

Abstract
Recently, several arylnitro-based fluorescent CO probes have been reported. The design was based on CO's ability to reduce an arylnitro group for fluorescence turn-on. In this work, we assessed the response of three published arylnitro-based fluorescent CO probes, namely COFP, LysoFP-NO2, and NIR-CO toward CO from various sources. We found that only ruthenium-based CO releasing molecules (CO-RMs) were able to turn on the fluorescence while pure CO gas and CO from other sources did not turn-on the probe in the absence of ruthenium. Further experiments with different ruthenium complexes indicate that the reduction of arylnitro group requires the ruthenium carbonyl complex as an essential ingredient. As further confirmation, we also conducted the reduction of the nitro group in a p-nitrobenzamide compound and came to the same conclusion. As such, COFP and related arynitro-based probes are able to sense CORM-2 and CORM-3, but not CO in general. Our findings also indicate the need to use CO from various sources in future assessment of new CO probes.
Graphical Abstract
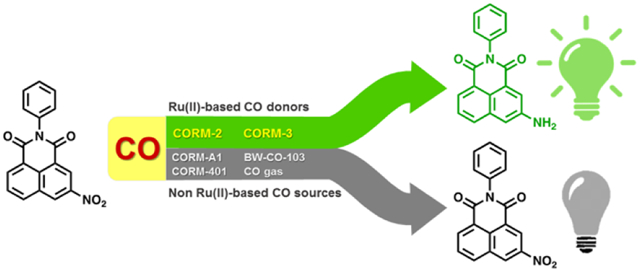
Nitro reduction-based fluorescent CO probes only sense ruthenium-based CO donors, CORM-2 and CORM-3, not CO in general.
Carbon monoxide (CO) has been firmly established as an endogenous signaling molecule along with nitric oxide (NO) and hydrogen sulfide (H2S).1, 2 The therapeutic effects of CO have been pharmacologically validated in multiple animal models including colitis, systemic inflammation and sepsis, drug induced cardiotoxicity and nephrotoxicity, and ischemia reperfusion injury of the liver, lung, heart, and kidney, among others.3-11 With the firmly established therapeutic effects, there are some important challenges in the field of CO research both in terms of mechanistic understandings and pharmaceutical development. On one hand, the pleiotropic effects of CO are not well understood at the molecular level. Tools are needed for facilitating such studies. On the other hand, there is a need for the development of pharmaceutically acceptable forms of CO delivery. Along this line, current delivery forms include CO gas, immobilized carbonyls that are referred to as metal-based CO-release molecules (CO-RMs) and photo-sensitive organic CO-RMs, CO in an oral formulation, organic and metal-free CO prodrugs.8, 12-25 Both for mechanistic studies and pharmaceutical development, there is a need for a thorough understanding of the pharmacokinetic profiles of each delivery form and their correlation with pharmacodynamics in a given indication. Therefore, tools for real-time and highly accurate measurement of CO levels in circulation and at the cellular and tissue levels are in great need. Currently, oximeters are the most commonly used tools for determining carboxy hemoglobin (COHb) levels as a surrogate indicator of CO exposures in the systemic circulation.26 There have been continuous efforts in developing molecular fluorescent probes and sensors for CO for cellular and tissue-based CO measurements and imaging work. After Chang’s palladium-based fluorescent probe for CO (COP-1),27 there have been extensive efforts along a similar line, leading to several analogous CO probes (Figure 1A).28-31 He and coworkers also reported a genetically encoded fluorescent protein as CO sensor based on the binding affinity of CO to the iron center on a heme cofactor.32 These probes all rely on the affinity of CO for a transition metal. Recently, there are exciting reports of metal-free fluorescent CO probes, which suggest the ability of CO to reduce a nitro group on an aromatic core, leading to fluorescent turn-on (Figure 1B).33-35 Such feasibility was demonstrated with two ruthenium-based CO-RMs, CORM-2 and CORM-3, as the source of CO.36, 37 The design is very innovative, and the results are very exciting. If broadly applicable, these studies would usher in the era of metal-free fluorescent probes for CO, which is the “Holy Grail” of CO sensing, especially for in vivo application. We were interested in borrowing from this exciting strategy for designing CO probes for pharmacokinetic studies. As a first step, we confirmed the reported results using COFP, and thus the reproducibility of the literature results. However, upon further examination of these probes using different delivery forms of CO, it was found that the fluorescent probe only worked when the CO donors were ruthenium-based. In order for a probe to be useful in “selective detection of CO,” they need to be able to sense CO from all sources. Herein, we discuss work in examining the general utility of the published nitro reduction-based CO probes. We conclude that the nitro reduction-based fluorescent CO probes rely on the ruthenium core for activity, and are only able to show fluorescent intensity increases in the presence of ruthenium-based CORM-2 and CORM-3, but are not broadly applicable to general CO detection.
