

Abstract
The genetic architecture of autism spectrum disorder (ASD) is itself a diverse allelic spectrum that consists of rare de novo or inherited variants in hundreds of genes and common polygenic risk at thousands of loci. ASD susceptibility genes are interconnected at the level of transcriptional and protein networks, and many function as genetic regulators of neurodevelopment or synaptic proteins that regulate neural activity. So that the core underlying neuropathologies can be further elucidated, we emphasize the importance of first defining subtypes of ASDs based on the phenotypic signatures of genes in model systems and humans.
Intro
The autism spectrum is a clinically-heterogeneous class of neurodevelopmental disorders that has a strong genetic basis. During the last decade a substantial proportion of the genetic architecture of ASD has come to light. These studies have delivered a trove of susceptibility genes. However, this growing “parts list” does not translate immediately into a mechanistic understanding of ASD. That mechanistic understanding could emerge once the specific effects of genes on neurodevelopment are understood. Here we review and discuss the current efforts to put gene discoveries into a mechanistic framework.
An emerging spectrum of genetic risk
At one extreme of the genetic architecture of ASD are the monogenic disorders, in which a major contributor to risk is a single gene mutation or CNV. At the other extreme is polygenic risk that is measured as the sum of thousands of common risk alleles with small effects. The gap between these two extremes spans a broad spectrum of alleles that, to date, have not been well characterized. Here we summarize three major components of genetic risk, including de novo mutations, rare inherited variants and common polygenic variation and illustrate these in Figure 1.
Figure 1. Components of the genetic basis of ASD.

(A) Genetic studies have found conclusive evidence for three categories of genetic risk including polygenic variation that is common in the human population, rare variants which have occurred relatively recently in the population, and de novo mutations which occur spontaneously in offspring. We illustrate all three within a single pedigree, but this depiction does not necessarily represent a typical family, because the contributions of de novo, inherited and polygenic risk varies between individuals. We highlight two specific examples of complex genetic inheritance that have been documented: (B) cases in which risk is attributable to multiple rare variants, for example a rare gene variant (+/−) and a large duplication; and (C) ases in which risk is attributable to a de novo gene mutation (+/−) and an increased load of polygenic risk inherited from both parents. The contribution of each to risk in offspring is represented by line thickness. Seventy percent of de novo mutations originate in the paternal germline and the paternal contribution to de novo mutation is shown to be greater than the maternal contribution. Variability of ASD symptom severity in offspring is represented by the tone of shading of pedigree symbols.
De novo mutations
Early studies established that de novo structural variants (SVs) (Brandler et al., 2016; Sebat et al., 2007) and protein-altering point mutations (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) contribute to risk for ASD. It is estimated that de novo mutations of genes contribute in approximately 30% of cases, including 25% of boys and 45% of girls (Iossifov et al., 2014). As would be expected for variants with large effects, subjects that carry de novo mutations have lower non-verbal IQs than subjects who do not (Iossifov et al., 2014).
More recently, statistical methods such as the transmission and de novo association (TADA) (He et al., 2013) have been developed for testing the disease association of genes based on the frequency of de novo and transmitted mutations observed in parent-offspring trios. By this method “high confidence” autism susceptibility genes are typically defined as those which meet a false discovery rate threshold of <10%. The application of this approach to exomes or whole genomes in large family samples has since become the most effective strategy for identifying ASD susceptibility genes (De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018).
As each new study is combined with the publicly available data from its predecessors, the number of ASD susceptibility genes grows. A recent study in a combined sample of 35,584 samples (11,986 with ASD), has brought the number of high confidence genes to 102 (Satterstrom et al., 2018). Fifty three genes had a greater frequency in ASD than in the broader category of Neurodevelopmental Disorders (NDDs), suggesting that genes such as GIGYF1, KDM6B, PTEN, ANK2, KMT5B, KMT2C, and CHD8 might have a more direct influence on social behavior. Here, unless otherwise specified, the term “ASD susceptibility genes” refers to the high confidence (FDR<0.1) genes reported in papers by Sanders et al. (Sanders et al., 2015) and Satterstrom et al (Satterstrom et al., 2018). For reference we provide a list of susceptibility genes and CNVs in Supplementary table 1.
Most germline mutations happen during mitotic cell division of spermatogonial cells, a process that occurs at a constant rate in the gonad (Crow, 2000). Thus, a majority (70%) of de novo mutations originate from the father and total rate of new mutations in offspring increase by 1–2 mutations per year with his age (Kong et al., 2012; Michaelson et al., 2012). (Fig 1A).
Rare inherited variants
A portion of genetic risk for ASD consists of rare variants that are inherited from a mother or father (Fig. 1A). Some of the first examples identified were large CNVs which occur as recurrent de novo mutations in the population, but due to variable level of cognitive impairments, can be transmitted from a parent with mild or no symptoms to their offspring. These include duplications of 15q11–13 and 16p11.2 and deletions of 15q11.2 (Doornbos et al., 2009). Recently, whole genome analysis of SVs in large samples has succeeded in capturing a broader array of inherited variants including deletions that disrupt individual genes or cis-regulatory elements (Brandler et al., 2018). Evidence for the contribution of inherited protein truncating variants to ASD has also been found by exome sequencing of families (Iossifov et al., 2015; Krumm et al., 2015) Thus, a portion of the genetic architecture consists of rare coding variants with incomplete penetrance that are inherited from parents who do not meet criteria for ASD.
Recessive variants account for a small fraction of idiopathic ASD (Gamsiz et al., 2013) and developmental delay, but they account for a larger proportion (up to 30%) of cases in consanguineous families (Martin et al., 2018). DNA sequencing in such families is an effective approach for identifying cases of ASD that are attributable to rare homozygous variants (Morrow et al., 2008; Scott et al., 2016). Sequencing of consanguineous families has identified novel candidate genes associated with autism (Yu et al., 2013). For example Novarino et al identified inactivating mutations in the gene Branched Chain Ketoacid Dehydrogenase Kinase (BCKDK) in consanguineous families with autism, epilepsy, and intellectual disability (Novarino et al., 2012). Bckdk knockout mice show abnormal brain amino acid profiles and neurobehavioral deficits that respond to dietary supplementation.
