol 74, 1973–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, and Sykes BD (1995). Structure-activity relationships of chemokines. J Leukoc Biol 57, 703–711. [DOI] [PubMed] [Google Scholar]
- Cowtan K (2010). Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr 66, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplewski LG, McKeating J, Craven CJ, Higgins LD, Appay V, Brown A, Dudgeon T, Howard LA, Meyers T, Owen J, et al. (1999). Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J Biol Chem 274, 16077–16084. [DOI] [PubMed] [Google Scholar]
- Deruaz M, Frauenschuh A, Alessandri AL, Dias JM, Coelho FM, Russo RC, Ferreira BR, Graham GJ, Shaw JP, Wells TN, et al. (2008). Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J Exp Med 205, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JM, Losberger C, Deruaz M, Power CA, Proudfoot AE, and Shaw JP (2009). Structural basis of chemokine sequestration by a tick chemokine binding protein: the crystal structure of the complex between Evasin-1 and CCL3. PLoS One 4, e8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson EJ, Winn M, and Ralph A (1997). Collaborative Computational Project, number 4: providing programs for protein crystallography. Methods Enzymol 277, 620–633. [DOI] [PubMed] [Google Scholar]
- Elbein AD, Kerbacher JK, Schwartz CJ, and Sprague EA (1991). Kifunensine inhibits glycoprotein processing and the function of the modified LDL receptor in endothelial cells. Arch Biochem Biophys 288, 177–184. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Epperson ML, Lee CA, and Fremont DH (2012). Subversion of cytokine networks by virally encoded decoy receptors. Immunol Rev 250, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esche C, Stellato C, and Beck LA (2005). Chemokines: key players in innate and adaptive immunity. J Invest Dermatol 125, 615–628. [DOI] [PubMed] [Google Scholar]
- Evans AG, Moorman NJ, Willer DO, and Speck SH (2006). The M4 gene of gammaHV68 encodes a secreted glycoprotein and is required for the efficient establishment of splenic latency. Virology 344, 520–531. [DOI] [PubMed] [Google Scholar]
- Fernandez EJ, and Lolis E (2002). Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol 42, 469–499. [DOI] [PubMed] [Google Scholar]
- Frauenschuh A, Power CA, Deruaz M, Ferreira BR, Silva JS, Teixeira MM, Dias JM, Martin T, Wells TN, and Proudfoot AE (2007). Molecular cloning and characterization of a highly selective chemokine-binding protein from the tick Rhipicephalus sanguineus. J Biol Chem 282, 27250–27258. [DOI] [PubMed] [Google Scholar]
- Gerard C, and Rollins BJ (2001). Chemokines and disease. Nat Immunol 2, 108–115. [DOI] [PubMed] [Google Scholar]
- Graham KA, Lalani AS, Macen JL, Ness TL, Barry M, Liu LY, Lucas A, Clark-Lewis I, Moyer RW, and McFadden G (1997). The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 229, 12–24. [DOI] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK, and Proudfoot AE (2005). Regulation of protein function by glycosaminoglycans--as exemplified by chemokines. Annu Rev Biochem 74, 385–410. [DOI] [PubMed] [Google Scholar]
- Handel TM, and Lau EK (2004). Chemokine structure and receptor interactions. Ernst Schering Res Found Workshop, 101–124. [DOI] [PubMed] [Google Scholar]
- Heidarieh H, Hernaez B, and Alcami A (2015). Immune modulation by virus-encoded secreted chemokine binding proteins. Virus Res S0168–1702(15)00118–5. [DOI] [PubMed] [Google Scholar]
- Holm L, and Sander C (1995). Dali: a network tool for protein structure comparison. Trends Biochem Sci 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Kim S, Jao S, Laurence JS, and LiWang PJ (2001). Structural comparison of monomeric variants of the chemokine MIP-1beta having differing ability to bind the receptor CCR5. Biochemistry 40, 10782–10791. [DOI] [PubMed] [Google Scholar]
- Koopmann W, Ediriwickrema C, and Krangel MS (1999). Structure and function of the glycosaminoglycan binding site of chemokine macrophage-inflammatory protein-1 beta. J Immunol 163, 2120–2127. [PubMed] [Google Scholar]
- Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, et al. (2005). NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Salanga CL, and Handel TM (2015). Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence JS, Blanpain C, Burgner JW, Parmentier M, and LiWang PJ (2000). CC chemokine MIP-1 beta can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 39, 3401–3409. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, and Colman PM (1993). Shape complementarity at protein/protein interfaces. J Mol Biol 234, 946–950. [DOI] [PubMed] [Google Scholar]
- Lee HR, Amatya R, and Jung JU (2015). Multi-step regulation of innate immune signaling by Kaposi’s sarcoma-associated herpesvirus. Virus Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, and Lanier LL (2004). The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med 200, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh J, Zhao G, Nelson CA, Coder P, Droit L, Handley SA, Johnson LS, Vachharajani P, Guzman H, Tesh RB, et al. (2011). Identification and sequencing of a novel rodent gammaherpesvirus that establishes acute and latent infection in laboratory mice. J Virol 85, 2642–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman OY, Cella M, Wang X, Monte K, Lenschow DJ, Huang YH, and Fremont DH (2014). Rodent herpesvirus Peru encodes a secreted chemokine decoy receptor. J Virol 88, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman OY, and Waksman G (2002). Dissection of the energetic coupling across the Src SH2 domain-tyrosyl phosphopeptide interface. J Mol Biol 316, 291–304. [DOI] [PubMed] [Google Scholar]
