

Summary:
Gut-dwelling Prevotella copri, the most prevalent Prevotella species in human gut, have been associated with diet and disease. However, our understanding of their diversity and function remains rudimentary, as studies have been limited to 16S and metagenomic surveys and experiments using a single type strain. Here, we describe the genomic diversity of 83 P. copri isolates from 11 human donors. We demonstrate that genomically distinct isolates, which can be categorized into different P. copri complex clades, utilize defined sets of polysaccharides. These differences are exemplified by variations in susC genes involved in polysaccharide transport as well as polysaccharide utilization loci (PULs) that were predicted in part from genomic and metagenomic data. Functional validation of these PULs showed that P. copri isolates utilize distinct sets of polysaccharides from dietary plant, but not animal, sources. These findings reveal both genomic and functional differences in polysaccharide utilization across human intestinal P. copri strains.
Graphical Abstract
eTOC:
Fehlner-Peach et al. describe 83 P. copri isolates from the stool of 11 donors. Isolates have extensive genome variation and differences in polysaccharide utilization genes. In vitro growth experiments confirm that P. copri isolates utilize distinct sets of polysaccharides from dietary plant, but not animal, sources.
Introduction:
Humans live in close association with a teeming bacterial ecosystem known as the gut microbiome. The diverse inhabitants of this extraordinary environment perform beneficial and remarkable services for their host, out-competing harmful bacteria and digesting the majority of dietary fiber (El Kaoutari et al., 2013; Martens et al., 2008). The relative abundance of two genera, Bacteroides and Prevotella, is inversely correlated in the intestine (Arumugam et al., 2011; Costea et al., 2018). Although a Bacteroides-dominated microbiome is more common, Prevotella are present at >10% relative abundance in the stool of 10–25% of healthy American and European individuals (Koren et al., 2013). Compared to at least 25 species of Prevotella in the oral cavity, and at least 17 species of intestinal Bacteroides, only five Prevotella species have been reported in human intestine, with Prevotella copri being the most abundant (Accetto and Avgustin, 2015; Ferrocino et al., 2015; Ibrahim et al., 2017; Li et al., 2009; Lin et al., 2013; Liu et al., 2018). Although mechanistic models of metabolism, growth, and colonization have been established for several intestinal Bacteroides and oral Prevotella spp., studies of intestinal Prevotella have lacked experimental tools and relied heavily on only two type strains, Prevotella copri and Prevotella stercorea (Hayashi et al., 2007).
Many researchers have observed Prevotella’s presence and abundance by 16S sequencing, but its genomic diversity and biological properties have not been described. Because Prevotella copri are highly sensitive to oxygen, and difficult to isolate and cultivate, previous studies have been limited to the single commercially available type strain, DSM 18205, to perform metagenomic comparisons and draw experimental conclusions. Prevotella spp. have extensive gene acquisition and loss (Gupta et al., 2015; Zhu et al., 2015). In fact, accessory genes within the P. copri DSM 18205 genome account for about 40% of all genes encoded within the genome, the highest proportion of accessory genes identified in a comparison of 11 gut-associated species (Zhu et al., 2015). This estimate should be revised, however, since a metagenomic study (supplemented by whole genome sequencing data from the present study) affirms that P. copri is diverse, comprising four genomically distinct clades (Tett et al., 2019). If species can be determined by average nucleotide identity (ANI) difference, these four clades of P. copri could be considered four distinct species, as they have >10% ANI difference (Tett et al., 2019). Further improvements in genomic comparisons of P. copri may be expected soon, since another study reports a closed genome of P. copri, generated from long-read sequencing of whole microbiomes (Moss and Bhatt, 2018). Whether the genomic diversity in human P. copri translates to functional diversity has not yet been fully explored. This is particularly relevant in light of elevated frequency of P. copri colonization in some cohorts of treatment-naïve rheumatoid arthritis patients (Maeda et al., 2016; Scher et al., 2013). A better understanding of whether there are functional differences in strains from patients compared to healthy subjects will contribute to determining whether there is a causal relationship between P. copri and this autoimmune disease.
Several studies have found associations between diets and the presence of intestinal Prevotella. Prevotella have been positively associated with diets high in plant fiber and carbohydrates, and negatively associated with fat and amino acid diets (De Filippo et al., 2010; Fragiadakis et al., 2019; Ruengsomwong S, 2016; Smits et al., 2017; Wu et al., 2011). Genes associated with carbohydrate catabolism were identified in P. copri metagenomes and correlated with vegan diets (De Filippis et al., 2019). The plant polysaccharide xylan has been used to select for growth of human-associated Prevotella (Tan et al., 2018). Prevotella isolates from livestock grow in the presence of plant polysaccharides and encode gene products capable of breaking down these polysaccharides (Accetto and Avgustin, 2019). Thus, diet may be an important factor promoting growth of intestinal Prevotella. Whether human-associated P. copri are uniquely suited to digest dietary components highly abundant in plant-based diets remains unsubstantiated by experimental and genomic evidence.
Intestinal commensal bacteria have a system of genes, often clustered, whose products collaborate to harness energy from carbohydrates present in the host’s intestinal lumen. Extensive and careful work in Bacteroides species has identified several polysaccharide utilization loci (PULs) that are specific for glycans derived from dietary plant and animal sources, as well as endogenous host sources (Martens et al., 2008; Martens et al., 2011). Upon exposure to the target polysaccharide, genes encoded in the PUL are upregulated. Bacteria lacking one of several critical genes in any polysaccharide-specific PUL fail to grow when the polysaccharide is provided as the sole carbon source (Martens et al., 2011; Raghavan et al., 2014). PULs have been linked to diet and subsequently exploited in experimental systems. Notably, a seaweed porphyran-utilizing PUL was horizontally transferred from marine bacteria to intestinal bacteria of seaweed-ingesting hosts (Hehemann et al., 2010). This PUL was experimentally introduced into B. thetaiotaomicron, and seaweed-selective growth was demonstrated in laboratory mice (Shepherd et al., 2018). Although by far the most extensive work on PULs has been performed in Bacteroides species, PULs have been annotated in Prevotella type strains and used to predict growth on plant polysaccharides in Prevotella isolates from livestock (Accetto and Avgustin, 2015, 2019). Despite their association with plant diet and PUL predictions, the functional capabilities of human-associated intestinal Prevotella have not been tested, due to a dearth of cultured isolates.
Previous studies have been limited to analysis and experimentation with the available genomes and strains of Prevotella species found in human, two of which were isolated from the same individual (Hayashi et al., 2007). Here, we report whole genome sequencing of 83 Prevotella copri isolates from the stool of eleven individuals. We observe extensive genomic diversity of P. copri isolates within and between hosts. This genomic variation was exemplified by specific gene clusters and linked to functional growth of isolates on predicted plant polysaccharides, explaining the association of colonization of Prevotella with a plant-rich diet.
Results:
Isolation of Prevotella from human stool
To investigate the genomic diversity of Prevotella copri and also identify strains for functional analysis, we developed a qPCR screening and isolation strategy for P. copri from human stool (Methods). P. copri was identified in 31/63 individuals by qPCR (Figure S1A). 16S rRNA gene sequencing of the hypervariable V3–V4 region found P. copri in 41/63 individuals, with 18/63 individuals harboring P. copri at levels >10% relative abundance (Figure S1B). From the V3–V4 16S rRNA gene data, five P. copri 16S variants were identified (Figure S1B, Supplemental file 1). The P. copri 16S variants displayed 2–6 single-nucleotide variants (SNVs) within the 253 bp region, corresponding to >97% sequence-sequence identity (Callahan et al., 2016). P. copri detection by qPCR positively correlated with P. copri detection by 16S sequencing (Figure S1C). We collected 83 isolates from eleven individuals harboring P. copri (Figure S1D), one of whom had <1% P. copri (Methods).
Variability in P. copri isolates
Our earlier study indicated differences in the P. copri genomes from new onset rheumatoid arthritis and healthy individuals (Scher et al., 2013). Other studies have since described genetic variation between strains from healthy individuals (De Filippis et al., 2019; Zhao et al., 2019). We assessed whether genomic diversity existed between P. copri isolates from different individuals, or, furthermore, between isolates from the same individual, by sequencing the full genomes of the isolates. Alignment of filtered isolate sequencing reads to the P. copri PanPhlAn database revealed that fewer than 60% of reads mapped to the reference genome, indicating unexpected and unexplored diversity of P. copri genomes in our isolates (Figure 1A) (Scholz et al., 2016).