Common polygenic risk
Current data from GWAS is consistent with a liability threshold model, in which many risk alleles contribute additively to the overall risk. Estimates of the heritability of ASD explained by common SNPs range from 17% (Autism Spectrum Disorders Working Group of The Psychiatric Genomics, 2017; Cross-Disorder Group of the Psychiatric Genomics et al., 2013) to 52% (Gaugler et al., 2014). A recent meta-analysis of 18,381 ASD cases and 27,969 controls has found credible evidence for five risk loci and seven additional loci shared with other psychiatric disorders (Grove et al., 2019). However, there are likely to be thousands of loci with very small effects. The contribution of all common SNPs to a trait can be summarized as a Polygenic Risk Score (PRS, (Dudbridge, 2013). A PRS constructed from previous GWAS of ASD has been shown to be significantly associated with cases in independent cohorts (Grove et al., 2017). Surprisingly, multiple studies have found that PRS for ASD correlates with higher educational attainment and IQ in the population (Brainstorm Consortium et al., 2018), opposite to what has been reported for PRS of other neurodevelopmental disorders (Niemi et al., 2018; Sniekers et al., 2017). This result has led to fascinating speculation as to the potential causative relationship of high intelligence to ASD (Crespi, 2016); but a rigorous dissection of the relationship of ASD PRS with intelligence in the general population cohorts is needed.
Complex genetic inheritance
In subjects who carry de novo CNVs or gene mutations, psychiatric diagnoses are variable and typically only a subset meet criteria for ASD (De Rubeis and Buxbaum, 2015; Malhotra and Sebat, 2012). This suggests that the determinants of psychiatric traits are multifactorial even in the context of a large-effect variant. Here, we summarize the evidence for the joint contributions of multiple rare or common genetic variants to psychiatric risk to the individual.
Oligogenic effects (≥2 hits)
Analysis of rare variants in ASD and NDD cohorts has found evidence for an oligogenic model in which variation in the clinical severity of known pathogenic mutations is influenced by the genomic burden of additional rare variants. A series of studies have demonstrated that the clinical outcomes of rare genetic disorders are influenced by rare variants in the genetic background. This was first observed for a large (500 kb) deletion of 16p12.1 that carries moderate risk for developmental disorders (Girirajan et al., 2010). Deletion carriers with developmental delay had an increased burden of secondary CNVs compared to deletion carriers in controls. Subsequent studies have shown that the clinical severity of subjects who carry CNVs at 1q21.1, 7q11.23 and 16p11.2 correlates with the number of secondary rare variants, and similar results have been observed for ASD subjects who carry gene disrupting de novo mutations (Pizzo et al., 2018). These results are consistent with a joint contribution of multiple rare variants to risk for ASD and other NDDs.
The joint effects of rare mutation and polygenic risk.
The clinical outcome of individuals that carry a rare variant of large effect can also be influenced by the background of common polygenic variation. A recent study reported that individuals with ASD that carry de novo mutations have significantly increased PRS for ASD compared to typically developing controls (Weiner et al., 2017). A second study examined the polygenic contribution to risk in a large cohort of NDDs (Niemi et al., 2018). This study reported that individuals with NDDs who carry a reportable clinical diagnostic variant have a significantly increased PRS. The joint contributions of rare CNVs and PRS are evident for other psychiatric disorders, such as schizophrenia. Bergen et al has shown that the polygenic contribution in CNV carriers is inversely proportional to the effect size of the CNV (Bergen et al., 2018), consistent with an attenuated contribution of common variation in subjects who carry a highly-penetrant rare variant.
The nature of gene action in ASDs
As will be discussed in detail in the following section, multiple ASD genes are connected within transcriptional and protein interaction networks. Consequently, a genetic effect that originates from a single gene mutation has the potential to influence the function of other ASD genes, and can thereby fan out broadly through a gene regulatory network. For example, a mutation that impacts a protein that regulates gene expression in the brain can have numerous downstream effects that are mediated through other regulatory genes in trans (Fig 2A). Other types of ASD risk alleles, such as CNV or common polygenic risk, may differ in how they directly impact gene function (Fig 2B-C). However, all 3 categories of genetic risk are similar in that their effects can also broadly distributed across a gene regulatory network.
Figure 2. Forms of gene action in ASD.
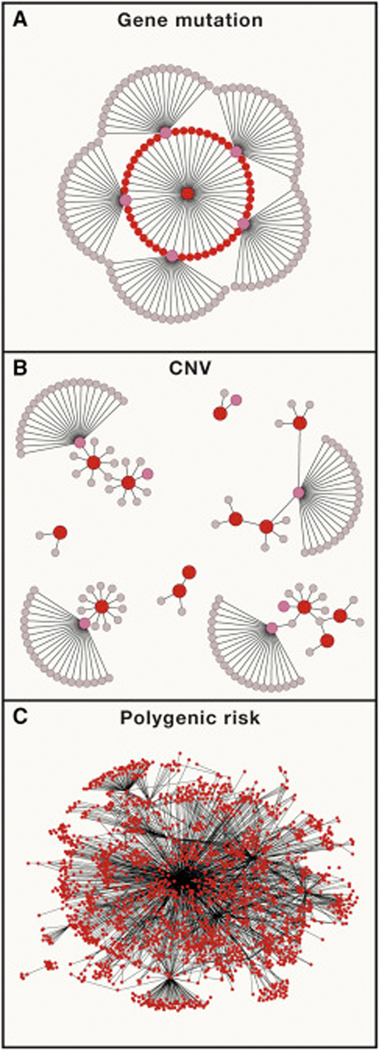
Network graphs represent interactions within a gene regulatory networks that are impacted by causal variants. Red nodes represent genes impacted directly by a risk variant in an individual case of ASD (de novo mutations, CNVs or common variants). Pink nodes represent other ASD susceptibility genes that are not mutated in the same individual but do interact closely with the primary gene mutation(s) in the network. (A) A de novo mutation in a key regulatory gene. Many ASD genes encode regulatory proteins that that control the expression of genes in the developing brain. Those target genes include other monogenic ASD genes. Thus, the effect of a single gene mutation can fan out quite broadly through a gene regulatory network. (B) Large CNVs directly alter the dosage of dozens of genes. Thus the network level effects of a CNV are distributed more broadly than the example shown in A. (C) Polygenic risk is very broadly distributed across the genome and throughout gene regulatory networks.
Large CNVs influence traits through the dosage effects of multiple genes
Large pathogenic CNVs typically affect the copy number of dozens of genes. There are a few known microdeletion syndromes where clinical features are attributable to a major driver gene within the region. For example, clinical features of Angelman, Phelan McDermid and Smith-Magenis Syndromes, which are caused by large deletions of multiple genes, can be recapitulated by point mutations in UBE3A (Fang et al., 1999) and SHANK3 (Bonaglia et al., 2006) and RAI1 (Slager et al., 2003) respectively. However, for the majority of large CNVs, studies have not found evidence for the existence of a single driver gene (De Rubeis et al., 2014; Iossifov et al., 2014). Sanders et al. examined rates of gene mutations within large CNVs and without and found no evidence for an increased rate of de novo protein truncation mutations inside of large CNVs that contained >7 genes, whereas there was a significantly increased rate of de novo mutation within smaller CNVs (Sanders et al., 2015).
