

Abstract
Despite a prevailing view that advances in cancer therapy will come through selective targeting of enzymes encoded by mutated oncogenes responsible for the neoplastic phenotype, recent advances in the treatment of patients with chronic lymphocytic leukemia (cLL) have instead exploited knowledge of its biology. indeed, cLL cells depend on interactions with cells and soluble factors present in the tumor microenvironment for proliferation and survival. B-cell receptor signaling and chemokine-receptor signaling play prominent roles. Elucidation of these signaling pathways has defined physiologic targets for drugs, such as ibrutinib, which inhibit Bruton tyrosine kinase and are therapeutically effective. The characteristic high-level expression of BCL2 in CLL that can enhance leukemia-cell survival has now become an Achilles heel targeted by clinically effective drugs such as venetoclax. Here we discuss advances in such targeted therapy and highlight other disease attributes, such as the distinctive expression of ROR1, which may be targeted for clinical benefit, alone or in combination with other targeted therapies.
Keywords: B-cell receptor signaling antagonists, BCL2 antagonist, cirmtuzumab, CLL, ibrutinib, leukemia microenvironment, ROR1, targeted therapy, venetoclax
Although chemotherapy1–3 and, more recently chemoimmunotherapy4–6 have been the mainstay for therapy of patients with chronic lymphocytic leukemia (CLL), the advent of targeted therapy has improved clinical outcomes and changed clinical practice. Such targeted therapies exploit our knowledge of the biology of CLL, which in some respects is a fastidious malignancy highly dependent on survival and growth factors elaborated by the leukemia microenvironment. Studies on such interactions have identified numerous targets for therapy (Fig. 1). Chronic lymphocytic leukemia cells also express distinctively high levels of BCL2, an antiapoptotic protein that plays an integral role in leukemia-cell survival. Targeting this protein also has proven to be highly effective in the treatment of patients with this disease.
FIGURE 1.

Cross-talk and survival-signaling pathways within the leukemia microenvironment.
The CLL Microenvironment
Chronic lymphocytic leukemia cells depend on interactions with cells and soluble factors present in the tumor microenvironment for proliferation and survival (Fig. 1).7 The migration of CLL cells into the lymphoid tissue primarily is mediated through CXCR4 in response to CXCL12,8 which is secreted mainly by nurse-like cells (NLCs, otherwise called lymphoma-associated macrophages) and mesenchymal-derived stromal cells.9,10 Chronic lymphocytic leukemia cells also are attracted to lymph nodes via CCR7 in response to the chemokines CCL19 and CCL21, which are produced by high endothelial venules.11,12 The basement membranes of high endothelial venules also express hyaluronan, which can interact with CD44, a signaling glycosaminoglycan expressed by CLL cells that can recruit membrane-associated receptor-tyrosine kinases or their substrates and thereby facilitate cell signaling.13,14 It also might enhance the production of active matrix metallopeptidase 9.15,16 Once in tissues, CLL cells can derive survival support from the same chemokines, as well as from additional factors elaborated by accessory cells in the CLL microenvironment. For example, CLL cells come in contact with NLCs that can promote CLL-cell survival through the production of CXCL12. Nurse-like cells also express BAFF (B cell–activating factor of the TNF family) and APRIL (a proliferation-inducing ligand),17 which can complement the survival stimulus afforded by CXCL12.9 Nurse-like cell–CLL interactions also can promote CLL-cell survival through cognate interactions between CD31 and CD38, which are expressed on NLC and CLL cells, respectively.18,19 In turn, CLL cells may secrete chemokines, such as CCL3 and CCL4,20 which can recruit T cells and NLC-precursor cells (monocytes) to the CLL microenvironment. T cells in that microenvironment might become activated and provide CLL cells with proliferative signals through CD154-CD40 interactions, as well as through the secretion of multiple cytokines, such as interleukin 2 (IL-2), IL-4, IL-10, and IL-21.21 In return, activated CLL cells secrete CCL3, CCL4, and CCL22, chemokines that can attract more CD4+ T cells into the CLL microenvironment. Stromal cells also can contribute to CLL-cell survival via the secretion of CXCL12 and CXCL1321 and. like NLCs, also via the production of various Wnt factors, including Wnt5a (wingless-type MMTV integration site family, member 5a),22 which can interact with ROR1 (receptor tyrosine kinase-like orphan receptor 1) expressed by CLL cells.23 Chronic lymphocytic leukemia-mesenchymal-derived stromal cells contact also can be established through vascular cell adhesion protein 1–CD49d interactions that contribute to CLL-cell survival.24 In tissues, CLL cells can be exposed to environmental and/or self-antigens that trigger cell activation through interactions with the surface immunoglobulin (sIg) expressed by CLL cells.25 Stimulation from ligation of sIg with antigen can amplify the responsiveness of CLL cells to the signals and factors provided by the CLL microenvironment.
Inhibitors of B-Cell Receptor and Chemokine-Receptor Signaling
Drugs that interfere with B-cell receptor (BCR) signaling have clinical activity in the treatment of patients with CLL. Drugs such as ibrutinib and idelalisib target intracellular kinases that are activated in response to ligation of sIg and thereby catalyze the cascade of intracellular events leading to B-cell stimulation (Fig. 2). The kinases targeted by these drugs also are involved in chemokine-receptor signaling.26–28 As such, use of these agents impairs the capacity of leukemia cells to recirculate between the blood and the protective microenvironmental niches present in lymphoid tissue. Because of this, treatment of CLL patients with these drugs generally results in rapid shrinkage of the lymph nodes and concomitant enhanced lymphocytosis, which abates over time as the circulating leukemia cells gradually succumb to depravation from the survival signals afforded by the lymphoid-tissue microenvironment (Fig. 1).