Figure 1. Hidden diversity in P. copri whole genome sequences.
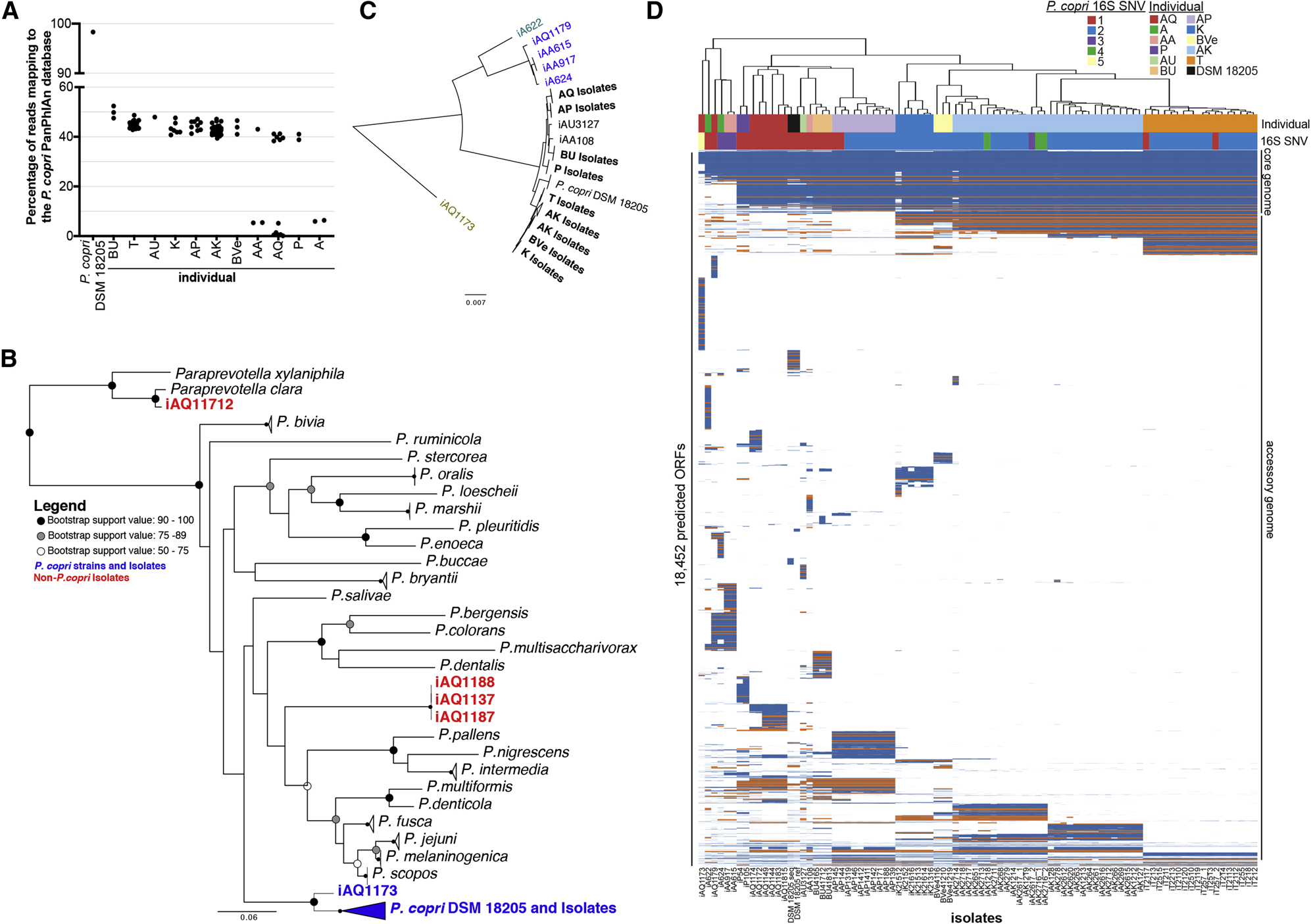
A. Alignment of whole genome sequencing reads of each isolate to the P. copri PanPhlAn database. Each point represents alignment of a single isolate genome. B. Phylogenetic tree based on the 16S rRNA gene sequences. P. copri isolates are shown in blue. Three Prevotella isolates and one Paraprevotella isolate that were not P. copri are shown in red. C. Phylogenetic tree based on the concatenated alignment of 30 conserved ribosomal protein gene sequences. Isolate names are colored by clade. D. Heatmap of P. copri isolate whole genomes showing presence (blue) and absence (white) of predicted ORFs, and highlighting susC genes (orange). Predicted proteins were clustered at 90% amino acid identity. Isolates from the same donor are indicated by color in line 1 above the heatmap, as well as by the first letters of the isolate name below the heatmap. See also Figure S1, Table S1, and File S1.
Evaluation of the de novo assembled genomes revealed low contamination and similar genome size among all isolates (Table S1) (Bankevich et al., 2012; Parks et al., 2015). Phylogenetic analysis based on the 16S rRNA gene revealed that most of the isolates clustered with Prevotella copri DSM 18205 (Figure 1B). One isolate clustered with Paraprevotella, and three isolates clustered with other species of Prevotella; these four isolates were excluded from the rest of the analysis. A separate phylogenetic analysis based on 30 conserved ribosomal proteins further confirmed that the isolates clustered with P. copri, and also revealed four distinct clades (Figure 1C). Comparison with a catalog of P. copri metagenome-assembled genomes is consistent with our conclusion that the majority of isolates belong to a single clade (clade A), four isolates are members of another clade (clade C), and one isolate each belongs to the two remaining clades (clades B and D) (Tett et al., 2019). Hosts AQ, A, and AA harbored isolates belonging to more than one clade (Figure 1C). Enzyme activity and acidification assays revealed functional differences between select isolates from different clades and within clade A (Table 1).
Table 1.
Differential characteristics of P. copri isolates
| isolates | Type strains | P. copri DSM 18205, reported by Hayashi, et al. (2007) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| P. copri complex clade | B | D | C | A | P. copri DSM 18205 (clade A) | B. thetaiotaomicron | B. ovatus | ||
| enzyme activities: | AQ1173 | A622 | AQ1179 | AA108 | AK1212 | ||||
| Alkaline phosphatase | + | + | + | + | + | + | + | + | NR |
| Esterase lipase (C8) | − | − | − | + | + | + | + | − | w |
| Leucine arylamidase | − | w | − | − | − | − | + | − | NR |
| alpha-galactosidase | + | + | + | + | + | w | w | w | + |
| beta-gal actosidase | + | + | + | + | + | + | + | + | − |
| beta-glucosidase | + | + | + | + | + | + | − | w | + |
| N-acetyl-beta-glucosaminidase | + | + | w | + | + | w | |||
| beta-galactosidase 6 phosphate | + | + | NR | ||||||
| alpha-arabinosidase | − | + | + | + | + | + | − | − | NR |
| alpha-fucosidase | − | w | + | − | − | − | − | − | − |
| gelatin hydrolysis | + | − | − | − | − | − | − | − | − |
| acid production from: | |||||||||
| glucose | ND | + | + | + | + | + | + | + | + |
| mannitol | ND | + | + | + | − | − | − | + | − |
| lactose | ND | + | + | + | + | + | + | + | + |
| sucrose | ND | + | + | + | + | + | + | + | + |
| maltose | ND | + | + | + | + | + | + | + | + |
| salicin | ND | + | + | + | − | − | − | + | + |
| xylose | ND | + | + | + | − | + | + | + | + |
| arabinose | ND | + | + | + | − | + | + | + | + |
| glycerol | ND | + | − | − | − | − | − | − | − |
| cellobiose | ND | + | + | + | + | + | − | + | + |
| mannose | ND | + | + | − | − | − | + | + | − |
| melezitose | ND | + | + | + | variable between 5 isolates | ||||
| raffinose | ND | + | + | + | + | + | + | + | + |
| sorbitol | ND | + | + | − | − | − | − | − | − |
| rhamnose | ND | + | − | − | − | + | + | + | + |
| trehalose | ND | + | − | − | − | − | + | + | − |
+, positive; −, negative; w, weak; ND, not detected; NR, not reported
P. copri genomic diversity between hosts
Despite close phylogenetic placement, most isolates exhibited striking genomic dissimilarities to the P. copri reference strain (Figure 1A). To better understand the differences in the P. copri genomes, we compared the presence of predicted ORFs in all the isolates (Figure 1D). This analysis revealed a core genome of approximately 1750 genes, suggesting accessory genomes ranging from approximately 1250–2250 genes per isolate.
