

Abstract
Glial signals are known to inhibit axonal regeneration and functional recovery after mammalian central nervous system trauma, including spinal cord injury. Such signals include membrane-associated proteins of the oligodendrocyte plasma membrane and astrocyte-derived, matrix-associated proteins. Here, using cell lines and primary cortical neuron cultures, recombinant protein expression, immunoprecipitation and immunoblot assays, transmission EM of exosomes, and axon regeneration assays, we explored the secretion and activity of the myelin-associated neurite outgrowth inhibitor Nogo-A and observed exosomal release of a 24-kDa C-terminal Nogo-A fragment from cultured cells. We found that the cleavage site in this 1192-amino-acid-long fragment is located between amino acids 961–971. We also detected a Nogo-66 receptor (NgR1)–interacting Nogo-66 domain on the exosome surface. Enzyme inhibitor treatment and siRNA knockdown revealed that β-secretase 1 (BACE1) is the protease responsible for Nogo-A cleavage. Functionally, exosomes with the Nogo-66 domain on their surface potently inhibited axonal regeneration of mechanically injured cerebral cortex neurons from mice. Production of this fragment was observed in the exosomal fraction from neuronal tissue lysates after spinal cord crush injury of mice. We also noted that, relative to the exosomal marker Alix, a Nogo-immunoreactive, 24-kDa protein is enriched in exosomes 2-fold after injury. We conclude that membrane-associated Nogo-A produced in oligodendrocytes is processed proteolytically by BACE1, is released via exosomes, and is a potent diffusible inhibitor of regenerative growth in NgR1-expressing axons.
Keywords: axon, regeneration, exosome (vesicle), β-secretase 1 (BACE1), oligodendrocyte, NgR1, Nogo, Nogo receptor, reticulon, spinal cord injury
Introduction
After traumatic spinal cord injury (SCI),3 neurological deficits are frequently profound and persistent below the level of damage. Dysfunction persists despite survival of nearly all neurons because of axonal damage and disconnection. Regrowth of injured axons is strongly inhibited in the CNS environment. Membrane-associated myelin-associated inhibitors (MAIs) and matrix-associated chondroitin sulfate proteoglycans have been identified as two major classes of inhibitors for axonal growth in the CNS.
There are multiple MAI proteins, most prominently Nogo-A (RTN4A), myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein (1–5). All three of these oligodendrocyte membrane–associated ligands bind to Nogo-66 receptor 1 (NgR1 or RTN4R) (6–9) and to paired immunoglobulin-like receptor B (PirB or LiLRB2) (10, 11). Nogo-A, the best-characterized MAI, is expressed by both oligodendrocytes and neurons and localizes to the endoplasmic reticulum and, to a lesser degree, to the oligodendrocyte plasma membrane at the innermost adaxonal and outermost myelin membranes in the spinal cord (12, 13). Axonal growth is inhibited by two regions of Nogo-A. One aa stretch, called amino-Nogo-A or Nogo-A-Δ20, acts via NgR1-independent mechanisms to prevent neural growth through integrins and S1PR2 (14, 15). The other region, Nogo-66, binds to NgR1 to inhibit axonal regeneration (1, 6). Further, Nogo-22, a 22-kDa fragment of Nogo-A that contains the Nogo-A-24 and Nogo-C39 regions in addition to the Nogo-66 domain, is a more potent inhibitor of axon regeneration than Nogo-66 (11). The mRNA and protein expression levels of Nogo-A in the spinal cord after injury are moderately up-regulated (12, 13). In contrast to Nogo-A, extracellular matrix–associated chondroitin sulfate proteoglycans are secreted by reactive astrocytes of the glial scar and are strongly up-regulated after injury (17, 18).
After spinal cord trauma, damaged axons are frequently demyelinated, so axonal cell surface contact with membrane-associated MAIs may be limited. Another injury-induced transmembrane MAI, Semaphorin 4D, is proteolyzed, and soluble protein fragments are released from the cell membrane after SCI (19–21). There are no direct mechanistic data regarding the release of Nogo-A, myelin-associated glycoprotein, or oligodendrocyte myelin glycoprotein, the MAIs secreted after injury.
Although evidence of diffusible glia signals limiting axonal regrowth after traumatic SCI has been limited, there is accumulating evidence of oligodendrocyte-derived exosomes participating in axonal and neuronal regulation. This includes firing rate and biochemical signaling (22–24). There is evidence that peripheral nervous system Schwann cell–derived exosomes enhance axon regeneration (25).
Here we report that cells proteolyze Nogo-A at C-terminal sites to produce a 24-kDa fragment. The C-terminal Nogo-A fragment is secreted into culture medium as an exosome component. Biochemical studies reveal that the inhibitory Nogo-66 region is exposed on the exosome surface. Alkalization decreases the level of C-terminal fragments, suggesting a role of lysosomal endopeptidases. Pharmacological and genetic studies indicate that BACE1 participates in Nogo-A C-terminal cleavage. We explored a role of the exosomal C-terminal fragment in axonal regeneration. Cortical neurons lacking NgR1 do not respond to the exosomal Nogo-A fragment, whereas WT neurons exhibit decreased axonal regeneration by low concentrations of exosomal Nogo-A. Moreover, the exosomal Nogo-A fragment level is increased after SCI in vivo. Thus, Nogo-A is proteolyzed and secreted as an exosome protein after injury to provide a potent diffusible inhibitor of axonal regeneration.
Results
The Nogo-A C-terminal region is secreted into culture supernatant as an exosome component
In C-terminally Myc-tagged Nogo-A–overexpressing HEK293T cells, both full-length Nogo-A and a number of proteolytic fragments were detected in the cell lysate fraction. Similar protein bands were detected when HEK293T cells overexpressed either Myc-tagged or untagged Nogo-A, excluding the possibility that addition of an epitope tag causes proteolysis (Fig. S1). In contrast, immunoprecipitation with anti-Myc antibody from culture supernatant predominantly detected a cleaved C-terminal fragment of ∼24 kDa and little or no full-length protein (Fig. 1A). When N-terminally FLAG-tagged Nogo-A was expressed in HEK293T cells, the lysate again showed full-length protein and several fragments. However, FLAG immunoprecipitates from the culture supernatant showed no specific bands (data not shown). Thus, the culture medium exclusively contains the C-terminal 24-kDa fragment of Nogo-A.