FIGURE 2.
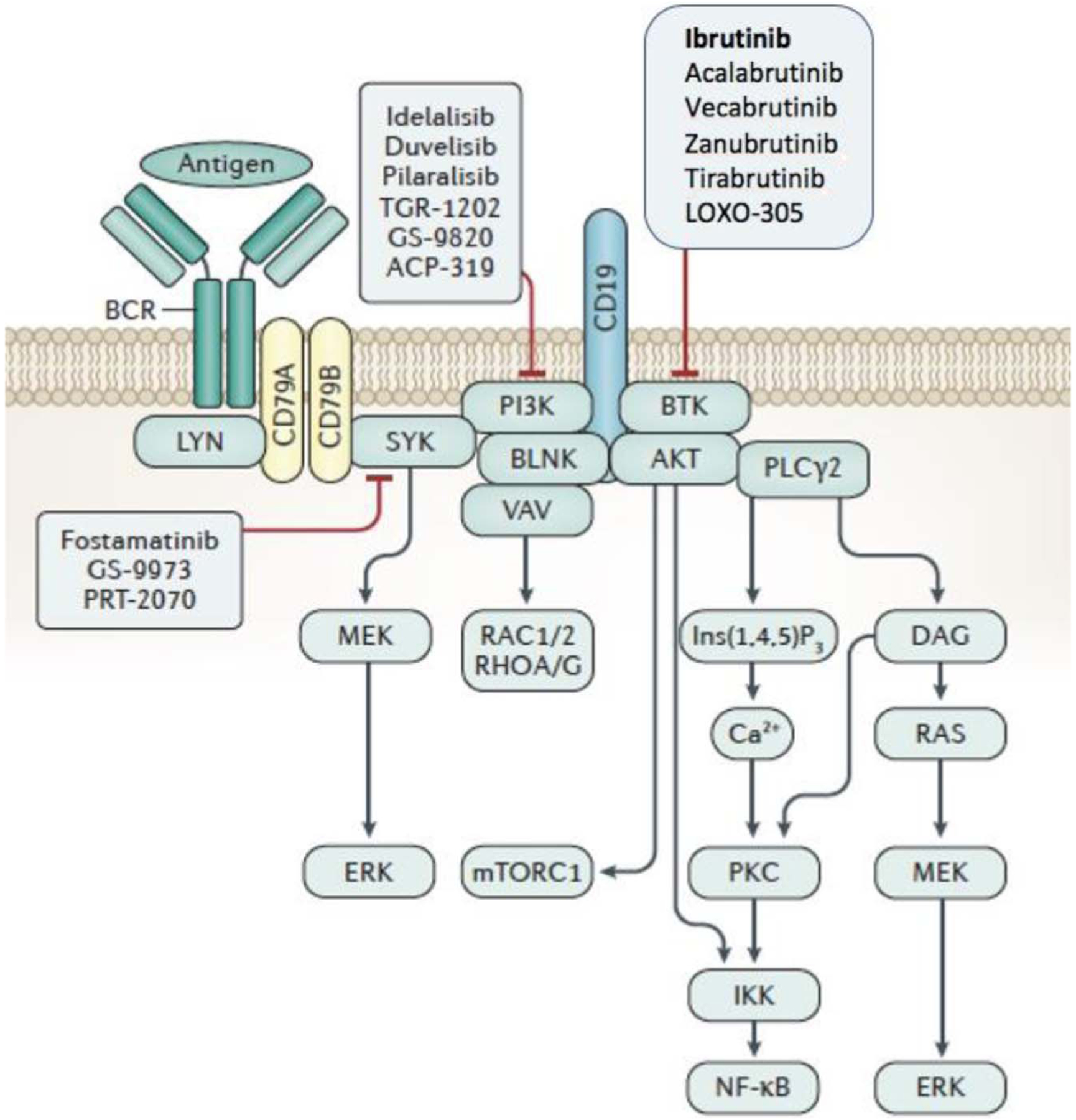
B-cell receptor signaling triggers formation of a multicomponent “signalosome,” which can activate BTK, AKT, PI3K, PLCγ2, and BLNK, and CD19, a coreceptor important for PI3K activation. Inhibitors target enzymes in this signaling pathway. Modified from Kipps et al.7
Ibrutinib and Second-Generation “Brutinibs” That Inhibit Bruton Tyrosine Kinase
Clinical trials have demonstrated that continuous ibrutinib therapy provides for progression-free survival (PFS) that is superior to that of chemotherapy-based regimens. Such trials also have shown improvements in overall survival and/or outcome for patients who were treated with ibrutinib compared with matched patients who were treated with anti-CD20 antibodies,29,30 chemotherapy,31 or chemoimmunotherapy.32,33 The improvement in survival is particularly apparent for patients who have CLL cells with del(17p), and/or inactivating mutations in TP53, which mitigate the efficacy of chemotherapy.34 A phase III trial showed that treatment-naive fit patients who were able to tolerate more intensive chemoimmunotherapy had an improved median PFS when treated with ibrutinib and rituximab rather than fludarabine, cyclophosphamide, and rituximab,35 long considered one of the most effective treatment regimens.36 Similarly, treatment-naive patients 65 years or older had a superior median PFS when treated with ibrutinib than with bendamustine and rituximab (BR).37 Patients treated with ibrutinib and rituximab in another arm of this same trial did not have a significant improvement in outcome,37 making it difficult to justify using anti-CD20 monoclonal antibodies (mAbs) in combination with ibrutinib.38 In view of the demonstrated clinical effectiveness of ibrutinib, the National Comprehensive Cancer Network committee on CLL/SLL has recommended primary consideration of single-agent ibrutinib as initial therapy for patients with CLL.38
Despite having excellent clinical activity, ibrutinib generally cannot eradicate the disease or induce durable responses in the absence of continuous therapy.39,40 Prognosis may be poor for patients with aggressive disease who discontinue ibrutinib soon after initiation of therapy, owing to the potential for rapid disease progression upon cessation of therapy. Moreover, the proportion of patients who achieve a complete response (CR) to ibrutinib appears consistently below 7%.29,41–44 As such, it generally is not recommended to discontinue therapy with ibrutinib unless there is demonstrated intolerance or resistance to therapy.
