

Abstract
The oncogenic receptor tyrosine kinase AXL is overexpressed in cancer and plays an important role in carcinomas of multiple organs. However, the mechanisms of AXL overexpression in cancer remain unclear. In this study, using HEK293T, Panc-1, and Panc-28 cells and samples of human pancreatic intraepithelial neoplasia (PanIN), along with several biochemical approaches and immunofluorescence microscopy analyses, we sought to investigate the mechanisms that regulate AXL over-expression in pancreatic ductal adenocarcinoma (PDAC). We found that AXL interacts with hematopoietic progenitor kinase 1 (HPK1) and demonstrate that HPK1 down-regulates AXL and decreases its half-life. The HPK1-mediated AXL degradation was inhibited by the endocytic pathway inhibitors leupeptin, bafilomycin A1, and monensin. HPK1 accelerated the movement of AXL from the plasma membrane to endosomes in pancreatic cancer cells treated with the AXL ligand growth arrest-specific 6 (GAS6). Moreover, HPK1 increased the binding of AXL to the Cbl proto-oncogene (c-Cbl); promoted AXL ubiquitination; decreased AXL-mediated signaling, including phospho-AKT and phospho-ERK signaling; and decreased the invasion capability of PDAC cells. Importantly, we show that AXL expression inversely correlates with HPK1 expression in human PanINs and that patients whose tumors have low HPK1 and high AXL expression levels have shorter survival than those with low AXL or high HPK1 expression (p < 0.001). Our results suggest that HPK1 is a tumor suppressor that targets AXL for degradation via the endocytic pathway. HPK1 loss of function may contribute to AXL overexpression and thereby enhance AXL-dependent downstream signaling and tumor invasion in PDAC.
Keywords: pancreatic cancer, endocytosis, receptor tyrosine kinase, ubiquitylation (ubiquitination), protein degradation, tumor suppressor gene, Axl, c-Cbl, Gas6, HPK1, kinase signaling
Introduction
Pancreatic ductal adenocarcinoma (PDAC)3 is one of the most lethal tumors among all malignancies and ranks as the third leading cause of cancer-related deaths in the United States. Pancreatic cancer is resistant to conventional chemotherapy and radiation therapies. The 5-year survival rate for patients with pancreatic cancer is ∼8.5% (1). The molecular mechanisms of pancreatic tumorigenesis remain unclear. Exploring the molecular mechanisms involved in the development, progression, and resistance to conventional therapies of pancreatic cancer may help to identify new therapeutic targets for this deadly disease.
Oncogenic receptor tyrosine kinase AXL plays a major role in cancer cell survival, proliferation, migration, invasion, metastasis, and immunosuppressive tumor microenvironment (2–4). AXL and its ligand Gas6 are overexpressed in pancreatic cancer and carcinomas of other organs, including lung, colon, prostate, breast, ovary, esophagus, stomach, and kidney (5–18). Our previous study showed that AXL was overexpressed in 70% of human pancreatic cancer samples and that overexpression of AXL correlated significantly with a higher rate of distant metastasis, shorter recurrence-free survival, and overall survival in patients with pancreatic cancer who underwent upfront pancreatectomy with curative intent (17). AXL silencing sensitizes pancreatic cancer cells to γ-irradiation and reduces their anchorage-independent growth, migration, as well as invasion potential, which has been attributed to the down-regulation of AKT signaling as well as transcription factors involved in epithelial–to–mesenchymal transition (EMT) such as slug, snail, and twist, etc. (17, 18). Subsequent studies targeting the Gas6–AXL-signaling pathways using either a small molecular inhibitor (BGB324), a high-affinity AXL decoy receptor, or neutralizing monoclonal antibodies (mABs) against AXL or Gas6 demonstrate that autocrine Gas6–AXL signaling is an important driver for therapeutic resistance, disease progression, and metastasis in pancreatic cancer and other human malignancies (2, 19–26). Ludwig et al. (2) showed that the selective AXL kinase inhibitor, BGB324, not only inhibits the aggressiveness of pancreatic cancer and sensitizes pancreatic cancer cells to gemcitabine, but also induces an immune stimulatory microenvironment. Neutralizing mAbs against AXL or Gas6 inhibits AKT signaling in pancreatic cancer and in vivo tumor growth in xenograft tumor models (2, 22). The results from these preclinical studies provide strong rationale for targeting the Gas6–AXL autocrine pathway to improve the treatment efficacies and clinical outcomes in patients with pancreatic cancer and cancers of other organs.
Previous studies by Leconet et al. (21, 22) showed that anti-AXL mAbs induced internalization and down-regulation of AXL in both pancreatic cancer and triple-negative breast cancer cells. Their results suggest that endocytosis may be one of the major mechanisms regulating AXL expression in cancer cells. Consistent with this notion, Valverde (27) reported that binding of Gas6 to AXL induces the phosphorylation, ubiquitination, and down-regulation of AXL in human lens epithelial cells through endocytosis/lysosomal degradation, but not through proteasomal degradation. However, the molecular mechanisms regulating the expression of AXL in pancreatic cancer or other human malignancies are unclear.
Hematopoietic progenitor kinase 1 (HPK1), also named MAP4K1, is a mammalian Ste20-related serine/threonine kinase, which has been shown to regulate NF-κB and c-Jun N-terminal kinase pathways in hematopoietic cells (28, 29). We previously showed that HPK1 protein is expressed in normal pancreatic ductal cells but is lost in pancreatic ductal adenocarcinomas (PDAC). Loss of HPK1 is strongly associated with the progression from early pancreatic intraepithelial neoplasia (PanIN) to PDAC. Restoring HPK1 expression in PDAC cells leads to cell cycle arrest and growth inhibition, which is due, in part, to the stabilization of p21 and p27 (30). Therefore, HPK1 may function as a novel tumor suppressor in pancreatic tumorigenesis. Our previous studies also demonstrated that loss of HPK1 in PDAC is mediated by CUL7/Fbxw8 ubiquitin ligase through 26S proteasome, which requires HPK1 kinase activity and autophosphorylation (31, 32). To further explore the mechanisms of the tumor suppressor functions of HPK1, we identified AXL as one of the major HPK1-interacting proteins in PDAC cells using antibody array-based screening, and we examined the role of HPK1 in regulating AXL signaling. Our study not only reveals a novel mechanism by which HPK1 down-regulates oncogenic AXL through the endocytic pathway, but also provides the new link between HPK1 and the oncogenic Gas6–AXL pathway in pancreatic cancer.
Results
AXL physically associates with HPK1
To identify the binding partners of HPK1 in PDAC cells, we performed an antibody array screening using Panc-1/HPK1 stable cells and identified AXL as one of the major binding partners of HPK1 in PDAC cells (Fig. 1A). To confirm the interaction between HPK1 and AXL, we performed reciprocal co-immunoprecipitation using either anti-AXL or anti-HPK1 and the lysates from HEK293T cells transfected with HPK1 and AXL plasmids. The AXL protein was effectively precipitated using an anti-HPK1 antibody (Fig. 1B). Similarly, the HPK1 protein was precipitated using an anti-AXL antibody (Fig. 1C). To demonstrate whether endogenous HPK1 interacts with endogenous AXL, reciprocal co-immunoprecipitation assays were formed using the cell lysate of Jurkat cells, which expressed both HPK1 and AXL. We found that endogenous HPK1 interacted with endogenous AXL in Jurkat cells (Fig. 1, D and E). These data demonstrate a novel interaction between AXL and HPK1 proteins.
