

Abstract
Besides being regulated by G-protein–coupled receptors, the activity of heterotrimeric G proteins is modulated by many cytoplasmic proteins. GIV/Girdin and DAPLE (Dvl-associating protein with a high frequency of leucine) are the best-characterized members of a group of cytoplasmic regulators that contain a Gα-binding and -activating (GBA) motif and whose dysregulation underlies human diseases, including cancer and birth defects. GBA motif–containing proteins were originally reported to modulate G proteins by binding Gα subunits of the Gi/o family (Gαi) over other families (such as Gs, Gq/11, or G12/13), and promoting nucleotide exchange in vitro. However, some evidence suggests that this is not always the case, as phosphorylation of the GBA motif of GIV promotes its binding to Gαs and inhibits nucleotide exchange. The G-protein specificity of DAPLE and how it might affect nucleotide exchange on G proteins besides Gαi remain to be investigated. Here, we show that DAPLE's GBA motif, in addition to Gαi, binds efficiently to members of the Gs and Gq/11 families (Gαs and Gαq, respectively), but not of the G12/13 family (Gα12) in the absence of post-translational phosphorylation. We pinpointed Met-1669 as the residue in the GBA motif of DAPLE that diverges from that in GIV and enables better binding to Gαs and Gαq. Unlike the nucleotide-exchange acceleration observed for Gαi, DAPLE inhibited nucleotide exchange on Gαs and Gαq. These findings indicate that GBA motifs have versatility in their G-protein–modulating effect, i.e. they can bind to Gα subunits of different classes and either stimulate or inhibit nucleotide exchange depending on the G-protein subtype.
Keywords: GTPase, G-protein–coupled receptor (GPCR), guanine nucleotide–exchange factor (GEF), G protein, protein–protein interaction, cell signaling, Gα-binding and -activating (GBA), guanine nucleotide dissociation inhibitor (GDI)
Introduction
Heterotrimeric G proteins are essential signaling molecules that relay extracellular signals acting on G-protein–coupled receptors (GPCRs)4 to intracellular effector proteins (1). They are involved in a plethora of physiological processes and mediate the effect of >30% of Food and Drug Administration–approved drugs (2). At the molecular level, heterotrimeric G proteins switch between “on” or “off” states depending on their guanine nucleotide–binding status. In the inactive state, GDP-bound Gα subunits associate tightly with obligate Gβγ dimers, which in turn serve to prevent spurious nucleotide exchange by working as guanine nucleotide–dissociation inhibitors (GDIs) (3, 4). The Gαβγ trimer is the substrate for the guanine nucleotide–exchange factor (GEF) activity of GPCRs, which promote the exchange of GDP for GTP on Gα and the subsequent dissociation of Gβγ. Both Gα-GTP and free Gβγ become active signaling species that engage their own set of effectors to propagate downstream signaling. Gα subunits are classified into four families depending on sequence conservation and the effector targets that they modulate: Gs, Gi/o, Gq/11, and G12/13 (5, 6). Signaling is terminated upon GTP hydrolysis by Gα, which leads to reassociation with Gβγ to form again an inactive Gαβγ trimer.
Based on the above, it is evident that the amplitude and duration of G-protein signaling is highly dependent on the regulation of nucleotide handling by Gα. In this regard, it has become increasingly clear that G-protein activity is controlled by a complex network of regulators that expands beyond GPCRs and Gβγ. The best-characterized ones are the regulators of G-protein signaling (RGS) proteins, which are GTPase-accelerating proteins (7–16). RGS proteins, as well as some effectors like PLC-β isoforms, enhance the rate of nucleotide hydrolysis on Gα, thereby facilitating the termination of G-protein signaling (16, 17). Another group of regulators, defined by the presence of a sequence called the GoLoco motif, locks Gα in the GDP-bound state by virtue of their GDI activity (18–26). There are also nonreceptor proteins that have the same biochemical activity as GPCRs, i.e. they are GEFs (27–32). Among them, it has been possible to link the GEF activity to a defined protein domain or sequence only for a subset of these nonreceptor G-protein regulators. This is for proteins that contain a Gα-binding and -activating (GBA) motif, an evolutionarily-conserved sequence of ∼30 amino acids with a well-defined mechanism of action at the structural level (32–37). Six GBA motif–containing proteins have been identified to date: GIV, DAPLE, Calnuc, NUCB2, PLCδ4b, and the C. elegans protein GBAS-1 (32, 33, 38–40).
Among the GBA motif–containing proteins, GIV (also known as Girdin) and DAPLE were the first ones to be identified and are the best-characterized ones to date (32, 34, 35, 39, 41). Early evidence indicated that both proteins bind to inactive, GDP-bound Gαi subunits (Gαi1, Gαi2, and Gαi3) to accelerate their rate of spontaneous nucleotide exchange in vitro, although they interacted poorly with other Gα subunits, including other members of the Gi/o family like Gαo (39, 42). This biochemical activity in vitro correlates well with an enhancement of G-protein signaling observed in cells. For example, it has been shown that the GBA motif of these proteins is required for signaling readouts that depend on Gαi (like inhibition of adenylyl cyclase or antibodies that specifically detect GTP-bound Gαi) (39, 40, 43–45), as well as on Gβγ (like free Gβγ biosensors or the PI3K–Akt effector pathway) (32, 39–41, 44, 46–48). It has also been recently shown that G-protein regulation by the GBA motif of DAPLE operates in vivo to control vertebrate development (41, 49). Overall, the biomedical relevance of this signaling mechanism is highlighted by multiple studies establishing the involvement of G-protein regulation by GIV and DAPLE in several human disorders like cancer, liver fibrosis, or embryonic defects (39, 41, 45–47, 50).
Although the seminal studies described above indicated that the GBA motif–containing proteins are GEFs that specifically work on Gαi subunits, more recent evidence has challenged this notion by suggesting that the biochemical activity and G-protein specificity of GBA motifs can vary from this. More specifically, it has been shown that upon sequential phosphorylation of the GBA motif of GIV at serine 1674 and serine 1689 by two different kinases, it does not bind to Gαi proteins anymore (51). Instead, such phosphoevents enhance the affinity of GIV's GBA motif for Gαs, which is poor in the absence of phosphorylation (51, 52). Interestingly, this interaction results in the inhibition of nucleotide exchange rather than in acceleration as observed for Gαi (51). Together, these findings suggest that GIV can switch from behaving as a GEF (for Gαi) to behaving as a GDI (for Gαs) upon phosphorylation. The term guanine nucleotide–exchange modulator has been proposed for GIV to convey that it can have different effects on nucleotide handling depending on context (51). However, it is not known whether any GBA motif present in a protein can have the simultaneous ability to work as a GEF or GDI or to interact efficiently with Gα subunits besides Gαi, in the absence of phosphomodifications. Here, we set out to characterize the previously unexplored G-protein selectivity of DAPLE, and we found that its GBA motif can efficiently bind to representative members of three different G-protein subfamilies (Gi/o, Gs, and Gq/11) without any phosphomodification. For Gαs and Gαq, this interaction results in the inhibition of nucleotide exchange, suggesting that DAPLE can at the same time work as a GEF or as a GDI depending on the G-protein substrate.
Results
DAPLE binds efficiently Gαs and Gαq in addition to Gαi3
We set out to characterize the G-protein specificity of DAPLE by investigating its binding to representative Gα subunits of each one of the four different families, i.e. Gαi3 from Gi/o, Gαs from Gs, Gαq from Gq/11, and Gα12 from G12/13. For this, we carried out protein–protein-binding experiments using lysates of HEK293T cells expressing each one of the G proteins and GST-fused DAPLE immobilized on resin. As an internal reference for these experiments, we used GST–GIV, which has been previously shown to have a marked preference for Gαi over other G proteins. The two GST-fused constructs consisted of C-terminal fragments of each protein containing the GBA motif (GST–DAPLE aa 1650–2028 and GST–GIV aa 1671–1755, see Fig. 1A). Purification of any of the two constructs based on affinity capture of their N-terminal GST tags resulted in proteins with degradation products that could not be avoided despite attempts to optimize expression conditions. However, all or most of the degradation products should contain G-protein–binding sites because the GBA motif is adjacent to the GST tag used for affinity capture (Fig. 1A). Based on this, we reasoned that using equal amounts of total GST-fused proteins would allow the direct comparison of G-protein binding to GST–DAPLE and GST–GIV because the number of binding sites should be similar. If so, the prediction was that we should see equivalent binding of Gαi3 to GST–DAPLE and GST–GIV because it has been previously reported that these two constructs have equivalent affinity for this G protein (39). We found that this is the case because the same amount of Gαi3–FLAG was detected in resin-bound complexes of GST–DAPLE and GST–GIV (Fig. 1B, left panel).
Figure 1.

DAPLE binds efficiently to Gαs and Gαq through its GBA motif. A, bar diagrams depicting the domains of DAPLE (left) and GIV (right) and the fragments of each one fused to GST used for experiments shown in this figure. B, DAPLE binds efficiently to Gαi3, Gαs, and Gαq but not to Gα12, whereas GIV only binds efficiently to Gαi3 among the G proteins tested. Lysates of HEK293T cells transfected with Gαi3–FLAG, Gαs, Gαq–HA, and Gα12–MYC were incubated with GST, GST–DAPLE, or GST–GIV immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. C, DAPLE WT but not DAPLE F1675A (FA) binds to purified Gαi3, Gαs, and Gαq. His–Gαi3, His–Gαs, or His–Gαq* were incubated with GST, GST–DAPLE (WT or FA mutant), or GST–GIV immobilized on GSH-agarose beads, and bead-bound proteins were detected by Ponceau S staining or IB as indicated. D, DAPLE WT, but not DAPLE FA mutant, co-immunoprecipitates with Gαi3, Gαs, and Gαq. Lysates of HEK293T cells co-expressing full-length MYC–DAPLE (WT or FA mutant) with the indicated FLAG-tagged G proteins (or no tagged G protein as negative control) were subjected to IP with a FLAG antibody, and bound proteins were detected by IB as indicated. The lower immunoblot panels (lysates) correspond to aliquots of the starting material used for IPs shown in the upper panels (IP: FLAG). All results presented in this figure are representative of at least three independent experiments (n ≥3).
Having established these experimental conditions to semi-quantitatively assess G-protein binding to DAPLE compared with GIV, we performed equivalent experiments with Gαs, Gαq, and Gα12. As expected, binding of Gαs, Gαq, or Gα12 to GST–GIV was undetectable or marginal relative to the binding observed for Gαi3 (the same proportion of input lysate was run in each experiment to facilitate the comparison of relative binding across G proteins) (Fig. 1B). In contrast, we detected robust binding of Gαs and Gαq, but not Gα12, to GST–DAPLE, which was comparable with the binding observed for Gαi3 based on comparison with their respective input lanes. To rule out that the observed binding of Gαs and Gαq to GST–DAPLE was due to the overexpression of G proteins, we performed analogous experiments to detect binding of Gαs or Gαq endogenously expressed in HEK293T cells. We found that endogenous Gαs and Gαq bind efficiently to GST–DAPLE but not to GST–GIV (Fig. S1). These results indicate that, in contrast to GIV, nonphosphorylated DAPLE can bind robustly to G proteins from three different subfamilies: Gi/o (Gαi3), Gs (Gαs), and Gq/11 (Gαq).