Figure 1.

The Nogo-A C-terminal region is secreted in culture supernatant as an exosome component. A, culture supernatants (culture sup) collected from vector- and Nogo-A–Myc–transfected HEK293T cells were immunoprecipitated (IP) with anti-Myc antibody and immunoblotted with anti-Myc antibody. Cell lysates were also immunoprecipitated and immunoblotted in the same membrane. B, schematic of exosome fractionation by centrifugation. C, each fraction from vector or Nogo-A–Myc–transfected HEK293T cells was immunoblotted with anti-Myc antibody. D, ratio of 24-kDa Nogo-A fragment to full-length (FL) Nogo-A in exosome and lysate fractions. Mean ± S.E., n = 12 for each group. ***, p < 0.005; Student's two-tailed t test.
To examine whether the culture medium Nogo-A 24-kDa fragment exists as a free protein or an extracellular vesicle component of the culture medium, we fractionated the medium (Fig. 1B). The C-terminal fragment was detected predominantly in the exosome fraction but not in the nonexosome fraction (Fig. 1C). Specifically, exosome enrichment of the 24-kDa fragment is such that the ratio for 24-kDa:full-length Nogo-A species shifts 100-fold, from 45:1 to 1:20, in exosomes versus cell lysates (Fig. 1D).
Transmission EM analysis of fractionated HEK293T culture medium confirmed the presence of exosomes in these preparations (Fig. 2A). Exosomes were identified by their particle size distribution of 30- to 100-nm diameter (26). The minor percentage of particles with a size distribution of more than 100 nm is likely due to microvesicles (26). We performed a sucrose density gradient analysis to compare the distribution of Nogo-A fractions with an exosome marker protein (Fig. 2B). The C-terminal 24-kDa fragment was detected in the same fractions as the exosome marker Alix. Furthermore, the levels of the C-terminal Nogo-A fragment in culture medium and the exosome fraction were significantly decreased after culture with the exocytic pathway inhibitor Exo1 (Fig. 2, C and D). These data indicate that the C-terminal 24-kDa Nogo-A fragment is secreted as an exosomal protein.
Figure 2.

The Nogo-A C-terminal region is enriched in exosomes. A, negative stain transmission EM images of the exosome fraction derived from HEK293T cell culture medium, with particle diameter distribution represented in a histogram. n = 546 particles. B, culture medium from Nogo-A–Myc–transfected HEK293T cells was separated by sucrose density centrifugation. Gradient fractions from the top were immunoblotted with anti-Alix and anti-Myc antibodies. C and D, HEK293T cells were transfected with vector (−) or Nogo-A–Myc. 24 h after transfection, media were changed to DMSO or Exo1 containing medium and cultured for another 12 h. Then the culture supernatants were immunoprecipitated (IP) with anti-Myc antibody or exosome-fractionated. Mean ± S.E., n = 3 independent experiments. *, p < 0.05; Student's two-tailed t test.
Proteolytic cleavage site in Nogo-A
To localize the cleavage site for the Nogo-A 24-kDa fragment, we expressed a truncated protein and compared the size with the fragment derived from the full-length construct (Fig. 3A). The exosomal fragment of Nogo-A migrated as a doublet by SDS-PAGE, with the Nogo-A 961–1192 and 971–1192 constructs showing similar migration in the same gel (Fig. 3B). This result suggests that proteolysis occurs around the 961 and 971 aa. We also created a series of 10-aa deletion mutant constructs from 891–1050 aa and assessed the secretion of a Nogo-A fragment into exosomes (data not shown). None of these 10-aa deletion mutant constructs strongly reduced the level of Nogo-A fragments in the exosome. In addition, we made a series of single aa substitution mutation constructs from 951–980 aa and examined the Nogo-A fragment in the exosome (data not shown). As for the 10-aa deletion constructs, the point mutation analysis failed to identify a single aa essential for exosomal secretion of the 24-kDa Nogo-A fragment. These data suggest that Nogo-A is cleaved for exosomal secretion in the vicinity of aa 961–971 immediately N-terminal to the first hydrophobic segment but that the topographic restriction may be more critical than a specific aa sequence.
Figure 3.
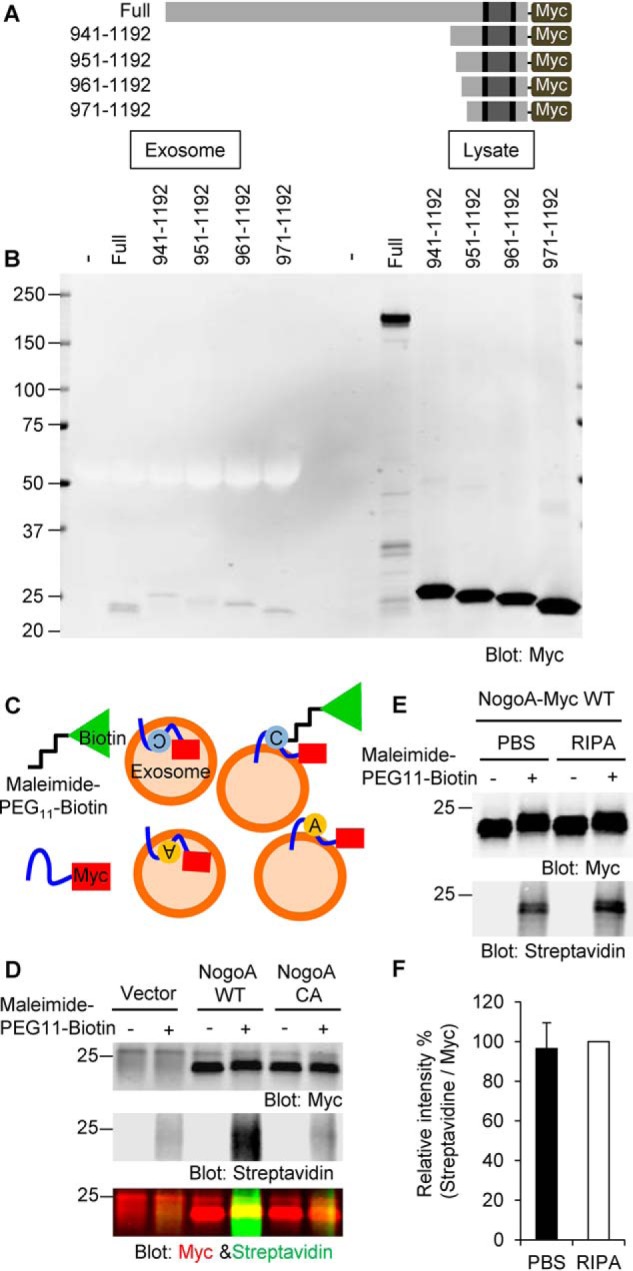
C-terminal cleavage site in Nogo-A. A, a series of Nogo-A–Myc C-terminally truncated constructs were generated. B, HEK293T cells were transfected with full-length Nogo-A–Myc or a series of C-terminal constructs of Nogo-A–Myc. 36 h after transfection, culture supernatants were collected, and exosomes were fractionated. Cell lysates were also collected. Samples were then immunoblotted with anti-Myc antibody. C, schematics of maleimide–PEG11–biotin labeling. A cysteine residue of Nogo-A exposed on the surface of exosomes can be labeled with nonpermeable maleimide–PEG11–biotin reagent. Cysteine is shown as a gray circle. Alanine substituted from cysteine is shown as a yellow circle. D, exosomes from vector-, Nogo-A WT–, or Nogo-A C1101A (NogoA CA)–transfected HEK293T cells were incubated with maleimide–PEG11–biotin. After the maleimide reaction, exosomes were lysed with RIPA buffer and immunoprecipitated with anti-Myc antibody. Immunoprecipitates were washed three times and blotted with anti-Myc antibody and Alexa Fluor 488 streptavidin. E, Nogo-A–Myc-exosomes resuspended in PBS or RIPA buffer were incubated with maleimide–PEG11–biotin. After the maleimide reaction, exosomes were resuspended in RIPA buffer and immunoprecipitated with anti-Myc antibody. Immunoprecipitates were washed three times and blotted with anti-Myc antibody and Alexa Fluor 488 streptavidin. F, streptavidin intensity was divided by Myc intensity under both PBS and RIPA conditions and expressed as normalized value as RIPA sample of 100%. Mean ± S.E., n = 9 independent experiments. p = 0.70, Student's two-tailed t test.