Figure 1.
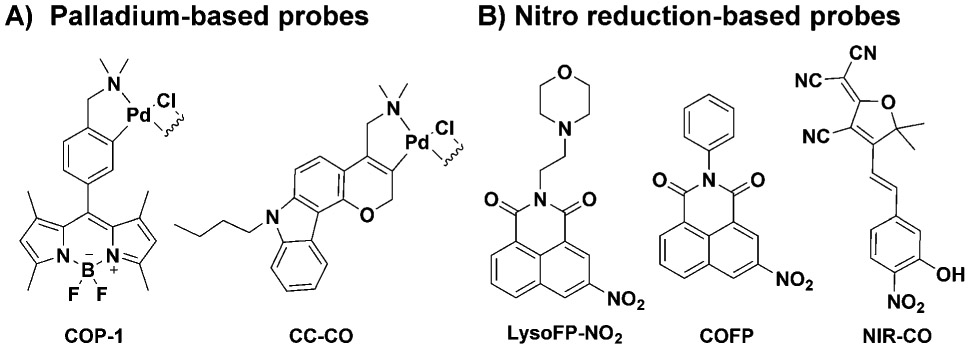
Structures of representative fluorescent CO probes for CO detection and in vitro imaging.
Following the exciting work of Dhara et al., we were interested in examining how well COFP would allow us to determine the quantity of CO release from different delivery forms. We first chose BW-CO-103 (CO-103), which belongs to the class of metal-free organic CO prodrugs widely validated in multiple pharmacological animal models including colitis, liver injury, systemic inflammation, and kidney ischemia reperfusion injury.5, 7, 38 An added advantage of using CO-103 for the initial study is the prodrug’s ability to concomitantly produce a fluorescent product, CP-103, together with CO release (Figure 2A).7 This would give a way to cross-validate the results from the fluorescent probe in a quantitative fashion in solution, in cell culture, and possibly in animal models. With these design ideas in mind, we first explored the ability for the 1,8-naphthalimide-based probe COFP to sense the CO produced from CO-103. Much to our dismay, no fluorescent turn-on was observed at around 522 nm, which is expected within 2 h from the fluorescent turn-on CO probe (Figure 2B). In contrast, we observed a significant fluorescent intensity increase due to the production of CP-103 after 2 h of incubation, suggesting CO production (Figure 2C). To further examine the generation of CO from CO-103, 1 μM COP-1 was incubated with CO-103 under the same conditions. A fluorescent signal increase at 507 nm was observed from COP-1 after 2 h (Figure S1) along with formation of CP-103 (Figure S2). Such results also confirmed CO production. As additional positive controls, we also examined the probe’s ability to detect CO released from CORM-2 and CORM-3 as reported in the literature (Figure S5, S7).33 As expected, it was reassuring that we were able to completely reproduce the solution studies of COFP as reported in the literature.
Figure 2.
