

Abstract
NADH-quinone oxidoreductase (complex I) couples electron transfer from NADH to quinone with proton translocation across the membrane. Quinone reduction is a key step for energy transmission from the site of quinone reduction to the remotely located proton-pumping machinery of the enzyme. Although structural biology studies have proposed the existence of a long and narrow quinone-access channel, the physiological relevance of this channel remains debatable. We investigated here whether complex I in bovine heart submitochondrial particles (SMPs) can catalytically reduce a series of oversized ubiquinones (OS-UQs), which are highly unlikely to transit the narrow channel because their side chain includes a bulky “block” that is ∼13 Å across. We found that some OS-UQs function as efficient electron acceptors from complex I, accepting electrons with an efficiency comparable with ubiquinone-2. The catalytic reduction and proton translocation coupled with this reduction were completely inhibited by different quinone-site inhibitors, indicating that the reduction of OS-UQs takes place at the physiological reaction site for ubiquinone. Notably, the proton-translocating efficiencies of OS-UQs significantly varied depending on their side-chain structures, suggesting that the reaction characteristics of OS-UQs affect the predicted structural changes of the quinone reaction site required for triggering proton translocation. These results are difficult to reconcile with the current channel model; rather, the access path for ubiquinone may be open to allow OS-UQs to access the reaction site. Nevertheless, contrary to the observations in SMPs, OS-UQs were not catalytically reduced by isolated complex I reconstituted into liposomes. We discuss possible reasons for these contradictory results.
Keywords: bioenergetics, mitochondria, complex I, ubiquinone, chemical biology, respiratory chain, proton pump
Introduction
Proton-translocating NADH-quinone oxidoreductase (complex I),2 which is the largest of the respiratory chain enzymes, couples electron transfer from NADH to quinone with the translocation of protons across the membrane. Energy-consuming reactions, such as ATP synthesis via oxidative phosphorylation and substrate transport, are driven by the electrochemical proton gradient produced by the enzyme (1–4). Although complex I has long been a “black box,” the recent rapid advances in X-ray crystallographic and single-particle cryo-EM studies (5–12) along with computational simulations (13–15) provided invaluable knowledge of the structure and functions of the enzyme. These outcomes have led to the consensus that structural and electrostatic rearrangements induced by quinone reduction, which takes place at the interface between the hydrophilic and membrane domains, transmit to the membrane domain through the link continuing over the domain as the central axis of potentially ionized or protonated residues to trigger proton translocation (four protons for each oxidation of NADH). Thus, the quinone reduction is a key part in the energy conversion processes; however, the mechanism responsible remains elusive.
Based on structural biology studies (5–12), it is considered that ubiquinones (UQs) of varying isoprenyl chain length (UQ1–UQ10) enter and transit the quinone-access channel to be reduced at the “top” of the channel. This channel extends from the membrane interior to the Fe-S cluster N2 (∼30 Å long) and is a completely enclosed tunnel with a narrow entry point (∼3 × 5Å diameter) that is framed by transmembrane helix 1 (TMH1), TMH6, and amphipathic α-helix 1 from the ND1 subunit (in the bovine enzyme) and TMH1 from the ND3 subunit. Because the planar UQ ring is wider (∼6 Å across) than the diameter of the entry point, the channel has been postulated to undergo structural rearrangement to allow UQs to move into and out of the channel (5, 11). The so-called quinone-site inhibitors, such as piericidin A and rotenone, are supposed to enter the channel's interior and block UQ's path (5, 6, 16). This proposal was originally derived from the electron density attributed to bound piericidin A, which was observed to overlay the electron density attributed to bound n-decyl benzoquinone (DB) in crystallographic maps from Thermus thermophilus complex I (5). However, it should be noted that these scenarios for the binding of UQs or inhibitors have yet to be confirmed experimentally.
To characterize structural features of the binding pocket for UQ or quinone-site inhibitors, we previously performed two varieties of chemical biology experiments using bovine heart submitochondrial particles (SMPs) (17). First, we synthesized unique UQs that are oversized (SF-UQs) or lipid-like (PC-UQs) (e.g. SF-UQ6 and PC-UQ4 in Fig. S1), which are unlikely to enter and transit the long narrow channel due to extensive physical restrictions. We examined whether complex I can catalyze the reduction of these quinones. As a result, SF-UQs and PC-UQs were catalytically reduced at the physiological reaction site for UQ, albeit at low rates of reaction. We note that a bulky tritylQ10 (Fig. S1), which is a neutral analog of MitoQ10 and is also unlikely to transit the channel, can be reduced by complex I in bovine heart mitochondria at a slower rate than that of UQ4 (18). Second, photoaffinity-labeling experiments using four amiloride-type inhibitors showed that they bind to the interfacial domain of multiple core subunits (49 kDa, ND1, and PSST) and the 39-kDa supernumerary subunit (17), although the latter does not make up the channel cavity in the current channel models. The binding of these amilorides to the multiple subunits was significantly suppressed by excess short-chain UQs or different types of inhibitors. Based on comprehensive interpretations of these results and previous findings (19–22), we questioned whether the current quinone/inhibitor-access channel models fully reflect physiologically relevant states.