The need for continuous therapy is problematic for patients who experience even mild adverse effects with ibrutinib, such as diarrhea, onychoschizia, myalgias, arthralgias, hypertension, or increased bruising due to drug-induced impairment in platelet function.45 The costs of continuous therapy also may poise a financial burden.46 Some patients may note improvement in drug-related symptoms over time with continued therapy, especially if they have resolution of lymphadenopathy and lymphocytosis. However, the incidence of other adverse effects, such as drug-related hypertension or atrial fibrillation, may increase over time on therapy. Also, despite noted improvement in some immunologic parameters with therapy,47 there are numerous reports of serious opportunistic infections occurring in patients on protracted ibrutinib therapy.48–54
Even with continuous therapy, remissions are not durable for all patients, particularly those with relapsed disease who have CLL cells with del(17p) and/or complex cytogenetics, for whom the estimated 30-month PFS on therapy is approximately 60%.55 Furthermore, the proportion of patients wanting or electing to discontinue therapy with these agents appears higher in community practice than reported in clinical trials,56,57 possibly due to the intolerance of patients facing lifelong therapy with even low-grade toxicity and/or the costs of therapy. In addition, even with continuous therapy, patients can develop drug resistance due to acquired mutations in genes encoding proteins involved in BCR signaling58 or Richter transformation.59,60
Second-generation small molecule inhibitors of Bruton tyrosine kinase (BTK) share with ibrutinib the same root designation “brutinib.” Among these are acalabrutinib,61 ONO/GS-4059,62 zanubrutinib (BGB-3111),63 SNS-062,64 and others. Each is touted to have a potentially higher therapeutic index than ibrutinib due to greater specificity for BTK and/or different mechanisms of action, potentially resulting in fewer “off-target” effects. Moreover, some of the drugs do not require covalent bonding with BTK and thus may be effective in patients who develop resistance to ibrutinib because of an acquired mutation in BTK, causing a C→S substitution at position 418 (BTKC481S), which precludes the covalent bonding of ibrutinib to BTK.58 Nonetheless, it does not appear that any one of the second-generation “brutinibs” will obviate continuous maintenance therapy.64,65
Idelalisib and Second-Generation “Lisibs” That Inhibit PI3K
Drugs that target BCR signaling by inhibiting the delta isoform of phosphoinositide 3 kinase (PI3Kδ) also have clinical activity (Fig. 2). As PI3Kδ also plays a critical role in chemokine-receptor signaling,66 treatment with drugs that inhibit PI3Kδ also can cause early and dramatic reduction in lymph node size and concomitant enhanced lymphocytosis, which can be mitigated when used in combination with rituximab.
Idelalisib was the first to receive US Food and Drug Administration (FDA) approval for treatment of patients with CLL in combination with the anti-CD20 mAb, rituximab. This was based on the demonstration that addition of idelalisib to therapy with rituximab improved PFS and overall survival,67 which was corroborated in long-term follow-up analyses.68 Although highly effective, widespread use of idelalisib has been compromised by concerns over toxicity related to its propensity for causing immune dysregulation, resulting in autoimmunity (e.g., pneumonitis,69 colitis70) or enhanced immune deficiency against opportunistic infection. The FDA recommended closure of clinical trials of frontline idelalisib due to the apparently higher incidence of hepatic toxicity observed in treatment-naive patients71 and a higher number of infections and deaths in treatment-naive patients who received idelalisib and rituximab relative to that of matched patients who received comparator-arm therapy. This has diminished the chance that “lisibs” will achieve approval for use in frontline therapy of patients with CLL.