DAPLE binds directly to Gαs and Gαq via its GBA motif
Next, we investigated whether the association of DAPLE with Gαs and Gαq described above is mediated by direct binding of the G proteins to the GBA motif of DAPLE as observed previously for Gαi (39). For this, we carried out experiments with purified G proteins instead of cell lysates, and we included a GST–DAPLE construct bearing a mutation in its GBA motif (F1675A) that has been previously shown to disrupt binding to Gαi3 (39). We also kept GST–GIV as an internal control in these experiments. The G proteins were His-tagged versions of Gαi3, Gαs, and Gαq purified from bacteria. For Gαq, we used a chimera containing partial sequences of Gαi (named here Gαq*) that can be expressed in bacteria and that have been previously validated to bind to a wide range of Gαq-specific partners (53). As observed with G proteins from cell lysates, both GST–DAPLE and GST–GIV bound similarly to Gαi3, but only GST–DAPLE bound efficiently to Gαs and Gαq* (Fig. 1C). DAPLE binding to any of the G proteins was disrupted by the F1675A mutation (Fig. 1C), indicating that the GBA motif of DAPLE is required for the direct binding of Gαs and Gαq.
To determine whether the binding of Gαs and Gαq to a fragment of DAPLE in vitro observed above occurs when the GBA motif is in the context of the full-length protein expressed in cells, we carried out co-immunoprecipitation experiments (co-IP) from HEK293T cells. FLAG-tagged Gαi3, Gαs, or Gαq was co-expressed with MYC-tagged full-length DAPLE, and IPs were carried out with FLAG antibodies. Cells expressing MYC–DAPLE in the absence of FLAG–Gα were used as negative control. We found that MYC–DAPLE WT was efficiently co-IPed by FLAG–Gαi3, FLAG–Gαs, or FLAG–Gαq (Fig. 1D). Moreover, we found that the GBA motif mutant F1675A prevents the co-IP of DAPLE with any of the three G proteins (Fig. 1D). These findings indicate that full-length DAPLE interacts with Gαs and Gαq through its GBA motif, much like Gαi proteins do.
DAPLE binds to inactive but not active Gαs and Gαq
A property of all previously characterized GBA motifs is that they bind preferentially to inactive (GDP-bound) but not to active (GTP-bound) conformations of Gαi (32, 33, 37–40). Thus, we set out to test whether this is also the case for the DAPLE–Gαs and DAPLE–Gαq interactions. For this, we first compared DAPLE binding to constitutively active, GTPase-deficient mutants of Gαs and Gαq (R201C and Q209L, respectively (54)) versus their WT G-protein counterparts. We found that both active mutants have diminished binding to DAPLE compared with WT (Fig. 2, A and C). As a complementary approach to address this point, we performed similar experiments with Gα subunits loaded with nucleotides that mimic different G-protein activation states. More specifically, G proteins were loaded with GDP (inactive conformation), GDP·AlF4− (which mimics the GTP-bound activation transition state) or GTPγS (a nonhydrolyzable GTP analog). The GTPγS condition was excluded for Gαq because it is known that this G protein exchanges nucleotide very slowly and loads GTPγS substoichiometrically even under experimental conditions that accelerate nucleotide exchange (55). Consistent with the results obtained with the constitutively-active mutants, loading of the G proteins with GDP·AlF4− and/or GTPγS markedly diminished binding to DAPLE compared with the GDP-loaded conditions (Fig. 2, B and D). Taken together, these results show that the interaction of DAPLE with Gαs or Gαq is G-protein state-dependent, having a marked preference for the inactive state. This feature resembles the previously characterized binding properties of GBA motifs to Gαi proteins (32, 33, 37–40).
Figure 2.

DAPLE binds preferentially to inactive versus active Gαs or Gαq. A, binding of DAPLE to the constitutively-active Gαs mutant R201C is diminished compared with Gαs WT. Lysates of HEK293T cells expressing Gαs WT, or Gαs R201C were incubated with GST or GST–DAPLE immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. B, binding of DAPLE to Gαs loaded with GDP·AlF4− or with GTPγS is diminished compared with binding to Gαs loaded with GDP. Lysates of HEK293T cells expressing Gαs were incubated with nucleotides as indicated under “Experimental procedures” and incubated with GST or GST–DAPLE immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. C, binding of DAPLE to the constitutively-active Gαq mutant Q209L is diminished compared with Gαq WT. Lysates of HEK293T cells expressing Gαq–HA WT or Gαq–HA Q209L were incubated with GST or GST–DAPLE immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. D, binding of DAPLE to Gαq loaded with GDP·4− is diminished compared with binding to Gαq loaded with GDP. Lysates of HEK293T cells expressing Gαq were incubated with nucleotides as indicated under Experimental procedures” and incubated with GST or GST–DAPLE immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. All results presented in this figure are representative of two independent experiments (n = 2).
Identification of a single amino acid in DAPLE that favors its binding to Gαs and Gαq
Our results so far indicate that the GBA motif of DAPLE interacts with Gαs or Gαq through a mechanism that resembles binding of Gαi to previously described GBA motifs (i.e. direct binding to inactive Gα subunits). Next, we set out to dissect the molecular determinants within the GBA motif of DAPLE that enable specific binding to Gαs and Gαq. We reasoned that this specificity would be encoded in the GBA motif itself and that certain amino acids within the GBA motif of DAPLE that are different in the GBA motif GIV might confer specificity in G-protein binding. To test this hypothesis, we constructed a chimera of DAPLE in which 26 amino acids of the GBA motif of DAPLE were replaced by the corresponding amino acids from the GBA motif of GIV (GST–DAPLE ch1). We found that DAPLE ch1, much like GIV, fails to bind Gαs and Gαq but retains binding to Gαi3 (Fig. 3A). This indicates that amino acids within the GBA motif of DAPLE different from those in the GBA motif of GIV are required for its improved binding to Gαs and Gαq. To map which one(s) of the differing amino acids is responsible for the observed difference in binding, we constructed six additional DAPLE/GIV GBA chimeras (GST–DAPLE ch2 to ch7) (Fig. 3B). These chimeras allowed us to reduce the number of amino acids that might confer increased binding to Gαs and Gαq to only two (Fig. 3B). Mutating each one of those two amino acids to the corresponding amino acid in GIV revealed that only M1669V reduces binding of DAPLE to Gαs and Gαq while still binding well to Gαi3 (Fig. 3B). These results indicate that one single amino acid of the GBA motif of DAPLE, the methionine in position 1669, confers increased binding to Gαs and Gαq and that its mutation to a valine impairs these interactions.
Figure 3.

Met-1669 in DAPLE is responsible for its enhanced binding to Gαs and Gαq compared with GIV. A, DAPLE binding to Gαs or Gαq, but not to Gαi3, is reduced upon replacing its GBA motif with that of GIV. Upper panel, diagram depicting the alignment of the GBA motifs of DAPLE and GIV and the sequence of the GBA motif of the DAPLE/GIV GBA chimera 1 (ch1) containing GIV's GBA motif residues (red) grafted into DAPLE's sequence (black). Lower panel, purified His–Gαi3, His–Gαs, or His–Gαq* was incubated with GST, GST–DAPLE (WT or ch1), or GST–GIV immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. The vertical dotted lines indicate that the images were assembled by splicing lanes from the same experiment and membrane. B, mapping of residues involved in the differential G-protein selectivity of DAPLE versus GIV. Upper panel, sequences of DAPLE/GIV GBA chimeras (ch1–7, M1669V, and S1666G), with the GIV residues replaced in DAPLE indicated in red. Lower panel, purified His–Gαi3, His–Gαs, or His–Gαq* was incubated with GST or GST–DAPLE (WT or mutants) immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. C, validation of the effects of DAPLE F1675A (FA) and M1669V (MV) mutations on binding to different G proteins using a shorter DAPLE-purified protein. Upper panel, diagram depicting the GST–DAPLE (short) construct used in this panel along with a diagram of the previously used GST–DAPLE construct. Lower panel, purified His–Gαi3, His–Gαs, or His–Gαq* was incubated with GST or GST–DAPLE (WT or mutants) immobilized on GSH-agarose beads. Bead-bound proteins were detected by Ponceau S staining or IB as indicated. All results presented in this figure are representative of at least three independent experiments (n ≥3).
In the course of our experiments, we observed that the different GST–DAPLE chimeras showed variable patterns of degradation products, which we reasoned might affect the validity of our conclusion that the M1669V mutation specifically impairs the interaction of DAPLE with Gαs or Gαq. To address this issue, we constructed a shorter fragment of DAPLE fused to GST that we named GST–DAPLE (short), which we anticipated to show less degradation. We introduced in this construct the mutation F1675A that abrogates binding to all Gα subunits, and the mutation M1669V, which, based on the results showed above, should disrupt binding to Gαs and Gαq but not to Gαi3. As expected, we found that the degradation products in GST–DAPLE (short) were greatly reduced and that the integrity of WT and mutant purified proteins were essentially the same. Moreover, we found that F1675A abolishes binding of DAPLE to all Gα subunits tested, whereas M1669V only disrupts binding to Gαs and Gαq but not to Gαi3 (Fig. 3C). These data confirm our results obtained with the longer fragment of DAPLE and indicate a key role for Met-1669 in mediating the binding to Gαs and Gαq.
Met-1669 in DAPLE is highly conserved in evolution, as is the corresponding Val-1679 in GIV (Fig. S2A). Next, we asked whether mutation of GIV Val-1679 to the corresponding methionine found in DAPLE would be sufficient to enhance its binding to Gαs and Gαq. We found that this is the case because GST–GIV V1679M bound to Gαs and to Gαq more efficiently than GST–GIV WT, whereas binding to Gαi3 was not significantly affected (Fig. S2). These observations strengthen the conclusion that Met-1669 in DAPLE is the key determinant that allows efficient binding to Gαs and Gαq.
Full-length DAPLE M1669V mutant displays impaired binding to Gαs and Gαq but not to Gαi3
To determine whether the different specificity of DAPLE WT, F1675A, or M1669V for binding to Gα observed in in vitro binding experiments with truncated proteins also occurs in the context of the full-length protein, we carried out co-IP experiments. FLAG-tagged Gαi3, Gαs, or Gαq was co-expressed with DAPLE WT, F1675A, or M1669V in HEK293T cells, and lysates were subjected to IP with FLAG antibodies. We found that DAPLE M1669V shows decreased binding to Gαs or Gαq but not to Gαi3 (Fig. 4A), whereas DAPLE F1675A displays diminished binding to all three G proteins (i.e. Gαi3, Gαs, or Gαq). These findings are in good agreement with our in vitro binding experiments, and they indicate that Met-1669 is required for binding of full-length DAPLE to Gαs or Gαq but is largely dispensable for interacting with Gαi3.
Figure 4.

M1669V mutation in full-length DAPLE disrupts binding to Gαs or Gαq but not to Gαi3. A, co-immunoprecipitation experiments comparing the effect of DAPLE M1669V and F1675A mutations on G-protein binding, which show that the former disrupts binding to Gαs and Gαq, but not to Gαi3, whereas the latter disrupts binding to all G proteins tested. Lysates of HEK293T cells co-expressing full-length MYC–DAPLE (WT or mutants) with the indicated FLAG-tagged G proteins (or no tagged G protein as negative control) were subjected to IP with a FLAG antibody, and bound proteins were detected by IB as indicated. The lower immunoblot panels (Lysates) correspond to aliquots of the starting material used for IPs shown in the upper panels (IP: FLAG). One representative experiment of four is shown for Gαs and Gαq (n = 4), or one representative experiment of two is shown for Gαi3 (n = 2). B, comparison of DAPLE Met-1669 and GIV Val-1679 in the context of their respective Gαi3/GBA motif complex structures. Left panel, homology model of DAPLE GBA motif (green, ribbon representation) in complex with Gαi3 (blue, space-filling representation) was generated using the X-ray crystal structure of the Gαi3/GIV GBA motif complex (PDB code 6MHF). The area of the Gαi3/DAPLE structure model within the dotted box is shown enlarged in the middle panel to illustrate that Met-1669 is largely solvent-exposed. Right panel, detail of the structure of Gαi3 in complex with GIV GBA motif (brown) showing that Val-1669 is also largely solvent-exposed. C, proposed model for the structural basis of DAPLE's G-protein selectivity. Much like GIV Val-1679, DAPLE Met-1669 does not make direct contact with Gαi3. In contrast, DAPLE Met-1669 is required for binding to Gαs or Gαq, suggesting that it makes a contact with these proteins that is not allowed by the shorter chain of the valine located in the corresponding position in GIV.