Figure 1.

HPK1 physically interacts with AXL. A, antibody array results and map showed that HPK1 interacts with AXL (marked by arrow). B and C, interaction between HPK1 and AXL was detected by reciprocal co-immunoprecipitation. HEK293T cells were transfected with pCMV-AXL alone or co-transfected with pCMV-AXL and Flag-HPK1 expression plasmids, and co-immunoprecipitation was performed as described under “Materials and methods.” D and E, endogenous HPK1 interacted with endogenous AXL in Jurkat cells. F, a schematic drawing of the domain structure of HPK1 protein. G, AXL binds to the C-terminal domain of HPK1. HEK293T cells were transfected with AXL alone or co-transfected with AXL and Flag-HPK1, Flag-HPK1 KD, or Flag-HPK1 CD expression plasmids. Cell lysates were collected for co-immunoprecipitation with M2 beads. Immunoprecipitated (IP) AXL was detected by immunoblotting. WB, Western blotting.
HPK1 possesses a kinase domain (KD, 1–274 amino acids) and a C-terminal domain (CD, 275–833 amino acids) (33). The C-terminal domain includes four proline-rich regions (PR1–PR4) and a citron homology domain (Fig. 1F). To identify whether a discrete domain of HPK1 interacts with AXL, Flag-tagged HPK1, HPK1-KD, or HPK1-CD plasmid was transfected into 293T cells alone or in combination with pCMV-AXL plasmid. Cells were collected 36 h after transfection, and co-immunoprecipitations were performed using M2 beads followed by immunoblotting using an anti-AXL antibody. The AXL protein was co-immunoprecipitated with full-length HPK1 and HPK1-CD, but not HPK1-KD (Fig. 1G), indicating that AXL binds to the C-terminal domain of HPK1.
Oncogenic AXL is down-regulated by HPK1, which requires HPK1 kinase activity
We noticed that AXL expression was consistently decreased when it was co-transfected with HPK1 (Fig. 1G, 4th and 5th lanes). To test whether AXL expression was down-regulated by HPK1, a fixed amount of pCMV-AXL was co-transfected with a different amount pCI-Flag-HPK1 into 293T cells. AXL protein levels decreased in a dose-dependent manner with the increase of HPK1 expression (Fig. 2, A and B). To test the specificity of HPK1-mediated AXL down-regulation, we performed similar co-transfection experiments using increasing amounts of pCI-Flag-HGK, which is also a member of MAP4K family but is overexpressed in PDAC (34). As shown in Fig. 2, C and D, increasing the expression levels of HGK protein did not affect AXL protein levels. To test whether HPK1-mediated AXL down-regulation required HPK1 kinase activity, we performed co-transfection experiments using fixed amounts of pCMV-AXL and increasing amounts of pCI-Flag-HPK1-M46, a dominant-negative HPK1 construct that has a methionine substituted for Lys-46, which abrogates ATP binding and HPK1 kinase activity (33, 35, 36). Increasing amounts of HPK1-M46 did not show a significant effect on the AXL protein levels (Fig. 2, E and F). These data suggest that HPK1 kinase activity is required for HPK1-mediated AXL down-regulation.
Figure 2.

AXL is down-regulated by HPK1, and HPK1 kinase activity is required for HPK1-mediated AXL down-regulation. A and B, overexpressed HPK1 down-regulates AXL expression in a dose-dependent manner when co-transfected with pCMV-AXL expression plasmid into HEK293T cells. C–F, co-transfection of HGK or HPK1-M46, a kinase-dead form of HPK1 with pCMV-AXL expression plasmid into HEK293T cells does not affect the AXL expression. The expression levels of AXL protein and actin were quantified using ImageJ software. Mean values of relative AXL protein expression from three independent experiments were calculated and plotted on bar graphs (B, D, and F). G, AXL mRNA expression levels measured by quantitative RT-PCR analysis. RPS6 was used as an internal control. The data are shown as bar graph from three independent experiments. H and I, HPK1 decreases the stability of AXL protein. HEK293T cells were transfected with pCMV-AXL alone or co-transfected with pCMV-AXL and pCI-Flag-HPK1 and then treated with 100 μg/ml CHX to inhibit protein translation for 0, 0.5, 1, 2, 4, and 6 h. The cell lysates were collected for immunoblotting to measure expression levels of AXL proteins. The expression levels of AXL protein and actin in each sample were quantified using ImageJ software. The average expression levels of AXL protein after adjusting to actin levels from multiple experiments were plotted. The estimated half-life of AXL protein was 4.7 h in the absence of HPK1 and 2.4 h in the presence of HPK1.
To examine the mechanisms of HPK1-mediated AXL down-regulation, we measured the half-life of AXL protein in the presence or absence of HPK1. When co-transfected with HPK1, the estimated half-life of AXL protein was 2.4 h, which was 2.3 h shorter than when AXL was transfected alone without HPK1 (Fig. 2, H and I). Overexpression of HPK1 had no significant effect on the expression levels of AXL mRNA (Fig. 2G), suggesting that HPK1 down-regulates AXL at the protein level, rather than by affecting AXL mRNA expression.
HPK1-mediated AXL down-regulation is blocked by the inhibitors of the endocytic pathway
Activated receptor tyrosine kinases are often regulated by internalization and endocytic trafficking that can target the activated receptors for degradation in lysosomes (37, 38). To examine whether HPK1-mediated AXL degradation requires lysosomal protease activity, AXL was transfected alone or with HPK1 into 293T cells. Eight hours after transfection, cells were treated with leupeptin for 16 h, which inhibits serine, cysteine, and threonine proteases in lysosome (39, 40). HPK1-mediated AXL down-regulation was blocked by leupeptin treatment (Fig. 3, A and B). Lysosomal degradation is a consequence of cargo entering endosomal sorting (41, 42). To determine whether HPK1-mediated AXL degradation requires that AXL proceeds through the endocytic pathway, we performed similar experiments using bafilomycin A1 (BA1) and monensin, which specifically inhibits vacuolar-type H+-ATPase and keeps a low pH environment in endosomal vesicles (43–45). Treatment with BA1 or monensin significantly inhibited HPK1-mediated AXL down-regulation (Fig. 3, C–F). These data indicate that HPK1-mediated AXL degradation occurs through endocytic trafficking and lysosomal degradation.
Figure 3.

HPK1 down-regulates AXL through endocytic/lysosomal pathway. A–F, lysosome inhibitors leupeptin, bafilomycin A1, and monensin blocked HPK1-mediated AXL degradation. HEK293T cells were transfected with pCMV-AXL alone or co-transfected with pCI-HPK1. The transfected cells were treated with 100 μg/ml leupeptin, 0.1 μm bafilomycin A1, or 100 μm monensin overnight. AXL and HPK1 protein levels were detected by immunoblotting using AXL and HPK1 antibodies (A, C, and E). The expression levels of AXL protein and actin were quantified using ImageJ software. Mean values of relative AXL protein expression from three independent experiments were calculated and plotted on bar graphs (B, D, and F). G, HPK1 accelerates endogenous AXL degradation in Panc-28/HPK1 stable cells. Panc-28 control cells and Panc-28/HPK1 stable cells were serum-starved for 16 h and then treated with 400 ng/ml Gas6 for the time lengths as indicated. The expression levels of AXL and HPK1 were detected by immunoblotting. H, HPK1 accelerates the movement of AXL protein from cytoplasmic membrane to late endosomes in pancreatic cancer cells as detected by immunofluorescence. Panc-28 control and HPK1 stable cells were treated with Gas6 (400 ng/ml) at 37 °C to observe AXL internalization or at 0 °C after serum starvation. The cells were fixed with methanol and incubated with antibody targeting AXL (green, Alexa Fluor 488) and LAMP1 (red, Alexa Fluor 564). Co-localization of AXL and LAMP1 (yellow) was visualized with mounting medium with DAPI (blue) using the Olympus IX73 inverted microscope.