Determination of the topology of the Nogo-66 loop region in the exosome
There is evidence that Nogo-A assumes several different topologies within lipid bilayers in different subcellular compartments (27). Because we could immunoprecipitate the vast majority of the 24-kDa Nogo-A fragment with an anti-Myc antibody in the absence of detergent, the C terminus is most likely exposed on the surface of the exosome (Fig. 3C, two models on the right). Based on prior evidence that the C-terminal hydrophobic segment assumes a hairpin topology (28), the Nogo-66 domain is also expected to be exposed on the exosome surface (Fig. 3C, the two models on the right). However, because of controversy regarding hairpin versus transmembrane topology, we investigated the Nogo-66 loop topology within the exosome fraction using a nonpermeable maleimide–PEG11–biotin reagent. There is one cysteine amino acid at 1101 aa in the Nogo-66 sequence, and maleimide reacts efficiently and specifically with free sulfhydryls. The C1101A point-mutated Nogo-A was generated and used as a negative control for this experiment. HEK293T cells were transfected with Nogo-A WT or the C1101A mutant, and exosome fractions were prepared from those culture media. The exosomes were resuspended in PBS and incubated with maleimide–PEG11–biotin, and then the reaction was stopped with DTT prior to lysis in RIPA buffer. Lysed exosomes were immunoprecipitated with anti-Myc antibody and blotted with anti-Myc antibody or streptavidin (Fig. 3C). Equal amounts of C-terminal fragments (in the anti-Myc blot) were detected in both the Nogo-A WT and CA mutant incubated with or without maleimide–PEG11–biotin (Fig. 3D). However, a streptavidin signal was detected only in a Nogo-A WT under the maleimide–PEG11–biotin condition, suggesting that the Nogo-66 loop is exposed on the exosome surface. To calculate how much of the Nogo-66 loop is exposed on the exosome surface, we compared the maleimide–PEG11–biotin binding ratio between exosomes suspended in PBS and RIPA. The streptavidin intensity in PBS-suspended exosomes was 96% of that detected in RIPA-suspended exosomes, indicating that nearly all of the Nogo-66 loop is exposed on the surface of the exosome (Fig. 3, E and F).
Identification of enzymes for Nogo-A proteolysis
To identify the proteolytic enzymes responsible for production of the 24-kDa fragment, we used several endopeptidase inhibitors. Nogo-A–Myc–overexpressing HEK293T cells were treated with inhibitors, and exosomes were collected and immunoblotted with anti-Myc and anti-CD9 (an exosome marker) antibodies (Fig. 4, A and B). A caspase inhibitor, Z-VAD-fmk, and a Cathepsin inhibitor, E64d, had no effect on the Nogo-A C-terminal fragment levels in exosomes compared with the DMSO control. The calpain inhibitor MG101 increased the fragment level, whereas the lysosomal proteinase inhibitor NH4Cl decreased the Nogo-A fragment level in exosomes. NH4Cl is known to reduce the acidification of lysosomes, which contributes to optimal lysosomal enzyme activity.
Figure 4.

The Nogo-66 loop region on the surface of exosomes. A and B, HEK293T cells were transfected with Nogo-A–Myc. 24 h after transfection, media were changed. DMSO, MG101 (20 μm), Z-VAD-fmk (20 μm), E64d (20 μm), and NH4Cl (20 mm) were added and cultured for further 12 h. Then culture supernatants were collected, and the exosome fraction was immunoblotted with anti-Myc and anti-CD9 antibodies (A). The graph shows Myc intensity divided by CD9 intensity compared to DMSO control (B). Mean ± S.E., n = 5–8 independent experiments. *, p < 0.05; ***, p < 0.005; one-way ANOVA followed by Dunnett's test. C and D, HEK293T cells were transfected with Nogo-A–Myc. 24 h after transfection, media were changed. DMSO, the indicated amounts of BACE inhibitors, and NH4Cl (20 mm) were added and cultured for further 12 h. Then culture supernatants were collected, and the exosome fraction was immunoblotted with anti-Myc and anti-CD9 antibodies (C). The graph shows Myc intensity divided by CD9 intensity compared to DMSO control (D). Mean ± S.E., n = 4 independent experiments. **, p < 0.01; ***, p < 0.005; one-way ANOVA followed by Dunnett's test. E, HEK293T cells were transfected with Nogo-A–Myc and siNC, siBACE1 #1, or siBACE1 #2. Exosomes were purified 36 h after transfection and immunoblotted with anti-Myc and anti-CD9 antibodies. F, quantification of Myc intensity divided by CD9 intensity compared to DMSO control. Mean ± S.E., n = 6 independent experiments. **, p < 0.01; ***, p < 0.005; one-way ANOVA followed by Dunnett's test. G, real-time PCR for replicates of siBACE-transfected HEK293T cells. BACE1 mRNA expression was normalized to Gapdh mRNA expression. Mean ± S.E., n = 4 independent experiments. ***, p < 0.005; one-way ANOVA followed by Dunnett's test.