To gain further insights into the structural basis for the role of Met-1669 in DAPLE and Val-1679 in the corresponding position of GIV in determining their different G-protein specificity, we leveraged the recently elucidated atomic resolution structure of the Gαi3/GIV GBA motif complex (35). We generated a homology model of the Gαi3/DAPLE GBA motif complex and compared the spatial localization of Met-1669 in DAPLE with that of Val-1679 in GIV (Fig. 4B). We observed that both GIV Val-1679 and DAPLE Met-1669 are largely solvent-exposed and do not make direct contact with Gαi3 (Fig. 4B). This is very consistent with our results above showing that the DAPLE–Gαi3 interaction tolerates well the replacement of Met-1669 in DAPLE by valine as determined in protein–protein-binding experiments above, and with previously published evidence showing that the GIV–Gαi3 interaction tolerates well the replacement of Val-1679 in GIV by almost any other amino acid as determined in peptide array binding experiments (34). Overall, these observations provide a reasonable structural explanation for the neutral role of the GIV V1679/DAPLE Met-1669 position in determining binding to Gαi3. Although we lack a reliable template to generate high-confidence homology models of DAPLE's GBA motif in complex with Gαs or Gαq, we propose that the larger side chain of DAPLE Met-1669 compared with GIV Val-1679 might allow for additional molecular contacts with Gαs and Gαq and thereby account for its ability to bind Gαs and Gαq better than GIV (Fig. 4C). Such contacts would be disrupted upon mutation of Met-1669 to valine, which in turn would have no effect on Gαi3 binding (Fig. 4C).
Peptide derived from the GBA motif of DAPLE recapitulates its GEF activity on Gαi3
Next, we set out to evaluate the consequences of DAPLE binding to Gαs and Gαq on G-protein activity. We reasoned that a peptide derived from the GBA motif of DAPLE could be used for this purpose, as previous observations suggest that GBA peptides recapitulate well the properties of their cognate proteins in that they regulate Gαi proteins (34, 35). We fully validated these previous observations by analyzing the dose-dependent effects of a 34-mer GBA peptide from DAPLE in two assays that monitor nucleotide exchange on Gαi: steady-state GTPase and GTPγS binding assays (56). Briefly, steady-state GTPase activity is a good proxy of nucleotide exchange rates for Gαi because hydrolysis is 1–2 orders of magnitude faster than nucleotide exchange, whereas GTPγS binding is a more direct readout of exchange (57). As expected, we found that the DAPLE GBA peptide, but not a control peptide (see “Experimental procedures”), increased the steady-state GTPase activity (Fig. S3, A and B) and GTPγS binding (Fig. S3C) of Gαi3 in a dose-dependent manner. The amplitude of the DAPLE GBA peptide effects (∼2–4-fold increases) and the corresponding EC50 values (∼5–10 μm) are similar to those previously reported for a very similar peptide from GIV whose molecular mechanism of action on Gαi has been extensively validated (34, 35). We conclude that the GBA peptide of DAPLE recapitulates the GEF activity of DAPLE protein in vitro.
DAPLE inhibits nucleotide exchange on Gαs
As for Gαi3, we determined the effects of DAPLE on Gαs nucleotide exchange using both steady-state GTPase assays and GTPγS-binding assays. We found that DAPLE GBA peptide decreases Gαs steady-state GTPase activity ∼40% (Fig. 5, A and B), which was in agreement with an ∼50% decrease in the rate of GTPγS binding (Fig. 5C). The half-maximal inhibitory concentration (IC50) was ∼3 μm in both assays (Fig. 5, B and D). These results indicate that DAPLE GBA peptide inhibits nucleotide exchange on Gαs and that the potency of this inhibition is similar to or higher than the potency it has for activating Gαi3 (∼5–10 μm, Fig. S3).
Figure 5.
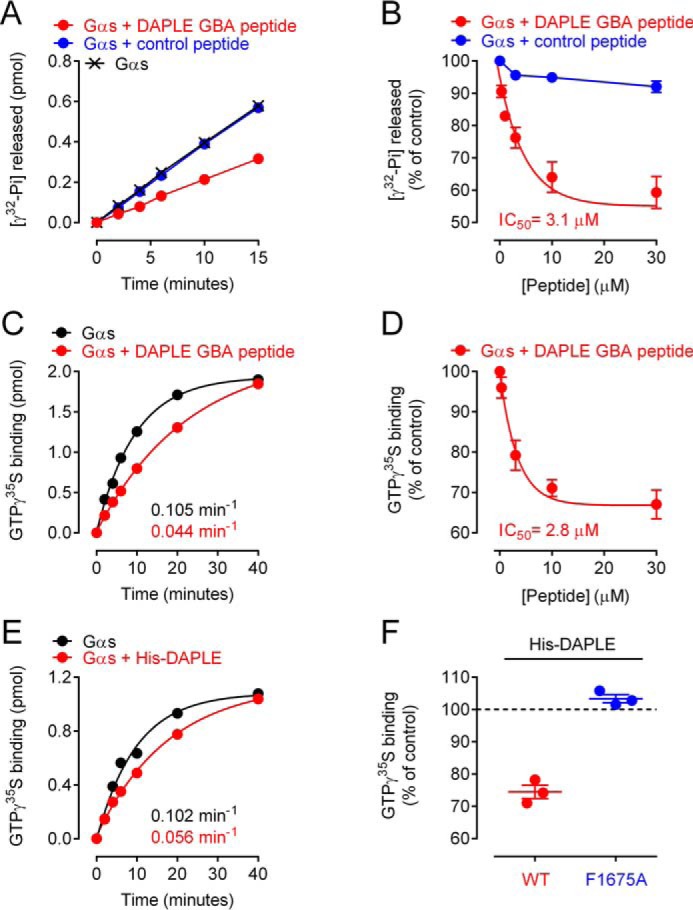
DAPLE inhibits nucleotide exchange on Gαs via its GBA motif. A and B, DAPLE GBA peptide decreases the steady-state GTPase activity of Gαs. A representative time course of the steady-state GTPase activity of His–Gαs alone (black), in the presence of DAPLE GBA peptide (30 μm, red), or control peptide (30 μm, blue) is shown in A, and quantification of the dose-dependent effect of the peptides is shown in B (mean ± S.E., n = 3). Results are presented as raw production of free [32P]Pi (pmol) in A or percent change relative to the production of free [32P]Pi by Gαs alone at 10 min (% of control) in B. Average IC50 value was determined as described under “Experimental procedures.” C and D, DAPLE GBA peptide decreases the rate of GTPγS binding to Gαs. A representative time course of [35S]GTPγS binding to His–Gαs in the absence (black) or presence of DAPLE GBA peptide (30 μm, red) is shown in C, and quantification of the dose-dependent effect of the DAPLE GBA peptide is shown in D (mean ± S.E., n = 3). Results are presented as raw [35S]GTPγS binding (picomoles) in C or percent change relative to [35S]GTPγS binding to Gαs alone at 10 min (% of control) in D. Rate constants and average IC50 values were determined as described under “Experimental procedures,” E and F, purified DAPLE WT (amino acids 1650–2028), but not F1675A mutant, decreases GTPγS binding to Gαs. A representative time course of [35S]GTPγS binding to His–Gαs in the absence (black) or presence of purified His–DAPLE (9 μm, red) is shown in E, and quantification of the effect of His–DAPLE WT (3.3 μm, red) compared with His–DAPLE F1675A (3.3 μm, blue) is shown in D (mean ± S.E., n = 3). Results are presented as raw [35S]GTPγS binding (picomoles) in E or percent change relative to [35S]GTPγS binding to Gαs alone at 10 min (% of control) in F. Rate constants determined as described under “Experimental procedures.”
To rule out that the observed effect on Gαs activity was related to the use of an isolated GBA motif out of protein context, we performed additional experiments with a larger fragment of DAPLE. For this, we used a purified protein consisting of the GBA-containing C-terminal region of DAPLE (DAPLE-CT, aa 1650–2028), which has been previously validated to preserve the G-protein regulatory functions of DAPLE on Gαi3 (39). We found that purified DAPLE-CT slows down GTPγS binding to Gαs to an extent similar to that observed with DAPLE GBA peptide (∼50%, Fig. 5E). Moreover, the inhibitory effect of DAPLE-CT on Gαs was blunted by the F1675A mutation that disrupts G-protein binding (Fig. 5F), confirming that this effect is mediated by the GBA motif of DAPLE. Altogether, these results indicate that the GBA motif of DAPLE can inhibit nucleotide-exchange activity of Gαs as potently as it activates Gαi and that this occurs in the absence of any phosphomodification because the experiments were performed with bacterially-expressed proteins or synthetic peptides.
DAPLE inhibits nucleotide exchange on Gαq
Next, we investigated the effect of DAPLE on the activity of Gαq. Instead of using the bacterially-expressed Gαq* chimera utilized in our protein-binding experiments described above, we used Gαq purified from Sf9 insect cells because its enzymatic properties have been more thoroughly characterized (55, 58), including its modulation by various regulators (17, 59). Gαq differs markedly from Gαi or Gαs in that its spontaneous exchange of nucleotide is very slow (55, 58). Thus, steady-state GTPase activity is not a good proxy for nucleotide exchange, so we exclusively measured GTPγS binding to assess the effects of DAPLE on nucleotide exchange. Moreover, we supplemented the assay buffer with 0.2 m ammonium sulfate, as described previously (55), to enhance nucleotide exchange to a faster rate that is more tractable experimentally. Under these conditions, we found that DAPLE GBA peptide slowed down GTPγS binding ∼40%, with an IC50 of ∼3 μm (Fig. 6, A and B). This effect was recapitulated by DAPLE-CT WT but not the G-protein binding-deficient F1675A mutant (Fig. 6C), indicating that Gαq inhibition is a bona fide action of the GBA motif of DAPLE. We ruled out that the observed inhibition was a consequence of performing the experiments under artificially accelerated nucleotide-exchange conditions by performing analogous experiments with a buffer not supplemented with ammonium sulfate (Fig. S3). As expected, the rate of nucleotide exchange was much slower, but the overall effects of DAPLE GBA peptide (Fig. S4, A and B) and DAPLE-CT (Fig. S4C) were analogous, i.e. DAPLE inhibited nucleotide exchange with similar potency and efficacy. Taken together, these results show that DAPLE inhibits nucleotide exchange on Gαq via its GBA motif.
Figure 6.

DAPLE inhibits nucleotide exchange on Gαq via its GBA motif. A and B, DAPLE GBA peptide decreases the rate of GTPγS binding to Gαq. A representative time course of [35S]GTPγS binding to Gαq in the absence (black) or presence of DAPLE GBA peptide (30 μm, red) using a buffer that contains 0.2 m (NH4)2SO4 is shown in A, and quantification of the dose-dependent effect of DAPLE GBA peptide (red) or control peptide (blue) in a buffer that contains 0.2 m (NH4)2SO4 is shown in B (mean ± S.E., n = 3). Results are presented as raw [35S]GTPγS binding (picomoles) in A or percent change relative to [35S]GTPγS binding to Gαq alone at 45 min (% of control) in B. Rate constants and average IC50 values were determined as described under “Experimental procedures.” C, purified DAPLE WT (amino acids 1650–2028), but not F1675A mutant, decreases GTPγS binding to Gαq. Quantification of the effect of His–DAPLE WT (3.3 μm, red) compared with His–DAPLE F1675A (3.3 μm, blue) in a buffer that contains 0.2 m (NH4)2SO4 is presented as percent change relative to [35S]GTPγS binding to Gαq alone at 45 min (% of control, mean ± S.E., n = 3).