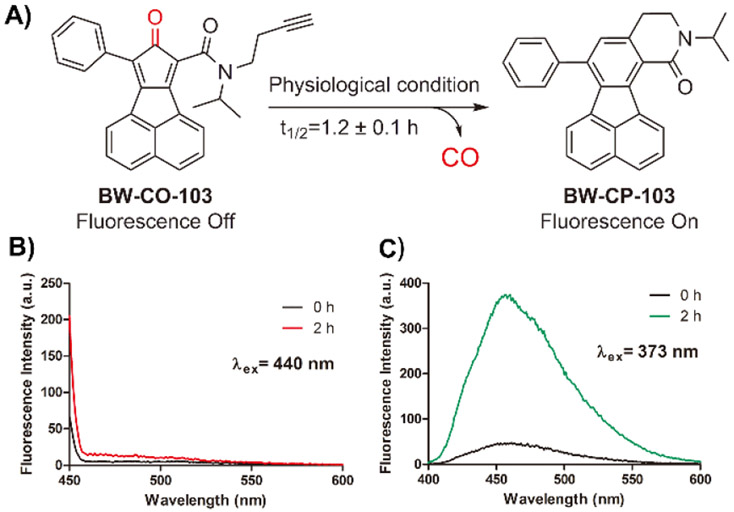
Responses of COFP upon treatment with CO-103. A) CO release from CO-103; B) Fluorescence spectra of COFP (10 μM) upon treatment with CO-103 (100 μM) over 2 h in DMSO/PBS (pH = 7.4) 5:1 at 37 °C (λex= 440 nm, slit widths: Wex = Wem = 10 nm); C) Fluorescence spectra of CP-103 formation after the incubation with COFP in DMSO/PBS (pH = 7.4) 5:1 at 37 °C. (λex = 373 nm, slit widths: Wex = Wem = 5 nm)
As additional controls, we also bubbled CO gas through the probe solution and did not see any fluorescent changes (Figure 3A, SI). We further examined the existence of CO in solution using Chang’s probe, COP-1,27 and observed strong fluorescent intensity changes at 507 nm (Figure 3B). Then, it became clear that COFP did not sense CO delivered in the form of CO gas or from CO-103. At this time, it is important to analyze CO’s chemistry in the context of CO sensing and CO’s reactivity in vivo. CO is a Lewis base with strong affinity for transition metals.39-41 However, it is a consensus in the CO field that in the body, CO undergoes no metabolism, despite the presence of a large number of organic molecules with reducing ability and enzymes capable of catalyzing redox reactions.42, 43 Administered CO largely eliminates through exhalation. There is a large body of literature along this line from studying the physiology and pharmacokinetic properties of CO in the smokers’ population.44-46 All such reports suggest that CO is very inert. A further search of the chemistry literature indicates that CO is also inert toward an arylnitro group under normal physiological conditions, as one would expect. However, there are ample precedents that CO is effective in reducing an arylnitro group in the presence of catalytic amounts of transition metal complexes such as that of Au, Ru, Se, and Rh among others.47-51 Coincidentally, CORM-2 and CORM-3 were the only ones examined as CO sources in the reported work.33-35 These nitro reduction-based CO probes were not tested on CO gas or other metal-free CO donors. In order to further examine the scope of the probe’s ability to detect CO from other CO-RMs, we also conducted studies using a boron-based CO donor CORM-A1 and a manganese-based CORM-401 with reported release half-life being 21 min and less than 4 min respectively.52, 53 Specifically, we incubated 10 μM COFP with 100 μM CORM-A1 and CORM-401 in PBS at 37 °C. However, we did not observe any fluorescent intensity changes from these CO-RMs within 60 min (Figure S13, S14). As positive controls, CORM-A1 and CORM-401 were also incubated with COP-1 under the same conditions; and fluorescent signal increases at 507 nm from COP-1 were observed after 60 min (Figure S3, S4).
Figure 3.