Evidence from transgenic model organisms suggests that developmental phenotypes associated with CNVs are influenced by the dosage effects of multiple genes. Oligogenic effects have been characterized in detail for one CNV that is frequently observed in ASD: the deletion and duplication of 16p11.2. Pizzo et al, tested the effect of RNA-knockdown of genes on >20 classic phenotypes used to assess development in Drosophila (Pizzo et al., 2018). This study found that combinations of multiple 16p11.2 genes could elicit a variety of effects on development of the eye, wing and nervous system. Our group has studied the effects of 16p11.2 genes on craniofacial development, and found that the CNV has significant effects on craniofacial structure that are conserved in human, mouse, zebrafish, and several genes have significant effects on growth of the skull (Qiu et al.). These observations are consistent with developmental phenotypes associated with a CNV being attributable to the dosage effects of multiple genes.
If the influence of a CNV on a trait is attributable to multiple genes, this suggests that effects of the CNV on genetic regulatory networks could be to some degree more complex than the monogenic example described above. In contrast to gene mutation in which the network level effects radiate outward from a single point, a large CNV contributes multiple rare “hits” that have direct effects that are distributed more broadly across a gene regulatory network (Fig 2B).
Polygenic effects
The nature of polygenic risk is fundamentally different from that of rare variants of large effect. Polygenic risk consist of thousands of small effects that segregate independently in the population. Thus a PRS that is defined for a specific trait represents genetic effects that originate from thousands of different points within a genetic regulatory network (Fig 2C) and propagate even more broadly through effects mediated in trans through downstream regulatory genes.
ASD susceptibility genes are interconnected within gene regulatory networks
Here we describe how ASD genes show significant functional convergence within the context of gene networks, synaptic function and signaling pathways, see Figure 3.
Figure 3. Points of convergence among ASD susceptibility genes.

Multiple ASD genes interact within the context of gene regulatory networks. These are highlighted as biological processes within a single neuron (cytoplasm in blue and nucleus in pink). Convergence is evident at multiple levels of interaction including DNA binding, RNA binding and Protein-Protein interactions. Biological processes that are associated with ASD genes include the regulation of gene expression and synaptic function. ASD genes are expressed preferentially in the developing brain. Rare gene mutations in ASD also converge upon specific signaling pathways involved in the regulation of ell proliferation and differentiation including mTOR, MAPK and Wnt signaling. [Note: the ideogram (white circle) in the center and the images depicting fetal brain development are stock photos and need to be replaced with original art]
Cis regulatory targets of ASD genes are enriched for other ASD genes
A subset of ASD susceptibility genes encode proteins that bind directly to DNA or RNA and are involved in the regulation of gene expression, referred to as DNA binding proteins (DBPs) and RNA binding proteins (RBPs). For example, TBR1 encodes a transcription factor that binds DNA and regulates gene expression in the developing brain (Bedogni et al., 2010; Hevner et al., 2001). Chromatin Immunoprecipitation Sequencing (ChIP-Seq) of TBR1 in the developing neocortex found that this protein binds next to high-confidence ASD genes more frequently than other transcription factors that are expressed in the brain (Notwell et al., 2016). Similarly, the chromatin remodeling factor CHD8, binds and positively regulates other ASD genes (Cotney et al.; Sugathan et al.).
Some ASD susceptibility genes encode RBPs that are involved in posttranscriptional regulation of messenger RNA abundance, splicing or translation. A seminal example is the Fragile X mental retardation protein (FMRP) (Darnell et al., 2011), and other RBPs that are associated with ASD include CELF4 (Wagnon et al., 2012), ELAVL3 (Ince-Dunn et al., 2012) and GIGYF1 (Peter et al., 2017). RNA targets of both FMRP (Darnell et al., 2011) and CELF4 (Wagnon et al., 2012) were reported to be significantly enriched for ASD genes, and ASD candidate genes were further enriched among the common targets of FMRP and CELF4 (Wagnon et al., 2012). By compiling the binding sites for 26 regulatory proteins associated with ASD, a recent study reported that the ASD genes that are enriched among these regulatory targets consist of other regulatory genes. By contrast, they did not find a significant enrichment for ASD genes involved in “neuronal communication” (Satterstrom et al., 2018).
ASD gene mutations lead to dysregulation of other ASD genes in trans
The studies described above suggest that gene mutations that disrupt DBPs or RBPs can directly affect the regulation of numerous target genes in cis. The effects of a rare gene mutation can be propagated further by regulatory proteins. Indeed, evidence for the enrichment of ASD genes amongi genes that are dysregulated in iPSC models of CHD8 (Sugathan et al., 2014) or FOXP1 (Araujo et al., 2015) was strongest for genes that were dysregulated in trans. Similarly transcriptomic analysis of developing brain in transgenic mouse models of SETD5 (Pizzo et al., 2018), FOXP1 (Araujo et al., 2015), TBR1 (Notwell et al., 2016) suggests that mutations in these genes leads to the dysregulation of ASD genes in trans.
Gene expression alterations from in idiopathic ASD overlap with developmentally regulated genes implicated on cortical patterning, cell cycle, proliferation and neural differentiation. Transcriptome analysis of NPCs derived from idiopathic ASD with macrocephaly found that differentially expressed genes were significantly enriched for ASD genes and genes within ASD-associated CNVs (Marchetto et al., 2017). Upregulation of immune genes and downregulation of synaptic genes was observed in postmortem brain from idiopathic ASD (Voineagu et al., 2011), and this finding has been replicated in a larger ASD sample (Gupta et al., 2014). Recent analyses of a much larger cohorts of postmortem brain collections from major psychiatric disorders such as autism, schizophrenia, bipolar disorder and depression assembled by the PsychEncode Consortium (Psych et al., 2015) have lent further support to previous observation by identifying upregulation of immune/microglia and mitochondrial modules, and downregulation of neuronal and synaptic modules in ASD and schizophrenia (Gandal et al., 2018).
Protein-Protein Interactions
Analysis of ASD genes within protein-protein interaction (PPI) networks (Corominas et al., 2014; Gilman et al., 2011; O’Roak et al., 2012; Pinto et al., 2014; Sakai et al., 2011) has shown that proteins encoded by ASD genes have “high connectivity”, meaning that these proteins are more closely connected in a PPI network compared to genes that are selected at random or genes that are mutated in controls. Studies of large CNVs in ASD have shown that genes within ASD CNVs are highly connected with each other (Corominas et al., 2014) and with other ASD genes (Pinto et al., 2014). Pathways implicated from above studies included synaptic transmission, chromatin remodeling, transcriptional regulation, translational regulation, ion transport, and cell adhesion.