Discussion
DAPLE belongs to a family of cytoplasmic G-protein regulators that are defined by the presence of a GBA motif. Although proteins with a GBA motif have been previously found to preferentially bind to Gα subunits of G proteins of the Gi/o family over Gα subunits of other G-protein families, the main discovery of the work presented here is that DAPLE binds to Gαs (Gs family) and to Gαq (Gq/11 family). Moreover, although GBA motifs have been characterized by their ability to exert GEF activity on Gαi subunits, here we found that DAPLE exerts GDI activity on Gαs and Gαq. Previously reported evidence indicates that GIV, another protein with a GBA motif, can bind to Gαs and exert GDI activity on it (51). However, this G-protein regulatory function on Gαs appeared only after sequential phosphorylation of two sites that flank the GBA motif, which also precluded Gαi binding and activation by GIV (51). This implies that GIV can work as either a GEF or a GDI depending on a switch of G-protein–binding preference determined by post-translational phosphorylation. In contrast, this work shows that, in the absence of post-translational phosphorylation, DAPLE binds to Gαs or Gαq and exerts GDI activity on them, while still retaining the ability to bind and regulate Gαi. This implies that DAPLE can work as a GEF for Gαi and a GDI for Gαs and Gαq (Fig. 7). To our knowledge, this is also the first description of a protein, other than Gβγ dimers, with GDI activity toward Gα subunits of the Gq/11 family. These findings expand on previous observations with synthetic peptides similar to GBA motifs that were shown to bind and regulate Gα proteins. For example, the synthetic peptide KB-752, based on which was the first GBA motif in GIV identified by similarity (32), was initially shown to be a GEF for Gαi (60) but later to also be a GDI for Gαs (61). This dual-specificity and bi-functional regulatory action on G proteins was also described for another GBA-like synthetic peptide, namely GSP, identified independently (62). Thus, although all other proteins with a GBA motif described to date have been shown to have GEF activity for Gαi in vitro (32, 33, 38–40), it still remains to be elucidated whether they can also work as GDIs depending on their post-translational modification status and/or the nature of their G-protein substrate.
Figure 7.
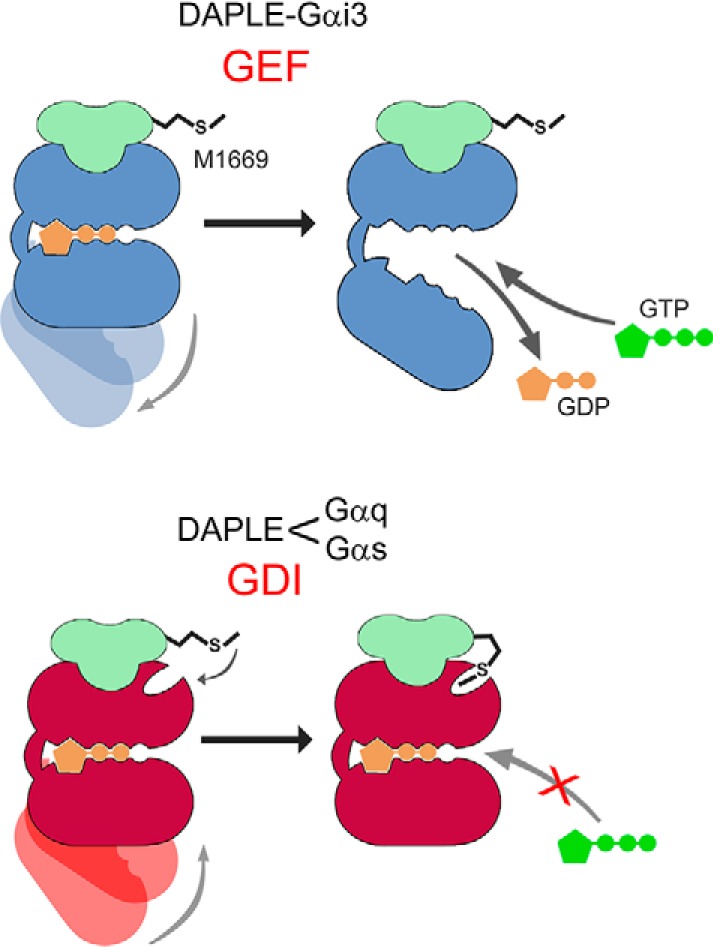
Proposed model. Top, DAPLE (green) binds to Gαi (blue) to stabilize a G-protein conformation that favors nucleotide exchange. DAPLE Met-1669 is not required for efficient binding to Gαi. Bottom, Met-1669 in DAPLE allows its efficient physical engagement to Gαs and Gαq (red), which in turn stabilizes a G-protein conformation that prevents nucleotide exchange.
Here, we also define the similarities and differences of the interaction between DAPLE and Gαs or Gαq compared with Gαi. As reported previously for the interaction of DAPLE with Gαi (and with GBA motifs of other proteins), DAPLE also binds preferentially to the inactive conformation of Gαs or Gαq. This interaction is ablated upon mutation of a hydrophobic amino acid (Phe-1675) that is highly conserved across GBA motifs of different proteins and is also essential for Gαi binding (37, 40, 47). This suggests that the overall binding mode of DAPLE's GBA motif on Gαs or Gαq probably resembles that observed for Gαi, i.e. docking onto a hydrophobic cleft formed by the switch II and α3 helix of Gα that becomes inaccessible upon G-protein activation due to conformational rearrangement of switch II (32, 37). Regarding the differences between DAPLE binding to Gαs/Gαq versus Gαi, we identified here a single amino acid in DAPLE (Met-1669) that is required for binding to Gαs and Gαq but not to Gαi. Although the existence of this additional molecular contact between DAPLE and Gαs/Gαq correlates with the ability to exert GDI activity, we can only speculate about the mechanism by which this additional contact might inhibit nucleotide exchange on Gα based on previously described observations in the literature. Essentially, there are two mechanisms of action described for proteins with GDI activity. One is that mediated by GoLoco motifs, which prevent nucleotide exchange by directly engaging GDP and securing it in the nucleotide-binding pocket through an “arginine finger” (18, 19). The other mechanism is that mediated by Gβγ subunits, which, instead of making direct contact with GDP, bind to the Ras-like domain of Gα and allosterically stabilize GDP in the nucleotide-binding pocket (4, 63, 64). For the GDI action of DAPLE, we favor a mechanism akin to the latter rather than to the former. This is because, based on the discussion above, it is unlikely that the overall docking pose of DAPLE on Gαs/Gαq differs markedly from that observed for Gαi, so the GBA motif would not make direct contact with the nucleotide. Instead, the additional G-protein contact established through Met-1669 might favor a conformation of Gα that is less prone to exchange nucleotide (Fig. 7). Elucidation of atomic resolution structures of DAPLE's GBA motif in complex with Gαs and/or Gαq would be required to shed light on this matter.
Finding that DAPLE has GDI activity toward Gαs/Gαq sets a new framework to discover and/or understand biological functions of DAPLE's GBA motif. From a traditional viewpoint of GPCR signaling, GDI activity exerted on Gαs and Gαq could be expected to suppress their ability to engage and modulate their respective effectors, such as adenylyl cyclase for Gαs or PLC-β and RhoGEFs for Gαq. However, preliminary evidence suggests that DAPLE's GBA motif does not influence GPCR-mediated modulation of some of these effector pathways.5 This is not entirely surprising because it is difficult to predict the context (e.g. specific GPCR) in which DAPLE might engage G proteins to regulate them. Previous reports with other GDIs, like the GoLoco motif-containing protein AGS3, have also found that GPCR/G-protein effector pathways are not necessarily impacted as one would predict based on their putative ability to prevent Gα-effector signaling (65). Instead, GoLoco motif-containing GDIs have been shown to be engaged into alternative modes of G-protein signaling (66, 67). One of them consists of forming complexes with Gα subunits that do not mediate signaling through GPCRs but are involved in the control of cell division in metazoans (68–71). In addition, the GoLoco motif containing GDIs was originally discovered in a yeast genetic screen based on their ability to enhance Gβγ-dependent signaling (72). It has been subsequently proposed that, while having inhibitory GDI activity on Gα subunits, GoLoco motifs promote Gβγ-dependent signaling by favoring Gα–Gβγ dissociation (73, 74). It is possible that the GDI activity of DAPLE on Gαs/Gαq results in a signaling mechanism in cells similar to that proposed for GoLoco motif-containing GDIs, as it has been reported that the physiological functions of DAPLE's GBA motif during embryonic development are Gβγ-dependent (41), and because it has been previously shown that GBA motifs can displace Gβγ subunits from Gα–Gβγ complexes (32, 39). It is also important to take into account that DAPLE is a modular multifunctional protein and that such modularity might have a significant impact on the context in which DAPLE's GDI function operates. Most notably, DAPLE contains a PDZ-binding motif (PBM) that is required for its localization and function at apical cell–cell junctions (41, 49). Without this PBM, DAPLE's GBA motif cannot promote actomyosin contractility and the subsequent effects on embryonic morphogenesis (41). Thus, the functional outcomes of DAPLE's GDI activity might be spatially restricted to apical cell junctions, where different G-protein subtypes can localize (75–78). Further investigation will be required to elucidate the biological consequences of DAPLE's GDI activity on Gαs and/or Gαq.
In summary, the findings reported here reveal that a protein with a GBA motif, namely DAPLE, can promiscuously bind to Gα subunits of different subfamilies and that, depending on the G protein subtype, it exerts opposing actions on nucleotide exchange: it inhibits nucleotide exchange on Gαs and Gαq, while it accelerates it on Gαi. These findings highlight the versatility of GBA motifs in modulating the activity of G proteins.
Experimental procedures
Reagents and antibodies
Unless otherwise indicated, all chemical reagents were obtained from Sigma or Thermo Fisher Scientific. Escherichia coli DH5α strain was purchased from New England Biolabs and the BL21(DE3) strain from Life Technologies, Inc. PfuUltra DNA polymerase was purchased from Agilent. [γ-32P]GTP and [35S]GTPγS were from PerkinElmer Life Sciences. Mouse monoclonal antibodies raised against α-tubulin (T6074), FLAG tag (F1804), or His tag (H1029) were from Sigma. Mouse mAb raised against hemagglutinin (HA) tag (clone 12CA5, catalog no. 11583816001) was obtained from Roche Applied Science. Mouse mAb raised against MYC tag (9B11, catalog no. 2276) was from Cell Signaling. Rabbit polyclonal antibodies raised against Gαs (C-18, sc-383) or Gαq (E-17, sc-393) were purchased from Santa Cruz Biotechnology. Goat anti-rabbit Alexa Fluor 680 (A21077) and goat anti-mouse IRDye 800 (catalog no. 926-32210) secondary antibodies were from Life Technologies, Inc., and LI-COR, respectively.