HPK1 accelerates the movement of AXL protein from cytoplasmic membrane to late endosomes and promotes AXL degradation in pancreatic cancer cells
To examine the role of HPK1 in endogenous AXL degradation in pancreatic cancer, the control Panc-28 cells and Panc-28/HPK1 stable cells were treated with Gas6 for different times, and the cell lysates were subjected to immunoblotting using anti-AXL antibody. As shown in Fig. 3G, Gas6 treatment accelerated endogenous AXL degradation in a time-dependent manner in Panc-28/HPK1 stable cells compared with the parental control cells. In Panc-28/HPK1 stable cells, AXL expression was markedly decreased at 4, 6, and 8 h after Gas6 stimulation compared with control cells. These results suggest that HPK1 is involved in AXL protein degradation in pancreatic cancer.
To investigate whether HPK1-mediated AXL degradation in pancreatic cancer cell requires trafficking through the endocytic pathway, we examined the localization of AXL protein by immunofluorescence microscopy. In unstimulated Panc-28 control cells and Panc-28/HPK1 stable cells, AXL was located predominantly on the cell membrane (green dot staining pattern, 0 °C, Fig. 3H). When treated with Gas6 at 37 °C for 40 min, we observed a significant shift of membranous AXL expression to perinuclear vesicles that co-localized with the lysosome marker, LAMP1 (red) in Panc-28/HPK1 stable cells (yellow dots). However, no significant shift of membranous AXL expression was observed in Panc-28 control cells (Fig. 3H, middle and lower panels). These results suggest that HPK1 accelerates AXL internalization and movement from the cell membrane to late endosomes in Panc-28 cells when stimulated with Gas6.
HPK1 enhances AXL and c-Cbl interaction and promotes AXL ubiquitination in vivo in pancreatic cancer cells
Ubiquitin ligase c-Cbl has been shown to play a role in epidermal growth factor receptor degradation through endocytosis and in GAS6-induced down-regulation of AXL. To further examine the role of c-Cbl in HPK1-mediated AXL degradation, the Panc-1/Tet-HPK1 stable cells were treated with doxycycline for 24 h to induce HPK1 expression. The cell lysates were subjected to immunoprecipitation using anti-AXL followed by immunoblotting using anti-AXL, c-Cbl, HPK1, and GAPDH antibodies. As expected, Panc-1/Tet-HPK1 stable cells had markedly increased HPK1 protein expression after treatment with doxycycline. HPK1 overexpression increased the binding of c-Cbl to endogenous AXL (Fig. 4A). To examine whether HPK1 affects AXL ubiquitination, control and Panc-28/HPK1 stable cells, which have higher levels of AXL protein than Panc-1/HPK1 stable cells after GAS6 treatment, were treated with GAS6 for 30 min after serum starvation for 16 h. Immunoprecipitations were performed using anti-AXL antibody followed by immunoblotting using anti-ubiquitin antibody. HPK1 enhanced the AXL ubiquitination in Panc-28/HPK1 stable cells after treatment with GAS6 (Fig. 4B).
Figure 4.
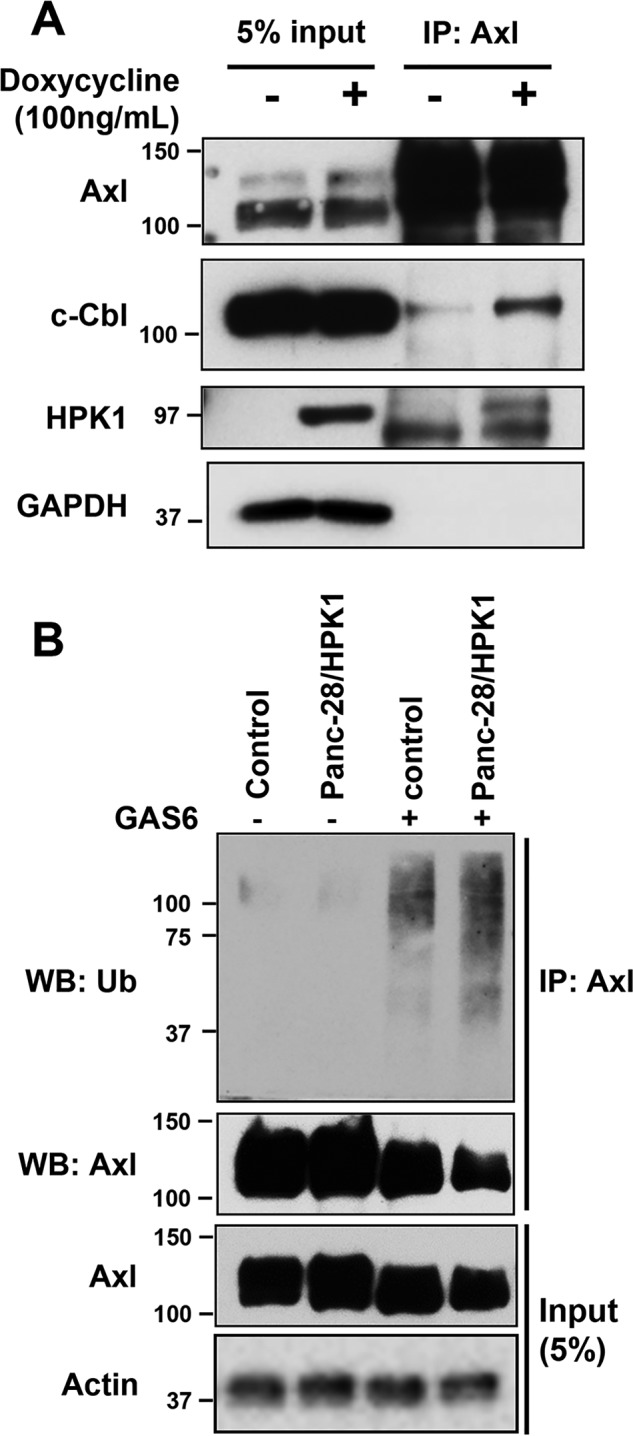
HPK1 enhances AXL and c-Cbl interaction and promotes AXL ubiquitination in pancreatic cancer cells. A, Panc-1/Tet-HPK1 stable cells were treated with 100 ng/ml doxycycline for 24 h. The cell lysates were subjected to immunoprecipitation (IP) using anti-AXL followed by immunoblotting using anti-AXL, Cbl, HPK1, and GAPDH antibodies. B, control and Panc-28/HPK1 stable cells were treated with 100 ng/ml GAS6 for 30 min after serum starvation for 16 h. Immunoprecipitations were performed using anti-AXL antibody followed by immunoblotting using anti-ubiquitin antibody. Ub, ubiquitin; WB, Western blotting.
HPK1-mediated AXL degradation decreases the invasion capability and downstream AKT and ERK signaling in pancreatic cancer
In a previous study, we showed that AXL overexpression correlates with distant metastasis in PDAC patients and that AXL promotes invasion (17). To determine whether HPK1-mediated AXL degradation affects the invasion capacity of Panc-28 cells, we performed Matrigel invasion assays. The number of cells invading the membrane was compared between the Panc-28/HPK1 stable cells and the control cells. The average cell numbers invading the Matrigel for Panc-28/HPK1 stable cell clones (HPK1 #1 and HPK1 #2) were significantly lower than for Panc-28 parental and vector controls (p < 0.01, Fig. 5, A and B).