The previous findings above provided new insights into the binding site for UQ or quinone-site inhibitors in complex I. However, to further advance our understanding of the coupling mechanism of the UQ reduction and proton translocation, there are some significant points to be improved to promote the use of these oversized UQs as quinone substrates (17). Taking the example of SF-UQ6 (the bulkiest substrate among the oversized UQs), the electron transfer activity of this quinone in SMPs was very poor (being comparable with UQ4) because of its extremely low solubility in water, thereby preventing homogeneous dissolving in the assay medium and, consequently, sufficient partitioning into SMPs (18, 23, 24). The low solubility of hydrophobic UQs in water is an experimental drawback in this type of assay (18, 25). For this reason, UQ1 and UQ2, but not UQ3 and UQ4, have been widely used as quinone substrates of various respiratory enzymes in in vitro assays. To further improve the reliability of analyzing the electron transfer activities of oversized UQs, their activities need to be increased by enhancing solubility in water as far as SMPs are used, while maintaining satisfactory bulkiness of the side chain. Alternatively, we should make use of another appropriate assay system that can evaluate the electron transfer activity of extremely hydrophobic UQs, as described hereafter.
Regarding the molecular solubility in water, we introduced a weakly acidic phenol part (pKa ≒ ∼8.0, Fig. S1) into the side chain of SF-UQ6 (also other oversized UQs) as the “block” to enhance its solubility in water as much as possible. This substructure has an amphiphilic character (being soluble both in water and hydrophobic medium) because a negative charge in the anionic form is delocalized over the long electron-conjugation system. However, SF-UQ6 functioned unexpectedly as a protonophoric uncoupler in the lipid membrane of SMPs despite its large molecular size relative to ordinary protonophores such as carbonyl cyanide p-trifluoromethoxyphenylhydrazone and SF6847 (17). The uncoupling activity hampered the measurement of membrane potential formed by proton translocation coupled with the SF-UQ6 reduction; therefore, we were unable to examine whether the proton-translocating efficiency of this extremely bulky quinone is identical to that of natural-type UQs such as UQ1 and UQ2. This assessment is necessary to elucidate how the UQ reduction is connected to the structural changes of its binding pocket, which is predicted to trigger proton translocation.
To improve the function of oversized UQs as quinone substrates, we synthesized a series of new oversized UQs (OS-UQ1−OS-UQ8, Fig. 1) through extensive modification of the block's structure by trial and error. Design concepts for OS-UQ1−OS-UQ5 and OS-UQ6−OS-UQ8 are different due to varying experimental purposes, and they are described at the beginning and in the latter part of “Results,” respectively. Any acid-dissociative substructure that has a pKa value in a physiological pH range was excluded as a candidate of the block for the reason mentioned above. We investigated the electron transfer activities of these OS-UQs and the proton-translocating efficiencies coupled with their reduction not only in bovine heart SMPs but also in complex I–reconstituted proteoliposomes (PLs), which is an excellent assay system for measuring the electron transfer activity of extremely hydrophobic UQs such as UQ8 and UQ10 (16, 25). The results obtained in SMPs not only strongly corroborated our previous proposition (the access path for UQ may be “open” to allow oversized UQs to access the reaction site (17)) but also revealed that the proton-translocating efficiencies coupled with reduction of OS-UQs significantly vary depending on the manner of their reaction, reflecting different side chain structures. Nevertheless, in contrast to the results obtained in SMPs, OS-UQs were not catalytically reduced by isolated complex I reconstituted into liposomes. We will discuss possible causes of the contradictory results.
Figure 1.

Structures of oversized ubiquinones (OS-UQ1−OS-UQ8) studied in this study. A stable conformation of OS-UQ2 is shown by a space-filling model (oxygen atoms are in red). The lengths of the models indicate the distances between the two corresponding (C)-H atoms. The structure of UQ10 is also shown as a reference, where a side-chain moiety located inside the predicted channel cavity is indicated (16).