Among endosomal/lysosomal proteases, we considered β-site amyloid precursor protein cleaving enzyme 1 (BACE1, β-secretase 1) as a candidate protease with a known acidic pH optimum. It has been reported that BACE1 interacts with Nogo-A–related Reticulon family proteins (29). Treatment of cells with a BACE1 inhibitor dose-dependently decreased the level of Nogo-A C-terminal fragments in the exosome fraction as completely as NH4Cl (Fig. 4, C and D). BACE inhibition acted selectively on the Nogo-A 24-kDa fragment in exosomes, whereas expression of full-length Nogo-A in cell lysates remained relatively constant (Fig. S2A). The very low to undetectable levels of the 24-kDa fragment in cell lysate preparations prevented detection of any effects of BACE inhibition in that fraction. Because the pharmacological study suggested that BACE1 might mediate Nogo-A proteolysis, we used silencing RNAs to suppress BACE1 levels in HEK293T cells. BACE1 knockdown with two unrelated siRNA species significantly reduced the level of Nogo-A fragment in exosomes compared with control siRNA but did not change the level of the exosome marker CD9 (Fig. 4, E and F). BACE1 knockdown was confirmed by quantitative PCR (Fig. 4G). Because the BACE1 inhibitor has the potential to inhibit other enzyme activities, such as Cathepsins, we also performed Cathepsin D (CTSD) knockdown experiment as a further control. HEK293T cells transfected with siCTSD did not alter the levels of either the Nogo-A fragment or CD9 in the exosome fraction (Fig. S2, B and C), even though CTSD expression was decreased significantly (Fig. S2, D and E).
Function of Nogo-A C-terminal fragment
Because the Nogo-A C-terminal fragment contains an axon-inhibitory region, Nogo-66, we sought to examine whether the exosome fraction inhibits axonal regeneration. For functional studies, we conducted an in vitro axon regeneration assessment with cultured cortical neurons. Neurons were cultured for 8 days, scraped with a metal pin tool for axotomy, and then incubated with exosome preparations for 3 days to allow regeneration. Axotomized WT neurons treated with exosomes secreted from Nogo-A–overexpressing HEK293T cells showed decreased axonal regeneration compared with the vector control, consistent with the exosomal 24-kDa Nogo-A fragment being an active inhibitor of regeneration (Fig. 5, A and B). Because Nogo-A signaling is transduced primarily through NgR1, we assessed regeneration with NgR1−/− neurons. In contrast to the case with WT neurons, regeneration of NgR1−/− axons was not suppressed by exosomes from Nogo-A–expressing HEK293T cells (Fig. 5C). Thus, the C-terminal fragment Nogo-A in exosomes limits axonal regeneration in an NgR1-dependent manner.
Figure 5.

Inhibitor function of the Nogo-A C-terminal fragment. A and B, cortical neurons were scraped and treated with the indicated amount of exosomes at 8 DIV for 3 days (A). The microphotographs show βIII-tubulin (in axons, green) and phalloidin (to stain F-actin, red). Scale bar = 200 μm. B, quantification of axonal regeneration. Mean ± S.E., n = 3 biological replicates. *, p < 0.05; ***, p < 0.005; one-way ANOVA followed by Tukey's test. C, cortical neurons from the WT or NgR1−/− were scraped and treated with the same amount of exosomes at 8 DIV for 3 days. The graph shows quantification of axon regeneration. *, p < 0.05; Student's two-tailed t test. #, not significant. D, cortical neurons were scraped and treated with exosomes (10, 20, 40, and 75 nm) or Nogo22 (0, 10, 30, 75, 100, 150, 300, and 600 nm) at 8 DIV for 3 days. The graph shows quantification of axonal regeneration. Mean ± S.E., n = 2–6 biological replicates. *, p < 0.05; Student's two-tailed t test. E, the IC80 from each separate experiment was calculated. Mean ± S.E., n = 15 (Nogo22) and n = 9 (exosomes) biological replicates. *, p < 0.05; Student's two-tailed t test.
A purified recombinant 22-kDa protein, Nogo22, containing the three known NgR1 binding domains of Nogo-A (Nogo-A-24, Nogo-66, and Nogo-C39) is a substantially more potent inhibitor of axonal regeneration than Nogo-66 alone (11). As shown above, the C-terminal fragment of Nogo-A includes all components of this Nogo22 domain associated with the exosome lipid bilayer, constituting a physiologically relevant version of Nogo22. We sought to compare the relative potency of Nogo22 versus the exosomal 24-kDa Nogo-A fragment. To measure the amount of Nogo-A fragments in the exosome, fractions were blotted with anti-Nogo22 antibody and titrated with purified Nogo22 (Fig. S3, A and B). Although high concentrations of Nogo in the exosomal preparation could not be tested, it is clear that, at lower concentrations, the exosomal Nogo-A fragment is more potent than purified Nogo22 (Fig. 5D). The Nogo protein concentration required for inhibition of regeneration to 80% (IC80) for Nogo22 and for Nogo in exosomes from each separate experiment was calculated and compared. The exosomal Nogo fragment exhibits a significantly lower IC80 (45 nm) than Nogo22 (97 nm) (Fig. 5E).