ASD genes are co-expressed during fetal brain development
Analysis of spatiotemporal expression of genes has revealed that ASD genes as a group are preferentially expressed in late mid-fetal prefrontal cortex, with a concentration in layer 5/6 cortical projection neurons (Willsey et al., 2013). Another study has identified several developmentally co-regulated modules of gene expression in fetal brain that are enriched in ASD risk genes, with layer 2–4 and glutamatergic projection neurons showing strongest enrichment (Parikshak et al., 2013). Likewise coexpression of genes within ASD CNVs was reported to be enriched in the developing cortex (Lin et al., 2015). Taken together, these observations highlight the relevance of early fetal brain development in the pathophysiology of ASD. Recently, common polygenic heritability of ASD was found to be significantly enriched in genes and enhancer marks that are expressed in the developing brain, with a similar enrichment of cortical cell types (Grove et al., 2017). Thus, common variation in ASD also preferentially impacts genes expressed in the fetal cortex during embryonic development.
Convergence of ASD Genes within Cell Signaling pathways
The regulation of cell-proliferation pathways in the developing brain has become another point of convergence in ASD (Ernst, 2016), specifically mammalian target of rapamycin (mTOR), mitogen activated protein kinase (MAPK) and Wnt signaling. Early studies made the observation that syndromes caused by the disruption of genes involved in mTOR signaling, including PTEN, TSC1, TSC2, and NF1 were frequently associated with brain overgrowth and ASD (Wang and Doering, 2013). Subsequently, inhibitors of mTOR have been proposed as treatments for such syndromes and one drug Everolimus has been approved for treatment of seizures in tuberous sclerosis (French et al., 2016).
Mutations in a set of genes involved in RAS/MAPK signaling form a set of syndromes known as “Rasopathies”, which are frequently associated with features of ASD (Adviento et al., 2014). MAPK (Pinto et al., 2010) and Rho GTPase (Lin et al., 2015) signaling has been implicated by gene set enrichment analysis of CNVs associated with ASD. The dysregulation of MAPK signaling has also been reported in transgenic mice carrying the ASD-associated microdeletion of 16p11.2 (Blizinsky et al., 2016; Pucilowska et al., 2015) and in other mouse models of social impairment (Faridar et al., 2014).
Dysregulation of Wnt signaling has been reported in multiple transgenic models of ASD genes. For example, knockdown of CHD8 in adult mice leads to a disruption of Wnt signaling and abnormal NPC proliferation (Durak et al., 2016). Mutations that disrupt ARID1B result in activation of Wnt signaling and dysregulation of B-catenin transcription (Vasileiou et al., 2015). Recent studies of SETD5 in mouse fetal tissue and human cells have reported that loss of SETD5 results in the activation of neuronal genes and Wnt signaling (Deliu et al., 2018).
Getting to the “core” of autism
Defining the core ASD genes
Our current knowledge of the neurobiological basis of ASD is derived almost exclusively from studies of rare variants. Some have cautioned that the genes and pathways that are implicated by rare variants may not represent the core biology of a complex trait (Wray et al.). However, it is not yet clear whether common variant associations in ASD will tell a different story. Indeed this seems unlikely. From the perspective of both rare variants and common variants, our current understanding of the genetics of ASD is generally compatible with an “omnigenic” model of gene action recently proposed by Boyle and Pritchard (Boyle et al.). This model posits that the genetic basis of complex traits is polygenic to such an extent that it becomes difficult to distinguish “core” genes that have direct effects on a trait from the many “peripheral” genes that act indirectly and which may have a distant relationship to the core biological processes. Core genes, according to Pritchard, are defined as those which have direct effects on a trait and which are not mediated through the regulation of other genes. For psychiatric traits, core genes might include those that have direct effects of neural activity such as neurotransmitter receptors and ion channels. By contrast, peripheral genes are defined as all genes which have effects that are mediated indirectly, for example through gene regulation. One of the major predictions of the omnigenic model is that a majority of trait heritability is explained by peripheral genetic effects that are propagated through genetic regulatory networks and genetic variation in core genes explains a small fraction of the overall heritability (Liu et al., 2018). The authors further assert that studies of rare variants might be more effective than GWAS for identifying core genes.
Critiques of the omnigenic model have been published elsewhere (Cox, 2017; Wray et al., 2018), but these have not addressed the model specifically with respect to ASDs or NDDs. From the perspective of ASD, major predictions of the model hold true. With respect to psychiatric traits, the most difficult aspect of the model to reconcile in our view is the rigid dichotomy of core and peripheral genes. Unlike some other complex traits, for example serum lipid levels which are controlled by a set of proteins involved in lipid transport and metabolism (Cox and Garcia-Palmieri, 1990), psychiatric traits cannot be easily reduced to dysfunction in a single biological pathway. Traits like social motivation, anxiety and aggression don’t emerge directly from a handful of gene products. Psychopathology emerges from dysfunction at the level of neural circuitry, and circuit function arises through a process of development in which multiple biological processes are tightly regulated (Polleux et al., 2007). Given that the regulation of neurodevelopment is central to ASD pathogenesis, we might argue that a master regulator of gene expression could be as near to the center of the hypothetical ASD gene network as any neurotransmitter receptor or voltage-gated sodium channel.
Defining the core neuropathologies of ASD
The diversity of clinical phenotypes across the autism spectrum is in part a reflection of the underlying genetic heterogeneity of ASD (McClellan and King, 2010). To the extent that there exist specific neuropathologies that are common among multiple ASD etiologies, identifying these could help to define the biological processes that influence social behavior. However, in conceptualizing the relationship between gene networks and cognitive dysfunction we must consider the hypothesis that the autism spectrum consists of multiple clinical subtypes each having characterisics that are driven by a subset of gene mutations and common risk alleles. To the extent that there exist multiple clinical entitities within ASD that have a distinct neurobiological basis, defining the genetic and clinical components of the autism spectrum is necessary to achieving a mechanistic understanding of it.