Second-generation small molecule inhibitors of PI3K also have been approved for treatment of patients with CLL. Duvelisib, which inhibits the gamma isoform of PI3K (PI3Kγ) and PI3Kδ, also has clinical activity. Clinical trials have demonstrated clinical activity similar to that of idelalisib with a favorable safety profile, suggesting this drug may have a higher therapeutic index than idelalisib.72–74 In September 2018, duvelisib was approved by the FDA for treatment of patients with CLL who have had at least 2 prior therapies.75 Umbralisib (TGR-12032) also has been shown to have clinical activity, particularly in combination with ibrutinib.76 However, combination of idelalisib with other kinase inhibitors (e.g., Syk) has resulted in unacceptable toxicity.77
Inhibitors of BCL2
Another category of drug recently approved for use in patients with relapsed CLL is venetoclax, a small molecule that functions as a BH3 mimetic to inhibit BCL2.78 BCL2 is an antiapoptotic protein that is expressed by CLL cells at high levels,79 thereby helping leukemia cells to mitigate spontaneous or drug-induced cell death by countering the activity of proapoptotic proteins, such as BAX, via their respective BH3 domains (Fig. 3). Cytotoxic drugs indirectly can tip the balance in favor of BAX and thereby induce apoptosis. Moreover, the cytotoxic activity of drugs, such as fludarabine, is largely due to their capacity to induce p53, which in turn induces expression of intrinsic inhibitors to BCL2, such as PUMA, freeing up proapoptotic proteins to induce cell death.80 Leukemia cells that harbor del(17p) and/or have inactivating mutations in p53 fail to make such intrinsic BCL2 inhibitors in response to cytotoxic drugs and therefore are relatively insensitive to chemotherapy. The importance of BCL2 in maintaining CLL-cell survival is underscored by the capacity of venetoclax to induce fulminant tumor lysis in patients initiating therapy with this drug.81
FIGURE 3.

Cartoon depicting a wrestler representing the antiapoptotic protein BCL2 holding in check the wrestler representing the proapoptotic protein BAX via handholds, which represent the BH3 domains of each protein. Inhibition of BCL2 by binding its BH3 domain frees up the proapoptotic proteins, allowing for release from mitochondria of cytochrome c, which results in apoptosis.
Clinical trials have demonstrated venetoclax to be highly effective in the therapy of patients with relapsed and/or refractory CLL,82 particularly those with CLL harboring del(17p),83 effecting an overall response rate of 79%, with 8% achieving a CR. Combination therapy of venetoclax with an anti-CD20 mAb, such as rituximab or obinutuzumab, achieved even higher CR rates in patients who were treatment-naive or who had relapsed after other therapies.84,85 Therapy eradicated detectable minimal residual disease (MRD) (at 10−4) in approximately 50% of cases. Some of these patients achieved only partial responses, per iwCLL guidelines (due to persistence of lymph nodes ≥1.5 cm in diameter),86 but had undetectable MRD in the blood or marrow; such patients also may have no detectable disease even in enlarged lymph nodes and have a median PFS comparable to that of patients who achieve a CR with undetectable MRD.87
The high rate of durable responses stimulated evaluation of fixed-duration therapy. The Murano study evaluated the outcome of patients with relapsed/refractory CLL who were treated for 24 months with venetoclax and rituximab (VR) or BR. A total of 389 patients were enrolled (194 in VR, 195 in BR). After a median follow-up time of 23.8 months, the PFS was much longer with VR (hazard ratio [HR], 0.19; P < 0.0001; median, not reached vs. 18.1 months). The benefit of VR was noted across all patient subgroups. The 2-year PFS was 82.8% for the VR group, which also enjoyed improved overall survival (HR, 0.48; P = 0.018). The improved outcome of patients treated with VR is even more apparent with longer-term follow-up.88 With a median of 9.9 months (1.4–22.5 months) after completion of venetoclax therapy, both PFS and overall survival remained superior for the VR-treated patients over that of patients treated with BR (HR, 0.16 [0.12–0.23] and 0.50 [0.30–0.85], respectively).
Upon demonstration that obinutuzumab could be administered safely to patients prior to the initiation of therapy with venetoclax,85 a randomized study was conducted to compare the activity of this combination with that of chlorambucil and obinutuzumab in 432 patients 65 years or older who had comorbidities, which precluded them from receiving more aggressive forms of chemoimmunotherapy.89 Patients received 6 cycles of obinutuzumab and twelve 28-day cycles of therapy with either venetoclax or chlorambucil. The percentage of patients with PFS at 24 months was significantly higher in the venetoclax-obinutuzumab treatment group (88.2% [95% confidence interval, 83.7–92.6]) than in the chlorambucil-obinutuzumab group (64% [95% confidence interval, 57.4–70.8]). Each treatment group had comparable rates of grade 3 or 4 neutropenia (52.8% vs. 48.1%, respectively). Based on these findings, the FDA and National Comprehensive Cancer Network guidelines committee recommended consideration of venetoclax and obinutuzumab as initial therapy.38
Despite the notable clinical activity of venetoclax, not all responses to this drug are durable, even with continuous therapy. The estimated 15-month PFS for patients with relapsed or refractory disease is 69%.82 Patients who achieve only a partial response, or a CR with detectable MRD, generally relapse after the drug is discontinued84 and/or develop drug resistance or even Richter transformation.90,91 Also, despite the aforementioned use of drug combinations of venetoclax with anti-CD20 mAb and/or ibrutinib,92 approximately a third of all patients fail to clear MRD even after 24 months of continuous therapy.