Figure 5.

Overexpression of HPK1 reduces the invasion potential and inhibits Gas6-mediated AKT and ERK activation in pancreatic cancer cells. A, representative micrographs showing the number of cells invaded through the membrane for Panc-28 control cells and two Panc-28/HPK1 stable clones (HPK1 #1 and HPK1 #2) in in vitro Matrigel invasion assays. B, cells invading through the membrane were counted under a microscope in five predetermined fields at ×200 magnification. The control and HPK1 stable cells were assayed in triplicate, and assays were repeated three times. The average number of cells invading through the membrane for the control and HPK1 stable cells was plotted. A significant decrease in the number of invading cells was observed in both HPK1 stable clones compared with the control cells (*, p < 0.001). C and D, HPK1 inhibits Gas6-mediated AKT and ERK activation in pancreatic cancer cells. Panc-28 control and Panc-28/HPK1 stable cells were serum-starved for 16 h and then treated with 400 ng/ml Gas6 for the time lengths as indicated. The expression levels of AXL, p-AKT (Ser-473), AKT, p-ERK, total ERK, HPK1, and tubulin control were detected by immunoblotting and were quantified using ImageJ software. Mean values of relative AXL protein expression (AXL/tubulin ratio), p-AKT/AKT ratio, and p-ERK/ERK ratio from three independent experiments were calculated and plotted on the bar graph (D).
We next examined the effect of HPK1 on AXL downstream mitogen-activated protein kinase and AKT signaling pathways in response to Gas6 stimulation. Control or Panc-28/HPK1 stable cells were stimulated with Gas6 for 15–120 min. We observed that HPK1 inhibited Gas6/AXL-mediated AKT activation throughout the entire stimulation period from 15 to 120 min and inhibited Gas6/AXL-mediated ERK activation at 30, 60, and 120 min compared with the parental Panc-28 cells (Fig. 5, C and D). Similar results were observed in Panc-1/Tet-HPK1 cells. HPK1 down-regulated endogenous AXL and inhibited ERK activation in Panc-1/Tet-HPK1 cells when treated with Gas6 (Fig. 6).
Figure 6.

HPK1 inhibits Gas6-mediated AXL degradation and ERK activation in Panc-1/Tet-HPK1 stable cells. Panc-1/Tet-HPK1 stable cells, untreated or treated with doxycycline (Dox), were serum-starved for 16 h and then treated with 400 ng/ml Gas6 for the time lengths as indicated. The expression levels of AXL, HPK1, p-ERK, total ERK, and GAPDH control were detected by immunoblotting.
Expression of HPK1 inversely correlated with AXL expression in human PanIN
To examine the significance of HPK1-mediated AXL degradation in pancreatic cancer, we examined the expression of HPK1 and AXL in 26 human PanIN lesions from 10 different patients by immunohistochemistry. Similar to our previous report, we found that HPK1 was expressed in normal pancreatic ductal cells in all patients and was expressed in 88.9% (8/9), 50% (5/10), and 28.6 (2/7) in PanIN1, PanIN2, and PanIN3 lesions, respectively (Fig. 7, A–D). In contrast, AXL was expressed in 20% (2/10), 22.2% (2/9), 70% (7/10), and 85.7% (6/7) in normal pancreatic ducts, PanIN1, PanIN2, and PanIN3 lesions, respectively (Fig. 7, E–H). Both the loss of HPK1 expression and AXL overexpression correlated with the progression from low-grade PanIN1 to high-grade PanIN3 (p < 0.05). More importantly, we found a significant inverse correlation between HPK1 expression and AXL expression in human PanIN lesions. Among the 15 PanIN lesions that were positive for HPK1, AXL was expressed only in 33% (5/15) compared with 91% (10/11) AXL expression in the PanIN lesions that were negative for HPK1 (p = 0.005). These data provided strong support that HPK1 mediates AXL down-regulation in pancreatic cancer.
Figure 7.

AXL protein expression inversely correlates with HPK1 expression in human PanIN. A–D, representative micrographs show the expression of HPK1 in normal pancreas (A), PanIN1 (B), PanIN2 (C), and PanIN3 (D). E–H, representative micrographs show the expression of AXL in normal pancreas (E), PanIN1 (F), PanIN2 (G), and PanIN3 (H). HPK1 is expressed in normal pancreatic ducts and PanIN1 but is not expressed in PanIN2 and PanIN3. In contrast, AXL is not expressed in normal pancreatic ducts and PanIN1 but is expressed in PanIN2 and PanIN3.
To further examine the clinical significance of HPK1-mediated AXL degradation in pancreatic cancer, we performed survival analysis using the RNA-Seq data of HPK1 and AXL from 176 patients with pancreatic ductal adenocarcinoma in the TCGA database. Patients, whose tumors were HPK1-low and high-AXL, had shorter survival (median survival 481 days) compared with those with AXL-low or HPK1-high tumors (median survival 1130 days, see Fig. 8, p < 0.001).
Figure 8.
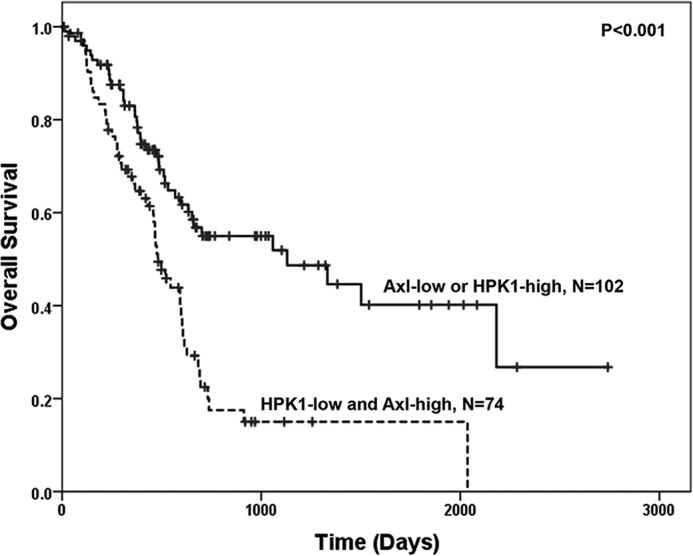
Kaplan-Meier survival curves stratified by the expression levels of HPK1 and AXL in 176 patients with pancreatic ductal adenocarcinoma. The expression data of HPK1 and AXL and survival data were downloaded from TCGA database (www.proteinatlas.org).4 Patients, whose tumors were HPK1-low and AXL-high, had the shorter survival (median survival, 481 days) compared with those with AXL-low or HPK1-high tumors (median survival, 1130 days, p < 0.001).