In principle, multiple core genes and regulators that are tightly connected in a genetic regulatory network might also have correlated phenotype profiles that underlie their predisposition to ASD (Fig. 4). Conversely, mutations of distantly related genes or reciprocal deletions or duplications of the same genes could have contrasting molecular and cellular phenotypes. Thus relationships between genes and biological processes could be defined based on the phenotype profiles of multiple genes across a common battery of assays. Modules of core genes could be defined based on their trait correlations in a manner not unlike the detection of modules based on gene co-expression (Langfelder and Horvath). Genotype-phenotype correlation has been proposed for clinical subtyping of common disease (Luo et al., 2018), and factor analysis of phenotype data has been proposed as a means to define homogenous clinical subtypes of ASD (Georgiades et al., 2013). We support the systematic phenotyping of a broad collection of ASD gene models as an experimental approach to defining subsets of interrelated genes and PRSs and to define how biological processes are influenced by these gene sets.
Figure 4. Defining core gene sets that regulate neurodevelopment based on trait correlations in human cell models.

We propose the characterization of genotype/trait correlations in cell based models as means to define sets of genes and CNVs that have common phenotype profiles which may reflect common effects on neuronal function. We illustrate an experimental pipeline where the effects of multiple gene mutations and CNVs are tested relative to isogenic controls across a series of cellular assays, Likewise, trait correlations with PRS could be tested in patient derived lines. In this manner genes, CNVs and common variants could be clustered into discrete groups. Comparing patterns of trait correlation in cell models and in clinical phenotype data (not shown) could help to identify clinical subtypes of ASD share common neuropathologies. (the heatmap in the lower right is a borrowed image. We need to create something similar from scratch and we need to increase the resolution of the 3rd image).
Recent efforts have been initiated to systematize the phenotyping of model systems and to apply a standard battery of assays to gene mutations on a single isogenic background (Deneault et al., 2018) and https://bit.ly/2Tc0lQ4). Such efforts are needed to compare the strength and directionality of effects of genes and CNVs on traits such as cell proliferation, dendritic arborization and synapse numbers, and electrophysiological network signals.
In order to characterize the effects of gene variants on neurodevelopment, model systems are needed that can elucidate the origins, migration, fate and activity of human cells. Induced pluripotent stem cells (iPSCs) are attractive human models for understanding complex diseases, and iPSC lines have been generated for a variety monogenic ASD diseases. A key advantage of human cellular models is the ability to model the effects of CNVs or gene mutations in isogenic cell lines (Deneault et al., 2019b) or to model naturally occurring variation in cell lines derived from patients (Chailangkarn et al.). Furthermore, results from recent studies highlight how different gene mutations can have different (sometimes opposite) effects on cellular phenotypes, which highlight how ASD might result from different types of neuropathology.
Cell proliferation and brain growth.
A robust finding from human clinical studies of ASD has been the observation of significantly accelerated brain overgrowth during the first three years of life in a subset of cases (Courchesne et al.). As genetic findings have emerged, it has become evident that macrocephaly is a characteristic of some genetic subtypes of ASD while the opposite phenotype (microcephaly) is associated with other genetic disorders within ASD. For example for CNVs at two loci, 1q21.1 and 16p11.2, reciprocal deletions and duplications have opposite effects on brain growth (Malhotra and Sebat, 2012). Among the ASD genes that have effects on head size, mutations in CHD8 (Bernier et al.) and PTEN (Butler et al., 2005) are associated with macrocephaly and mutations of DYRK1A (Courcet et al., 2012) and CDKL5 (Archer et al., 2006) are associated with microcephaly.
Cellular models have the potential to elucidate the basis for these different effects on cell proliferation. IPSC models of rare microcephaly syndromes have been shown to undergo loss of neuronal progenitor cells and premature neural differentiation (Lancaster et al., 2013). Human iPSC-derived Neural progenitor cells (NPCs) from subjects with ASD and early developmental brain enlargement displayed rapid proliferation, consistent with altered cell-cycle regulation underlying neuroanatomical traits (Marchetto et al., 2017). Neurons derived from this cohort formed fewer excitatory synapses and matured into defective neuronal networks with less bursting. Cellular models of reciprocal deletions and duplications of 16p11.2 recapitulate mirror effects on cell proliferation and reveal mirror effects on synaptic density (Deshpande et al., 2017). These results suggest that standard assays for cell proliferation and differentiation could detect genetic effects on development in specific gene models.
Dendritic arborization and synapse number.
Early studies of human cellular models were performed on neural cell lines derived from Rett Syndrome patients with loss-of-function mutations in the gene MECP2. Human cortical neurons displayed reduced arborization and lower glutamatergic synaptic puncta, leading to defects on neural networks (Marchetto et al., 2010). Reductions in neurite outgrowth, dendritic arborization and excitatory synapses are neuronal phenotypes that have been commonly observed in cellular models of gene mutation, including models of SHANK3 (Shcheglovitov et al., 2013; Yi et al., 2016), FMR1 (Doers et al., 2014) and CACNA1C (Krey et al., 2013). Importantly, cellular models of the reciprocal MECP2 gain of function (MECP2 duplication syndrome, (Lugtenberg et al., 2009) exhibit the opposite effects of increased synapses and dendrites (Nageshappa et al., 2016). An increase in dendrite length and synaptogenesis has also been reported in neuron models of Williams syndrome (Chailangkarn et al., 2016) and SHANK2 (Zaslavsky et al., 2019), further highlight how different mutations of the same gene or different genes can have opposing effects on a cellular phenotype
Electrophysiology.
In a systematic screen of ten ASD susceptibility genes (AFF2/FMR2, ANOS1, ASTN2, ATRX, CACNA1C, CHD8, DLGAP2, KCNQ2, SCN2A, TENM1) using CRISPR/Cas9-created iPSC lines and isogenic controls (Deneault et al., 2018), patch-clamp recordings detected reduced excitatory postsynaptic potentials (EPSPs) in a subset of gene models including ATRX, AFF2, KCNQ2, SCN2A, and ASTN2. Other studies have reported that neuronal models of SHANK2 (Zaslavsky et al., 2019), CNTNAP5 and EHMT1 (Deneault et al., 2019a) display hyperconnectivity. Electrophysiological behavior of neurons thus represent a readout that could potentially distinguish genes with different effects on neural activity.
Models of early fetal brain development.
An attractive approach to systematically record multiple phenotypes within a single system is to use brain organoids, three-dimensional self-assembled multi-cellular structures that mimic the organization, transcriptional and epigenetic signature of a developing human brain (Lancaster et al.). Brain organoids can be used to dynamically study neural proliferation of progenitor cells, migration, differentiation and maturation (Trujillo et al., 2018). Brain organoid models have been shown to recapitulate cell proliferation effects in microcephaly (Thomas et al., 2017) and have been used to study neural migration defects in Timothy syndrome, a rare ASD form caused by mutations in the Cav1.2 calcium channel gene (Birey et al., 2017). Long-term maturation of the neural networks on brain organoids can lead to EEG-like oscillatory waves that could be directly compared to EEG signals from ASD individuals. However, It is currently unknown if brain organoids can functionally mature to a stage in which network effects could be modeled. As methodologies improve, we may be able to identify additional or more specific in vitro correlates of behaviors, and network‐level changes that accompany them, in human iPSC‐derived neurons.