Increased Nogo-A fragment levels after spinal cord trauma in vivo
The evidence that a 24-kDa Nogo-A fragment is secreted as an exosomal protein from HEK293T cells and inhibits axon regeneration after axotomy prompted us to assess production of the Nogo-A fragment in vivo. To evaluate injured spinal cord tissue, we chose a midthoracic spinal cord crush model because the injury is severe but the dura matter remains intact, preventing a cerebrospinal fluid leak from the lesion that might release local exosome accumulation. Spinal cords from sham and injured (SCI) animals were homogenized in TBS, and fractions were separated as in Fig. 1B. A Nogo-immunoreactive 24-kDa protein was detected in the exosome fraction from injured animals but not from the sham surgery group (Fig. 6A). Quantification of the Nogo-immunoreactive 24-kDa protein level relative to the exosomal marker Alix showed a significant increase in the injured spinal cord (Fig. 6B).
Figure 6.
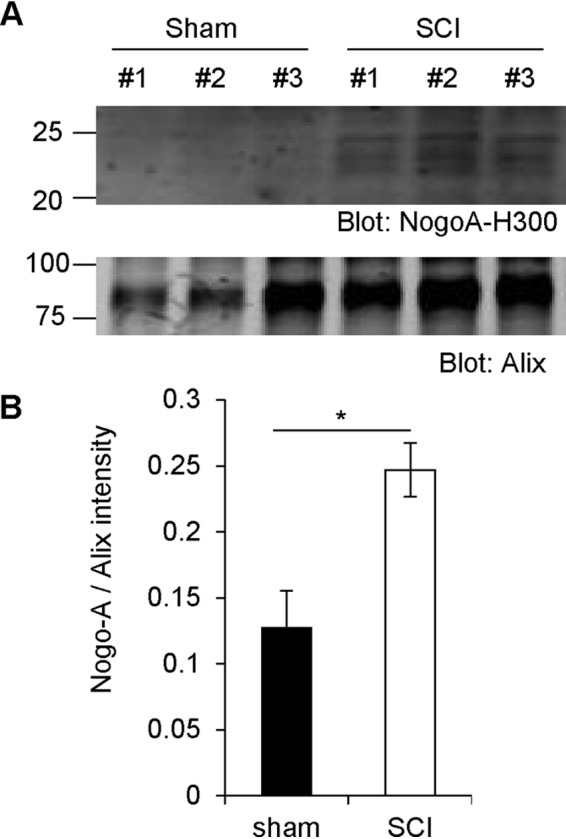
Increased Nogo-A fragment levels after spinal cord trauma in vivo. A, WT mice had their spinal cord crushed and were sacrificed 3 days after surgery. The spinal cords were then taken out and homogenized in TBS. Exosome fractions were immunoblotted with anti-Nogo-A and anti-Alix antibodies. B, quantification of Nogo-A divided by Alix intensity. Mean ± S.E., n = 3 animals. *, p < 0.05; Student's two-tailed t test.
Discussion
The major finding of this study is that exosomes contain a proteolytic fragment of the MAI Nogo-A. A role of exosomes as diffusible inhibitors of axon regeneration after SCI is supported by several observations. A specific fragment of Nogo-A containing the three NgR1-interacting domains is present in exosomes, and the Nogo-66 domain is exposed to medium. Functional tests showed that exosomes containing the 24-kDa Nogo-A fragment are as active as the previously described NgR1 ligand Nogo22 in blocking axon regeneration in vitro. Furthermore, the levels of exosomal Nogo-immunoreactive 24-kDa protein are increased in the spinal cord after SCI in mice.
The exosome fraction is highly enriched in the 24-kDa Nogo-A fragment but not full-length Nogo-A. Thus, proteolysis of Nogo-A is invariably associated with secretion in exosomes. This may imply that production of Nogo-A–containing exosomes requires passage of the protein through a subcellular compartment in which this cleavage is robust, such as the endo-lysosome pathway. It is also possible that this proteolytic processing plays an instructive role in targeting Nogo-A to the exosomal pathway.
The BACE1 enzyme is required for Nogo-A cleavage to the exosomal 24-kDa fragment. There are data demonstrating a direct interaction of BACE1 with reticulon family members, most prominently Reticulon3 (RTN3) and, to a lesser extent, Nogo-A (RTN4) (29–31). In this context, the focus has been on reticulon inhibition of BACE1 activity with respect to amyloid precursor protein rather than reticulons serving as BACE1 substrates or participating in exosomal processing. Our data show that BACE1 inhibitors reduce the release of potent Nogo-A fragments into exosomes. Administration of BACE1 inhibitors is therefore predicted to shift the balance of Nogo-A inhibition from membrane-associated to secreted exosomes. Therefore, BACE1 inhibition might alter the regional distribution of Nogo-A action through NgR1 on neurons.
The exosome fraction contains all of the NgR1-interacting domains of Nogo-A that have been linked previously in Nogo22 (11). However, localization to the surface of exosomes appears to further enhance the potency of NgR1-mediated axon growth inhibition. In this context, the increase in Nogo-immunoreactive 24-kDa protein after SCI implies greater Nogo-A-related inhibitory activity post-trauma. Further studies will be required to define the mechanism of this increase and the regional distribution of exosomal Nogo-A in the injured nervous system.
Overall, the existence of the proteolyzed exosomal Nogo-A fragment broadens the action of the MAI Nogo-A from cellular membrane–associated to secreted exosomes. Further, BACE1 activity may titrate axonal growth inhibition after CNS trauma independent of amyloid precursor protein.
Experimental procedures
Cell lines and primary cortical neuron culture
HEK293T cells were maintained in DMEM containing 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Mouse E17 cortical neurons were dissected in ice-cold Hibernate E medium (catalog no. HE-Ca, BrainBits) and incubated in digestion Hank's Balanced Salt Solution medium containing 30 units/ml papain (catalog no. LS003127, Worthington Biochemical), 1.5 mm CaCl2, 2.5 mm EDTA, and 2 mg/ml DNaseI (catalog no. DN25, Sigma) at 37 °C for 20 min. Digested tissues were triturated and suspended in Neurobasal A medium supplemented with B-27, GlutaMAX, and penicillin–streptomycin (all from Invitrogen). Neurons were plated on tissue culture plates coated with poly-d-lysine.