Discussion
In previous studies, we and others showed that AXL is overexpressed in 70% of human PDAC samples and pancreatic cancer cell lines (17, 18). Overexpression of AXL correlated with higher frequency of distance metastasis and poor prognosis in patients with PDAC who underwent surgical resection (17, 18). AXL silencing decreases invasion/migration potential and increases apoptosis of PDAC cells in response to radiation (17). In addition, previous studies have shown that AXL is overexpressed in carcinomas of lung, breast, brain, colon, skin, prostate, ovary, liver, and other types of malignancies and contributes to the tumor progression in these malignancies (5, 7, 9, 14, 46–51). However, the mechanisms resulting in AXL overexpression in cancers are largely unknown. In this study, we showed for the first time that HPK1 physically associates with AXL using an antibody array assay and reciprocal co-immunoprecipitation assays. We observed that HPK1 down-regulated AXL protein expression and its downstream signaling pathways through endocytic trafficking coupled with lysosomal degradation. More importantly, our study demonstrated a strong inverse correlation between AXL protein expression and HPK1 expression in human PanIN lesions. Thus, our results reveal a novel tumor suppressor function of HPK1 by targeting an important oncogenic receptor tyrosine kinase AXL for degradation. Consistent with this notion, we found that low expression of HPK1 and high expression of AXL correlated significantly with poor survival in patients with pancreatic ductal adenocarcinoma. Given the fact that HPK1 protein expression was lost in over 90% of human PDAC samples, our results suggest that loss of HPK1 expression may contribute to the overexpression of AXL in pancreatic cancer and thus reveal a new mechanism of AXL overexpression in cancer.
One of the major mechanisms that controls the signaling of receptor tyrosine kinases is through internalization, early and late endosomes, and then delivery to lysosome for degradation (38, 52, 53). In this study, we showed that HPK1-mediated AXL degradation can be completely blocked by the lysosomal proteinase inhibitor, leupeptin. HPK1-mediated AXL degradation was also significantly blocked by inhibitors of endocytosis, bafilomycin A1, and monensin. Thus, we concluded that HPK1-mediated AXL degradation occurs by targeting AXL for endocytic trafficking and lysosomal degradation. Our results are consistent with previous reports that binding of Gas6 to AXL induces the phosphorylation and down-regulation of AXL in human lens epithelial cells through endocytosis/lysosomal degradation (27).
AXL trafficking and down-regulation have been proposed to significantly influence downstream signaling. Treatment using mABs against AXL induced down-regulation of AXL by internalization and inhibited its downstream AKT activation, which lead to inhibition of proliferation and migration of pancreatic cancer cells in vitro and growth inhibition in vivo in pancreatic cancer xenograft models (22). Similar findings have also been reported in triple-negative breast cancer cells, in which AXL mAB induces internalization and degradation of AXL and inhibited AXL/Gas6 signaling, EMT, and cell migration/invasion (21). Consistent with these reports, we found that HPK1-mediated AXL down-regulation through endocytosis-coupled lysosomal degradation led to decreased activation of AKT and ERK and inhibited the invasion potential of pancreatic cancer cells. Our data suggest that targeting the HPK1-Gas6/AXL pathway may represent a new therapeutic strategy for pancreatic cancer.
In this study, we found that HPK1 interacted with AXL preferentially through its C-terminal domain and resulted in down-regulation of the AXL protein. It is interesting that HPK1-mediated AXL degradation depends on HPK1 kinase activity. In addition, we found that HPK1 increased the binding of AXL to c-Cbl and increased AXL protein ubiquitination. It is possible that HPK1 phosphorylates AXL at unidentified serine/threonine residue(s), which subsequently led to the binding of AXL to c-Cbl, which promotes AXL ubiquitination and accelerates AXL internalization, and trafficking through endosomes to lysosomes for degradation.
HPK1 has been shown to be a negative regulator in the activation of T lymphocytes (54). A recent study using HPK1-M46 transgenic mice showed that HPK1 kinase activity is required for the immunosuppressive functions of HPK1. Inactivation of the kinase domain enhanced the anti-tumor immune responses and anti-PD-L1 efficacy (36). These findings suggested that targeting HPK1 kinase activity in immune cells using select small molecular inhibitors in combination with the immune checkpoint inhibitor therapy may be an alternative strategy for effective cancer treatment (36). However, our study demonstrated that HPK1 kinase activity is required in the HPK1-mediated degradation of oncogenic tyrosine receptor AXL and inhibition of its downstream signaling pathways in pancreatic cancer. The function of HPK1 in the interplay between tumor cells and activation of immune cells in tumor microenvironment needs to be investigated.
Materials and methods
Cell lines and transfection
HEK293T, Panc-1, and Panc-28 cells were grown in DMEM supplemented with 10% fetal bovine serum and 100 units/ml streptomycin/penicillin. HEK293T cells were transfected with different amounts of plasmids expressing AXL, HPK1, and its mutants, as indicated in the figure legends. To establish HPK1 stable cell lines using Panc-28 cells, a retrovirus-based expression system pBABE–Flag-tagged HPK1 was used. To establish an inducible HPK1 expression system in Panc-1 cells (Panc-1/Tet-HPK1), a lenti-XTM Tet-On® advanced inducible expression system (Clontech) was used. The cells were subsequently selected in the presence of puromycin (1–2 μg/ml). The stable clones expressing HPK1 were screened by RT-PCR and immunoblotting. Panc-1 and Panc-28 were chosen for this study because both cell lines expressed high levels of AXL and Gas6 proteins.
Antibody array-based screening for HPK1-interacting proteins in pancreatic cancer cells
The cell lysates from Panc-1/HPK1 stable cells were incubated with a membrane filter arrayed with 400 antibodies (Hypromatrix, Worcester, MA), which were pre-blocked in a buffer containing 5% nonfat milk. After overnight incubation at 4 °C, the antibody–antigen–HPK1 complex was detected by horseradish peroxidase–conjugated anti-Flag antibody, followed by chemiluminescence according to the manufacturer's protocol.
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously (30). For each co-immunoprecipitation, 500 μg of total proteins were mixed with 2 μg of either antibody or normal serum IgG. The precipitated proteins were analyzed by 10% SDS-PAGE, which was then electroblotted onto polyvinylidene difluoride membranes (Novex, Grand Island, NY), blocked in 5% skim milk in 1× TBST, and probed with the primary antibodies indicated in the figure legends. The following antibodies were used for immunoprecipitation or immunoblotting: anti-β-actin and anti-Flag (M2) were purchased from Sigma; anti-HPK1 (N-19) and anti-AXL were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX), and anti-p-AKTSer-473 and anti-phospho-ERK were from Cell Signaling (Danvers, MA).
Half-life of AXL protein
The half-life of AXL protein was measured in HEK293T cells co-transfected with AXL and HPK1 constructs compared with those transfected with AXL alone. The cells were treated with 100 μg of cycloheximide (CHX) for 0 and 30 min and for 1, 2, 4, and 6 h. The AXL protein levels were measured by immunoblotting and quantified using ImageJ software. The ratio between AXL and the loading control, actin, was calculated. The average value after three repeats was calculated and plotted.
Quantitative RT-PCR analysis of AXL mRNA
Total RNA from 293T cells was extracted using TRIzol (Invitrogen). The cDNA was synthesized using a reverse transcription system (Promega). RT-PCR was performed using a thermal cycler (Bio-Rad) with the primers (AXL forward, 5′-GGTGGCTGTGAAGACGATGA-3′, and reverse, 5′-CTCAGATACTCCATGCCACT-3′; ribosomal protein small subunit 6 (RPS6) forward, AAGGAGAGAAGGATATTCCTGGAC-3′, and reverse, 5′-AGAGAGATTGAAAAGTTTGCGGAT-3′).