Clustering of the isolates by presence of predicted ORFs revealed that isolates often clustered by donor. When isolates from the same donor fell into the same clade, accessory genomes were often unique to isolates from individual hosts, as visualized by blocks of predicted ORFs in the accessory genomes in Figure 1D. Host AK had isolates with distinct accessory genomes within the same clade (Figures 1C and 1D). Annotation of the genomes revealed that, among other functions, many of the host-specific genes were involved in polysaccharide transport, as indicated by the frequency of susC genes (Figure 1D).
Polysaccharide utilization prediction
SusC is the bacterial transmembrane protein that transports polysaccharides into the periplasm. SusC collaborates with the outer-membrane glycan-binding protein SusD, as well as other proteins, for uptake and breakdown of polysaccharides. Conveniently, the genes for this polysaccharide utilization machinery usually occur grouped in close proximity to one another in the genome in polysaccharide utilization loci (PULs) in Bacteroidetes, although occasionally genes encoding polysaccharide catabolism enzymes are located elsewhere in the genome (Sonnenburg et al., 2010). We hypothesized that the genetic diversity of susC genes might indicate functional diversity in polysaccharide utilization by the isolates. We surveyed the susC and surrounding annotated genes in the isolate genomes to predict the repertoire of PULs and, by extension, candidate polysaccharide substrates for the P. copri isolates.
By clustering susC genes at 90% identity, and examining the surrounding gene products within each genome, we categorized 87 susC gene clusters that had (1) a neighboring susD gene and (2) annotated genes encoding glycan-degrading functions, which we called predicted PULs (pPULs) (Figure 2A, Methods, Table S2). Genes annotated to encode similar carbohydrate-active enzymes were found in pPULs with distinct susC clusters in different isolate genomes. For example, xylanases were found in close proximity to susC clusters 2, 65, and the adjacent clusters 100 and 140. SusC cluster 2 was only in isolate genomes from individual BU, susC cluster 65 was only in the genome of isolate A622, and susC clusters 100 and 140 were only in isolate genomes from individual AA. In contrast with several Bacteroides species, which encode PULs that catabolize polysaccharides from both animal and plant dietary glycans, all annotated enzymes in the P. copri isolate pPULs were predicted to catabolize dietary plant glycans and endogenous host mucin, but none were predicted to catabolize dietary animal glycans (Martens et al., 2011; Salyers AA, 1982). Consistent with this prediction, the V1–V3 16S sequences of select isolates were most closely related to two previously reported non-omnivore-associated P. copri 16S sequences, P10 and P19 (De Filippis et al., 2016) (Figure S2A, Supplemental file 2).
Figure 2. Categorization of polysaccharide utilization loci in P. copri isolates.

A. pPULs were categorized by presence of annotated genes with polysaccharide catabolism activity (see also Table S2, Methods). Presence of a pPUL is indicated by a blue box. Stars indicate susC genes that have different flanking annotated enzyme genes in different isolates. B. Schematic of select pPULs containing xylan catabolism genes. PATRIC annotations are indicated above the PUL. Genes with the same annotation are the same color. Genes annotated as hypothetical proteins are gray. Genes with annotations unlikely to be involved in carbohydrate metabolism are colored dark blue. See also Figure S2.
Consistent with the 17 predicted PULs for P. copri DSM 18205, the number of pPULs per isolate ranged from 14–28, with a median of 23 (Terrapon et al., 2018). Intestinal Prevotella have been reported to utilize xylan and, as expected, pPULs containing xylanases were ubiquitously represented throughout the isolate genomes (Figure 2A) (Tan et al., 2018). A survey of the pPULs containing annotated xylan catabolism enzymes revealed additional features: all isolates contained pPULs with xylan catabolism enzymes; each genome contained 4–5 xylan catabolism enzymes that were present in pPULs (Figure S2B); and susC/D pairs in pPULs containing xylan catabolism enzymes often occurred in tandem (Figure 2B).
Polysaccharide utilization in vitro
We predicted that P. copri isolates would grow in the presence of polysaccharides targeted by the enzymes encoded by the annotated genes in the pPULs (candidate substrates in Figure 2A). We therefore tested the ability of P. copri isolates to grow when polysaccharides were provided as the sole carbon source in vitro (Methods). Growth on individual polysaccharide substrates revealed functional diversity between isolates (Figure 3A, 3B, Figure S3A). For example, isolate AA108 reached a high OD when pullulan was provided as the sole carbon source, but isolates P54 and T2112 did not. Interestingly, isolates from the same individual grew in the presence of different polysaccharides: compared to isolate AK1212, isolate AK2718 grew to a higher OD in the presence of polygalacturonic acid and rhamnogalacturonan I, and a lower OD in the presence of beta-glucan and arabinan. None of the isolates grew in the presence of animal polysaccharides chondroitin or heparin or the proteoglycan mucin (Figure 3A, Figure S3A), indicating that Prevotella isolates could be cultured in the presence of glycans from dietary plant sources, but not dietary animal or endogenous host sources. Growth on particular polysaccharides distinguished isolates from each clade (Figure 3B, Table S3). In particular, arabinan, levan, inulin, xyloglucan, beta-glucan, and glucomannan growth distinguished isolates from each clade. All isolates except AQ1173 grew on arabinoxylan and arabinan (Figures 3A and 3B). Isolate AQ1173 also failed to grow on xyloglucan and glucomannan. Isolate A622 failed to grow on both levan and inulin. Tested isolates from clade C failed to grow on beta-glucan, xyloglucan, glucomannan, and rhamnogalacturonan I. Tested isolates from clade A grew well on most substrates, although there was a range of growth among clade A isolates on several plant polysaccharide sources.
Figure 3. Growth of P. copri isolates on polysaccharides.