Expression plasmids, antibodies, and reagents
The C-terminally Myc-tagged WT human Nogo-A plasmid has been described previously (1) and was used to generate mutant constructs by PCR methods using KOD Hot start DNA polymerase (TOYOBO) and sequencing. Anti-Myc (M4439 or C3956) and anti-β-actin (A1978) antibodies (Sigma); anti-βIII-tubulin antibody (G7121, Promega); anti-Alix (2171), CD9 (13174) and Cathepsin D (2284) antibodies (Cell Signaling Technology); anti-Nogo-A-H300 antibody (sc-25660) (Santa Cruz Biotechnology); and anti-Nogo-A-M (AF3515) and anti-Nogo-A-N (AF3098) antibodies (R&D Systems) were used for the following experiments. Rabbit polyclonal antibody against Nogo22 was generated using purified Nogo22 as an antigen (Covance). Nogo22 protein has been described previously (11). Maleimide–PEG11–Biotin was purchased from Thermo Scientific. MG101 (R&D Systems), Z-VAD-FMK (Promega), E64d (Santa Cruz Biotechnology), and β-secretase inhibitor IV (Merck) were used for inhibitor experiments.
Transfection and transduction
HEK293T cells were transfected with Myc-tagged or untagged Nogo-A plasmids using Lipofectamine 2000 (Invitrogen) and siRNA Non-Targeting Control (sense, rCrGrUrUrArArUrCrGrCrGrUrArUrArArUrArCrGrCrUrUAT; antisense, rArUrArCrGrCrGrUrArUrUrArUrArCrGrCrGrArUrUrArArCrGrArC) or targeting human BACE1 (#1 sense, rCrUrArArCrArUrUrArCrUrGrCrCrUrUrCrArGrUrArUrCAA; antisense, rGrArGrArUrUrGrUrArArUrGrArCrGrGrArArGrUrCrArUrArGrUrU; #2 sense, rArGrCrArUrGrArUrCrArUrUrGrGrArGrGrUrArUrCrGrACC; antisense, rCrCrUrCrGrUrArCrUrArGrUrArArCrCrUrCrCrArUrArGrCrUrGrG), and human Cathepsin D (#1 sense, rArGrUrArUrUrArCrArArGrGrGrUrUrCrUrCrUrGrUrCrCTA; antisense, rUrArGrGrArCrArGrArGrArArCrCrCrUrUrGrUrArArUrArCrUrUrG; #2 sense, rGrUrUrUrGrArCrCrGrUrGrArCrArArCrArArCrArGrGrGTG; antisense, rCrArCrCrCrUrGrUrUrGrUrUrGrUrCrArCrGrGrUrCrArArArCrArC; #3 sense, rArUrUrCrArGrGrGrCrGrArGrUrArCrArUrrGrArUrCrCrCCT; antisense, rArGrGrGrGrArUrCrArUrGrUrArCrUrCrGrCrCrCrUrGrArArUrCrA) using Lipofectamine RNAiMAX (Invitrogen).
Immunoprecipitation and immunoblotting
HEK293T cells were lysed with RIPA buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X-100) and centrifuged at 20,000 × g for 20 min at 4 °C. An antibody and protein G–Sepharose mixture was added to supernatants or lysates and incubated for 2 h at 4 °C with gentle rotation. The beads were washed three times, and the immune complexes were then resolved by SDS-PAGE. After transfer, nitrocellulose membranes were incubated in blocking buffer (Blocking Buffer for Fluorescent Western Blotting, Rockland, MB-070-010) for 1 h at room temperature and immunoblotted with the appropriate primary antibodies. Following primary antibody incubation, secondary antibodies (Odyssey IRDye 680 or 800) were applied for 1 h at room temperature. Membranes were then washed and visualized using the LI-COR Odyssey IR imaging system.
Exosome purification and sucrose density gradient experiment
Exosomes from culture supernatant of HEK293T cells were prepared by differential centrifugation. HEK293Tcells were transfected with vector or Nogo-A–Myc (3 μg) for 24 h, and then the medium was changed to Neurobasal A and cultured for another 24 h. The culture medium was centrifuged at 2000 × g for 10 min to remove the large cell debris and dead cells and at 20,000 × g for 20 min to eliminate small debris. Then exosomes were pelleted by ultracentrifugation at 120,000 × g for 70 min. All procedures were performed at 4 °C. Exosome pellets were resuspended in 100 μl of PBS, protein concentration was measured with a Bradford assay for cortical axon regeneration, and then the exosome pellets were resuspended in 10 μl of Laemmli buffer for immunoblot analysis.
For further separation by differential sedimentation velocity, the culture supernatant of Nogo-A–overexpressing HEK293T cells was placed on top of the sucrose gradient (0.25–2.0 M sucrose, PBS) and ultracentrifuged at 140,000 × g for 16 h. Gradient fractions (500 μl) were collected from the top and analyzed by immunoblot analysis.
Transmission EM of exosomes
Exosomes from HEK293T cell culture supernatant were isolated by ultracentrifugation as described above. The exosome pellet was resuspended in 10 μl of PBS and submitted to the Yale University Center for Cellular and Molecular Imaging Electron Microscopy Facility. 5 μl of exosome solution was placed on freshly glow-discharged copper grids (carbon-coated, 300 mesh, Electron Microscopy Services). Following 2 min of incubation, the grids were rinsed twice on deionized water droplets and then stained for 2 min with 2% aqueous uranyl acetate. The excess staining solution was blotted off with Whatman #1 filter paper, and the grids were allowed to air-dry for 20 min. Samples were examined using a FEI Tecnai TF20 transmission electron microscope operated at 200 kV of accelerating voltage. Images were acquired using an AMT NanoSprint 1200 CMOS camera. Particle diameter was measured using ImageJ, and size distribution was represented as the percentage of the total number of particles analyzed.
Maleimide–PEG11–biotin labeling
Exosomes prepared from HEK293T cells were resuspended in PBS or RIPA buffer and incubated with membrane nonpermeable maleimide–PEG11–biotin for 2 h. Then DTT was added to terminate the reaction, and exosomes were lysed with RIPA buffer for immunoprecipitation with anti-Myc antibody. The immune complexes were washed three times, resolved by SDS-PAGE, and blotted with anti-Myc antibody and Alexa Fluor 488 streptavidin (Thermo Fisher Scientific),
RT-PCR and quantitative PCR
Total RNA was prepared using TRIzol (Sigma) and subjected to RT-PCR using M-MuLV reverse transcriptase (New England Biolabs). Complementary DNA was used for real-time quantitative PCR with iQ Supermix (Bio-Rad) and a TaqMan gene expression assay (Hs06633371 for huBACE1 and Hs02758991 for hGapdh from Applied Biosystems) on a Bio-Rad CFX Connect real-time PCR detection system using standard cycles. Each sample was analyzed in triplicate.