Some patients who develop resistance to venetoclax are found to have mutations in BCL2 that impede the binding of venetoclax to the mutated BCL2 protein.93 Other mutations affecting the capacity of venetoclax to inhibit BCL2 have been identified in the lymphoma cells of patients or lymphoma cell lines with acquired resistance to venetoclax.94,95 Such mutations compromise the cytotoxic activity of venetoclax or second-generation BCL2 antagonists under development.96
ROR1
Targeting other survival-signaling pathways in CLL may allow for development of therapies that may be clinically effective, either alone and/or in combination with newly approved targeted therapies (e.g., ibrutinib, idelalisib, venetoclax).7 One such survival-signaling pathway is triggered by activation of ROR1.
ROR1 is an oncoembryonic surface antigen, which is expressed by CLL cells23,97,98 and by the neoplastic cells of many other types of cancer,99 but not by virtually all normal adult tissues.23,100,101 ROR1 can serve as a receptor for Wnt5a (Fig. 4),23 which is found at high levels in the plasma of patients with CLL relative to that of healthy adults. Wnt5a induces ROR1 to recruit and activate Rho GTPases and enhance chemokine-directed migration, proliferation, and survival of CLL cells.103 Furthermore, ROR1 signaling may promote development and progression of CLL.104,105 Such signaling could be blocked by cirmtuzumab,101 a humanized IgG1 mAb with high affinity and specificity for ROR1 that was generated and selected based on its capacity to inhibit the survival-promoting effects of Wnt5a on CLL cells. A limited-duration phase I study of cirmtuzumab in patients with relapsed CLL showed this antibody had a long half-life, lacked dose-limiting toxicity, and was effective in blocking ROR1 signaling in vivo.102 Transcriptome analyses revealed that treatment reversed cancer stem-cell gene expression signatures noted in the leukemia cells of patients prior to therapy.
FIGURE 4.

ROR1 signaling in CLL. Adapted from Choi et al.102
Studies indicate that the survival-signaling pathway triggered by Wnt5a via ROR1 is active in patients undergoing therapy with ibrutinib.106 Although ibrutinib may mitigate the capacity of CLL cells to enter the protective leukemia microenvironment of lymphoid tissues, the factor triggering ROR1 signaling, namely, Wnt5a, can be found at high levels in the plasma of patients with CLL relative to healthy adults.102,103 As such, stimulation of ROR1 signaling may transcend the leukemia microenvironment. Although ibrutinib can block BCR signaling because of its capacity to inhibit BTK, ibrutinib is not able to inhibit Wnt5a-induced ROR1-dependent activation of Rho GTPases, such as Rac1.106 Such ancillary survival-signaling pathways could provide a lifeline to leukemia cells of patients undergoing therapy, thereby mitigating the capacity of BTK inhibitors to eradicate the disease. Consistent with this notion, treatment with ibrutinib and cirmtuzumab appeared more effective than treatment with either agent alone in clearing leukemia cells in preclinical studies.106 This has fostered phase Ib/II clinical studies evaluating the combination of cirmtuzumab and ibrutinib in patients with CLL (ClinicalTrials.gov identifier ).
Other studies indicate that targeting ROR1 may enhance the efficacy of venetoclax. The most common genetic lesion in CLL is loss or down-regulation of 2 microRNA, miR-15/16,107,108 which were found to target BCL2109 and, more recently, ROR1.110 Deletion or down-regulation of miR-15/16 is conducive to high-level expression of BCL2 and ROR1. Furthermore, leukemia cells that express the highest levels of surface ROR1 also are found to have the highest levels of cytoplasmic BCL2.110 The anti-ROR1 mAb cirmtuzumab was found to enhance the cytotoxic activity of venetoclax for CLL cells in vitro,110 indicating that targeting ROR1 and BCL2 may have additive, if not synergistic, activity in patients with this disease.
Because of its specificity, in vivo stability, long serum half-life, and potential capacity to concentrate conjugated drugs into lysosomal compartments, cirmtuzumab also appears suited to serve as the targeting moiety in anti-ROR1 antibody-drug conjugates (ADCs). Cirmtuzumab has been conjugated with monomethyl auristatin E to generate an ADC that preserves its high-affinity binding specificity for ROR1 and allows for ROR1-targeted intracellular release of monomethyl auristatin E.111 This ADC is selectively cytotoxic for CLL cells that express ROR1 and can affect clearance of adoptively transferred ROR1-positive leukemia cells in preclinical models. This noted preclinical activity has fostered initiation of clinical studies evaluating this ROR1-specific ADC in patients with CLL or mantle cell lymphoma.
CONCLUSIONS
There have been tremendous improvements in therapy for patients with CLL with novel agents that target distinctive facets of its biology. Research defining the importance of the CLL microenvironment and BCR/chemokine-receptor signaling has ushered development of drugs such as ibrutinib, which has changed clinical practice. Research into mechanisms that promote survival of CLL cells has led to development of venetoclax, which also has potent clinical activity. Despite the success of these agents, challenges persist. The BCR-associated kinase inhibitors generally cannot eradicate the disease and thus generally mandate continuous therapy. Although venetoclax may effect deep remissions that allow for fixed-duration therapy, sizeable proportions of patients fail to clear detectable MRD even when treated with venetoclax in combination with anti-CD20 mAbs and/or other targeted therapies. Such patients may experience disease progression even in the setting of venetoclax therapy. Research on agents that can hit other survival factors produced by cells within the leukemia microenvironment may define novel targeted therapies that ensure a successful outcome for all patients requiring treatment for this disease.