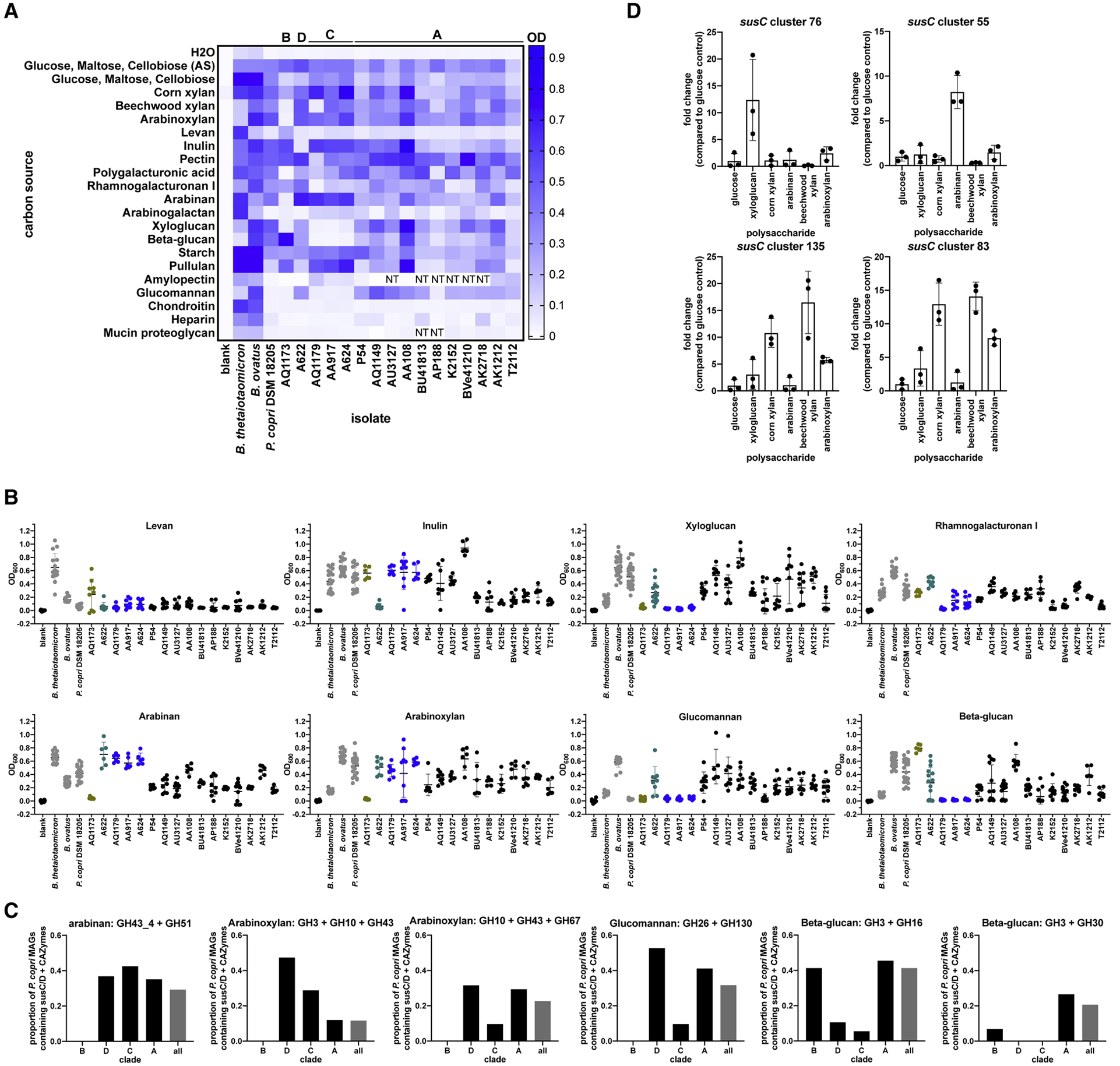
Isolates were cultured anaerobically at 37 °C in YCFA media supplemented with individual polysaccharides as the sole carbon source. OD600 was measured at 24, 48, and 72h. Data indicate mean ± SD and represent 2–3 experiments, with each condition tested in duplicate or triplicate. The highest OD from 24, 48, or 72h for each condition for each experiment is shown. Heatmap (A) shows the mean of the highest OD for each condition of each experiment. P. copri complex clades are indicated above the heatmap. AS: media prepared by Anaerobe Systems. NT: not tested. Graphs (B) show the highest measured OD on select polysaccharide substrates. Isolates from the same P. copri complex clade are plotted in the same color (Tett et al., 2019). C. PULs were predicted in P. copri MAGs. Proportion of P. copri MAGs within each clade containing pPULs for select polysaccharides is shown. D. P. copri DSM 18205 was grown to mid-log phase in the presence of the indicated polysaccharide substrates. Levels of susC transcripts were quantified by qRT-PCR. Graphs show fold change compared to a glucose control and indicate mean ± SD. See also Figure S3 and Table S3.
Isolates AQ1173 and A622 were the only isolates representing clades B and D, respectively. Because growth data from single isolates limited our ability to generalize to those clades, we used P. copri metagenome-assembled genomes (MAGs) reconstructed from metagenomic datasets to ask whether pPULs containing enzymes targeting relevant polysaccharides were present in these clades (Tett et al., 2019) (STAR Methods; Figure 3C). Consistent with our observations that isolate AQ1173 (clade B) lacked pPULs containing enzymes targeting arabinan, arabinoxylan, and glucomannan and also did not grow on those polysaccharides (Figures 2A, 3A, and 3B), the clade B P. copri MAGS lacked pPULs with CAZymes capable of degrading those polysaccharides (Figure 3C). The clade B MAGs contained pPULs with CAZymes capable of degrading beta-glucan, however. pPULs with CAZymes capable of degrading glucomannan and arabinan were found in the clade D P. copri MAGs, consistent with our predictions and growth observations for isolate A622 (Clade D).
To determine which pPULs respond to specific polysaccharides, we asked which susC genes were upregulated in P. copri DSM 18205 during growth on arabinan, xyloglucan, corn xylan, beechwood xylan, and arabinoxylan. A screen for all 22 susC genes in P. copri DSM 18205 revealed upregulation of distinct susC genes in each condition (Methods, Figures 3D and Figure S3B). Upregulation of the susC gene is considered a proxy for expression of the whole PUL, therefore, the pPULs associated with the susC gene likely targeted the tested polysaccharides. Consistent with the presence of arabinosidase genes in close proximity to the susC gene, susC cluster 55 was upregulated in the presence of arabinan. Similarly, the tandem susC clusters 135 and 83, which are flanked by genes encoding xylanases, were upregulated in the presence of both corn and beechwood xylans, and to a lesser extent in the arabinoxylan condition. Analysis of pPUL structure revealed synteny between genes flanking susC clusters 83, 135, 100, 140, 71, and 138 (Figure 2B). Surprisingly, susC cluster 76 was upregulated in the presence of xyloglucan. The susC cluster 76 was flanked by genes encoding two mannosidases and several hypothetical proteins. Further examination of the locus revealed a hypothetical protein with homology to GH5. GH5 has endo-beta-glucanase activity, which could potentially target xyloglucan. Synteny was also observed between susC clusters 76 and 73, and surrounding genes, suggesting that the cluster 73 pPUL may also target xyloglucan (Figure S3C).
Polysaccharide growth predictions
Using a combination of genomic predictions, gene expression data, and homology, we evaluated how well the pPULs analysis predicted in vitro growth on candidate polysaccharides: 181 isolate/polysaccharide conditions matched predictions while 55 did not (Figure S3D). We observed 24 instances in which we did not predict particular polysaccharide utilization by an isolate, yet we observed growth. For 19 of these cases of unpredicted growth, we found genes encoding relevant polysaccharide breakdown enzymes elsewhere in the genome. Perhaps the genes are part of fragmented PULs (as has been observed in Bacteroides fructan PULs (Sonnenburg et al., 2010)), or are simply located on a contig separate from the rest of the PUL. More puzzlingly, we observed isolates that failed to grow on a particular substrate, despite presence of a corresponding intact pPUL in the genome. For example, we predicted PULs with arabinogalactan catabolism genes in most of genomes of clade A, yet most failed to grow or grew poorly on this substrate. Perhaps P. copri isolates utilize a different form or source of arabinogalactan. Additionally, substrate predictions were based on gene annotations that may be incomplete or inaccurate. Overall, the pPUL analysis performed well in predicting growth. Welch’s t-test on predicted growth or unpredicted growth had a p-value < 0.0001 (Methods).
Discussion:
In this study, we observed genomic and functional diversity of human intestinal P. copri isolates. Whole genome sequencing exposed diversity of P. copri isolates, demonstrated by the low percentage of isolate whole genome reads that mapped to the P. copri PanPhlAn database. Within-species genomic variation, both between individuals and within individuals, was demonstrated by genetic diversity of susC genes and pPULs and linked to functional growth of isolates on plant polysaccharides. PULs predicted to target several polysaccharides in large metagenomic datasets support our predictions and growth data for representative isolates, indicating that the results of this study may be generalizable beyond the isolates shown here.
Variability in P. copri genomes
Despite 97% sequence identity in the 16S rRNA gene and conservation of ribosomal protein sequences, the P. copri isolates vary in their genomes, enzyme activities, and polysaccharide utilization profiles, suggesting that they could be considered distinct species by some definitions and the same species by others. ORF presence among isolates revealed host-specific genes in the accessory genome, several of which were susC genes. The differences in SusC proteins encoded within the P. copri isolates derived from different hosts (Figure 1D) could be the result of several factors, including selective pressures from the environment. SusC proteins from different isolates could also transport slightly different substrates, as has been observed in two closely related B. thetaiotaomicron strains (Joglekar et al., 2018). Hosts harboring multiple strains of P. copri suggest that each strain may inhabit a distinct niche. Hosts AQ, A, and AA harbored isolates from different clades. Isolates from host AK all fell into the same clade and possessed two distinct accessory genomes. Perhaps one type of isolate inhabits the cecum, the other the colon; alternatively, polysaccharide cross-feeding may occur between P. copri isolates, as was demonstrated in vitro with Bacteroides species (Rakoff-Nahoum et al., 2014). In hosts with more than one strain of P. copri, representative isolates often complemented each other to grow in the presence of more polysaccharides than any individual isolate. This point was illustrated best by isolates from donor AQ: isolates AQ1173 and AQ1179 did not grow on xyloglucan or glucomannan, but AQ1149 grew on both polysaccharides (Figure 3A). These results suggest that the total P. copri population in hosts with multiple strains was capable of catabolizing a greater diversity of polysaccharides than any individual strain. Indeed, a separate study found that non-Western microbiomes contained multiple P. copri clades, and that some carbohydrate active enzymes were differentially represented between clades (Tett et al., 2019). Whether microbiomes of individuals with only one P. copri strain completely lack the ability to break down additional polysaccharides or rely on other bacterial species to maintain a full complement of polysaccharide digestion should be the subject of future metagenomic studies.