Cortical axon regeneration assay
The Ngr1−/− mouse line has been described previously (32, 33) and was backcrossed for more than 10 generations to C57BL/6 WT (Ngr1+/+) mice. Primary neuron cultures were obtained from these mice. The regeneration assay was performed as described previously (34). Primary cortical cultures were established from E17 C57BL/6 mice. Digested cells were plated on 96-well poly-d-lysine–coated plates at a density of 25,000 cells/well in 200 μl of plating medium. After 8 DIV, 96-well cultures were scraped using a custom-fabricated 96-pin array, and the indicated amount of Nogo22 or exosomes was added. Neurons were allowed to regenerate for another 72 h before fixation with 4% paraformaldehyde. Regenerating axons in the scrape zone were visualized using an antibody against βIII-tubulin (1:2000). Growth cones were visualized by staining for F-actin using rhodamine-conjugated phalloidin (1:2000, R415, Life Technologies). Images were taken on a ×10 objective in an automated high-throughput imager (ImageXpress Micro XLS, Molecular Devices) under identical conditions. Regeneration zone identification, image thresholding, skeletonizing, and quantitation were performed in ImageJ to score the extent of axon regeneration. Measurements from different wells for the same condition in any one experiment were averaged together for one n value, and statistical differences were calculated between cultures from n embryos.
Spinal cord injury
WT female mice were subjected to spinal cord crush surgery as described previously (16, 35). Animals received a subcutaneous injection of Buprenex (0.01 mg/kg) 30 min before surgery and were deeply anesthetized with ketamine (100 mg/kg) and xylazine (15 mg/kg). To expose the dorsal spinal cord at T7 and T8, a laminectomy was performed. Then the spinal cord was fully crushed with forceps for 3 s. Forceps (Dumont no. 5) had been filed to a width of 0.2 mm at the tips. The tips were inserted to include the whole spinal cord across the ventral bone to avoid any spare tissue ventrally and laterally. Sham animals were laminectomized but not crushed. Muscle and skin overlying the lesion were sutured. Animals received subcutaneous injections of 100 mg/kg ampicillin and 0.1 mg/kg Buprenex twice a day for the first 2 days after surgery. Procedures and postoperative care were performed in accordance with the guidelines of the Institutional Animal Use and Care Committee at Yale University. Three days after injury, animals were sacrificed, spinal cords were homogenized with TBS, and exosomes were fractionated.
Statistics
Statistical comparisons included one-way ANOVA and Student's t test as specified in the figure legends using Excel and Prism software. Statistical significance was set at p < 0.05. All data are mean ± S.E. No statistical methods were used to calculate sample size estimates.
Author contributions
Y. S. and S. M. S. conceptualization; Y. S. and S. M. S. data curation; Y. S., J. A. L., and S. M. S. formal analysis; Y. S. and J. A. L. investigation; Y. S. and J. A. L. methodology; Y. S. and S. M. S. writing-original draft; Y. S., J. A. L., and S. M. S. writing-review and editing; S. M. S. supervision; S. M. S. funding acquisition; S. M. S. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Xinran Liu (Yale University Center for Cellular and Molecular Imaging Electron Microscopy Facility) for sample preparation and image acquisition.
This work was supported by NINDS, National Institutes of Health Grant R35NS097283 and a Falk Medical Research Trust Transformative Research Award (to S. M. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.M.S. is a cofounder of ReNetX Bio, which seeks to develop NgR1-based therapeutics.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S3.
- SCI
- spinal cord injury
- MAI
- myelin-associated inhibitor
- CNS
- central nervous system
- aa
- amino acid(s)
- RIPA
- radioimmune precipitation assay
- CTSD
- Cathepsin D
- TBS
- Tris-buffered saline
- ANOVA
- analysis of variance
- DIV
- days in vitro
- Z-VAD
- fmk, benzyloxycarbonyl-VAD-fluoromethyl ketone.
References
- 1. GrandPré T., Nakamura F., Vartanian T., and Strittmatter S. M. (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444 10.1038/35000226 [DOI] [PubMed] [Google Scholar]
- 2. Chen M. S., Huber A. B., van der Haar M. E., Frank M., Schnell L., Spillmann A. A., Christ F., and Schwab M. E. (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439 10.1038/35000219 [DOI] [PubMed] [Google Scholar]
- 3. Mukhopadhyay G., Doherty P., Walsh F. S., Crocker P. R., and Filbin M. T. (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13, 757–767 10.1016/0896-6273(94)90042-6 [DOI] [PubMed] [Google Scholar]
- 4. McKerracher L., David S., Jackson D. L., Kottis V., Dunn R. J., and Braun P. E. (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13, 805–811 10.1016/0896-6273(94)90247-X [DOI] [PubMed] [Google Scholar]
- 5. Kottis V., Thibault P., Mikol D., Xiao Z. C., Zhang R., Dergham P., and Braun P. E. (2002) Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J. Neurochem. 82, 1566–1569 10.1046/j.1471-4159.2002.01146.x [DOI] [PubMed] [Google Scholar]
- 6. Fournier A. E., GrandPre T., and Strittmatter S. M. (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409, 341–346 10.1038/35053072 [DOI] [PubMed] [Google Scholar]
- 7. Liu B. P., Fournier A., GrandPré T., and Strittmatter S. M. (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297, 1190–1193 10.1126/science.1073031 [DOI] [PubMed] [Google Scholar]
- 8. Wang K. C., Koprivica V., Kim J. A., Sivasankaran R., Guo Y., Neve R. L., and He Z. (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417, 941–944 10.1038/nature00867 [DOI] [PubMed] [Google Scholar]
- 9. Laurén J., Hu F., Chin J., Liao J., Airaksinen M. S., and Strittmatter S. M. (2007) Characterization of myelin ligand complexes with neuronal Nogo-66 receptor family members. J. Biol. Chem. 282, 5715–5725 10.1074/jbc.M609797200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atwal J. K., Pinkston-Gosse J., Syken J., Stawicki S., Wu Y., Shatz C., and Tessier-Lavigne M. (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322, 967–970 10.1126/science.1161151 [DOI] [PubMed] [Google Scholar]