Plasmids
Plasmids for expression of GST–DAPLE (human aa 1650–2028, pGEX-4T-DAPLE-CT) and GST–GIV (human aa 1671–1755, pGEX-4T-GIV-CT) in bacteria were described previously (39). GST–DAPLE (short) (human aa 1650–1745) was generated by introduction of a stop codon in position 1746 of pGEX-4T-DAPLE-CT using site-directed mutagenesis. The plasmid for expression of His–DAPLE (human, aa 1650–2028, pET28b-DAPLE) in bacteria was described previously (39). Plasmids for expression of FLAG–Gαi3 (rat, p3XFLAG-CMV10-Gαi3, N-terminal 3XFLAG tag), Gαi3–FLAG (rat, p3XFLAG-CMV14-Gαi3, C-terminal 3XFLAG tag), or Gαs (human, pcDNA3.1(+)-Gαs) in mammalian cells were described previously (32, 52). Plasmids for the expression of Gαq–HA (mouse, pcDNA3-Gαq–HA, internally tagged) or Gα12–MYC (mouse, pcDNA3.1-Gα12–MYC, internally tagged) in mammalian cells were kindly provided by P. Wedegaertner (Thomas Jefferson University) (79) and T. Meigs (University of North Carolina, Asheville) (80), respectively. Plasmid for the expression of His–Gαi3 (rat, pET28b-Gαi3) was described previously (32). Plasmids for the expression of His–Gαs (bovine, pHis6–Gαs) and His–Gαq* (mouse, pET28a–Gαq*) in bacteria were kindly provided by N. Artemyev (University of Iowa) and S. Sprang (University of Montana), respectively (53, 81). Plasmids for expression of Gαq, Gβ1–His, and Gγ2–His in Sf9 insect cells were described previously (59). Plasmid for the expression of full-length MYC–DAPLE (human, pCS2–6XMYC–DAPLE) in mammalian cells was described previously (41). All point mutations, including DAPLE/GIV GBA chimeras, were generated by site-directed mutagenesis following the manufacturer's instructions (QuikChange II, Agilent).
Expression and purification of proteins
GST, GST–DAPLE, GST–GIV, GST–DAPLE (short), and His–Gαi3 proteins were purified from bacteria as described previously (32, 39). Briefly, proteins were expressed in BL21(DE3) E. coli (Life Technologies, Inc.) transformed with the corresponding plasmids by overnight induction at 23 °C with 1 mm isopropyl β-d-1-thio-galactopyranoside (IPTG) when the OD600 reached ∼0.7. Bacteria were pelleted and resuspended at 4 °C in lysis buffer: 50 mm NaH2PO4, pH 7.4, 300 mm NaCl, 10 mm imidazole, and 1% (v/v) Triton X-100 supplemented with protease inhibitor mixture (1 μm leupeptin, 2.5 μm pepstatin, 0.2 μm aprotinin, and 1 mm PMSF). For Gαi3, this buffer was also supplemented with 25 μm GDP and 5 mm MgCl2. After sonication (30-s bursts, four times), lysates were cleared by centrifugation at 12,000 × g for 30 min at 4 °C. The supernatant was used for affinity purification on HisPur Cobalt or GSH-agarose resins (Thermo Fisher Scientific) for 90 min at 4 °C with rotation. Resins were washed four times with lysis buffer, and proteins were eluted with lysis buffer supplemented with 250 mm imidazole or with 50 mm Tris-HCl, pH 8, 100 mm NaCl, and 30 mm reduced GSH for HisPur Cobalt and GSH-agarose resins, respectively. The eluted fractions were dialyzed overnight at 4 °C against PBS, except for Gαi3, which was buffer exchanged into 20 mm Tris-HCl, pH 7.4, 20 mm NaCl, 1 mm MgCl2, 1 mm, DTT, 10 μm GDP, and 5% (v/v) glycerol using a HiTrap desalting column (GE Healthcare). All protein samples were aliquoted and stored at −80 °C.
His–Gαs was purified from bacteria as described previously (82). Briefly, His–Gαs was expressed in BL21(DE3) E. coli transformed with the corresponding plasmid by overnight induction at 23 °C with 0.1 mm IPTG when OD600 reached ∼0.5. Bacteria were pelleted and resuspended at 4 °C in lysis buffer (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 5 mm MgCl2, 50 μm GDP, and 5 mm β-mercaptoethanol) supplemented with protease inhibitor mixture (1 μm leupeptin, 2.5 μm pepstatin, 0.2 μm aprotinin, and 1 mm PMSF). After sonication (30-s bursts, four times), the lysate was cleared by centrifugation at 12,000 × g for 30 min at 4 °C. The supernatant was adjusted to 500 mm NaCl and 20 mm imidazole before affinity purification by incubation with nickel-nitrilotriacetic acid resin (Qiagen) for 90 min at 4 °C. Resin was washed four times with lysis buffer, and protein was eluted with lysis buffer supplemented with 100 mm imidazole. The eluted fraction was adjusted to 50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 5 mm MgCl2 using a protein concentrator with a 10-kDa cutoff (Pierce) before loading onto a HiTrap Q HP column (GE Healthcare) connected to an ÄKTA FPLC. Proteins were eluted by applying a 50–500 mm NaCl gradient, and fractions containing His–Gαs were pooled and supplemented with 10 μm GDP and 5 mm β-mercaptoethanol before concentration in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm MgCl2, 5% (w/v) glycerol, 10 μm GDP, and 5 mm β-mercaptoethanol, and storage at −80 °C.
His–Gαq* was purified from bacteria as described previously (53). His–Gαq* was expressed in BL21(DE3) E. coli transformed with the corresponding plasmid by overnight induction at 23 °C with 1 mm IPTG when OD600 reached ∼0.7. Bacteria were pelleted and then resuspended at 4 °C in lysis buffer: 20 mm HEPES, pH 8.0, 300 mm NaCl, 5 mm MgCl2, 10 mm β-mercaptoethanol, 15 mm imidazole, 10% (v/v) glycerol, 50 μm GDP, 30 μm AlCl3, and 10 mm NaF supplemented with protease inhibitor mixture (1 μm leupeptin, 2.5 μm pepstatin, 0.2 μm aprotinin, and 1 mm PMSF). After sonication (30-s bursts, four times), the lysate was cleared by centrifugation at 12,000 × g for 30 min at 4 °C. The supernatant was used for affinity purification on HisPur Cobalt resin (Pierce) for 90 min at 4 °C with rotation. The resin was washed four times with lysis buffer, and the protein was eluted with lysis buffer supplemented with 500 mm imidazole. The eluted fraction was supplemented with 100 mm EDTA and incubated on ice for 20 min before loading onto a Superdex 200 HR10/30 gel-filtration column using a running buffer consisting of 20 mm HEPES, pH 8.0, 100 mm NaCl, 1 mm MgCl2, 2 mm DTT, 2% (v/v) glycerol, and 10 μm GDP. Fractions containing His–Gαq* were concentrated and stored at −80 °C. Gαq was purified from Sf9 insect cells as described previously (83).
In vitro protein-binding assays with GST-fused proteins
The following GST-fused proteins were immobilized on GSH-agarose beads for 90 min at room temperature in PBS (amounts per condition are indicated in parentheses): GST (30 μg), GST–DAPLE (30 μg), GST–GIV (30 μg), and GST–DAPLE (short) (30 μg). Beads were washed twice with PBS and resuspended in 300 μl of binding buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 0.4% (v/v) Nonidet P-40, 5 mm EDTA, 2 mm DTT) supplemented with 30 μm GDP unless indicated otherwise.
For experiments using cell lysates as a source of soluble binding ligands, HEK293T cells (ATCC CRL-3216) were grown at 37 °C, 5% CO2 in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1% l-glutamine. Approximately two million HEK293T cells were seeded on 10-cm dishes and transfected the day after using the calcium phosphate method with plasmids encoding the following constructs (DNA amounts in parentheses): Gαi3–FLAG (3 μg), Gαs (3 μg), Gαq–HA (6 μg), or Gα12–MYC (3 μg). Cell medium was changed 6 h after transfection. Thirty two hours after transfection, cells were lysed at 4 °C with 700 μl of lysis buffer (20 mm HEPES, pH 7.2, 125 mm K(CH3COO), 0.4% (v/v) Triton X-100, 1 mm DTT, 10 mm β-glycerophosphate, and 0.5 mm Na3VO4 supplemented with a protease inhibitor mixture (SigmaFAST, catalog no. S8830)). Cell lysates were cleared by centrifugation at 14,000 × g for 10 min. For experiments using nucleotide-loaded Gαs, the lysates of HEK293T were incubated at 30 °C for 30 min with nucleotides as follows: 125 μm GDP (GDP condition), or 125 μm GDP, 125 μm AlCl3, 10 mm NaF (GDP·AlF4− condition), or 125 μm GTPγS (GTPγS condition). For experiments using nucleotide-loaded Gαq, the lysates were incubated at 4 °C for 30 min with nucleotides as follows: 30 μm GDP (GDP condition), or 30 μm GDP, 30 μm AlCl3, 10 mm NaF (GDP·AlF4− condition).
One hundred microliters (∼400 μg) of cell lysate prepared as described above were added to the GST-fused proteins immobilized on resin beads and incubated 4 h at 4 °C with constant rotation. Beads were washed four times with 1 ml of wash buffer (4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.4, 137 mm NaCl, 2.7 mm KCl, 0.1% (v/v) Tween 20, 5 mm EDTA, 1 mm DTT) supplemented with 30 μm GDP, except for experiments using Gα loaded with GDP·AlF4− or GTPγS, in which the wash buffer was supplemented with 30 μm GDP, 30 μm AlCl3, and 10 mm NaF, or with 30 μm GTPγS, respectively. Resin-bound proteins were eluted by boiling for 5 min in Laemmli sample buffer, and proteins were separated by SDS-PAGE and immunoblotted with antibodies as indicated under “Immunoblotting.”
For experiments using purified proteins as source of soluble binding ligands, purified proteins were prepared as described under “Expression and purification of proteins.” Aliquots of protein stored at −80 °C were quickly thawed and cleared by centrifugation at 14,000 × g for 2 min before addition to GST-fused proteins immobilized on GSH-agarose beads in 300 μl of binding buffer supplemented with 30 μm GDP. The amount of purified proteins used were as follows: 1.7 μg of His–Gαi3, 1.65 μg of His–Gαs, and 1.7 μg of His–Gαq*. Tubes were incubated for 4 h at 4 °C with constant rotation, and washes and elution were performed as described above. Proteins were separated by SDS-PAGE and immunoblotted with antibodies as indicated under “Immunoblotting.”
Immunoprecipitation
Approximately three million HEK293T cells were seeded in 10-cm dishes and transfected the day after using polyethyleneimine (PEI; Polysciences, Inc.; catalog no. 23966, 1 mg/ml solution reconstituted in water). For each 10-cm dish, a total of 9 μg of DNA were transfected, consisting of 3 μg of plasmids encoding FLAG–Gαi3, FLAG–Gαs, FLAG–Gαq, or an empty vector, along with 6 μg of MYC–DAPLE (WT or mutants). Plasmid DNA was added to 500 μl of DMEM and immediately mixed with PEI reagent (DNA/PEI reagent ratio of 1:3) by vortexing for 2 s. Tubes were incubated at room temperature for 15 min before adding to cells, and media were changed 6 h later. Twenty four hours after transfection, cells were lysed on ice with 750 μl of lysis buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 0.4% (v/v) Triton X-100, 1 mm DTT, 10 mm β-glycerophosphate, 0.5 mm Na3VO4, and 30 μm GDP supplemented with a protease inhibitor mixture (Sigma S8830)) and cleared (14,000 × g, 10 min). Cleared lysates were incubated with 2 μg of FLAG antibodies (Sigma F1804) for 2.5 h at 4 °C with constant rotation. Forty μl of an ∼50% protein G-agarose beads suspension, pre-blocked with 5% (w/v) BSA in PBS for 2 h at room temperature, were added to the lysate/antibody mixture and incubated for 90 min at 4 °C. Beads were washed three times with wash buffer (4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.4, 137 mm NaCl, 2.7 mm KCl, 0.1% (v/v) Tween 20, 10 mm MgCl2, 5 mm EDTA, 1 mm DTT, and 30 μm GDP), and proteins were eluted by boiling in Laemmli sample buffer for 5 min. Proteins were separated by SDS-PAGE and immunoblotted with antibodies as indicated under “Immunoblotting.”