Acknowledgments
TJ.K. has received research funding and/or has served as an advisor to Ascerta/AstraZeneca, Celgene, Genentech/Roche, Gilead, Janssen, Loxo Oncology, Octernal Therapeutics, Pharmacyclics/AbbVie, TG Therapeutics, VelosBio, and Verastem. Cirmtuzumab was developed by TJ.K. and licensed by the University of California to Oncternal Therapeutics, Inc., which has provided stock/options to the university and T.J.K.
REFERENCES
- 1.Robak T Therapy of chronic lymphocytic leukaemia with purine nucleoside analogues: facts and controversies. DrugsAging. 2005;22:983–1012. [DOI] [PubMed] [Google Scholar]
- 2.Chang JE, Kahl BS. Bendamustine for treatment of chronic lymphocytic leukemia. Expert Opin Pharmacother. 2012;13:1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukenbill J, Kalaycio M. Fludarabine: a review of the clear benefits and potential harms. LeukRes. 2013;37:986–994. [DOI] [PubMed] [Google Scholar]
- 4.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376: 1164–1174. [DOI] [PubMed] [Google Scholar]
- 5.Goede, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370: 1101–1110. [DOI] [PubMed] [Google Scholar]
- 6.Hillmen, Robak T, Janssens A, et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet. 2015;385:1873–1883. [DOI] [PubMed] [Google Scholar]
- 7.Kipps T, Stevenson FK, Wu CJ, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Primers. 2017;3:16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43:461–466. [DOI] [PubMed] [Google Scholar]
- 9.Burger JA, Tsukada N, Burger M, et al. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96: 2655–2663. [PubMed] [Google Scholar]
- 10.Tsukada N, Burger JA, Zvaifler NJ, et al. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood. 2002;99:1030–1037. [DOI] [PubMed] [Google Scholar]
- 11.Stein J, Soriano SF, M’Rini C, et al. CCR7-mediated physiological lymphocyte homing involves activation of a tyrosine kinase pathway. Blood. 2003;101:38–44. [DOI] [PubMed] [Google Scholar]
- 12.Calpe E, Codony C, Baptista MJ, et al. ZAP-70 enhances migration of ma-lignant B lymphocytes toward CCL21 by inducing CCR7 expression via IgM-ERK1/2 activation. Blood. 2011;118:4401–4410. [DOI] [PubMed] [Google Scholar]
- 13.Till K, Zuzel M, Cawley JC. The role of hyaluronan and interleukin 8 in the migration of chronic lymphocytic leukemia cells within lymphoreticular tissues. Cancer Res. 1999;59:4419–4426. [PubMed] [Google Scholar]
- 14.Zhang S, Wu CC, Fecteau JF, et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc Natl AcadSci U S A. 2013;110:6127–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redondo-Munoz J, Ugarte-Berzal E, Garcia-Marco JA, et al. Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood. 2008;112:169–178. [DOI] [PubMed] [Google Scholar]
- 16.Matas-Cespedes A, Vidal-Crespo A, Rodriguez V et al. Thehuman CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia-microenvironment interactions in chronic lymphocytic leukemia. Clin CancerRes. 2017;23:1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106:1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zucchetto A, Benedetti D, Tripodo C, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009;69:4001–4009. [DOI] [PubMed] [Google Scholar]
- 19.Deaglio S, Aydin S, Grand MM, et al. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med. 2010;16:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113:3050–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahearne MJ, Willimott S, Pinon L, et al. Enhancement of CD 154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling Tfh cells in chronic lymphocytic leukaemia. Br JHaematol. 2013;162:360–370. [DOI] [PubMed] [Google Scholar]
- 22.Chen, Chen L, Yu J, et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF-κB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood. 2019;134:1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda T, Chen L, Endo T, et al. Antisera induced by infusions of autol-ogous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen andreceptorforWnt5a. Proc Natl Acad Sci USA. 2008;105:3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plander M, Ugocsai P, Seegers S, et al. Chronic lymphocytic leukemia cells induce anti-apoptotic effects of bone marrow stroma. Ann Hematol. 2011; 90: 1381–1390. [DOI] [PubMed] [Google Scholar]
- 25.Ghia, Chiorazzi N, Stamatopoulos K Microenvironmental influences in chronic lymphocytic leukaemia: the role of antigen stimulation. J Intern Med. 2008;264:549–562. [DOI] [PubMed] [Google Scholar]
- 26.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011; 118:3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Rooij M, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–2594. [DOI] [PubMed] [Google Scholar]
- 28.Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012; 119:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown JR, Hillmen P, O’Brien S, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia. 2018;32:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robak T, Burger JA, Tedeschi A, et al. Single-agent ibrutinib versus chemoimmunotherapy regimens for treatment-naïve patients with chronic lymphocytic leukemia: a cross-trial comparison of phase 3 studies. Am J Hematol. 2018;93:1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser G, Cramer P Demirkan F, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2019;33:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones J, Mato A, Coutre S, et al. Evaluation of 230 patients with relapsed/refractory deletion 17p chronic lymphocytic leukaemia treated with ibrutinib from 3 clinical trials. Br J Haematol. 2018;182:504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shanafelt TD, Wang XV Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wierda WG, Byrd JC, Abramson JS, et al. NCCN guidelines insights: chronic lymphocytic leukemia/small lymphocytic lymphoma, version 2.2019. J Natl Compr Canc Netw. 2019;17:12–20. [DOI] [PubMed] [Google Scholar]