P. copri presence in the human intestine may be diet-dependent
In humans, differences in P. copri 16S sequences and metagenomes have been correlated with host diet (De Filippis et al., 2019; De Filippis et al., 2016; Fragiadakis et al., 2019; Smits et al., 2017). Although our study lacks host diet information, the pPUL analysis predicted and in vitro growth experiments confirmed that the P. copri isolates used only plant polysaccharides. A previous study found that particular P. copri V1–V3 16S sequences were associated with diet (De Filippis et al., 2016). The V1–V3 sequences of select isolates from different clades in this study were most closely related to two non-omnivore-associated sequences, consistent with their growth on plant polysaccharides (Figure S2A). Although the dynamics of dietary and intestinal growth of P. copri within a complex microbiome require experimental validation, in vitro data presented here suggest that dietary plant polysaccharides promote P. copri growth, but dietary animal polysaccharides do not. These data provide a possible mechanism for the correlation between vegetable-rich diets and abundance of intestinal Prevotella: perhaps dietary plant polysaccharides are continuously required to maintain Prevotella in the gut. Their absence may eliminate Prevotella from the intestinal microbiome.
Prevotella polysaccharide utilization
Although the pPUL analysis predicted growth surprisingly well, incorrect predictions may indicate interesting biology or areas warranting technical improvements. For cases in which PULs were predicted but growth on a corresponding candidate polysaccharide was not observed, perhaps mutations in the genes encoding the enzymes or regulators rendered the gene products non-functional. Future studies should aim to evaluate the frequency and rate of such mutations in intestinal commensals. Open genomes may account for some cases in which polysaccharide utilization was not predicted by the presence of a PUL, yet growth was observed. If a PUL were split across contigs, it would be impossible to predict a candidate substrate. Recent closure of a P. copri genome from metagenomic analysis may reduce such instances in future studies (Moss and Bhatt, 2018).
Utilization of xylan, which is found in cereal grains, has been repeatedly established in Prevotella species and in P. copri specifically (Accetto and Avgustin, 2019; da Silva et al., 2012; Dodd et al., 2011; Tan et al., 2018). The numerous xylan-degrading enzymes identified in pPULs in P. copri isolates in this study suggest that P. copri may have an expanded xylan degrading enzyme repertoire, and possibly a superior ability to target xylans compared to other intestinal bacteria.
Xyloglucan is an abundant polysaccharide in vascularized plants, seeds, and food additives, yet its utilization by known intestinal microbes is relatively rare, demonstrated only in a low proportion of Bacteroides species (Larsbrink et al., 2014; Nishinari et al., 2007). Despite its abundance in the human diet, xyloglucan utilization was difficult to predict from P. copri genome sequence alone (Figure 2A, Figure S3D, Table S2). Gene expression analysis of susC genes in P. copri grown on xyloglucan suggests that gene products in the pPUL catabolize xyloglucan. Given the rarity of xyloglucan catabolism genes reported to date in Bacteroides, perhaps P. copri is responsible for xyloglucan degradation in hosts lacking Bacteroides species possessing xyloglucan-breakdown genes.
Implications
The collection of P. copri isolates and their corresponding polysaccharide utilization repertoire described herein enable future experimentation in vitro and in vivo with isolates whose cultivation was previously elusive perhaps due to the absence of required plant polysaccharides. The human genome encodes enzymes that break down only starch, sucrose, and lactose (El Kaoutari et al., 2013). Carbohydrates make up 45–65% of typical human diets (Board., 2005). Therefore, the ability of intestinal microbes to break down polysaccharides is critical for human survival. Whether P. copri strain-type and presence is meaningful to human disease, or simply an indication of diet, requires further investigation. Future studies that aim to assess human microbiome interactions with disease will require dietary intervention or, at minimum, collection of patient dietary habits or records. To avoid confounding effects of diet, future metagenomic analyses may better illuminate disease-associated genes by excluding genes known to interact with host diet. If P. copri does exacerbate or ameliorate disease in specific populations, diet may be readily manipulated to modulate P. copri growth. Establishment of polysaccharide substrate preferences for genetically diverse human P. copri isolates allows for informed experimentation in vitro and in vivo, as well as clinical study design and interpretation.
STAR Methods:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dan R. Littman (Dan.Littman@med.nyu.edu). All unique reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Subjects enrolled in this study included new onset rheumatoid arthritis (NORA) patients and healthy controls, as described in STAR methods. The study included 63 individuals: 33 healthy controls and 30 NORA patients. None were treated with antibiotics in the previous six months. The 33 healthy controls (individuals C, D, F, H, I, J, K, L, M, N, O, AD, AE, AF, AI, AJ, AK, AL, AM, AN, AO, AR, AS, AT, AU, AV, AW, AX, AY, AZ, BA, BD, and BE) included both males and females, with an average age of 40. The 30 NORA patients (individuals A, B, P, Q, R, S, T, U, V, W, X, Y, Z, AA, AB, AC, AP, AQ, BB, BC, BF, BK, BM, BN, BO, BQ, BS, BT, BU, BVe) included both males and females, with an average age of 49 years. NORA patients were previously untreated, presenting with symptoms, seropositive for anti-citrullinated protein antibodies, and not treated with NSAIDs or DMARDs. Ethical approval was granted by the Institutional Review Board of New York University School of Medicine (NYU IRB protocol #i14–00487).
Bacterial culture
Bacteria not isolated in this study were purchased or obtained from other laboratories: P. copri DSM 18205 was purchased from DSMZ. Bacteroides ovatus ATCC 8483 and Bacteroides thetaiotaomicron VPI-5482 were gifts from Michael Fischbach. Frozen glycerol stocks of all bacteria were prepared to minimize the need to passage them. Glycerol stocks were struck out on BRU plates (Anaerobe Systems) and incubated anaerobically for 24–48h at 37 degrees C. Culturing conditions for specific procedures were consistent for all strains and isolates and are described in the Method Details.
METHOD DETAILS
Sample collection
Stool was collected into anaerobic collection tubes (Anaerobe Systems) from healthy controls and new onset rheumatoid arthritis patients, deidentified, and frozen at −80°C.