Immunoblotting
Proteins were separated by SDS-PAGE and transferred to PVDF membranes, which were blocked with 5% (w/v) nonfat dry milk and sequentially incubated with primary and secondary antibodies. For protein–protein-binding experiments with GST-fused proteins, PVDF membranes were stained with Ponceau S and scanned before blocking. The primary antibodies used were the following: MYC (1:1000); His (1:2500); FLAG (1:2000); α-tubulin (1:2500); HA (1:1000); Gαs (1:500); and Gαq (1:500). The secondary antibodies were goat anti-rabbit Alexa Fluor 680 (1:10,000) and goat anti-mouse IRDye 800 (1:10,000). IR imaging of immunoblots was performed using an Odyssey IR Imaging System (Li-Cor Biosciences). Images were processed using ImageJ software (National Institutes of Health) and assembled for presentation using Photoshop and Illustrator softwares (Adobe).
Homology modeling
A model of Gαi3 bound to the GBA motif of DAPLE was generated by homology in combination with protein–protein docking. First, a model of the DAPLE GBA motif (residues 1663–1680) was built via homology to GIV residues 1673–1690 using an X-ray co-crystal structure of rat Gαi3 bound to a human GIV peptide (PDB code 6MHF) as a template (35). The position of hydrogens and the isomeric/tautomeric state and positioning of side chains were energetically optimized on the parent structure prior to modeling. The binding of the GBA motif of DAPLE to the Gαi3 structure (PDB code 6MHF, chain A) was then simulated with a rigid body two-stage fast Fourier transform protein–protein docking procedure. The solution with the lowest predicted energy was selected, and the side-chain positions of DAPLE residues were energetically minimized with respect to the binding site on Gαi3. Homology modeling and protein docking were performed with ICM version 3.8–3 (Molsoft LLC., San Diego) (84, 85). Model images were generated with PyMOL Molecular Graphics System, version 2.3.0 (Schrodinger, LLC.).
Peptide synthesis
Peptides were synthesized as described previously (20, 24). The sequences corresponding to the GBA motif of human DAPLE (residues 1662–1695, SASPSSEMVTLEEFLEESNRSSPTHDTPSCRDDL) or control peptide, corresponding to the GBA motif of GIV 1671–1705 containing the mutation F1685A (KTGSPGSEVVTLQQALEESNKLTSVQIKSSSQENL) (48), were synthesized using the in situ neutralization protocol for t-butoxycarbonyl-solid–phase peptide synthesis on a p-methylbenzhydrylamine resin (Novabiochem, 0.67 mmol/g, 100–200 mesh). Following chain elongation, peptides were cleaved using a solution of hydrofluoric acid containing 5% anisole for 1 h at 0 °C. Next, the hydrofluoric acid solution containing the peptides was removed under vacuum, and the solution was crushed out with Et2O and filtered. The collected solids were redissolved in a 50% CH3CN/H2O solution containing 0.05% of trifluoroacetic acid (TFA), frozen down, and lyophilized. Crude peptides were purified by reverse phase-HPLC using a Jupiter Proteo (90 Å, 10 μm, 100 × 21.2 mm) at a flow rate of 16 ml/min using H2O (A, 0.1% TFA) and CH3CN (B, 0.05% TFA) as eluents following a linear gradient: from 5% B to 70% B in 80 min. The identity and final purity (>97%) of the peptides were determined by analytical RP-HPLC and MS (ESI-TOF).
Steady-state GTPase assays
Steady-state GTPase assays of Gαi3 and Gαs were performed as described previously (42, 82). Briefly, His–Gαi3 (100 nm) or His–Gαs (100 nm) was diluted in assay buffer (20 mm Na-HEPES, pH 8, 100 mm NaCl, 1 mm EDTA, 2 mm MgCl2, 1 mm DTT, 0.05% (w/v) C12E10) and preincubated with peptides or purified proteins (as indicated in the figures) for 15 min at 30 °C for Gαi3 or 30 min at 4 °C for Gαs. Reactions were initiated at 30 °C by mixing (1:1) (v/v) the mixture prepared above with 1 μm [γ-32P]GTP (∼50 cpm/fmol) diluted in assay buffer. Duplicate aliquots (25 μl) were removed at the times indicated in the figures or figure legends, and the reaction was stopped by the addition of 975 μl of ice-cold 5% (w/v) activated charcoal in 20 mm H3PO4, pH 3. Samples were centrifuged for 10 min at 10,000 × g, and 500 μl of the resultant supernatants were scintillation counted to quantify the amount of [32P]Pi released. Background [32P]Pi detected in the absence of G protein was subtracted from each reaction. Data are expressed as picomoles of [γ-32P]Pi released or percentage of [γ-32P]Pi released compared with control. EC50/IC50 values were estimated from sigmoidal dose-response curve fits generated using the GraphPad software.
GTPγS-binding assays
GTPγS-binding assays for Gαi3 and Gαs were performed essentially as described previously (42, 82). Purified His–Gαi3 (100 nm) and His–Gαs (100 nm) were diluted in assay buffer (20 mm Na-HEPES, pH 8, 100 mm NaCl, 1 mm EDTA, 25 mm MgCl2, 1 mm DTT, 0.05% (w/v) C12E10) and preincubated with peptides or purified proteins (as indicated in the figure) for 15 min at 30 °C for Gαi3 or 30 min at 4 °C for Gαs. Reactions were initiated by adding an equal volume of assay buffer containing 1 μm [35S]GTPγS (∼50 cpm/fmol) at 30 °C for Gαi3 or 20 °C for Gαs. Duplicate aliquots (25 μl) were removed at different time points as described in the figures, and binding of radioactive nucleotide was stopped by addition of 3 ml of ice-cold wash buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 25 mm MgCl2). The quenched reactions were rapidly passed through BA-85 nitrocellulose filters (GE Healthcare) and washed with 4 ml of cold wash buffer. Filters were dried and subjected to liquid scintillation counting. Background [35S]GTPγS detected in the absence of G protein was subtracted from each reaction. Data are expressed as picomoles of [35S]GTPγS bound or percentage of [35S]GTPγS binding relative to control (% of control).
GTPγS-binding assays with Gαq were performed using two different buffers: assay buffer A, containing (NH4)2SO4 (20 mm Na-HEPES, pH 7.5, 100 mm NaCl, 0.1 mm EDTA, 0.3 mm MgCl2, 0.2 m (NH4)2SO4, 1 mm DTT, 0.05% (w/v) C12E10), and assay buffer B, not containing (NH4)2SO4 (20 mm Na-HEPES, pH 7.5, 100 mm NaCl, 1 mm EDTA, 10 mm MgCl2, 1 mm DTT, 0.05% (w/v) Genapol). Purified Gαq (100 nm) was diluted in either assay buffer A or assay buffer B and preincubated at 4 °C for 30 min with peptides or purified proteins, as indicated in the figure legends. Reactions were initiated by adding an equal volume of assay buffer containing 0.5 μm [35S]GTPγS (∼50 cpm/fmol) at 20 °C. Single aliquots (25 μl) were removed at different time points (as indicated in the figures), and reactions were stopped and analyzed as described above. EC50/IC50 values were estimated from sigmoidal dose-response curve fits using the GraphPad software. The rate constants of time-course experiments were estimated by fitting the data to a one-phase exponential association curve using GraphPad.
Author contributions
A. M. data curation; A. M., M. M., V. D., E. M. R., P. G., and M. G.-M. formal analysis; A. M., M. M., V. D., I. O. C., E. A. M., J. E., P. G., and M. G.-M. investigation; A. M., P. G., and M. G.-M. writing-original draft; A. M., P. G., and M. G.-M. writing-review and editing; J. Z. methodology; V. D. visualization; J. B. B.-C., E. M. R., P. G., and M. G.-M. resources; P. G. and M. G.-M. conceptualization; P. G. and M. G.-M. supervision; P. G. and M. G.-M. funding acquisition; P. G. and M. G.-M. project administration.
Supplementary Material
Acknowledgments
We thank Phil Wedegaertner (Thomas Jefferson University), Ted Meigs (University of North Carolina, Asheville), Nikolai Artemyev (University of Iowa), and Stephen Sprang (University of Montana) for providing plasmids. We thank Jason Casler and Anthony Cheung for their technical help.
This work was supported by National Institutes of Health Grants R01GM108733 and R01GM136132 (to M. G.-M.), American Cancer Society–Funding Hope Postdoctoral Fellowship PF-19-084-01-CDD (to M. M.), a postdoctoral fellowship from the Hartwell Foundation (to V. D.), National Institutes of Health Grant R01GM030355 (to E. M. R.), and National Institutes of Health Grants CA100768 and CA160911 (to P. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
A. Marivin, M. Maziarz, and M. Garcia-Marcos, unpublished observations.
- GPCR
- G-protein–coupled receptor
- GBA
- Gα-binding and -activating
- GEF
- guanine–nucleotide exchange factor
- GDI
- guanine nucleotide–dissociation inhibitor
- RGS
- regulator of G-protein signaling
- GIV
- Gα-interacting, vesicle-associated protein
- DAPLE
- Dvl-associating protein with a high frequency of leucine
- PLC
- phospholipase C
- GST
- glutathione S-transferase
- IP
- immunoprecipitation
- IB
- immunoblotting
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- DMEM
- Dulbecco's modified Eagle's medium
- IPTG
- isopropyl β-d-1-thio-galactopyranoside
- PMSF
- phenylmethylsulfonyl fluoride
- PVDF
- polyvinylidene difluoride
- PDB
- Protein Data Bank
- PBM
- PDZ-binding motif
- aa
- amino acid.