Ubiquitination of AXL in pancreatic cancer cells
Control and Panc-28/HPK1 stable cells were treated with 100 ng/ml GAS6 for 30 min after serum starvation for 16 h. Immunoprecipitations were performed at 4 °C overnight using anti-AXL antibody (Santa Cruz Biotechnology, Inc., sc-1097). Immunoprecipitated and input samples were run on SDS-PAGE, transferred to nitrocellulose membranes, and blotted using anti-ubiquitin (P4D1, sc-1097) and anti-AXL (sc-1097) antibodies.
Immunofluorescence microscopy
Panc-28 control and Panc-28/HPK1 stable cells were plated onto glass coverslips in 24-well plates to about 70% confluence. Prior to fixation, the cells were serum-starved for 16 h and then incubated with Gas6 (400 ng/ml) for 30 min on ice. Plates either remained on ice (0 °C) or were transferred to 37 °C for 40 min to allow AXL internalization. The cells were then fixed with methanol for 5 min at −20 °C and incubated at 4 °C overnight in 1% BSA-containing antibodies directed against AXL (1:200). Following PBS washes, cells were incubated with secondary antibodies at room temperature for 45 min. Subsequently, cells were fixed with 4% paraformaldehyde for 5 min and permeabilized with methanol for 2 min at −20 °C. Following PBS washes, the cells were incubated in 1% BSA-containing antibodies directed against LAMP1 (1:100) for 1 h at room temperature. After PBS washes, the cells were incubated with secondary antibodies at room temperature for 45 min and then washed with PBS, and coverslips were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Inc., Burlingame, CA). The cell images were obtained using Olympus IX73 inverted microscope.
Treatment of pancreatic cancer cells with Gas6
Cells were subjected to serum starvation for 16 h and then treated with recombinant human Gas6 (400 ng/ml, R&D Systems Inc., Minneapolis, MN) for the time length indicated in the figures. The cells were harvested, and the expression of phospho-AKT, phospho-ERK, and total ERK was detected by immunoblotting.
In vitro chemoinvasion assay
Chemoinvasion assays were performed using 24-well BioCoat Matrigel invasion chambers as described previously (17). Briefly, the lower compartment contained 0.6 ml of DMEM with 5.0% fetal bovine serum as chemoattractants or serum-free DMEM as a control. In the upper compartment, 2.5 × 104 cells/well were placed in triplicate wells and incubated for 24 h at 37 °C in a humidified incubator with 5% CO2. Then the cells that passed through the filter into the lower wells were stained with Giemsa (Thermo Fisher Scientific, Orangeburg, NY) and counted under a microscope in five predetermined fields. All assays were repeated at least three times. The differences in the invasion rates between the parental PDAC cells, vector control cells, and the PDAC cells with stable HPK1 expression were analyzed by one-way analysis of variance tests.
Immunohistochemical analysis for HPK1 and AXL expression in human pancreatic intraepithelial neoplasia
The use of human tissue in this study was approved by the Institutional Review Board of MD Anderson Cancer Center. Whole-tissue sections, which contained histologically verified PanIN of different grades, from 10 patients were used for immunohistochemistry. Immunohistochemical staining for HPK1 and AXL was performed on 4-μm unstained sections using the antibodies and conditions as described previously (17, 30). The expression of AXL and HPK1 in PanIN lesions and adjacent normal pancreas was evaluated by a pathologist who specializes in pancreatic cancer.
Survival analysis
The RNA-Seq data of HPK1 and AXL expression and survival data of 176 pancreatic cancer patients in the Cancer Genome Atlas (TCGA) database were downloaded from the Human Protein Atlas. The expression of HPK1 and AXL was categorized as low or high using the cutoff values set by TCGA (3.42 for HPK1 and 14.09 for AXL). Survival curves were constructed using the Kaplan-Meier method, and the log-rank test was used to evaluate the statistical significance of differences.
Author contributions
X. S., Hua Wang, R. A., C. D. L., A. M., A. J. B., and Huamin Wang conceptualization; X. S., H. A., Hua Wang, R. A., J.-h. S., F. Z., J. C., C. D. L., and Huamin Wang data curation; X. S., H. A., Hua Wang, J.-h. S., F. Z., J. C., A. J. B., and Huamin Wang formal analysis; X. S. and Huamin Wang supervision; X. S. and Huamin Wang funding acquisition; X. S., H. A., Hua Wang, R. A., J.-h. S., F. Z., J. C., C. D. L., A. M., A. J. B., and Huamin Wang investigation; X. S., H. A., Hua Wang, R. A., J.-h. S., C. D. L., A. M., A. J. B., and Huamin Wang methodology; X. S., H. A., Hua Wang, R. A., and Huamin Wang writing-original draft; X. S. and Huamin Wang project administration; X. S., H. A., Hua Wang, R. A., J.-h. S., C. D. L., A. M., A. J. B., and Huamin Wang writing-review and editing; C. D. L., A. M., A. J. B., and Huamin Wang resources.
This work was supported by National Institutes of Health Grants 1R01 CA196941 and 1R01CA195651 and the Khalifa Bin Zayed Al Nahyan Foundation Institute for Pancreatic Cancer Research at University of Texas M. D. Anderson Cancer Center. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PDAC
- pancreatic ductal adenocarcinoma
- HPK1
- hematopoietic progenitor kinase 1
- PanIN
- pancreatic intraepithelial neoplasia
- EMT
- epithelial–to–mesenchymal transition
- CD
- C-terminal domain
- KD
- kinase domain
- CHX
- cycloheximide
- DAPI
- 4,6-diamidino-2-phenylindol
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DMEM
- Dulbecco's modified Eagle's medium
- BA1
- bafilomycin A1
- RNA-Seq
- RNA-sequencing
- ERK
- extracellular signal-regulated kinase.