- 11. Huebner E. A., Kim B. G., Duffy P. J., Brown R. H., and Strittmatter S. M. (2011) A multi-domain fragment of Nogo-A protein is a potent inhibitor of cortical axon regeneration via Nogo receptor 1. J. Biol. Chem. 286, 18026–18036 10.1074/jbc.M110.208108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huber A. B., Weinmann O., Brösamle C., Oertle T., and Schwab M. E. (2002) Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 22, 3553–3567 10.1523/JNEUROSCI.22-09-03553.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X., Chun S. J., Treloar H., Vartanian T., Greer C. A., and Strittmatter S. M. (2002) Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J. Neurosci. 22, 5505–5515 10.1523/JNEUROSCI.22-13-05505.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu F., and Strittmatter S. M. (2008) The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J. Neurosci. 28, 1262–1269 10.1523/JNEUROSCI.1068-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kempf A., Tews B., Arzt M. E., Weinmann O., Obermair F. J., Pernet V., Zagrebelsky M., Delekate A., Iobbi C., Zemmar A., Ristic Z., Gullo M., Spies P., Dodd D., Gygax D., et al. (2014) The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS Biol. 12, e1001763 10.1371/journal.pbio.1001763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sekine Y., Siegel C. S., Sekine-Konno T., Cafferty W. B. J., and Strittmatter S. M. (2018) The nociceptin receptor inhibits axonal regeneration and recovery from spinal cord injury. Sci. Signal. 11, eaao4180 10.1126/scisignal.aao4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snow D. M., Lemmon V., Carrino D. A., Caplan A. I., and Silver J. (1990) Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp. Neurol. 109, 111–130 10.1016/S0014-4886(05)80013-5 [DOI] [PubMed] [Google Scholar]
- 18. Shen Y., Tenney A. P., Busch S. A., Horn K. P., Cuascut F. X., Liu K., He Z., Silver J., and Flanagan J. G. (2009) PTPσ is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326, 592–596 10.1126/science.1178310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreau-Fauvarque C., Kumanogoh A., Camand E., Jaillard C., Barbin G., Boquet I., Love C., Jones E. Y., Kikutani H., Lubetzki C., Dusart I., and Chédotal A. (2003) The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J. Neurosci. 23, 9229–9239 10.1523/JNEUROSCI.23-27-09229.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Basile J. R., Holmbeck K., Bugge T. H., and Gutkind J. S. (2007) MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J. Biol. Chem. 282, 6899–6905 10.1074/jbc.M609570200 [DOI] [PubMed] [Google Scholar]
- 21. Raissi A. J., Staudenmaier E. K., David S., Hu L., and Paradis S. (2013) Sema4D localizes to synapses and regulates GABAergic synapse development as a membrane-bound molecule in the mammalian hippocampus. Mol. Cell Neurosci. 57, 23–32 10.1016/j.mcn.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fröhlich D., Kuo W. P., Frühbeis C., Sun J. J., Zehendner C. M., Luhmann H. J., Pinto S., Toedling J., Trotter J., and Kramer-Albers E. M. (2014) Multifaceted effects of oligodendroglial exosomes on neurons: effect on neuronal firing rate, signal transduction and gene regulation. Philos. Trans. R Soc. Lond. B Biol. Sci. 369, 20130510 10.1098/rstb.2013.0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakhti M., Winter C., and Simons M. (2011) Inhibition of myelin membrane sheath formation by oligodendrocyte-derived exosome-like vesicles. J. Biol. Chem. 286, 787–796 10.1074/jbc.M110.190009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frühbeis C., Fröhlich D., Kuo W. P., Amphornrat J., Thilemann S., Saab A. S., Kirchhoff F., Möbius W., Goebbels S., Nave K. A., Schneider A., Simons M., Klugmann M., Trotter J., and Krämer-Albers E. M. (2013) Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11, e1001604 10.1371/journal.pbio.1001604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lopez-Verrilli M. A., Picou F., and Court F. A. (2013) Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia 61, 1795–1806 10.1002/glia.22558 [DOI] [PubMed] [Google Scholar]
- 26. Raposo G., and Stoorvogel W. (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383 10.1083/jcb.201211138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teng F. Y., and Tang B. L. (2008) Cell autonomous function of Nogo and reticulons: the emerging story at the endoplasmic reticulum. J. Cell Physiol. 216, 303–308 10.1002/jcp.21434 [DOI] [PubMed] [Google Scholar]
- 28. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., and Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 10.1016/j.cell.2005.11.047 [DOI] [PubMed] [Google Scholar]
- 29. He W., Lu Y., Qahwash I., Hu X. Y., Chang A., and Yan R. (2004) Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat. Med. 10, 959–965 10.1038/nm1088 [DOI] [PubMed] [Google Scholar]
- 30. He W., Hu X., Shi Q., Zhou X., Lu Y., Fisher C., and Yan R. (2006) Mapping of interaction domains mediating binding between BACE1 and RTN/Nogo proteins. J. Mol. Biol. 363, 625–634 10.1016/j.jmb.2006.07.094 [DOI] [PubMed] [Google Scholar]
- 31. Sharoar M. G., and Yan R. (2017) Effects of altered RTN3 expression on BACE1 activity and Alzheimer's neuritic plaques. Rev. Neurosci. 28, 145–154 [DOI] [PubMed] [Google Scholar]
- 32. Kim J. E., Liu B. P., Park J. H., and Strittmatter S. M. (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44, 439–451 10.1016/j.neuron.2004.10.015 [DOI] [PubMed] [Google Scholar]
- 33. Sekine Y., Lin-Moore A., Chenette D. M., Wang X., Jiang Z., Cafferty W. B., Hammarlund M., and Strittmatter S. M. (2018) Functional genome-wide screen identifies pathways restricting central nervous system axonal regeneration. Cell Rep. 23, 415–428 10.1016/j.celrep.2018.03.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sekine Y., Algarate P. T., Cafferty W. B. J., and Strittmatter S. M. (2019) Plexina2 and CRMP2 signaling complex is activated by Nogo-A-liganded Ngr1 to restrict corticospinal axon sprouting after trauma. J. Neurosci. 39, 3204–3216 10.1523/JNEUROSCI.2996-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zukor K., Belin S., Wang C., Keelan N., Wang X., and He Z. (2013) Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J. Neurosci. 33, 15350–15361 10.1523/JNEUROSCI.2510-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.