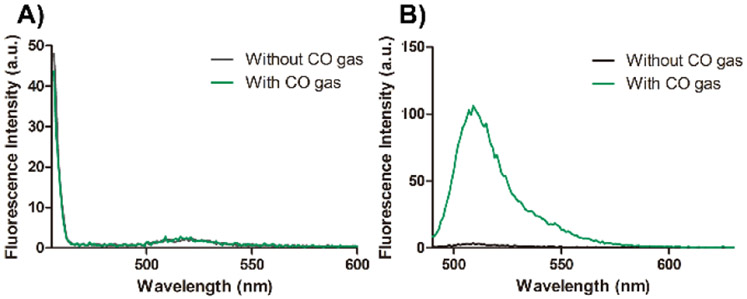
Fluorescence spectra of A) COFP (10 μM) and B) COP-1 (1 μM) upon treatment with CO gas in PBS (2% DMSO) at 37 °C for 1 h. (COFP: λex= 440 nm, slit widths: Wex = 15 nm, Wem = 10 nm; COP-1: λex = 475 nm, slit widths: Wex = 5 nm, Wem = 3 nm)
As further assessment, we also studied the other two published nitro-based CO probes, LysoFP-NO234 and NIR-CO35 (Figure 2, and see SI for synthesis details), for their response toward CO from ruthenium-based CO-RMs and pure CO gas. As shown in Figure S11, CO gas did not turn on LysoFP-NO2 after 1 h incubation at 37 °C. However, consistent with the original report, we did observe fluorescent signal increase from LysoFP-NO2 upon the incubation with CORM-3 (Figure S10). Indeed, the results are consistent with what the authors of the LysoFP-NO2 probe claim i.e. LysoFP-NO2 is a CORM-3 probe. Next, NIR-CO was assessed by using various concentrations of CORM-2 following the reported procedure.35 After incubation for 15 min at room temperature, it was found the absorption peak of NIR-CO at 400 nm decreased in a concentration-dependent fashion while the absorption peak at around 625 nm increased concomitantly, indicating the formation of the reduced amino product (Figure S12). In contrast, no spectroscopic change was observed for NIR-CO after treatment with pure CO gas (Figure S12). Such results indicate that NIR-CO does not sense CO delivered in the gas form and is not a general CO probe. Since the biology experiments are beyond the scope of the chemistry question on hand, we did not assess the ability for NIR-CO to sense CO in cell culture and in zebrafish as reported in the original study.
To further investigate the ability for ruthenium-based CO-RMs to reduce an arylnitro group, we also studied a p-nitrobenzamide compound, PNB, as a substrate (see SI for details). The HPLC results showed that PNB was completely consumed within 30 min after CORM-3 addition (Figure S16), accompanied by the formation of a new peak corresponding to the reduced product, p-aminobenzamide (PAB). We also performed LC-MS experiments as a secondary verification of the formation of PAB (Figure S17). After confirmation of CORM-3’s ability to reduce PNB to PAB, CO gas was used to conduct the same experiments. It was found that bubbling CO gas into a PNB solution for 2 h at 37 °C did not lead to either changes to the PNB peak nor formation of PAB as studied using HPLC and LC-MS. Such results again indicate that CO alone does not reduce an arylnitro group in the absence of a metal complex such as CORM-3 (Figure S17). As such, it is reasonable for us to conclude that CO alone does not reduce an arylnitro group to turn on the fluorescence of the probes in question. Likely, the ruthenium core in CORM-2 or CORM-3 played a catalytic role in enabling the reduction of the nitro group by CO. Then we looked into whether the ruthenium core without CO would turn on COFP by itself. Complex D was obtained without carbonyl groups attached to the ruthenium core (Figure 4B, and see SI for details).54 Upon incubation of Complex D with COFP in PBS at 37 °C, no fluorescent increase was observed within 1 h (Figure S15). Such results suggest that the ruthenium core alone cannot reduce the aryl nitro group. Then, it is reasonable to assume that the reducing ability of CORM-2 and CORM-3 might come from the ruthenium-carbonyl complex.
Figure 4.