qPCR Screen for Prevotella
Genomic fecal DNA was extracted from fecal samples using the MoBio PowerSoil DNA extraction kit. Fecal DNA was screened for the presence of P. copri using qPCR primers designed for six conserved regions of the P. copri genome (Scher et al., 2013), as well as two conserved regions of the Prevotella 16S gene (see Key Resources Table). Universal primers specific for the 16S rRNA gene were used to quantify total bacterial load in each sample. qPCR was performed with the SYBR green master mix on the Roche Lightcycler 480, with the following cycle conditions: 90°C for 5 minutes, then 40 cycles of 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. Genomic DNA from P. copri DSM 18205 was used to generate a standard curve. The standard curve was used to determine the absolute quantity of Prevotella and 16S in a sample. Fold change was calculated by dividing absolute quantity of Prevotella by absolute quantity of 16S. Feces with a fold change > 0.1 for any Prevotella-specific primer set were considered positive and used to isolate P. copri, with the exception of sample AU, which had a lower fold change.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Fecal samples from new onset rheumatoid arthritis patients and healthy controls | This study | N/A |
| Critical commercial assays | ||
| PowerSoil DNA isolation kit | Qiagen | CAT: 12888–100 |
| TruSeq DNA PCR-free Library Prep Kit | Illumina, California, USA | CAT: 20015962 |
| Wheaton™ CryoELITE™ Cryogenic Storage Vials | Fisher Scientific | CAT: 02-912-737 |
| FALCON 14 ml Polypropylene Round-Bottom Tubes | Fisher Scientific | CAT: 352059 |
| Titertube® Micro Test Tubes | Bio-Rad | CAT: 2239391 |
| RNeasy Mini Kit | Qiagen | CAT: 74104 |
| RNA Protect Bacteria Reagent | Qiagen | CAT: 76506 |
| SuperScript™ IV First-Strand Synthesis System | ThermoFisher | CAT: 18091050 |
| API® ZYM | BioMerieux | CAT: 25200 |
| API® Rapid ID 32 A Microbial Identification Kit | BioMerieux | CAT: 32300 |
| API® 20 A Microbial Identification Kit | BioMerieux | CAT: 20300 |
| Deposited Data | ||
| P. copri isolate genomes | This study | NCBI-BioProject: PRJNA559898 and PATRIC: https://www.patricbrc.org/ |
| 16S sequencing | This study | NCBI-BioProject: PRJNA559898 |
| Experimental Models: Organisms/Strains | ||
| Prevotella copri DSM 18205 | DSMZ | CAT: DSM 18205 |
| Bacteroides ovatus ATCC 8483 | ATCC | CAT: 8483 |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | CAT: 29148 |
| P. copri isolates | This study | N/A |
| Oligonucleotides | ||
| Primers for P. copri screen, see Table S4 | This study and Scher et al., 2013 | N/A |
| Primers for susC screen, see Table S4 | This study | N/A |
| Software and Algorithms | ||
| DADA2 | Callahan et al., 2016 | https://benjjneb.github.io/dada2/ |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| SPAdes | Bankevich et al., 2012 | http://cab.spbu.ru/software/spades/ |
| PanPhlAn | Scholz et al., 2016 | https://bitbucket.org/CibioCM/panphlan/src/default/ |
| CheckM | Parks et al., 2015 | https://ecogenomics.github.io/CheckM/ |
| PATRIC | (Wattam et al., 2014) | https://www.patricbrc.org/ |
| MUSCLE | Edgar, 2004 | https://www.drive5.com/muscle/ |
| RAxML | Stamatakis, 2014 | https://cme.h-its.org/exelixis/web/software/raxml/index.html |
| Silva SINA aligner | (Pruesse et al., 2012) | https://www.arb-silva.de/aligner/ |
| Other | ||
| Anaerobic Tissue Transport Medium Surgery Pack – ATTMSP | Anaerobe Systems | AS-915 |
| Brucella Blood Agar - BRU | Anaerobe Systems | AS-141 |
| Laked Brucella Blood Agar w/ Kanamycin and Vancomycin – LKV | Anaerobe Systems | AS-112 |
| Peptone Yeast Broth with Glucose - PYG | Anaerobe Systems | AS-822 |
| Yeast Casitone Fatty Acids Broth with Carbohydrates – YCFAC Broth | Anaerobe Systems | AS-680 |
| Amylopectin from maize | Sigma | 10120–250G |
| Arabinan (Sugar Beet) | Megazyme | P-ARAB |
| Arabinogalactan (Larch Wood) | Megazyme | P-ARGAL |
| Arabinoxylan (Wheat Flour; Low Viscosity ~ 10 cSt) | Megazyme | P-WAXYL |
| Beta-glucan (Barley; High Viscosity) | Megazyme | P-BGBH |
| Chondroitin sulfate sodium salt (from shark cartilage) | Sigma | C4384–5G |
| Glucomannan (Konjac; Low Viscosity) | Megazyme | P-GLCML |
| Heparin sodium salt (from porcine intestinal mucosa) | Sigma | H3393–100KU |
| Inulin (chicory) | Megazyme | P-INUL |
| Levan (from Erwinia herbicola) | Sigma | L8647–1G |
| Pectin (from apple) | Sigma | 93854–100G |
| Polygalacturonic Acid (PGA) | Megazyme | P-PGACT |
| Pullulan (from Aureobasidium pullulans) | Sigma | P4516–1G |
| Rhamnogalacturonan (Soy Bean) | Megazyme | P-RHAGN |
| Starch | VWR | 1.01252.0100 |
| Xylan (Beechwood; purified) | Megazyme | P-XYLNBE-10G |
| Xylan (from Corn Core, TCI America™) | Fisher | X007825G |
| Xyloglucan (Tamarind) | Megazyme | P-XYGLN |
| Mucin from porcine stomach, Type III, bound sialic acid 0.5–1.5 %, partially purified powder (100g) | Sigma | M1778–100G |
Isolation of Prevotella
P. copri was isolated from Prevotella-positive feces by anaerobic quadrant streaking of PBS-diluted and undiluted stool on BRU and LKV plates (Anaerobe Systems), then incubated anaerobically at 37°C for 24–48 h. Isolate colonies were picked, enumerated, streaked on a fresh plate, and the inoculation loop dipped into PCR grade water. The loop-dipped PCR water was used with the qPCR primer sets (Table S4) to screen for P. copri. Prevotella-positive-screened isolates were confirmed by Sanger sequencing of the V3–V4 hypervariable region of the 16S rRNA gene, and finding >97% identity to known Prevotella species. Glycerol stocks of Prevotella isolates were frozen at −80°C.
16S rRNA gene sequencing
Genomic fecal DNA was extracted from fecal samples using the MoBio PowerSoil DNA extraction kit. The V3–V4 region of the 16S rRNA gene was amplified, purified, and sequenced on Illumina MiSeq (Caporaso et al., 2012). Sequences generated from amplicon sequencing were trimmed (maxN=0, truncQ=2) and denoised using DADA2 (Callahan et al., 2016). Sequences were annotated using the Silva nr v128 database.
Whole genome sequencing
Isolates were grown on BRU plates for 24–48 hours, until a healthy lawn was present. The lawn was suspended in PYG media (Anaerobe Systems), pelleted, and DNA was isolated with the MoBio PowerSoil DNA isolation kit. Libraries were prepared for sequencing with the TruSeq PCR-free library preparation kit and sheared to 500bp fragment length. Prepared libraries were sequenced on Illumina HiSeq2500 2×100 bp. Paired-end sequences were trimmed using Trimmomatic (Bolger et al., 2014), then whole genomes were assembled using SPAdes (Bankevich et al., 2012). Trimmed reads were also mapped to the panphlan_pcopri_16 database (https://bitbucket.org/CibioCM/panphlan/wiki/Pangenome%20databases) using PanPhlAn (default parameters, Figure 1A) (Scholz et al., 2016). Quality and completeness of the assemblies were evaluated using CheckM (Parks et al., 2015). Genomes were annotated in PATRIC and are available at https://www.patricbrc.org/ and under BioProject PRJNA559898. Presence and absence of predicted proteins can be visualized at https://flatironinstitute.github.io/genome_cluster_array_visualization/.
Phylogenetic Trees
Representative genomes for the Prevotella and Bacteroides genera were downloaded from NCBI. For each genome, a set of 30 ribosomal protein genes (LSU ribosomal proteins L1, L2, L3, L4, L5, L6, L10, L13, L14, L15, L18, L22, L23, L24, L29, and SSU ribosomal proteins S2, S3, S4, S5, S7, S8, S9, S10, S11, S12, S13, S14, S15, S17, S19) were identified and aligned using MUSCLE (Edgar, 2004). A maximum-likelihood tree was calculated using RAxML (-m GTRCAT; (Stamatakis, 2014)) from a concatenated alignment of the 30 ribosomal protein genes. A 16S rRNA gene tree was also estimated from the alignment of full length 16S rRNA gene sequences using Silva’s SINA aligner (https://www.arb-silva.de/aligner/) and RAxML (-m GTRCAT).
Enzyme activity and acid production assays
Isolates and type strains were grown anaerobically for 18–24, harvested, and plated as per manufacturer’s instructions on API® ZYM, API® Rapid ID 32 A Microbial Identification Kit, and API® 20 A Microbial Identification Kit test strips (BioMerieux). Test strips were incubated at 37 degrees C. Enzyme reactions were developed after 4.5 h, and acid production was assessed after 24h.
susC analysis and candidate substrate prediction
All annotated susC genes in the isolate genomes were clustered at >90% identity. This analysis revealed 168 susC gene clusters throughout all the genomes. For each susC gene cluster, ten flanking genes on either side were pulled from the genome and annotated using the PATRIC genome annotation tool (Table S2) (Wattam et al., 2014). Presence of paired susC and susD genes constituted a predicted PUL (pPUL). The flanking region was examined to locate adjacent encoding carbohydrate catabolizing enzymes. Candidate polysaccharide(s) were determined based on proximity of susCD to either a single hallmark glycosyl hydrolase or polysaccharide lyase gene, or a combination of enzymatically active gene products encoded in the pPUL, and analogous to known gene activities in Bacteroides species PULs. The susC clusters lacking a companion susD gene, as well as those comprised of genes unrelated to polysaccharide catabolism, were excluded from the pPUL repertoire analysis.