References
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 10.1146/annurev.bi.56.070187.003151 [DOI] [PubMed] [Google Scholar]
- 2. Hauser A. S., Attwood M. M., Rask-Andersen M., Schiöth H. B., and Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 10.1038/nrd.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brandt D. R., and Ross E. M. (1985) GTPase activity of the stimulatory GTP-binding regulatory protein of adenylate cyclase, Gs. Accumulation and turnover of enzyme-nucleotide intermediates. J. Biol. Chem. 260, 266–272 [PubMed] [Google Scholar]
- 4. Higashijima T., Ferguson K. M., Sternweis P. C., Smigel M. D., and Gilman A. G. (1987) Effects of Mg2+ and the βγ-subunit complex on the interactions of guanine nucleotides with G proteins. J. Biol. Chem. 262, 762–766 [PubMed] [Google Scholar]
- 5. Wettschureck N., and Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 10.1152/physrev.00003.2005 [DOI] [PubMed] [Google Scholar]
- 6. Neves S. R., Ram P. T., and Iyengar R. (2002) G protein pathways. Science 296, 1636–1639 10.1126/science.1071550 [DOI] [PubMed] [Google Scholar]
- 7. Watson N., Linder M. E., Druey K. M., Kehrl J. H., and Blumer K. J. (1996) RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature 383, 172–175 10.1038/383172a0 [DOI] [PubMed] [Google Scholar]
- 8. Berman D. M., Wilkie T. M., and Gilman A. G. (1996) GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell 86, 445–452 10.1016/S0092-8674(00)80117-8 [DOI] [PubMed] [Google Scholar]
- 9. Soundararajan M., Willard F. S., Kimple A. J., Turnbull A. P., Ball L. J., Schoch G. A., Gileadi C., Fedorov O. Y., Dowler E. F., Higman V. A., Hutsell S. Q., Sundström M., Doyle D. A., and Siderovski D. P. (2008) Structural diversity in the RGS domain and its interaction with heterotrimeric G protein α-subunits. Proc. Natl. Acad. Sci. U.S.A. 105, 6457–6462 10.1073/pnas.0801508105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siderovski D. P., Hessel A., Chung S., Mak T. W., and Tyers M. (1996) A new family of regulators of G-protein–coupled receptors? Curr. Biol. 6, 211–212 10.1016/S0960-9822(02)00454-2 [DOI] [PubMed] [Google Scholar]
- 11. Kimple A. J., Bosch D. E., Giguère P. M., and Siderovski D. P. (2011) Regulators of G-protein signaling and their Gα substrates: promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 63, 728–749 10.1124/pr.110.003038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tesmer J. J., Berman D. M., Gilman A. G., and Sprang S. R. (1997) Structure of RGS4 bound to AlF4–activated G(iα1): stabilization of the transition state for GTP hydrolysis. Cell 89, 251–261 10.1016/S0092-8674(00)80204-4 [DOI] [PubMed] [Google Scholar]
- 13. Dohlman H. G., Song J., Ma D., Courchesne W. E., and Thorner J. (1996) Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein α subunit). Mol. Cell. Biol. 16, 5194–5209 10.1128/MCB.16.9.5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Vries L., Mousli M., Wurmser A., and Farquhar M. G. (1995) GAIP, a protein that specifically interacts with the trimeric G protein Gi3, is a member of a protein family with a highly conserved core domain. Proc. Natl. Acad. Sci. U.S.A. 92, 11916–11920 10.1073/pnas.92.25.11916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koelle M. R., and Horvitz H. R. (1996) EGL-10 regulates G-protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84, 115–125 10.1016/S0092-8674(00)80998-8 [DOI] [PubMed] [Google Scholar]
- 16. Ross E. M., and Wilkie T. M. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G-protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827 10.1146/annurev.biochem.69.1.795 [DOI] [PubMed] [Google Scholar]
- 17. Chidiac P., and Ross E. M. (1999) Phospholipase C-β1 directly accelerates GTP hydrolysis by Gαq and acceleration is inhibited by Gβγ subunits. J. Biol. Chem. 274, 19639–19643 10.1074/jbc.274.28.19639 [DOI] [PubMed] [Google Scholar]
- 18. Kimple R. J., Kimple M. E., Betts L., Sondek J., and Siderovski D. P. (2002) Structural determinants for GoLoco-induced inhibition of nucleotide release by Gα subunits. Nature 416, 878–881 10.1038/416878a [DOI] [PubMed] [Google Scholar]
- 19. Peterson Y. K., Bernard M. L., Ma H., Hazard S. 3rd., Graber S. G., and Lanier S. M. (2000) Stabilization of the GDP-bound conformation of Giα by a peptide derived from the G-protein regulatory motif of AGS3. J. Biol. Chem. 275, 33193–33196 10.1074/jbc.C000509200 [DOI] [PubMed] [Google Scholar]
- 20. De Vries L., Fischer T., Tronchère H., Brothers G. M., Strockbine B., Siderovski D. P., and Farquhar M. G. (2000) Activator of G-protein signaling 3 is a guanine dissociation inhibitor for Gαi subunits. Proc. Natl. Acad. Sci. U.S.A. 97, 14364–14369 10.1073/pnas.97.26.14364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blumer J. B., Oner S. S., and Lanier S. M. (2012) Group II activators of G-protein signalling and proteins containing a G-protein regulatory motif. Acta Physiol. 204, 202–218 10.1111/j.1748-1716.2011.02327.x [DOI] [PubMed] [Google Scholar]
- 22. Cao X., Cismowski M. J., Sato M., Blumer J. B., and Lanier S. M. (2004) Identification and characterization of AGS4: a protein containing three G-protein regulatory motifs that regulate the activation state of Giα. J. Biol. Chem. 279, 27567–27574 10.1074/jbc.M312786200 [DOI] [PubMed] [Google Scholar]
- 23. Willard F. S., Kimple R. J., and Siderovski D. P. (2004) Return of the GDI: the GoLoco motif in cell division. Annu. Rev. Biochem. 73, 925–951 10.1146/annurev.biochem.73.011303.073756 [DOI] [PubMed] [Google Scholar]
- 24. Kimple R. J., De Vries L., Tronchère H., Behe C. I., Morris R. A., Gist Farquhar M., and Siderovski D. P. (2001) RGS12 and RGS14 GoLoco motifs are Gα(i) interaction sites with guanine nucleotide dissociation inhibitor activity. J. Biol. Chem. 276, 29275–29281 10.1074/jbc.M103208200 [DOI] [PubMed] [Google Scholar]
- 25. Peterson Y. K., Hazard S. 3rd., Graber S. G., and Lanier S. M. (2002) Identification of structural features in the G-protein regulatory motif required for regulation of heterotrimeric G-proteins. J. Biol. Chem. 277, 6767–6770 10.1074/jbc.C100699200 [DOI] [PubMed] [Google Scholar]
- 26. Bernard M. L., Peterson Y. K., Chung P., Jourdan J., and Lanier S. M. (2001) Selective interaction of AGS3 with G-proteins and the influence of AGS3 on the activation state of G-proteins. J. Biol. Chem. 276, 1585–1593 10.1074/jbc.M005291200 [DOI] [PubMed] [Google Scholar]
- 27. Cismowski M. J., Ma C., Ribas C., Xie X., Spruyt M., Lizano J. S., Lanier S. M., and Duzic E. (2000) Activation of heterotrimeric G-protein signaling by a ras-related protein. Implications for signal integration. J. Biol. Chem. 275, 23421–23424 10.1074/jbc.C000322200 [DOI] [PubMed] [Google Scholar]
- 28. Tall G. G., Krumins A. M., and Gilman A. G. (2003) Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 10.1074/jbc.M211862200 [DOI] [PubMed] [Google Scholar]
- 29. Chan P., Gabay M., Wright F. A., and Tall G. G. (2011) Ric-8B is a GTP-dependent G protein αs guanine nucleotide exchange factor. J. Biol. Chem. 286, 19932–19942 10.1074/jbc.M110.163675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee M. J., and Dohlman H. G. (2008) Coactivation of G-protein signaling by cell-surface receptors and an intracellular exchange factor. Curr. Biol. 18, 211–215 10.1016/j.cub.2008.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Natochin M., Campbell T. N., Barren B., Miller L. C., Hameed S., Artemyev N. O., and Braun J. E. (2005) Characterization of the Gα(s) regulator cysteine string protein. J. Biol. Chem. 280, 30236–30241 10.1074/jbc.M500722200 [DOI] [PubMed] [Google Scholar]
- 32. Garcia-Marcos M., Ghosh P., and Farquhar M. G. (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183 10.1073/pnas.0900294106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coleman B. D., Marivin A., Parag-Sharma K., DiGiacomo V., Kim S., Pepper J. S., Casler J., Nguyen L. T., Koelle M. R., and Garcia-Marcos M. (2016) Evolutionary conservation of a GPCR-independent mechanism of trimeric G protein activation. Mol. Biol. Evol. 33, 820–837 10.1093/molbev/msv336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Opakua A. I., Parag-Sharma K., DiGiacomo V., Merino N., Leyme A., Marivin A., Villate M., Nguyen L. T., de la Cruz-Morcillo M. A., Blanco-Canosa J. B., Ramachandran S., Baillie G. S., Cerione R. A., Blanco F. J., and Garcia-Marcos M. (2017) Molecular mechanism of Gαi activation by non-GPCR proteins with a Gα-binding and activating motif. Nat. Commun. 8, 15163 10.1038/ncomms15163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalogriopoulos N. A., Rees S. D., Ngo T., Kopcho N. J., Ilatovskiy A. V., Sun N., Komives E. A., Chang G., Ghosh P., and Kufareva I. (2019) Structural basis for GPCR-independent activation of heterotrimeric Gi proteins. Proc. Natl. Acad. Sci. U.S.A. 116, 16394–16403 10.1073/pnas.1906658116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garcia-Marcos M., Kietrsunthorn P. S., Pavlova Y., Adia M. A., Ghosh P., and Farquhar M. G. (2012) Functional characterization of the guanine nucleotide exchange factor (GEF) motif of GIV protein reveals a threshold effect in signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1961–1966 10.1073/pnas.1120538109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DiGiacomo V., Marivin A., and Garcia-Marcos M. (2018) When heterotrimeric G proteins are not activated by G protein-coupled receptors: structural insights and evolutionary conservation. Biochemistry 57, 255–257 10.1021/acs.biochem.7b00845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garcia-Marcos M., Kietrsunthorn P. S., Wang H., Ghosh P., and Farquhar M. G. (2011) G-protein–binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of G(α)i-regulatory motifs. J. Biol. Chem. 286, 28138–28149 10.1074/jbc.M110.204099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aznar N., Midde K. K., Dunkel Y., Lopez-Sanchez I., Pavlova Y., Marivin A., Barbazán J., Murray F., Nitsche U., Janssen K. P., Willert K., Goel A., Abal M., Garcia-Marcos M., and Ghosh P. (2015) Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. Elife 4, e07091 10.7554/eLife.07091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maziarz M., Broselid S., DiGiacomo V., Park J. C., Luebbers A., Garcia-Navarrete L., Blanco-Canosa J. B., Baillie G. S., and Garcia-Marcos M. (2018) A biochemical and genetic discovery pipeline identifies PLCδ4b as a nonreceptor activator of heterotrimeric G-proteins. J. Biol. Chem. 293, 16964–16983 10.1074/jbc.RA118.003580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marivin A., Morozova V., Walawalkar I., Leyme A., Kretov D. A., Cifuentes D., Dominguez I., and Garcia-Marcos M. (2019) GPCR-independent activation of G proteins promotes apical cell constriction in vivo. J. Cell Biol. 218, 1743–1763 10.1083/jcb.201811174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia-Marcos M., Ghosh P., Ear J., and Farquhar M. G. (2010) A structural determinant that renders Gα(i) sensitive to activation by GIV/girdin is required to promote cell migration. J. Biol. Chem. 285, 12765–12777 10.1074/jbc.M109.045161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin C., Ear J., Midde K., Lopez-Sanchez I., Aznar N., Garcia-Marcos M., Kufareva I., Abagyan R., and Ghosh P. (2014) Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol. Biol. Cell 25, 3654–3671 10.1091/mbc.e14-05-0978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Midde K. K., Aznar N., Laederich M. B., Ma G. S., Kunkel M. T., Newton A. C., and Ghosh P. (2015) Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 112, E937–E946 10.1073/pnas.1420140112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lopez-Sanchez I., Dunkel Y., Roh Y. S., Mittal Y., De Minicis S., Muranyi A., Singh S., Shanmugam K., Aroonsakool N., Murray F., Ho S. B., Seki E., Brenner D. A., and Ghosh P. (2014) GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat. Commun. 