References
- 1. Are M., McIntyre A., and Reddy S. (2017) Global disparities in cancer pain management and palliative care. J. Surg. Oncol. 115, 637–641 10.1002/jso.24585 [DOI] [PubMed] [Google Scholar]
- 2. Ludwig K. F., Du W., Sorrelle N. B., Wnuk-Lipinska K., Topalovski M., Toombs J. E., Cruz V. H., Yabuuchi S., Rajeshkumar N. V., Maitra A., Lorens J. B., and Brekken R. A. (2018) Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. 78, 246–255 10.1158/0008-5472.CAN-17-1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skinner H. D., Giri U., Yang L. P., Kumar M., Liu Y., Story M. D., Pickering C. R., Byers L. A., Williams M. D., Wang J., Shen L., Yoo S. Y., Fan Y. H., Molkentine D. P., Beadle B. M., et al. (2017) Integrative analysis identifies a novel AXL–PI3 kinase–PD–L1 signaling axis associated with radiation resistance in head and neck cancer. Clin. Cancer Res. 23, 2713–2722 10.1158/1078-0432.CCR-16-2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang G., Wu L. W., Mender I., Barzily-Rokni M., Hammond M. R., Ope O., Cheng C., Vasilopoulos T., Randell S., Sadek N., Beroard A., Xiao M., Tian T., Tan J., Saeed U., et al. (2018) Induction of telomere dysfunction prolongs disease control of therapy-resistant melanoma. Clin. Cancer Res. 24, 4771–4784 10.1158/1078-0432.CCR-17-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutterer M., Knyazev P., Abate A., Reschke M., Maier H., Stefanova N., Knyazeva T., Barbieri V., Reindl M., Muigg A., Kostron H., Stockhammer G., and Ullrich A. (2008) Axl and growth arrest–specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 130–138 10.1158/1078-0432.CCR-07-0862 [DOI] [PubMed] [Google Scholar]
- 6. O'Bryan J. P., Frye R. A., Cogswell P. C., Neubauer A., Kitch B., Prokop C., Espinosa R. 3rd., Le Beau M. M., Earp H. S., and Liu E. T. (1991) Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 11, 5016–5031 10.1128/MCB.11.10.5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vajkoczy P., Knyazev P., Kunkel A., Capelle H.-H., Behrndt S., von Tengg-Kobligk H., Kiessling F., Eichelsbacher U., Essig M., Read T.-A., Erber R., and Ullrich A. (2006) Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. U.S.A. 103, 5799–5804 10.1073/pnas.0510923103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung B. I., Malkowicz S. B., Nguyen T. B., Libertino J. A., and McGarvey T. W. (2003) Expression of the proto-oncogene Axl in renal cell carcinoma. DNA Cell Biol. 22, 533–540 10.1089/10445490360708946 [DOI] [PubMed] [Google Scholar]
- 9. Craven R. J., Xu L. H., Weiner T. M., Fridell Y. W., Dent G. A., Srivastava S., Varnum B., Liu E. T., and Cance W. G. (1995) Receptor tyrosine kinases expressed in metastatic colon cancer. Int. J. Cancer 60, 791–797 10.1002/ijc.2910600611 [DOI] [PubMed] [Google Scholar]
- 10. Meric F., Lee W. P., Sahin A., Zhang H., Kung H. J., and Hung M. C. (2002) Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 8, 361–367 [PubMed] [Google Scholar]
- 11. Nakano T., Tani M., Ishibashi Y., Kimura K., Park Y. B., Imaizumi N., Tsuda H., Aoyagi K., Sasaki H., Ohwada S., and Yokota J. (2003) Biological properties and gene expression associated with metastatic potential of human osteosarcoma. Clin. Exp. Metastasis 20, 665–674 10.1023/A:1027355610603 [DOI] [PubMed] [Google Scholar]
- 12. Nemoto T., Ohashi K., Akashi T., Johnson J. D., and Hirokawa K. (1997) Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology 65, 195–203 10.1159/000164123 [DOI] [PubMed] [Google Scholar]
- 13. Shieh Y. S., Lai C. Y., Kao Y. R., Shiah S. G., Chu Y. W., Lee H. S., and Wu C. W. (2005) Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 7, 1058–1064 10.1593/neo.05640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Ginkel P. R., Gee R. L., Shearer R. L., Subramanian L., Walker T. M., Albert D. M., Meisner L. F., Varnum B. C., and Polans A. S. (2004) Expression of the receptor tyrosine kinase Axl promotes ocular melanoma cell survival. Cancer Res. 64, 128–134 10.1158/0008-5472.CAN-03-0245 [DOI] [PubMed] [Google Scholar]
- 15. Wu C. W., Li A. F., Chi C. W., Lai C. H., Huang C. L., Lo S. S., Lui W. Y., and Lin W. C. (2002) Clinical significance of AXL kinase family in gastric cancer. Anticancer Res. 22, 1071–1078 [PubMed] [Google Scholar]
- 16. Zhang Y. X., Knyazev P. G., Cheburkin Y. V., Sharma K., Knyazev Y. P., Orfi L., Szabadkai I., Daub H., Kéri G., and Ullrich A. (2008) AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 68, 1905–1915 10.1158/0008-5472.CAN-07-2661 [DOI] [PubMed] [Google Scholar]
- 17. Song X., Wang H., Logsdon C. D., Rashid A., Fleming J. B., Abbruzzese J. L., Gomez H. F., Evans D. B., and Wang H. (2011) Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer 117, 734–743 10.1002/cncr.25483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koorstra J. B., Karikari C. A., Feldmann G., Bisht S., Rojas P. L., Offerhaus G. J., Alvarez H., and Maitra A. (2009) The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 8, 618–626 10.4161/cbt.8.7.7923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kariolis M. S., Miao Y. R., Diep A., Nash S. E., Olcina M. M., Jiang D., Jones D. S. 2nd, Kapur S., Mathews I. I., Koong A. C., Rankin E. B., Cochran J. R., and Giaccia A. J. (2017) Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Invest. 127, 183–198 10.1172/JCI85610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirane A., Ludwig K. F., Sorrelle N., Haaland G., Sandal T., Ranaweera R., Toombs J. E., Wang M., Dineen S. P., Micklem D., Dellinger M. T., Lorens J. B., and Brekken R. A. (2015) Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 75, 3699–3705 10.1158/0008-5472.CAN-14-2887-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leconet W., Chentouf M., du Manoir S., Chevalier C., Sirvent A., Aït-Arsa I., Busson M., Jarlier M., Radosevic-Robin N., Theillet C., Chalbos D., Pasquet J. M., Pèlegrin A., Larbouret C., and Robert B. (2017) Therapeutic activity of anti-AXL antibody against triple-negative breast cancer patient-derived xenografts and metastasis. Clin. Cancer Res. 23, 2806–2816 10.1158/1078-0432.CCR-16-1316 [DOI] [PubMed] [Google Scholar]
- 22. Leconet W., Larbouret C., Chardès T., Thomas G., Neiveyans M., Busson M., Jarlier M., Radosevic-Robin N., Pugnière M., Bernex F., Penault-Llorca F., Pasquet J. M., Pèlegrin A., and Robert B. (2014) Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene 33, 5405–5414 10.1038/onc.2013.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moody G., Belmontes B., Masterman S., Wang W., King C., Murawsky C., Tsuruda T., Liu S., Radinsky R., and Beltran P. J. (2016) Antibody-mediated neutralization of autocrine Gas6 inhibits the growth of pancreatic ductal adenocarcinoma tumors in vivo. Int. J. Cancer 139, 1340–1349 10.1002/ijc.30180 [DOI] [PubMed] [Google Scholar]
- 24. Antony J., Zanini E., Kelly Z., Tan T. Z., Karali E., Alomary M., Jung Y., Nixon K., Cunnea P., Fotopoulou C., Paterson A., Roy-Nawathe S., Mills G. B., Huang R. Y., Thiery J. P., et al. (2018) The tumour suppressor OPCML promotes AXL inactivation by the phosphatase PTPRG in ovarian cancer. EMBO Rep. 19, e45670 10.15252/embr.201745670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goyette M. A., Duhamel S., Aubert L., Pelletier A., Savage P., Thibault M. P., Johnson R. M., Carmeliet P., Basik M., Gaboury L., Muller W. J., Park M., Roux P. P., Gratton J. P., and Côté J. F. (2018) The receptor tyrosine kinase AXL is required at multiple steps of the metastatic cascade during HER2-positive breast cancer progression. Cell Rep. 23, 1476–1490 10.1016/j.celrep.2018.04.019 [DOI] [PubMed] [Google Scholar]