Mapping core genes onto human psychiatric traits
The last and arguably the most important piece to the precision psychiatry puzzle is to relate sets of core genes to specific dimensions of psychopathology in humans. This is not a task to be taken lightly. During the past decade, significant efforts were undertaken to define the genetic underpinnings of psychiatric “endophenotypes” (Iacono et al., 2014). These studies, which were large for studies that apply biometrics like electroencephalography (EEG) but small by GWAS standards, were largely unsuccessful in achieving the goal of mapping genes to endophenotypes. This is unsurprising in retrospect given that the endophenotypes themselves were complex traits.
With hundreds of ASD susceptibility genes now identified, this problem can now be revisited with a reverse-genetic approach: starting with the genotype and determining how genes influence clinical phenotypes. Large cohort studies of neuroimaging (Franke et al., 2016) and medical health information (Collins and Varmus, 2015) and efforts to define dimensional psychiatric traits in large samples (https://bit.ly/2qTsged) and in rare genetic disorders (https://bit.ly/2VYSk1O) are providing opportunities conduct such studies. For rare mutations, a key challenge is in obtaining a large sample of subjects with a specific genetic disorder. Recruitment of large cohorts is being achieved for the most frequent genetic disorders (D’Angelo et al.; Gur et al.; Simons VIP Consortium). For instance, the International Brain Behavior Consortium (IBBC) has begun to apply a battery of dimensional measures of psychopathology systematically across multiple genetic disorders (Gur et al., 2017), with a particular focus on CNVs that carry significant risk. Results suggest that the developmental course of clinical phenotypic expression of CNV carriers resembles that of idiopathic populations (Monks et al., 2014). At the same time there are measurable differences in phenotypic expression between different genetic disorders, which can serve as windows into distinct molecular subtypes of ASD. For example, reciprocal deletions of duplications of multiple loci associate with opposing brain phenotypes in neural circuits involved in social cognition, language and reward (Deshpande and Weiss, 2018; Lin et al., 2017; Maillard et al., 2015). Cognitive and language studies in carriers of the reciprocal deletions and duplication of 16p11.2 have reported mirror effects on specific linguistic skills, deletion carriers having reduced performance and and duplication carriers having greater performance than IQ-matched controls (Hippolyte et al., 2016). This finding highlights the importance of fine-grained dimensional phenotyping, and further suggests that the specificity of genotype-phenotype relationships, which are evident at the level of pathways and molecular and cellular phenotypes can be extended to human psychiatric traits.
Conclusion
Within the last decade, ASD has gone from being one of the most mysterious and misunderstood common disorders to one of the success stories of the post-genomics era. We attribute this success to the arrival of game-changing sequencing technologies and the creation of large genomic datasets via National Institutes of Health and privately funded resources (Fischbach and Lord, 2010; RK et al., 2017; Spark Consortium, 2018). Bridging the gap between genes, neurodevelopment and cognitive function will likely require adoption of similar big data approaches in the fields of translational neuroscience and clinical psychiatry. In this review we have not addressed the formidable technical challenges to doing translational science at scale, including the number of cellular assays or clinical measures that could be evaluated across a significant number of genes, and the number of samples or subjects required for adequate statistical power. Making precision psychiatric medicine a reality is a task for the next decade. Gene discoveries have accelerated this process, by providing a foundation of knowledge that will form the basis for clinical sequencing and the development of new treatment strategies for ASD.
Supplementary Material
Acknowledgments
The Beyster Center for Psychiatric Genomics is supported by a gift from the Beyster Family Foundation. JS is supported by grants from the NIH (R21MH113179, U01MH109501, R01MH113715) and the Simons Foundation (SFARI #606768). LI is supported by R01MH109885; R01MH108528; R21MH104766 and the Simons Foundation (SFARI #345469). AM is supported by SFARI (#345469), and a NARSAD Independent Investigator Grant.
Footnotes
Conflicts of Interest
Dr. Muotri is a co-founder and has equity interest in TISMOO, a company dedicated to genetic analysis focusing on therapeutic applications customized for autism spectrum disorder and other neurological disorders with genetic origins. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adviento B, Corbin IL, Widjaja F, Desachy G, Enrique N, Rosser T, Risi S, Marco EJ, Hendren RL, Bearden CE, et al. (2014). Autism traits in the RASopathies. J Med Genet 51, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, Rieger MA, Huang HC, Rajkovich K, Loerwald KW, et al. (2015). FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 29, 2081–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R, O’Callaghan F, Huyton M, O’Regan M, Tolmie J, Sampson J, et al. (2006). CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 43, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Autism Spectrum Disorders Working Group of The Psychiatric Genomics, C. (2017). Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RA, Beyer RP, Bammler TK, Rubenstein JL, and Hevner RF (2010). Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A 107, 13129–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergen SE, Ploner A, Howrigan D, Group CNVA, the Schizophrenia Working Group of the Psychiatric Genomics, C., O’Donovan MC, Smoller JW, Sullivan PF, Sebat J, Neale B, et al. (2018). Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia. Am J Psychiatry, appiajp201817040467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N, et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blizinsky KD, Diaz-Castro B, Forrest MP, Schurmann B, Bach AP, Martin-de-Saavedra MD, Wang L, Csernansky JG, Duan J, and Penzes P (2016). Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc Natl Acad Sci U S A 113, 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonaglia MC, Giorda R, Mani E, Aceti G, Anderlid BM, Baroncini A, Pramparo T,and Zuffardi O (2006). Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet 43, 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle EA, Li YI, and Pritchard JK (2017). An Expanded View of Complex Traits: FromPolygenic to Omnigenic. Cell 169, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Consortium Brainstorm, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J,Duncan L, Escott-Price V, Falcone GJ, Gormley P, et al. (2018). Analysis of shared heritability in common disorders of the brain. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, et al. (2018). Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandler WM, Antaki D, Gujral M, Noor A, Rosanio G, Chapman TR, Barrera DJ, Lin GN, Malhotra D, Watts AC, et al. (2016). Frequency and Complexity of De Novo Structural Mutation in Autism. Am J Hum Genet 98, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, et al. (2016). A human neurodevelopmental model for Williams syndrome. Nature 536, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins FS, and Varmus H (2015). A new initiative on precision medicine. N Engl J Med 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L, Broly M, Rodriguez M, Tam S, Trigg SA, et al. (2014). Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat Commun 5, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. (2015). The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courcet JB, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P, Lambert L,Lemesle M, Thevenon J, Gigot N, et al. (2012). The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet 49, 731–736. [DOI] [PubMed] [Google Scholar]