5, 4451 10.1038/ncomms5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leyme A., Marivin A., Perez-Gutierrez L., Nguyen L. T., and Garcia-Marcos M. (2015) Integrins activate trimeric G proteins via the nonreceptor protein GIV/Girdin. J. Cell Biol. 210, 1165–1184 10.1083/jcb.201506041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leyme A., Marivin A., Maziarz M., DiGiacomo V., Papakonstantinou M. P., Patel P. P., Blanco-Canosa J. B., Walawalkar I. A., Rodriguez-Davila G., Dominguez I., and Garcia-Marcos M. (2017) Specific inhibition of GPCR-independent G-protein signaling by a rationally engineered protein. Proc. Natl. Acad. Sci. U.S.A. 114, E10319–E10328 10.1073/pnas.1707992114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parag-Sharma K., Leyme A., DiGiacomo V., Marivin A., Broselid S., and Garcia-Marcos M. (2016) Membrane recruitment of the non-receptor protein GIV/Girdin (Gα-interacting, vesicle-associated protein/Girdin) is sufficient for activating heterotrimeric G-protein signaling. J. Biol. Chem. 291, 27098–27111 10.1074/jbc.M116.764431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marivin A., and Garcia-Marcos M. (2019) DAPLE and MPDZ bind to each other and cooperate to promote apical cell constriction. Mol. Biol. Cell 30, 1900–1910 10.1091/mbc.E19-02-0091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ear J., Dunkel Y., Mittal Y., Lim B. B. C., Liu L., Holda M. K., Nitsche U., Barbazán J., Goel A., Janssen K. P., Aznar N., and Ghosh P. (2019) Two isoforms of the guanine nucleotide exchange factor, Daple/CCDC88C cooperate as tumor suppressors. Sci. Rep. 9, 12124 10.1038/s41598-019-48420-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupta V., Bhandari D., Leyme A., Aznar N., Midde K. K., Lo I. C., Ear J., Niesman I., López-Sánchez I., Blanco-Canosa J. B., von Zastrow M., Garcia-Marcos M., Farquhar M. G., and Ghosh P. (2016) GIV/Girdin activates Gαi and inhibits Gαs via the same motif. Proc. Natl. Acad. Sci. U.S.A. 113, E5721–E5730 10.1073/pnas.1609502113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beas A. O., Taupin V., Teodorof C., Nguyen L. T., Garcia-Marcos M., and Farquhar M. G. (2012) Gαs promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Mol. Biol. Cell 23, 4623–4634 10.1091/mbc.e12-02-0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maziarz M., Leyme A., Marivin A., Luebbers A., Patel P. P., Chen Z., Sprang S. R., and Garcia-Marcos M. (2018) Atypical activation of the G protein Gαq by the oncogenic mutation Q209P. J. Biol. Chem. 293, 19586–19599 10.1074/jbc.RA118.005291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Hayre M., Vázquez-Prado J., Kufareva I., Stawiski E. W., Handel T. M., Seshagiri S., and Gutkind J. S. (2013) The emerging mutational landscape of G proteins and G-protein–coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424 10.1038/nrc3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chidiac P., Markin V. S., and Ross E. M. (1999) Kinetic control of guanine nucleotide–binding to soluble Gα(q). Biochem. Pharmacol. 58, 39–48 10.1016/S0006-2952(99)00080-5 [DOI] [PubMed] [Google Scholar]
- 56. Ross E. M. (2002) Quantitative assays for GTPase-activating proteins. Methods Enzymol. 344, 601–617 10.1016/S0076-6879(02)44743-X [DOI] [PubMed] [Google Scholar]
- 57. Mukhopadhyay S., and Ross E. M. (2002) Quench-flow kinetic measurement of individual reactions of G-protein–catalyzed GTPase cycle. Methods Enzymol. 344, 350–369 10.1016/S0076-6879(02)44727-1 [DOI] [PubMed] [Google Scholar]
- 58. Hepler J. R., Kozasa T., Smrcka A. V., Simon M. I., Rhee S. G., Sternweis P. C., and Gilman A. G. (1993) Purification from Sf9 cells and characterization of recombinant Gqα and G11α. Activation of purified phospholipase C isozymes by Gα subunits. J. Biol. Chem. 268, 14367–14375 [PubMed] [Google Scholar]
- 59. Biddlecome G. H., Berstein G., and Ross E. M. (1996) Regulation of phospholipase C-β1 by Gq and m1 muscarinic cholinergic receptor. Steady-state balance of receptor-mediated activation and GTPase-activating protein-promoted deactivation. J. Biol. Chem. 271, 7999–8007 10.1074/jbc.271.14.7999 [DOI] [PubMed] [Google Scholar]
- 60. Johnston C. A., Willard F. S., Jezyk M. R., Fredericks Z., Bodor E. T., Jones M. B., Blaesius R., Watts V. J., Harden T. K., Sondek J., Ramer J. K., and Siderovski D. P. (2005) Structure of Gα(i1) bound to a GDP-selective peptide provides insight into guanine nucleotide exchange. Structure 13, 1069–1080 10.1016/j.str.2005.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnston C. A., Ramer J. K., Blaesius R., Fredericks Z., Watts V. J., and Siderovski D. P. (2005) A bifunctional Gαi/Gαs modulatory peptide that attenuates adenylyl cyclase activity. FEBS Lett. 579, 5746–5750 10.1016/j.febslet.2005.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Austin R. J., Ja W. W., and Roberts R. W. (2008) Evolution of class-specific peptides targeting a hot spot of the Gαs subunit. J. Mol. Biol. 377, 1406–1418 10.1016/j.jmb.2008.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wall M. A., Coleman D. E., Lee E., Iñiguez-Lluhi J. A., Posner B. A., Gilman A. G., and Sprang S. R. (1995) The structure of the G protein heterotrimer Giα1β1γ2. Cell 83, 1047–1058 10.1016/0092-8674(95)90220-1 [DOI] [PubMed] [Google Scholar]
- 64. Ueda N., Iñiguez-Lluhi J. A., Lee E., Smrcka A. V., Robishaw J. D., and Gilman A. G. (1994) G protein βγ subunits. Simplified purification and properties of novel isoforms. J. Biol. Chem. 269, 4388–4395 [PubMed] [Google Scholar]
- 65. Sato M., Gettys T. W., and Lanier S. M. (2004) AGS3 and signal integration by Gα(s)- and Gα(i)-coupled receptors: AGS3 blocks the sensitization of adenylyl cyclase following prolonged stimulation of a Gα(i)-coupled receptor by influencing processing of Gα(i). J. Biol. Chem. 279, 13375–13382 10.1074/jbc.M312660200 [DOI] [PubMed] [Google Scholar]
- 66. Blumer J. B., and Lanier S. M. (2014) Activators of G-protein signaling exhibit broad functionality and define a distinct core signaling triad. Mol. Pharmacol. 85, 388–396 10.1124/mol.113.090068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wilkie T. M., and Kinch L. (2005) New roles for Gα and RGS proteins: communication continues despite pulling sisters apart. Curr. Biol. 15, R843–R854 10.1016/j.cub.2005.10.008 [DOI] [PubMed] [Google Scholar]
- 68. Gotta M., Dong Y., Peterson Y. K., Lanier S. M., and Ahringer J. (2003) Asymmetrically distributed C. elegans homologs of AGS3/PINS control spindle position in the early embryo. Curr. Biol. 13, 1029–1037 10.1016/S0960-9822(03)00371-3 [DOI] [PubMed] [Google Scholar]
- 69. Afshar K., Willard F. S., Colombo K., Johnston C. A., McCudden C. R., Siderovski D. P., and Gönczy P. (2004) RIC-8 is required for GPR-1/2–dependent Gα function during asymmetric division of C. elegans embryos. Cell 119, 219–230 10.1016/j.cell.2004.09.026 [DOI] [PubMed] [Google Scholar]
- 70. Du Q., and Macara I. G. (2004) Mammalian Pins is a conformational switch that links NuMA to heterotrimeric G proteins. Cell 119, 503–516 10.1016/j.cell.2004.10.028 [DOI] [PubMed] [Google Scholar]
- 71. Blumer J. B., Kuriyama R., Gettys T. W., and Lanier S. M. (2006) The G-protein regulatory (GPR) motif-containing Leu–Gly–Asn-enriched protein (LGN) and Giα3 influence cortical positioning of the mitotic spindle poles at metaphase in symmetrically dividing mammalian cells. Eur. J. Cell Biol. 85, 1233–1240 10.1016/j.ejcb.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 72. Cismowski M. J., Takesono A., Ma C., Lizano J. S., Xie X., Fuernkranz H., Lanier S. M., and Duzic E. (1999) Genetic screens in yeast to identify mammalian nonreceptor modulators of G-protein signaling. Nat. Biotechnol. 17, 878–883 10.1038/12867 [DOI] [PubMed] [Google Scholar]
- 73. Ghosh M., Peterson Y. K., Lanier S. M., and Smrcka A. V. (2003) Receptor- and nucleotide exchange-independent mechanisms for promoting G protein subunit dissociation. J. Biol. Chem. 278, 34747–34750 10.1074/jbc.C300271200 [DOI] [PubMed] [Google Scholar]
- 74. Webb C. K., McCudden C. R., Willard F. S., Kimple R. J., Siderovski D. P., and Oxford G. S. (2005) D2 dopamine receptor activation of potassium channels is selectively decoupled by Gα-specific GoLoco motif peptides. J. Neurochem. 92, 1408–1418 10.1111/j.1471-4159.2004.02997.x [DOI] [PubMed] [Google Scholar]
- 75. Saha C., Nigam S. K., and Denker B. M. (2001) Expanding role of G proteins in tight junction regulation: Gα(s) stimulates TJ assembly. Biochem. Biophys. Res. Commun. 285, 250–256 10.1006/bbrc.2001.5154 [DOI] [PubMed] [Google Scholar]
- 76. Denker B. M., Saha C., Khawaja S., and Nigam S. K. (1996) Involvement of a heterotrimeric G protein α subunit in tight junction biogenesis. J. Biol. Chem. 271, 25750–25753 10.1074/jbc.271.42.25750 [DOI] [PubMed] [Google Scholar]
- 77. Acharya B. R., Nestor-Bergmann A., Liang X., Gupta S., Duszyc K., Gauquelin E., Gomez G. A., Budnar S., Marcq P., Jensen O. E., Bryant Z., and Yap A. S. (2018) A mechanosensitive RhoA pathway that protects epithelia against acute tensile stress. Dev. Cell 47, 439–452.e6 10.1016/j.devcel.2018.09.016 [DOI] [PubMed] [Google Scholar]
- 78. Meyer T. N., Schwesinger C., and Denker B. M. (2002) Zonula occludens-1 is a scaffolding protein for signaling molecules. Gα(12) directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J. Biol. Chem. 277, 24855–24858 10.1074/jbc.C200240200 [DOI] [PubMed] [Google Scholar]
- 79. Wedegaertner P. B., Chu D. H., Wilson P. T., Levis M. J., and Bourne H. R. (1993) Palmitoylation is required for signaling functions and membrane attachment of Gqα and Gsα. J. Biol. Chem. 268, 25001–25008 [PubMed] [Google Scholar]
- 80. Ritchie B. J., Smolski W. C., Montgomery E. R., Fisher E. S., Choi T. Y., Olson C. M., Foster L. A., and Meigs T. E. (2013) Determinants at the N and C termini of Gα12 required for activation of Rho-mediated signaling. J. Mol. Signal. 8, 3 10.1186/1750-2187-8-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Natochin M., and Artemyev N. O. (1998) A single mutation Asp229 → Ser confers upon Gsα the ability to interact with regulators of G-protein signaling. Biochemistry 37, 13776–13780 10.1021/bi981155a [DOI] [PubMed] [Google Scholar]
- 82. Leyme A., Marivin A., Casler J., Nguyen L. T., and Garcia-Marcos M. (2014) Different biochemical properties explain why two equivalent Gα subunit mutants cause unrelated diseases. J. Biol. Chem. 289, 21818–21827 10.1074/jbc.M114.549790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Navaratnarajah P., Gershenson A., and Ross E. M. (2017) The binding of activated Gαq to phospholipase C-β exhibits anomalous affinity. J. Biol. Chem. 292, 16787–16801 10.1074/jbc.M117.809673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Abagyan R., and Totrov M. (1994) Biased probability Monte-Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 10.1006/jmbi.1994.1052 [DOI] [PubMed] [Google Scholar]
- 85. Abagyan R., Totrov M., and Kuznetsov D. (1994) Icm–a new method for protein modeling and design–applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 10.1002/jcc.540150503 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.