- 39.Byrd JC, O’Brien S, James DF. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:1278–1279. [DOI] [PubMed] [Google Scholar]
- 40.Komarova NL, Burger JA, Wodarz D. Evolution of ibrutinib resistance in chronic lymphocytic leukemia (CLL). Proc NatlAcadSci USA. 2014;111: 13906–13911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brien S, Jones JA, Coutre SE, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17:1409–1418. [DOI] [PubMed] [Google Scholar]
- 44.Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones JA, Hillmen P, Coutre S, et al. Use of anticoagulants and antiplatelet in patients with chronic lymphocytic leukaemia treated with single-agent ibrutinib. Br J Haematol. 2017;178:286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shanafelt TD, Borah BJ, Finnes HD, et al. Impact of ibrutinib and idelalisib on the pharmaceutical cost of treating chronic lymphocytic leukemia at the individual and societal levels. J Oncol Pract. 2015;11:252–258. [DOI] [PubMed] [Google Scholar]
- 47.Barrientos JC, O’Brien S, Brown JR, et al. Improvement in parameters of hematologic and immunologic function and patient well-being in the phase III RESONATE study of ibrutinib versus ofatumumab in patients with previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18:803–13.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamoto K, Proia LA, Demarais PL. Disseminated cryptococcal disease in a patient with chronic lymphocytic leukemia on ibrutinib. Case Rep In-fectDis. 2016;2016:4642831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arthurs B, Wunderle K, Hsu M, et al. Invasive aspergillosis related to ibrutinib therapy for chronic lymphocytic leukemia. Respir Med Case Rep. 2017;21:27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kreiniz N, Bejar J, Polliack A, et al. Severe pneumonia associated with ibrutinib monotherapy for CLL and lymphoma. Hematol Oncol. 2018; 36:349–354. [DOI] [PubMed] [Google Scholar]
- 51.Voshtina E, Huang H, Raj R, et al. Amebic encephalitis in a patient with chronic lymphocytic leukemia on ibrutinib therapy. Case Rep Hematol. 2018;2018:6514604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stein MK, Karri S, Reynolds J, et al. Cutaneous mucormycosis following a bullous pemphigoid flare in a chronic lymphocytic leukemia patient on ibrutinib. World J Oncol. 2018;9:62–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teh BW, Tam CS, Handunnetti S, et al. Infections in patients with chronic lymphocytic leukaemia: mitigating risk in the era of targeted therapies. Blood Rev 2018;32:499–507. [DOI] [PubMed] [Google Scholar]
- 54.Williams AM, Baran AM, Meacham PJ, et al. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk Lymphoma. 2018;59:625–632. [DOI] [PubMed] [Google Scholar]
- 55.Coutre SE, Furman RR, Flinn IW, et al. Extended treatment with single-agent ibrutinib at the 420 mg dose leads to durable responses in chronic lymphocytic leukemia/small lymphocytic lymphoma. Clin Cancer Res. 2017;23:1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mato AR, Nabhan C, Barr PM, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016;128:2199–2205. [DOI] [PubMed] [Google Scholar]
- 57.Jain, Thompson PA, Keating M, et al. Long-term outcomes for patients with chronic lymphocytic leukemia who discontinue ibrutinib. Cancer. 2017;123:2268–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370: 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015; 125: 2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan KL, Blombery P, Jones K, et al. Plasmablastic Richter transformation as a resistance mechanism for chronic lymphocytic leukaemia treated with BCR signalling inhibitors. Br J Haematol. 2017;177:324–328. [DOI] [PubMed] [Google Scholar]
- 61.Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walter HS, Rule SA, Dyer MJ, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thompson PA, Burger JA. Bruton’s tyrosine kinase inhibitors: first and second generation agents for patients with chronic lymphocytic leukemia (CLL). Expert Opin Investig Drugs. 2018;27:31–2. [DOI] [PubMed] [Google Scholar]
- 65.Walter HS, Jayne S, Rule SA, et al. Long-term follow-up of patients with CLL treated with the selective Bruton’s tyrosine kinase inhibitor ONO/GS-4059. Blood. 2017;129:2808–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Hayre M, Salanga CL, Kipps TJ, et al. Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PLoS One. 2010;5:e11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharman J, Coutre SE, Furman RR, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37:1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gupta A, Li HC. Idelalisib-induced pneumonitis. BMJ Case Rep. 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weidner AS, Panarelli NC, Geyer JT, et al. Idelalisib-associated colitis: histologic findings in 14 patients. Am JSurgPathol. 2015;39:1661–1667. [DOI] [PubMed] [Google Scholar]
- 71.Lampson BL, Kasar SN, Matos TR, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-me-diated hepatotoxicity. Blood. 2016;128:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flinn RV, O’Brien S, Kahl B, et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131:877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vangapandu Hv, Jain N, Gandhi V Duvelisib: a phosphoinositide-3 kinase δ/γ inhibitor for chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017;26:625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Frustaci AM, Tedeschi A, Deodato M, et al. Duvelisib: a new phosphoinositide-3-kinase inhibitor in chronic lymphocytic leukemia. Future Oncol. 2019;15:2227–2239. [DOI] [PubMed] [Google Scholar]