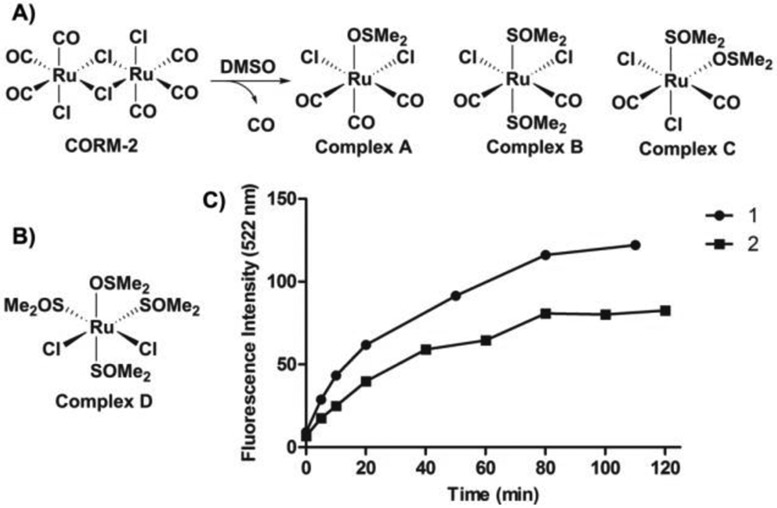
The reactivities of CORM-2 and its analogs toward COFP. A) reaction between CORM-2 and DMSO; B) chemical structure of Complex D; C) fluorescence intensity changes from COFP (10 μM) upon treatment with (1) CORM-2 and (2) reaction products from mixing CORM-2 and DMSO in PBS (pH = 7.4, 4% DMSO) at 37 °C. (λex = 440 nm, λem= 522 nm, slit widths: Wex = Wem = 10 nm)
Previous NMR studies showed that one CORM-2 molecule can dissociate into tri-carbonyl (Complex A) and di-carbonyl monomers (Complex B and C) by DMSO during the solubilization process (Figure 4A).37 One carbonyl group can be displaced by DMSO to form complex B or C. With this in mind, we were interested in comparing the reducing ability of CORM-2 and its products from ligand substitution with DMSO with the aim of examining if the ruthenium-coordinated CO is involved in the reduction mechanism. Following a reported procedure, CORM-2 was dissolved in DMSO and incubated at room temperature for 30 min. Gas bubbles from the solution were observed during the incubation, which is consistent with previously published results, and indicate the transformation from CORM-2 to Complexes B and C with the release of one CO molecule. The resulting solution was further examined with COFP by monitoring the fluorescence signal (Figure S6), which should be indicative of the reduction reaction. As shown in Figure 4C, both of the reduction rate and final fluorescence intensity were lower when the DMSO-CORM-2 solution was used when compared to the fresh CORM-2 treated control group. Such results were presumably due to the loss of CO from the ruthenium complex prior to exposure to COFP. Additionally, we also examined the responses of COFP upon treatment with inactive CORM-3 (iCORM-3). According to previous infrared spectroscopy studies, iCORM-3 is a dicarbonyl species formed by loss of one CO molecule from CORM-3.36 Similar to the case of CORM-2 and DMSO-CORM-2 solution, the reduction rate and final fluorescence intensity were lower when compared with the CORM-3 treatment group (Figure S8, S9). Such results suggest that, with the loss of one carbonyl ligand, the reducing ability of the ruthenium complex also decreased. Such results also further indicate that the coordinated CO is critical for the ability to reduce an arylnitro group by the ruthenium carbonyl complex. Previously, a ruthenium (II) carbonyl complex, Ru3(CO)12, was extensively reported to quantitatively reduce nitrobenzene to aniline in the presence of an amine under high CO pressure (20 to 50 bar) and at high temperature (150 to 180 °C).55-57 The mechanism was interpreted as involving nitrene formation from the nitro moiety via ruthenium carbonyl complex-mediated metallacyclization and extrusion of CO2, followed by reaction with CO/H2O to yield the aromatic amine.56 In this case, it might be possible that CORM-2 and CORM-3 would lead to the reduction of an arylnitro group through a similar mechanism as presented in Figure S18. However, detailed mechanistic studies remain scarce. It should be noted that the most important aspect of all these studies is the demonstration of a ruthenium complex as a prerequisite for probe reduction, not necessarily in the detailed steps of the reaction. Such findings suggest that COFP and other related nitro-based CO probes are capable of sensing only CO from ruthenium-based CO-RMs, as described in the original papers, but is not a general CO probe. Figure 5 summarizes the key findings of this study.
Figure 5.