In vitro polysaccharide growth assays
B. thetaiotaomicron, B. ovatus (both from Michael Fischbach), Prevotella copri DSM 18205 (DSMZ), and P. copri isolates were grown anaerobically on BRU plates (Anaerobe Systems) for 48h at 37 degrees C. Strains were passaged to YCFAC liquid media (Anaerobe Systems). 48h later, cultures were passaged to pre-reduced YCFA +/− 0.5% (w/v) individual polysaccharides or mucin proteoglycan (Key Resources Table) at a dilution of 1:100 in a total volume of 1ml (Browne et al., 2016). Cultures were incubated anaerobically in sterile polypropylene tubes at 37 degrees C. Importantly, P. copri grew in Wheaton™ CryoELITE™ Cryogenic Storage Vials, FALCON 14 ml Polypropylene Round-Bottom Tubes, and Titertube® Micro Test Tubes (see Key Resources Table), but not in Axygen™ Storage Microplates from Fisher Scientific (Catalog no. 14-222-224). Every 24h, 100 μl of each culture was transferred to a flat-bottom 96-well plate, and OD600 was measured with the Envision plate reader. OD600 for each culture condition was subtracted from the average blank OD600 for the corresponding polysaccharide. Cultures were grown in duplicate or triplicate. For each condition, the maximum average OD600 from the 24h, 48h, or 72h time point was determined and duplicate or triplicate values were used in Figure 3A, 3B, and Figure S3A.
susC gene expression analysis
P. copri DSM 18205 was grown anaerobically on BRU plates and passaged to YCFAC liquid media (Anaerobe Systems), as for in vitro polysaccharide growth assays (above). 48h later, cultures were passaged to pre-reduced YCFA +/− 0.5% (w/v) individual polysaccharides (Key Resources Table) or glucose as a negative control at a dilution of 1:100 in a total volume of 50 ml. The OD of cultures was monitored and when it indicated mid-log phase, 10 ml of each culture was harvested, pelleted, stabilized in RNA Protect Bacteria Reagent (Qiagen), and frozen at −80 degrees C. RNA was extracted with the RNeasy kit (Qiagen), and cDNA was generated with the Superscript IV kit. qPCR was performed with the SYBR green master mix on the Roche Lightcycler 480, with the following cycle conditions: 90°C for 5 minutes, then 40 cycles of 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. mRNA levels of susC genes were measured with primers specific for each susC gene (Table S4), and normalized to the 16S rRNA gene. Genomic DNA from P. copri DSM 18205 was used to generate a standard curve.
PUL prediction in P. copri MAGs
SusC and susD homologs in P. copri MAGs (Tett et al., 2019) were identified by blastp (Identity>75% , Bitscore > 300) using susC and susD predicted proteins from the P. copri isolate genomes. Only MAG contigs containing both susC and susD homologs within 10 gene products were considered further. For each susC centroid, the PUL operon was extended to include the 10 gene products upstream and downstream of the first and last occurrence of a susC or SusD homolog. Each PUL was checked for CAZy annotations (Lombard et al., 2014) predicted using HMMSEARCH (version 3.1b2) (Eddy, 2011) against dbCAN HMMs v6 (Yin et al., 2012) with an E-value < 1e–18 and coverage > 0.35. PULs were predicted by searching for the presence of CAZymes known to act on known polysaccharides. Predictions are presented as the proportion of P. copri MAGs containing at least one pPUL out of all P. copri MAGs within each P. copri complex clade.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prediction analysis
For each isolate used in the in vitro polysaccharide growth assay, a one-way ANOVA was performed on the maximum OD for each polysaccharide. A post hoc Dunnet’s test was performed with the H2O condition (no carbon source) as the negative control. Isolate-polysaccharide conditions with OD values that were statistically significant compared to the H2O control were considered positive for growth. Isolate-polysaccharide conditions that were not significant were considered negative for growth.
Statistical analysis of prediction
Isolate-polysaccharide conditions were separated according to whether growth on a candidate polysaccharide was predicted by susC gene expression or genome sequence. Isolate pPULs sharing synteny with P. copri DSM 18205 pPULs confirmed by susC gene expression were grouped together. If a pPUL was present in the genome of an isolate, the isolate-polysaccharide condition was considered “growth predicted.” If a pPUL was absent in the genome of an isolate, the isolate-polysaccharide condition was considered “growth not predicted.” For xylan, arabinan, and xyloglucan, for which there were both gene expression or genome sequence predictions, gene expression predictions were used in instances of discrepancies between the predictions. Isolate-polysaccharide conditions that were statistically significant as determined by a one-way ANOVA with a post hoc Dunnet’s test using the H2O condition (no carbon source) as the negative control were given a score of 1 and isolate-polysaccharide conditions that were not significant were given a score of 0. Welch’s t test was performed on the data.
Supplementary Material
Highlights:
Human P. copri isolates have diverse genomes making up four distinct clades
Genome diversity is exemplified by differences in susC genes and predicted PULs
P. copri isolates utilize distinct sets of plant polysaccharides
Acknowledgements:
We thank Paul Zappile and Adriana Heguy at the NYU Genome Technology Center for sequencing support. We thank Rhina Medina and Lucy Alvarado for sample collection, Michael Fischbach for Bacteroides strains, Gretchen Diehl for critical reading of the manuscript, and Dawn Hershey for helpful discussions about dietary fiber. We thank Curtis Huttenhower for helpful discussions and for connecting us with NS and AT. The graphical abstract was created with https://biorender.com.
The NYU Genome Technology Center was partially supported by the Cancer Center Support Grant P30CA016087. HF-P was supported by an NYU-HCC CTSI grant (1TL1TR001447). JUS is funded through NIH/NIAMS R01AR074500. JUS is further supported by The Riley Family Foundation, The Beatriz Snyder Foundation, the Rheumatology Research Foundation and The Judith and Stewart Colton Center for Autoimmunity. LMC was supported by NIH Grant 1UL1RR029893. NS was supported by European Research Council (ERC-STG project MetaPG-716575). RB was supported by the Simons Foundation. DRL was supported by the Howard Hughes Medical Institute, the Colton Center for Autoimmunity, and the Rainin Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
All 16S sequencing data and whole genome sequencing data are available under BioProject PRJNA559898.