- 75.Blair HA. Duvelisib: first global approval. Drugs. 2018;78:1847–1853. [DOI] [PubMed] [Google Scholar]
- 76.Davids MS, Kim HT, Nicotra A, et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: a multicentre phase 1–1b study. Lancet Haematol. 2019;6:e38–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barr PM, Saylors GB, Spurgeon SE, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127:2411–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. [DOI] [PubMed] [Google Scholar]
- 79.Tracey L, Perez-Rosado A, Artiga MJ, et al. Expression of the NF-kappaB targets BCL2 and BIRC5/survivin characterizes small B-cell and aggressive B-cell lymphomas, respectively. J Pathol. 2005;206:123–134. [DOI] [PubMed] [Google Scholar]
- 80.Mackus WJ, Kater AP, Grummels A, et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced PUMA upregulation. Leukemia. 2005;19:427–34. [DOI] [PubMed] [Google Scholar]
- 81.Howard SC, Trifilio S, Gregory TK, et al. Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review. Ann Hematol. 2016;95:563–573. [DOI] [PubMed] [Google Scholar]
- 82.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778. [DOI] [PubMed] [Google Scholar]
- 84.Seymour J, Ma S, Brander DM, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18:230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flinn I, Gribben JG, Dyer MJS, et al. Phase 1b study of venetoclax-obinutuzumab in previously untreated and relapsed/refractory chronic lymphocytic leukemia. Blood. 2019;133:2765–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–2760. [DOI] [PubMed] [Google Scholar]
- 87.Choi MY, Wang HY, Kipps TJ. SOHO state of the art updates and next questions: the conundrum in assessing the therapy response of patients with chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2019;19:321–325. [DOI] [PubMed] [Google Scholar]
- 88.Kater A, Seymour JF, Hillmen P, et al. A fixed duration of venetoclax-rituximab in relapsed/refractory CLL eradicates minimal residual disease and prolongs survival: long-term outcomes of the MURANO phase III study. J Clin Oncol. 2019;37:269–277. [DOI] [PubMed] [Google Scholar]
- 89.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380: 2225–2236. [DOI] [PubMed] [Google Scholar]
- 90.Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017;129:3362–3370. [DOI] [PubMed] [Google Scholar]
- 91.Rossi D, Gaidano G. Richter syndrome: pathogenesis and management. Semin Oncol. 2016;43:311–319. [DOI] [PubMed] [Google Scholar]
- 92.Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380:2095–2103. [DOI] [PubMed] [Google Scholar]
- 93.Blombery, Anderson MA, Gong JN, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9: 342–353. [DOI] [PubMed] [Google Scholar]
- 94.Blombery, Birkinshaw RW, Nguyen T, et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br J Haematol. 2019;186:e188–e191. [DOI] [PubMed] [Google Scholar]
- 95.Fresquet, Rieger M, Carolis C, et al. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123:4111–4119. [DOI] [PubMed] [Google Scholar]
- 96.Reed JC. Bcl-2 on the brink of breakthroughs in cancer treatment. Cell Death Differ. 2018;25:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baskar S, Kwong KY, Hofer T, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14:396–404. [DOI] [PubMed] [Google Scholar]
- 98.Daneshmanesh AH, Mikaelsson E, Jeddi-Tehrani M, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int J Cancer. 2008;123:1190–1195. [DOI] [PubMed] [Google Scholar]
- 99.Zhang S, Chen L, Wang-Rodriguez J, et al. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol. 2012; 181: 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Broome HE, Rassenti LZ, Wang HY, et al. ROR1 is expressed on hematogones (non-neoplastic human B-lymphocyte precursors) and a minority of precursor-B acute lymphoblastic leukemia. Leuk Res. 2011;35: 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Choi M, Widhopf GF 2nd, Wu CC, et al. Pre-clinical specificity and safety of UC-961, a first-in-class monoclonal antibody targeting ROR1. Clin Lymphoma Myeloma Leuk. 2015;(15 suppl):S167–S169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choi MY, Widhopf GF 2nd, Ghia EM, et al. Phase I trial: cirmtuzumab inhibits ror1 signaling and stemness signatures in patients with chronic lymphocytic leukemia. Cell Stem Cell. 2018;22:951–9.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu, Chen L, Cui B, et al. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J Clin Invest. 2016;126: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Widhopf GF 2nd, Cui B, Ghia EM, et al. ROR1 can interact with TCL1 and enhance leukemogenesis in Eμ-TCL1 transgenic mice. Proc Natl AcadSci USA. 2014;111:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cui B, Ghia EM, Chen L, et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood. 2016; 128:2931–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu J, Chen L, Cui B, et al. Cirmtuzumab inhibits Wnt5a-induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia. 2017;31:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16–1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Palamarchuk A, Efanov A, Nazaryan N, et al. 13q14 Deletions in CLL involve cooperating tumor suppressors. Blood. 2010;115:3916–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rassenti LZ, Balatti V Ghia EM, et al. MicroRNA dysregulation to identify therapeutic target combinations for chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2017;114:10731–10736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mian YA, Widhopf Ii GF, Vo T-T, et al. Development of cirmtuzumab antibody-drug conjugates (ADCs) targeting receptor tyrosine kinase-like orphan receptor1 (ROR1). Blood. 2018;132:1862. [Google Scholar]