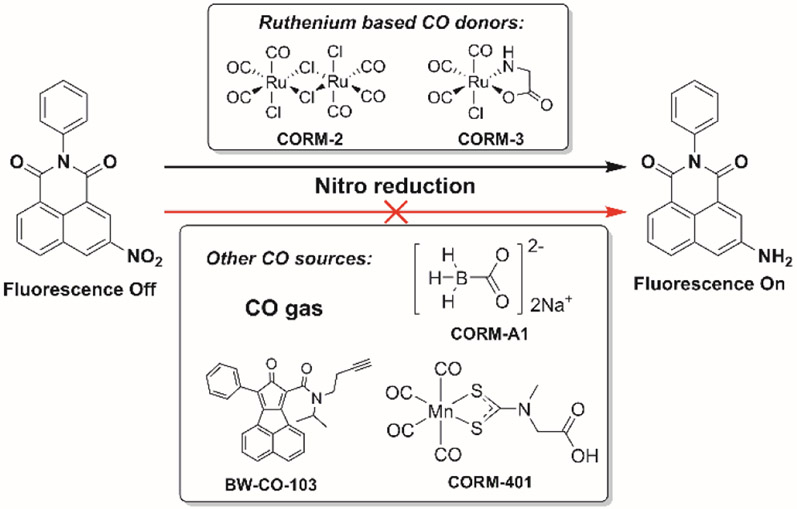
The requirement of the ruthenium carbonyl complex in CO sources for probe reduction.
In conclusion, we found that arylnitro reduction-based CO probes do not respond to CO in general. CO alone cannot reduce the arylnitro group, which is the chemical event necessary for turning on the fluorescent probe. Our studies coupled with literature precedents revealed the requirement for a ruthenium carbonyl moiety for the reduction of an arylnitro group. As a result, nitro reduction-based CO probes only sense ruthenium-based CO donors, CORM-2 and CORM-3, not CO in general. The new insights into the nitro-based CO probes define their scope of applications and lend a mechanistic understanding of the reduction of an arylnitro group by CO in the context of biological applications. Additionally, such studies also indicate the need to use CO from various sources in future assessment of new CO probes.
Supplementary Material
Acknowledgments
BW and LKDLC are partially supported by a grant from the National Institutes of Health (R01DK119202) on CO-related work. We also acknowledge the financial support from Georgia State University and the Georgia Research Alliance through an Eminent Scholar endowment (BW), a Brains & Behavior Fellowship to Z.Y. and a CDT Fellowship to LKDLC.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Mustafa AK, Gadalla MM and Snyder SH, Sci. Signal, 2009, 2, re2–re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R, Antioxid. Redox Signal, 2003, 5, 493–501. [DOI] [PubMed] [Google Scholar]
- 3.Motterlini R and Otterbein LE, Nat. Rev. Drug Discov, 2010, 9, 728–728. [DOI] [PubMed] [Google Scholar]
- 4.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA and Choi AMK, Nat. Med, 2000, 6, 422–428. [DOI] [PubMed] [Google Scholar]
- 5.Correa-Costa M, Gallo D, Csizmadia E, Gomperts E, Lieberum J-L, Hauser CJ, Ji X, Wang B, Câmara NOS, Robson SC and Otterbein LE, Proc. Natl. Acad. Sci. U.S.A, 2018, 115, E2302–E2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tayem Y, Johnson TR, Mann BE, Green CJ and Motterlini R, Am. J. Physiol. Renal Physiol, 2006, 290, F789–F794. [DOI] [PubMed] [Google Scholar]
- 7.Ji X, Zhou C, Ji K, Aghoghovbia RE, Pan Z, Chittavong V, Ke B and Wang B, Angew. Chem. Int. Ed, 2016, 55, 15846–15851. [DOI] [PubMed] [Google Scholar]
- 8.Steiger C, Uchiyama K, Takagi T, Mizushima K, Higashimura Y, Gutmann M, Hermann C, Botov S, Schmalz H-G, Naito Y and Meinel L, J. Control. Release, 2016, 239, 128–136. [DOI] [PubMed] [Google Scholar]
- 9.Piantadosi Claude A, Carraway Martha S, Babiker A and Suliman Hagir B, Circ. Res, 2008, 103, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soni H, Pandya G, Patel P, Acharya A, Jain M and Mehta AA, Toxicol. Appl. Pharmacol, 2011, 253, 70–80. [DOI] [PubMed] [Google Scholar]