Supplemental Information:
Table S1. CheckM summary, related to Figure 1
Table S2. SusC gene clusters, related to Figure 2
Data file 1. P. copri 16S SNV sequences, related to Figure 1
Data file 2. P. copri 16S V1–V3 sequences aligned to P. copri 16S sequences from De Filippis et al., 2016, related to Figure 2
References
- Accetto T, and Avgustin G (2015). Polysaccharide utilization locus and CAZYme genome repertoires reveal diverse ecological adaptation of Prevotella species. Syst Appl Microbiol. 38(7), 453–461. Published online 2015/09/30 DOI: 10.1016/j.syapm.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Accetto T, and Avgustin G (2019). The diverse and extensive plant polysaccharide degradative apparatuses of the rumen and hindgut Prevotella species: A factor in their ubiquity? Syst Appl Microbiol. 42(2), 107–116. Published online 2019/03/12 DOI: 10.1016/j.syapm.2018.10.001. [DOI] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. (2011). Enterotypes of the human gut microbiome. Nature. 473(7346), 174–180. Published online 2011/04/22 DOI: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5), 455–477. Published online 2012/04/18 DOI: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Board I.o.M.F.a.N. (2005). Dietary Reference Intakes: Recommended Intakes for Individuals of Macronutrients, 1–5. [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15), 2114–2120. Published online 2014/04/04 DOI: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 13(7), 581–583. Published online 2016/05/24 DOI: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME Journal. 6(8), 1621–1624. DOI: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costea PI, Hildebrand F, Arumugam M, Backhed F, Blaser MJ, Bushman FD, de Vos WM, Ehrlich SD, Fraser CM, Hattori M, et al. (2018). Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 3(1), 8–16. Published online 2017/12/20 DOI: 10.1038/s41564-017-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva AE, Marcelino HR, Gomes MCS, Oliveira EE, Nagashima-Jr T, and Egito EST (2012). Xylan, a Promising Hemicellulose for Pharmaceutical Use. Products and Applications of Biopolymers. [Google Scholar]
- De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. (2019). Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe. 25(3), 444–453 e443. Published online 2019/02/26 DOI: 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- De Filippis F, Pellegrini N, Laghi L, Gobbetti M, and Ercolini D (2016). Unusual sub-genus associations of faecal Prevotella and Bacteroides with specific dietary patterns. Microbiome. 4(1), 57 Published online 2016/10/23 DOI: 10.1186/s40168-016-0202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 107(33), 14691–14696. Published online 2010/08/04 DOI: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Mackie RI, and Cann IK (2011). Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol. 79(2), 292–304. Published online 2011/01/12 DOI: 10.1111/j.1365-2958.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (2011). Accelerated Profile HMM Searches. PLoS Computational Biology. 7(10), e1002195 DOI: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5), 1792–1797. Published online 2004/03/23 DOI: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, and Henrissat B (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 11(7), 497–504. Published online 2013/06/12 DOI: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Ferrocino I, Di Cagno R, De Angelis M, Turroni S, Vannini L, Bancalari E, Rantsiou K, Cardinali G, Neviani E, and Cocolin L (2015). Fecal Microbiota in Healthy Subjects Following Omnivore, Vegetarian and Vegan Diets: Culturable Populations and rRNA DGGE Profiling. PLOS ONE. 10(6), e0128669 DOI: 10.1371/journal.pone.0128669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragiadakis GK, Smits SA, Sonnenburg ED, Van Treuren W, Reid G, Knight R, Manjurano A, Changalucha J, Dominguez-Bello MG, Leach J, et al. (2019). Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes. 10(2), 216–227. DOI: 10.1080/19490976.2018.1494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VK, Chaudhari NM, Iskepalli S, and Dutta C (2015). Divergences in gene repertoire among the reference Prevotella genomes derived from distinct body sites of human. BMC Genomics. 16, 153 Published online 2015/04/19 DOI: 10.1186/s12864-015-1350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Shibata K, Sakamoto M, Tomita S, and Benno Y (2007). Prevotella copri sp. nov. and Prevotella stercorea sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 57(Pt 5), 941–946. Published online 2007/05/03 DOI: 10.1099/ijs.0.64778-0. [DOI] [PubMed] [Google Scholar]
- Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, and Michel G (2010). Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 464(7290), 908–912. Published online 2010/04/09 DOI: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Subramanian A, and Anishetty S (2017). Comparative pan genome analysis of oral Prevotella species implicated in periodontitis. Functional & Integrative Genomics. 17(5), 513–536. DOI: 10.1007/s10142-017-0550-3. [DOI] [PubMed] [Google Scholar]
- Joglekar P, Sonnenburg ED, Higginbottom SK, Earle KA, Morland C, Shapiro-Ward S, Bolam DN, Sonnenburg JL, Abbott W, and Koropatkin N (2018). Genetic Variation of the SusC/SusD Homologs from a Polysaccharide Utilization Locus Underlies Divergent Fructan Specificities and Functional Adaptation in Bacteroides thetaiotaomicron Strains. mSphere. 3(3), e00185–00118. DOI: 10.1128/mspheredirect.00185-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, and Ley RE (2013). A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLoS Computational Biology. 9(1), e1002863 DOI: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, et al. (2014). A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 506(7489), 498–502. Published online 2014/01/28 DOI: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou H, Hua W, Wang B, Wang S, Zhao G, Li L, Zhao L, and Pang X (2009). Molecular diversity of Bacteroides spp. in human fecal microbiota as determined by group-specific 16S rRNA gene clone library analysis. Systematic and Applied Microbiology. 32(3), 193–200. DOI: 10.1016/j.syapm.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Lin A, Bik EM, Costello EK, Dethlefsen L, Haque R, Relman DA, and Singh U (2013). Distinct Distal Gut Microbiome Diversity and Composition in Healthy Children from Bangladesh and the United States. PLoS ONE. 8(1), e53838 DOI: 10.1371/journal.pone.0053838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cui L, Yan X, Zhao X, Cheng J, Zhou L, Gao J, Cao Z, Xinhua, and Hu S (2018). Analysis of Oral Microbiota Revealed High Abundance of Prevotella Intermedia in Gout Patients. Cellular Physiology and Biochemistry. 49(5), 1804–1812. DOI: 10.1159/000493626. [DOI] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, and Henrissat B (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Research. 42(D1), D490–D495. DOI: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, Hirota K, Matsushita M, Furuta Y, Narazaki M, et al. (2016). Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis & Rheumatology. 68(11), 2646–2661. DOI: 10.1002/art.39783. [DOI] [PubMed] [Google Scholar]
- Martens EC, Chiang HC, and Gordon JI (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 4(5), 447–457. Published online 2008/11/11 DOI: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, et al. (2011). Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9(12), e1001221 Published online 2011/12/30 DOI: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EL, and Bhatt AS (2018). Generating closed bacterial genomes from long-read nanopore sequencing of microbiomes. bioRxiv. 489641 DOI: 10.1101/489641. [DOI] [Google Scholar]
- Nishinari K, Takemasa M, Zhang H, and Takahashi R (2007). Storage Plant Polysaccharides: Xyloglucans, Galactomannans, Glucomannans. (Elsevier), pp. 613–652. [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research. 25(7), 1043–1055. DOI: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan V, Lowe EC, Townsend GE, Bolam DN, and Groisman EA (2014). Tuning transcription of nutrient utilization genes to catabolic rate promotes growth in a gut bacterium. Mol Microbiol. 93(5), 1010–1025. DOI: 10.1111/mmi.12714. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Michael, and Laurie(2014). An Ecological Network of Polysaccharide Utilization among Human Intestinal Symbionts. 24(1), 40–49. DOI: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruengsomwong S, L.-O. O, Jiang J, Wannissorn B, Nakayama J, Nitisinprasert S (2016). Microbial community of healthy Thai vegetarians and non-vegetarians, their core gut microbiota and pathogens risk. Journal of microbiology and biotechnology. 26(20), 1723–1735. [DOI] [PubMed] [Google Scholar]
- Salyers AA, O.B. M, Kotarski SF (1982). Utilization of Chondroitin Sulfate by Bacteroides thetaiotaomicron Growing in Carbohydrate-Limited Continuous Culture. J Bacteriol. 150(3), 1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2, e01202 Published online 2013/11/07 DOI: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, and Segata N (2016). Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods. 13(5), 435–438. Published online 2016/03/22 DOI: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature. 557(7705), 434–438. Published online 2018/05/11 DOI: 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 357(6353), 802–806. DOI: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, and Sonnenburg JL (2010). Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 141(7), 1241–1252. Published online 2010/07/07 DOI: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9), 1312–1313. Published online 2014/01/24 DOI: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Zhao J, Zhang H, Zhai Q, and Chen W (2018). Isolation of Low-Abundant Bacteroidales in the Human Intestine and the Analysis of Their Differential Utilization Based on Plant-Derived Polysaccharides. Front Microbiol. 9, 1319 Published online 2018/07/05 DOI: 10.3389/fmicb.2018.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrapon N, Lombard V, Drula E, Lapebie P, Al-Masaudi S, Gilbert HJ, and Henrissat B (2018). PULDB: the expanded database of Polysaccharide Utilization Loci. Nucleic Acids Res. 46(D1), D677–D683. Published online 2017/11/01 DOI: 10.1093/nar/gkx1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tett A, Huang KD, Asnicar F, Fehlner-Peach H, Pasolli E, Karcher N, Armanini F, Manghi P, Bonham K, Zolfo M, et al. (2019). The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host & Microbe. DOI: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. 42(D1), D581–D591. DOI: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science. 334(6052), 105–108. DOI: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Mao X, Yang J, Chen X, Mao F, and Xu Y (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. 40(W1), W445–W451. DOI: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lieberman TD, Poyet M, Kauffman KM, Gibbons SM, Groussin M, Xavier RJ, and Alm EJ (2019). Adaptive Evolution within Gut Microbiomes of Healthy People. Cell Host Microbe. 25(5), 656–667 e658. Published online 2019/04/28 DOI: 10.1016/j.chom.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Sunagawa S, Mende DR, and Bork P (2015). Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 16, 82 Published online 2015/04/22 DOI: 10.1186/s13059-015-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.