- 21.Courchesne E, Campbell K, and Solso S (2011). Brain growth across the life span inautism: age-specific changes in anatomical pathology. Brain Res 1380, 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox NJ (2017). Comments on Pritchard Paper. Journal of Psychiatry and Brain Science 2, S5. [Google Scholar]
- 23.Cox RA, and Garcia-Palmieri MR (1990). Cholesterol, Triglycerides, and AssociatedLipoproteins. In Clinical Methods: History The, Physical, and Laboratory Examinations, rd, Walker HK, Hall WD, and Hurst JW, eds. (Boston: ). [Google Scholar]
- 24.Crespi BJ (2016). Autism As a Disorder of High Intelligence. Front Neurosci 10, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cross-Disorder Group of the Psychiatric Genomics, C., Lee SH, Ripke S, Neale BM,Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, et al. (2013). Genetic relationship between five psychiatric disorders estimated from genomewide SNPs. Nat Genet 45, 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crow JF (2000). The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1, 40–47. [DOI] [PubMed] [Google Scholar]
- 27.D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, Hanson E, Maillard AM, Faucett WA, Mace A, et al. (2016). Defining the Effect of the 16p11.2 Duplication on Cognition, Behavior, and Medical Comorbidities. JAMA Psychiatry 73, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Rubeis S, and Buxbaum JD (2015). Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet 24, R24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deliu E, Arecco N, Morandell J, Dotter CP, Contreras X, Girardot C, Kasper EL, Kozlova A, Kishi K, Chiaradia I, et al. (2018). Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat Neurosci 21, 1717–1727. [DOI] [PubMed] [Google Scholar]
- 32.Deneault E, Faheem M, White SH, Rodrigues DC, Sun S, Wei W, Piekna A, Thompson T, Howe JL, Chalil L, et al. (2019a). CNTN5(−)(/+)or EHMT2(−)(/+)human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, Wang Z, Alexandrova R, Pellecchia G, Wei W, et al. (2018). Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Reports 11, 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, Wang Z, Alexandrova R, Pellecchia G, Wei W, et al. (2019b). Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Reports 12, 427–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshpande A, and Weiss LA (2018). Recurrent reciprocal copy number variants: Roles and rules in neurodevelopmental disorders. Dev Neurobiol 78, 519–530. [DOI] [PubMed] [Google Scholar]
- 36.Deshpande A, Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, Wiita AP, Jan YN, Ullian EM, and Weiss LA (2017). Cellular Phenotypes in Human iPSC-Derived Neurons from a Genetic Model of Autism Spectrum Disorder. Cell Rep 21, 2678–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, and Bhattacharyya A (2014). iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev 23, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CAL, Dijkhuizen T, Bijlsma EK, Gijsbers ACJ, Hilhorst-Hofstee Y, Hordijk R, Verbruggen KT, Kerstjens-Frederikse WS, et al. (2009). Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. European Journal of Medical Genetics 52, 108–115. [DOI] [PubMed] [Google Scholar]
- 39.Dudbridge F (2013). Power and predictive accuracy of polygenic risk scores. PLoS Genet 9, e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, and Tsai LH (2016). Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ernst C (2016). Proliferation and Differentiation Deficits are a Major Convergence Pointfor Neurodevelopmental Disorders. Trends Neurosci 39, 290–299. [DOI] [PubMed] [Google Scholar]
- 42.Fang P, Lev-Lehman E, Tsai TF, Matsuura T, Benton CS, Sutcliffe JS, Christian SL,Kubota T, Halley DJ, Meijers-Heijboer H, et al. (1999). The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 8, 129–135. [DOI] [PubMed] [Google Scholar]
- 43.Faridar A, Jones-Davis D, Rider E, Li J, Gobius I, Morcom L, Richards LJ, Sen S, andSherr EH (2014). Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol Autism 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischbach GD, and Lord C (2010). The Simons Simplex Collection: a resource foridentification of autism genetic risk factors. Neuron 68, 192–195. [DOI] [PubMed] [Google Scholar]
- 45.Franke B, Stein JL, Ripke S, Anttila V, Hibar DP, van Hulzen KJE, Arias-Vasquez A,Smoller JW, Nichols TE, Neale MC, et al. (2016). Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat Neurosci 19, 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, et al. (2016). Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388, 2153–2163. [DOI] [PubMed] [Google Scholar]
- 47.Gamsiz ED, Viscidi EW, Frederick AM, Nagpal S, Sanders SJ, Murtha MT, Schmidt M, Simons Simplex Collection Genetics, C., Triche EW, Geschwind DH, et al. (2013).Intellectual disability is associated with increased runs of homozygosity in simplex autism. Am J Hum Genet 93, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, et al. (2018). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M,Manaa D, Pawitan Y, Reichert J, et al. (2014). Most genetic risk for autism resides with common variation. Nat Genet 46, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Georgiades S, Szatmari P, Boyle M, Hanna S, Duku E, Zwaigenbaum L, Bryson S,Fombonne E, Volden J, Mirenda P, et al. (2013). Investigating phenotypic heterogeneity in children with autism spectrum disorder: a factor mixture modeling approach. J Child Psychol Psychiatry 54, 206–215. [DOI] [PubMed] [Google Scholar]
- 51.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare denovo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, et al. (2010). A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 42, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grove J, Ripke S, Als TD, Mattheisen M, Walters R, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. (2017). Common risk variants identified in autism spectrum disorder. bioRxiv. [Google Scholar]
- 54.Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, and Arking DE (2014). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gur RE, Bassett AS, McDonald-McGinn DM, Bearden CE, Chow E, Emanuel BS, Owen M, Swillen A, Van den Bree M, Vermeesch J, et al. (2017). A neurogenetic model for the study of schizophrenia spectrum disorders: the International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol Psychiatry 22, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. (2013). Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 9, e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, and Rubenstein JL (2001). Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29, 353–366. [DOI] [PubMed] [Google Scholar]
- 59.Hippolyte L, Maillard AM, Rodriguez-Herreros B, Pain A, Martin-Brevet S, Ferrari C, Conus P, Mace A, Hadjikhani N, Metspalu A, et al. (2016). The Number of Genomic Copies at the 16p11.2 Locus Modulates Language, Verbal Memory, and Inhibition. Biol Psychiatry 80, 129–139. [DOI] [PubMed] [Google Scholar]