- 11.Nakao A, Yamada T, Kohama K, Yoshie N, Fujisaki N and Kotani J, Acute Medicine & Surgery, 2014, 1, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji X, Damera K, Zheng Y, Yu B, Otterbein LE and Wang B, J. Pharm. Sci, 2016, 105, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji X and Wang B, Acc. Chem. Res, 2018, 51, 1377–1385. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty I, Carrington SJ and Mascharak PK, Acc. Chem. Res, 2014, 47, 2603–2611. [DOI] [PubMed] [Google Scholar]
- 15.Diring S, Carné-Sánchez A, Zhang J, Ikemura S, Kim C, Inaba H, Kitagawa S and Furukawa S, Chem. Sci, 2017, 8, 2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palao E, Slanina T, Muchová L, Šolomek T, Vítek L and Klán P, J. Am. Chem. Soc, 2016, 138, 126–133. [DOI] [PubMed] [Google Scholar]
- 17.Antony LAP, Slanina T, Šebej P, Šolomek T and Klán P, Org. Lett, 2013, 15, 4552–4555. [DOI] [PubMed] [Google Scholar]
- 18.Peng P, Wang C, Shi Z, Johns VK, Ma L, Oyer J, Copik A, Igarashi R and Liao Y, Org. Biomol. Chem, 2013, 11, 6671–6674. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Viennois E, Ji K, Damera K, Draganov A, Zheng Y, Dai C, Merlin D and Wang B, Chem. Commun, 2014, 50, 15890–15893. [DOI] [PubMed] [Google Scholar]
- 20.Ji X, De La Cruz LKC, Pan Z, Chittavong V and Wang B, Chem. Commun, 2017, 53, 9628–9631. [DOI] [PubMed] [Google Scholar]
- 21.Ji X, Ji K, Chittavong V, Yu B, Pan Z and Wang B, Chem. Commun, 2017, 53, 8296–8299. [DOI] [PubMed] [Google Scholar]
- 22.Pan Z, Zhang J, Ji K, Chittavong V, Ji X and Wang B, Org. Lett, 2018, 20, 8–11. [DOI] [PubMed] [Google Scholar]
- 23.Popova M, Soboleva T, Ayad S, Benninghoff AD and Berreau LM, J. Am. Chem. Soc, 2018, 140, 9721–9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soboleva T, Esquer HJ, Benninghoff AD and Berreau LM, J. Am. Chem. Soc, 2017, 139, 9435–9438. [DOI] [PubMed] [Google Scholar]
- 25.Mann BE, Organometallics, 2012, 31, 5728–5735. [Google Scholar]
- 26.Hess DR, Respir. Care, 2017, 62, 1333–1342. [DOI] [PubMed] [Google Scholar]
- 27.Michel BW, Lippert AR and Chang CJ, J. Am. Chem. Soc, 2012, 134, 15668–15671. [DOI] [PubMed] [Google Scholar]
- 28.Zheng K, Lin W, Tan L, Chen H and Cui H, Chem. Sci, 2014, 5, 3439–3448. [Google Scholar]
- 29.Li Y, Wang X, Yang J, Xie X, Li M, Niu J, Tong L and Tang B, Anal. Chem, 2016, 88, 11154–11159. [DOI] [PubMed] [Google Scholar]
- 30.Feng W, Liu D, Feng S and Feng G, Anal. Chem, 2016, 88, 10648–10653. [DOI] [PubMed] [Google Scholar]
- 31.Pal S, Mukherjee M, Sen B, Mandal SK, Lohar S, Chattopadhyay P and Dhara K, Chem. Commun, 2015, 51, 4410–4413. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Karpus J, Zhao BS, Luo Z, Chen PR and He C, Angew. Chem. Int. Ed, 2012, 51, 9652–9656. [DOI] [PubMed] [Google Scholar]
- 33.Das B, Lohar S, Patra A, Ahmmed E, Mandal SK, Bhakta JN, Dhara K and Chattopadhyay P, New J. Chem, 2018, 42, 13497–13502. [Google Scholar]