- Luster AD (1998). Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med 338, 436–445. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Molina H, Holers VM, Li B, Fung Y, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, and Chaplin DD (1996). Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci U S A 93, 3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nelson CA, Lee CA, and Fremont DH (2014). Oxidative refolding from inclusion bodies. Methods Mol Biol 1140, 145–157. [DOI] [PubMed] [Google Scholar]
- Nemerow GR, Mold C, Schwend VK, Tollefson V, and Cooper NR (1987). Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J Virol 61, 1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flaherty BM, Soni T, Wakeman BS, and Speck SH (2014). The murine gammaherpesvirus immediate-early Rta synergizes with IRF4, targeting expression of the viral M1 superantigen to plasma cells. PLoS Pathog 10, e1004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AH, Gaffney DF, Subak-Sharpe JH, and Stow ND (1990). DNA sequence of the gene encoding a major secreted protein of vaccinia virus, strain Lister. J Gen Virol 71 (Pt 9), 2013–2021. [DOI] [PubMed] [Google Scholar]
- Pease JE, Wang J, Ponath PD, and Murphy PM (1998). The N-terminal extracellular segments of the chemokine receptors CCR1 and CCR3 are determinants for MIP-1alpha and eotaxin binding, respectively, but a second domain is essential for efficient receptor activation. J Biol Chem 273, 19972–19976. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Bonvin P, and Power CA (2015). Targeting chemokines: Pathogens can, why can’t we? Cytokine. [DOI] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, et al. (2015). Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TR, and Sheldrick GM (2002). Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr 58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2008). A short history of SHELX. Acta Crystallogr A 64, 112–122. [DOI] [PubMed] [Google Scholar]
- Smith CA, Smith TD, Smolak PJ, Friend D, Hagen H, Gerhart M, Park L, Pickup DJ, Torrance D, Mohler K, et al. (1997). Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits beta chemokine activity yet lacks sequence homology to known chemokine receptors. Virology 236, 316–327. [DOI] [PubMed] [Google Scholar]
- Smith P, Fallon RE, Mangan NE, Walsh CM, Saraiva M, Sayers JR, McKenzie AN, Alcami A, and Fallon PG (2005). Schistosoma mansoni secretes a chemokine binding protein with antiinflammatory activity. J Exp Med 202, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson PG, and Efstathiou S (2005). Immune mechanisms in murine gammaherpesvirus-68 infection. Viral Immunol 18, 445–456. [DOI] [PubMed] [Google Scholar]
- Szakonyi G, Klein MG, Hannan JP, Young KA, Ma RZ, Asokan R, Holers VM, and Chen XS (2006). Structure of the Epstein-Barr virus major envelope glycoprotein. Nat Struct Mol Biol 13, 996–1001. [DOI] [PubMed] [Google Scholar]
- Teng MS, Shadbolt P, Fraser AG, Jansen G, and McCafferty J (2008). Control of feeding behavior in C. elegans by human G protein-coupled receptors permits screening for agonist-expressing bacteria. Proc Natl Acad Sci U S A 105, 14826–14831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkel V, Barrett J, Tiffany HL, Fremont DH, Murphy PM, McFadden G, Speck SH, and Virgin HI (2000). Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J Virol 74, 6741–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Bresnahan W, and Shenk T (2004). Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci U S A 101, 16642–16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Natarajan K, Revilleza MJ, Boyd LF, Zhi L, Zhao H, Robinson H, and Margulies DH (2012). Structural basis of mouse cytomegalovirus m152/gp40 interaction with RAE1gamma reveals a paradigm for MHC/MHC interaction in immune evasion. Proc Natl Acad Sci U S A 109, E3578–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Jiang N, Fang YF, Xu C, Mao D, Singh J, Fu YX, and Molina H (2000). Impaired affinity maturation in Cr2−/− mice is rescued by adjuvants without improvement in germinal center development. J Immunol 165, 3119–3127. [DOI] [PubMed] [Google Scholar]
- Yogavel M, Gill J, Mishra PC, and Sharma A (2007). SAD phasing of a structure based on cocrystallized iodides using an in-house Cu Kalpha X-ray source: effects of data redundancy and completeness on structure solution. Acta Crystallogr D Biol Crystallogr 63, 931–934. [DOI] [PubMed] [Google Scholar]
- Yogavel M, Gill J, and Sharma A (2009). Iodide-SAD, SIR and SIRAS phasing for structure solution of a nucleosome assembly protein. Acta Crystallogr D Biol Crystallogr 65, 618–622. [DOI] [PubMed] [Google Scholar]
- Zhang L, Derider M, McCornack MA, Jao SC, Isern N, Ness T, Moyer R, and LiWang PJ (2006). Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1beta. Proc Natl Acad Sci U S A 103, 13985–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi L, Mans J, Paskow MJ, Brown PH, Schuck P, Jonjic S, Natarajan K, and Margulies DH (2010). Direct interaction of the mouse cytomegalovirus m152/gp40 immunoevasin with RAE-1 isoforms. Biochemistry 49, 2443–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.