- 60.Iacono WG, Vaidyanathan U, Vrieze SI, and Malone SM (2014). Knowns and unknowns for psychophysiological endophenotypes: integration and response to commentaries. Psychophysiology 51, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ince-Dunn G, Okano HJ, Jensen KB, Park WY, Zhong R, Ule J, Mele A, Fak JJ, Yang C, Zhang C, et al. (2012). Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron 75, 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iossifov I, Levy D, Allen J, Ye K, Ronemus M, Lee YH, Yamrom B, and Wigler M (2015). Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci U S A 112, E5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, andDolmetsch RE (2013). Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci 16, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat Genet 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlationnetwork analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin A, Ching CRK, Vajdi A, Sun D, Jonas RK, Jalbrzikowski M, Kushan-Wells L, Pacheco Hansen L, Krikorian E, Gutman B, et al. (2017). Mapping 22q11.2 Gene Dosage Effects on Brain Morphometry. J Neurosci 37, 6183–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin GN, Corominas R, Lemmens I, Yang X, Tavernier J, Hill DE, Vidal M, Sebat J, and Iakoucheva LM (2015). Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 85, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu X, Li YI, and Pritchard JK (2018). Trans effects on gene expression can driveomnigenic inheritance. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J, et al. (2009). Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet 17, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo Y, Mao C, Yang Y, Wang F, Ahmad FS, Arnett D, Irvin MR, and Shah SJ(2018). Integrating hypertension phenotype and genotype with hybrid non-negative matrix factorization. Bioinformatics. [Google Scholar]
- 75.Maillard AM, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S, Dukart J, Ferrari C, Conus P, Mannik K, et al. (2015). The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry 20, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malhotra D, and Sebat J (2012). CNVs: harbingers of a rare variant revolution inpsychiatric genetics. Cell 148, 1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, Beltrao-Braga P, Trujillo CA, Mendes APD, Padmanabhan K, et al. (2017). Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 22, 820–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, and Muotri AR (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin HC, Jones WD, McIntyre R, Sanchez-Andrade G, Sanderson M, Stephenson JD, Jones CP, Handsaker J, Gallone G, Bruntraeger M, et al. (2018). Quantifying the contribution of recessive coding variation to developmental disorders. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McClellan J, and King MC (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. [DOI] [PubMed] [Google Scholar]
- 81.Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. (2012). Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Monks S, Niarchou M, Davies AR, Walters JT, Williams N, Owen MJ, van den Bree MB, and Murphy KC (2014). Further evidence for high rates of schizophrenia in 22q11.2 deletion syndrome. Schizophr Res 153, 231–236. [DOI] [PubMed] [Google Scholar]
- 83.Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E,Vanderhaeghen P, Verfaillie CM, Raitano S, Kumar A, et al. (2016). Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry 21, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Niemi MEK, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, et al. (2018). Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Notwell JH, Heavner WE, Darbandi SF, Katzman S, McKenna WL, Ortiz-Londono CF, Tastad D, Eckler MJ, Rubenstein JL, McConnell SK, et al. (2016). TBR1 regulates autism risk genes in the developing neocortex. Genome Res 26, 1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, Silhavy JL, Kara M, Khalil RO, Ben-Omran T, et al. (2012). Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, andGeschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peter D, Weber R, Sandmeir F, Wohlbold L, Helms S, Bawankar P, Valkov E, Igreja C, and Izaurralde E (2017). GIGYF½ proteins use auxiliary sequences to selectively bind to 4EHP and repress target mRNA expression. Genes Dev 31, 1147–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, McCready E, Pichon O, Le Caignec C, Van Dijck A, et al. (2018). Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Polleux F, Ince-Dunn G, and Ghosh A (2007). Transcriptional regulation of vertebrate axon guidance and synapse formation. Nat Rev Neurosci 8, 331–340. [DOI] [PubMed] [Google Scholar]
- 96.Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, et al. (2015). The PsychENCODE project. Nat Neurosci 18, 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, and Landreth GE (2015). The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci 35, 3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qiu Y, Arbogast T, Martin Lorenzo S, Li H, Shih T, Ellen R, Hong O, Cho S, Shanta O, Timothy P, et al. (2019). Oligogenic effects of 16p11.2 copy number variation on craniofacial development. bioRxiv, 540732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.RK CY, Merico D, Bookman M, J, L.H., Thiruvahindrapuram, B., Patel, R.V., Whitney, J., Deflaux, N., Bingham, J., Wang, Z., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, and Zoghbi HY (2011). Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med 3, 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins RL, Grove J, Klei L, et al. (2018). Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. bioRxiv. [Google Scholar]
- 104.Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, Gabriel SB, Belkadi A, Boisson B, Abel L, et al. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet 48, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simons VIP Consortium (2012). Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron 73, 1063–1067. [DOI] [PubMed] [Google Scholar]
- 108.Slager RE, Newton TL, Vlangos CN, Finucane B, and Elsea SH (2003). Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 33, 466–468. [DOI] [PubMed] [Google Scholar]
- 109.Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, Taskesen E, Hammerschlag AR, Okbay A, Zabaneh D, et al. (2017). Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence. Nat Genet 49, 1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Consortium Spark (2018). SPARK: A US Cohort of 50,000 Families to Accelerate Autism Research. Neuron 97, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A 111, E4468–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, Macia A, Crow YJ, and Muotri AR (2017). Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 21, 319–331 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Trujillo CA, Gao R, Negraes PD, Chaim IA, Domissy A, Vandenberghe M, Devor A, Yeo GW, Voytek B, and Muotri AR (2018). Nested oscillatory dynamics in cortical organoids model early human brain network development. bioRxiv, 358622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, Behrens J, Reis A, and Hadjihannas MV (2015). Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. Am J Hum Genet 97, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wagnon JL, Briese M, Sun W, Mahaffey CL, Curk T, Rot G, Ule J, and Frankel WN (2012). CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. PLoS Genet 8, e1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang H, and Doering LC (2013). Reversing autism by targeting downstream mTOR signaling. Front Cell Neurosci 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, et al. (2017). Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 49, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wray NR, Wijmenga C, Sullivan PF, Yang J, and Visscher PM (2018). Common Disease Is More Complex Than Implied by the Core Gene Omnigenic Model. Cell 173, 1573–1580. [DOI] [PubMed] [Google Scholar]
- 121.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, and Sudhof TC (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, et al. (2013). Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, Zhao M, Ross PJ, El Hajjar J, Romm A, et al. (2019). SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci 22, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.