- 34.Dhara K, Lohar S, Patra A, Roy P, Saha SK, Sadhukhan GC and Chattopadhyay P, Anal. Chem, 2018, 90, 2933–2938. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Liu C, Wang X, Duan Q, Jia P, Zhu H, Li Z, Zhang X, Ren X, Zhu B and Sheng W, Sens. Actuators B Chem, 2019, 291, 329–336. [Google Scholar]
- 36.Clark James E, Naughton P, Shurey S, Green Colin J, Johnson Tony R, Mann Brian E, Foresti R and Motterlini R, Circ. Res, 2003, 93, e2–e8. [DOI] [PubMed] [Google Scholar]
- 37.Motterlini R, Clark James E, Foresti R, Sarathchandra P, Mann Brian E and Green Colin J, Circ. Res, 2002, 90, e17–e24. [DOI] [PubMed] [Google Scholar]
- 38.Ji X, Pan Z, Li C, Kang T, De La Cruz LKC, Yang L, Yuan Z, Ke B and Wang B, J. Med. Chem, 2019, 62, 3163–3168. [DOI] [PubMed] [Google Scholar]
- 39.Bennett B, Lemon BJ and Peters JW, Biochemistry, 2000, 39, 7455–7460. [DOI] [PubMed] [Google Scholar]
- 40.Piantadosi CA, Antioxid. Redox Signal , 2002, 4, 259–270. [DOI] [PubMed] [Google Scholar]
- 41.Bagley KA, Van Garderen CJ, Chen M, Woodruff WH, Duin EC and Albracht SPJ, Biochemistry, 1994, 33, 9229–9236. [DOI] [PubMed] [Google Scholar]
- 42.Ernst A and Zibrak JD, N. Engl. J. Med, 1998, 339, 1603–1608. [DOI] [PubMed] [Google Scholar]
- 43.Ryter SW and Otterbein LE, BioEssays, 2004, 26, 270–280. [DOI] [PubMed] [Google Scholar]
- 44.Zevin S, Saunders S, Gourlay SG, Jacob P and Benowitz NL, J. Am. Coll. Cardiol, 2001, 38, 1633–1638. [DOI] [PubMed] [Google Scholar]
- 45.Aronow WS, Dendinger J and Rokaw SN, Ann. Intern. Med, 1971, 74, 697–702. [DOI] [PubMed] [Google Scholar]
- 46.Cohen SI, Perkins NM, Ury HK and Goldsmith JR, Arch. Environ. Occup. Health, 1971, 22, 55–60. [DOI] [PubMed] [Google Scholar]
- 47.He L, Wang L-C, Sun H, Ni J, Cao Y, He H-Y and Fan K-N, Angew.Chem. Int. Ed, 2009, 48, 9538–9541. [DOI] [PubMed] [Google Scholar]
- 48.Liu X and Lu S, J. Mol. Catal. A: Chem, 2004, 212, 127–130. [Google Scholar]
- 49.Nomura K, Ishino M and Hazama M, Bull. Chem. Soc. Jpn, 1991, 64, 2624–2628. [Google Scholar]
- 50.Shvo Y and Czarkie D, J. Organomet. Chem, 1989, 368, 357–365. [Google Scholar]
- 51.Tafesh AM and Weiguny J, Chem. Rev, 1996, 96, 2035–2052. [DOI] [PubMed] [Google Scholar]
- 52.Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R and Green CJ, FASEB J., 2004, 19, 284–286. [DOI] [PubMed] [Google Scholar]
- 53.Crook SH, Mann BE, Meijer AJHM, Adams H, Sawle P, Scapens D and Motterlini R, Dalton Trans., 2011, 40, 4230–4235. [DOI] [PubMed] [Google Scholar]
- 54.Rauchfuss TB, in Inorganic Syntheses, ed. Rauchfuss TB, John Wiley & Sons, Hoboken N,J, 1 edn., 2010, vol. 35, ch. 8, pp. 148–163. [Google Scholar]
- 55.Nomura K, J. Mol. Catal, 1992, 73, L1–L1. [Google Scholar]
- 56.Nomura K, J. Mol. Catal. A: Chem, 1995, 95, 203–210. [Google Scholar]
- 57.Nomura K, Chem. Lett , 1991, 20, 1679–1682. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.