- 26. Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., Jamieson A. M., Langdon W. Y., Ikeda F., Fededa J. P., Cronin S. J., Nitsch R., Schultz-Fademrecht C., Eickhoff J., Menninger S., et al. (2014) The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 10.1038/nature12998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valverde P. (2005) Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem. Biophys. Res. Commun. 333, 180–185 10.1016/j.bbrc.2005.05.086 [DOI] [PubMed] [Google Scholar]
- 28. Arnold R., Liou J., Drexler H. C., Weiss A., and Kiefer F. (2001) Caspase-mediated cleavage of hematopoietic progenitor kinase 1 (HPK1) converts an activator of NFκB into an inhibitor of NFκ. J. Biol. Chem. 276, 14675–14684 10.1074/jbc.M008343200 [DOI] [PubMed] [Google Scholar]
- 29. Chen Y. R., Meyer C. F., Ahmed B., Yao Z., and Tan T. H. (1999) Caspase-mediated cleavage and functional changes of hematopoietic progenitor kinase 1 (HPK1). Oncogene 18, 7370–7377 10.1038/sj.onc.1203116 [DOI] [PubMed] [Google Scholar]
- 30. Wang H., Song X., Logsdon C., Zhou G., Evans D. B., Abbruzzese J. L., Hamilton S. R., Tan T.-H., and Wang H. (2009) Proteasome-mediated degradation and functions of hematopoietic progenitor kinase 1 in pancreatic cancer. Cancer Res. 69, 1063–1070 10.1158/0008-5472.CAN-08-1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang H., Chen Y., Lin P., Li L., Zhou G., Liu G., Logsdon C., Jin J., Abbruzzese J. L., Tan T. H., and Wang H. (2014) The CUL7/F-box and WD repeat domain containing 8 (CUL7/Fbxw8) ubiquitin ligase promotes degradation of hematopoietic progenitor kinase 1. J. Biol. Chem. 289, 4009–4017 10.1074/jbc.M113.520106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H., Maitra A., and Wang H. (2016) The emerging roles of F-box proteins in pancreatic tumorigenesis. Semin. Cancer Biol. 36, 88–94 10.1016/j.semcancer.2015.09.004 [DOI] [PubMed] [Google Scholar]
- 33. Hu M. C., Qiu W. R., Wang X., Meyer C. F., and Tan T. H. (1996) Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 10, 2251–2264 10.1101/gad.10.18.2251 [DOI] [PubMed] [Google Scholar]
- 34. Liang J. J., Wang H., Rashid A., Tan T. H., Hwang R. F., Hamilton S. R., Abbruzzese J. L., Evans D. B., and Wang H. (2008) Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clin. Cancer Res. 14, 7043–7049 10.1158/1078-0432.CCR-08-0381 [DOI] [PubMed] [Google Scholar]
- 35. Ling P., Meyer C. F., Redmond L. P., Shui J. W., Davis B., Rich R. R., Hu M. C., Wange R. L., and Tan T. H. (2001) Involvement of hematopoietic progenitor kinase 1 in T cell receptor signaling. J. Biol. Chem. 276, 18908–18914 10.1074/jbc.M101485200 [DOI] [PubMed] [Google Scholar]
- 36. Hernandez S., Qing J., Thibodeau R. H., Du X., Park S., Lee H. M., Xu M., Oh S., Navarro A., Roose-Girma M., Newman R. J., Warming S., Nannini M., Sampath D., Kim J. M., et al. (2018) The kinase activity of hematopoietic progenitor kinase 1 is essential for the regulation of T cell function. Cell Rep. 25, 80–94 10.1016/j.celrep.2018.09.012 [DOI] [PubMed] [Google Scholar]
- 37. Grandal M. V., and Madshus I. H. (2008) Epidermal growth factor receptor and cancer: control of oncogenic signalling by endocytosis. J. Cell. Mol. Med. 12, 1527–1534 10.1111/j.1582-4934.2008.00298.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wiley H. S., and Burke P. M. (2001) Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic 2, 12–18 10.1034/j.1600-0854.2001.020103.x [DOI] [PubMed] [Google Scholar]
- 39. Hyman C., and Froehner S. C. (1983) Degradation of acetylcholine receptors in muscle cells: effect of leupeptin on turnover rate, intracellular pool sizes, and receptor properties. J. Cell Biol. 96, 1316–1324 10.1083/jcb.96.5.1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moore R. H., Tuffaha A., Millman E. E., Dai W., Hall H. S., Dickey B. F., and Knoll B. J. (1999) Agonist-induced sorting of human β2-adrenergic receptors to lysosomes during downregulation. J. Cell Sci. 112, 329–338 [DOI] [PubMed] [Google Scholar]
- 41. Pryor P. R., and Luzio J. P. (2009) Delivery of endocytosed membrane proteins to the lysosome. Biochim. Biophys. Acta 1793, 615–624 10.1016/j.bbamcr.2008.12.022 [DOI] [PubMed] [Google Scholar]
- 42. Seaman M. (2008) Membrane traffic in the secretory pathway. Cell. Mol. Life Sci. 65, 2842–2858 10.1007/s00018-008-8354-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oda K., Nishimura Y., Ikehara Y., and Kato K. (1991) Bafilomycin A1 inhibits the targeting of lysosomal acid hydrolases in cultured hepatocytes. Biochem. Biophys. Res. Commun. 178, 369–377 10.1016/0006-291X(91)91823-U [DOI] [PubMed] [Google Scholar]
- 44. Yoshimori T., Yamamoto A., Moriyama Y., Futai M., and Tashiro Y. (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712 [PubMed] [Google Scholar]
- 45. Tveten K., Ranheim T., Berge K. E., Leren T. P., and Kulseth M. A. (2009) The effect of bafilomycin A1 and protease inhibitors on the degradation and recycling of a Class 5-mutant LDLR. Acta Biochim. Biophys. Sin. 41, 246–255 10.1093/abbs/gmp008 [DOI] [PubMed] [Google Scholar]
- 46. Gjerdrum C., Tiron C., Høiby T., Stefansson I., Haugen H., Sandal T., Collett K., Li S., McCormack E., Gjertsen B. T., Micklem D. R., Akslen L. A., Glackin C., and Lorens J. B. (2010) Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. U.S.A. 107, 1124–1129 10.1073/pnas.0909333107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L., Greger J., Shi H., Liu Y., Greshock J., Annan R., Halsey W., Sathe G. M., Martin A.-M., and Gilmer T. M. (2009) Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 69, 6871–6878 10.1158/0008-5472.CAN-08-4490 [DOI] [PubMed] [Google Scholar]
- 48. Tsou A.-P., Wu K.-M., Tsen T.-Y., Chi C.-W., Chiu J.-H., Lui W.-Y., Hu C.-P., Chang C., Chou C.-K., and Tsai S.-F. (1998) Parallel hybridization analysis of multiple protein kinase genes: identification of gene expression patterns characteristic of human hepatocellular carcinoma. Genomics 50, 331–340 10.1006/geno.1998.5338 [DOI] [PubMed] [Google Scholar]
- 49. Wimmel A., Glitz D., Kraus A., Roeder J., and Schuermann M. (2001) Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur. J. Cancer 37, 2264–2274 10.1016/S0959-8049(01)00271-4 [DOI] [PubMed] [Google Scholar]
- 50. Macleod K., Mullen P., Sewell J., Rabiasz G., Lawrie S., Miller E., Smyth J. F., and Langdon S. P. (2005) Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 65, 6789–6800 10.1158/0008-5472.CAN-04-2684 [DOI] [PubMed] [Google Scholar]
- 51. Wu Y.-M., Robinson D. R., and Kung H.-J. (2004) Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. 64, 7311–7320 10.1158/0008-5472.CAN-04-0972 [DOI] [PubMed] [Google Scholar]
- 52. Benke D. (2010) in Advances in Pharmacology (Thomas P. B., ed) pp. 93–111, Academic Press, Orlando [Google Scholar]
- 53. Sorkin A., and Goh L. K. (2008) Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 314, 3093–3106 10.1016/S0014-4827(08)00404-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shui J.-W., Boomer J. S., Han J., Xu J., Dement G. A., Zhou G., and Tan T.-H. (2007) Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 8, 84–91 10.1038/ni1416 [DOI] [PubMed] [Google Scholar]