Results
Design and syntheses of new oversized UQs
We synthesized a series of new oversized UQs (OS-UQ1−OS-UQ8, Fig. 1) by the procedures described in the supporting information (Schemes S1–S5). In these UQs, 1-methoxy-2,6-di(3-methoxy-3-methyl-1-butynyl)benzene was attached to a terminal of the side chain as the block to prevent the quinone ring from accessing the reaction site located deep in the channel. The diameter of this rigid block (∼4 × 13 Å) is much wider than the diameter of the channel of bovine complex I (the entry point is ∼3 × 5 Å) (7). To produce oversized UQs that have both satisfactory bulkiness and efficient electron transfer activity, we synthesized numerous UQs and, consequently, chose the block through trial-and-error tests in SMPs. On considering the current channel model (∼30 Å long), the whole molecule of these OS-UQs needs to transit to the top of the channel to be reduced. Then, even though the quinone head-ring of OS-UQs enters the channel entrance, the ring is highly unlikely to reach the deep reaction site because of crucial steric hindrance arising from the block at the entrance and/or somewhere along the narrow channel. We will examine this point by atomistic molecular dynamics (MD) simulations in the next section.
We previously proposed that there is an “open” access path for oversized UQs to the reaction site, which is not identical to the currently predicted channel cavity (17). In either case, it is clear that reduction of the quinone ring takes place near the Fe-S cluster N2, a direct electron donor to UQ (5–11). To deduce the length of the access path (the depth of the quinone reaction site from the enzyme surface), we changed the length of the spacer linking the quinone ring and the block (Fig. 1). Unless the reaction site is exposed to the enzyme surface, there should be a threshold length of the spacer that prevents the quinone ring from accessing the reduction site due to steric hindrance caused by the block.
Modeling and simulations of UQ binding to complex I
We modeled OS-UQ2 and OS-UQ3 in the reaction site near the Fe-S cluster N2 of bovine complex I based on an earlier MD simulation study (26). Modeling and simulations showed that both UQs are fully buried in the site (Fig. 2A and Fig. S2, respectively), and their bulky substituted-butynyl groups are partly stabilized in grooves, whereas their binding causes structural perturbations in protein backbone relative to the binding of natural UQ10. The areas of higher fluctuations comprise the loop connecting TMHs 5–6 of the ND1 subunit, the central loop of the PSST subunit, and a segment of 49-kDa subunit carrying Asp160 (Fig. 2B), all of which are critical for the catalytic reaction of the enzyme (5–7). Thus, the bulky UQs may perturb the key coupling elements of proton-pumping machinery if they bind at this reaction site in the channel.
Figure 2.

Modeling and simulations of UQ binding to bovine complex I. A, simulation snapshot of OS-UQ2 molecule at the reaction site near the Fe-S cluster N2. B, higher root mean square fluctuation of the backbone of central subunits ND1, 49 kDa, and PSST that occur upon binding of OS-UQ3 (relative to natural UQ10) at the reaction site is shown in red on selected segments surrounding the compound. Blue color corresponds to higher fluctuations in UQ10 simulation. It is noteworthy that many of these regions are critical for the proton-translocating function of complex I. C, steered MD simulation snapshot showing OS-UQ3 en route to the catalytic site with substituted-butynyl chains bending due to constriction in the cavity. Distortion in the rodlike substituted-butynyl tails is displayed in plots of C–C distance (d) and C-C-C angle (a) with respect to SMD simulation time (equilibrium values of d and a are 9.5 Å and 150°, respectively). In the panels, oxygen and carbon atoms are shown in red and cyan, respectively, and the subunits 49 kDa, PSST, ND1, and ND3 are shown in green, light blue, pink, and yellow, respectively.
To accept electrons from the N2 cluster to be reduced, OS-UQs would have to move in and out of the main channel during the catalytic reaction; however, the movement would not occur without significant structural perturbations. To test this, we performed steered MD simulations by pulling OS-UQ3 from the entry site of the channel to the reaction site near the N2 cluster. We found that during transition, the rodlike substituted-butynyl groups undergo marked deformation (bending) due to the significant steric hinderance caused by network of residues in the channel (Fig. 2C). Altogether, the MD simulations suggest that OS-UQs can occupy the reaction site near the N2 cluster but not without perturbing the critical elements of the proton-pumping machinery. Notably, their transition through the main narrow channel would be energetically much more difficult.
Electron transfer activity of OS-UQs in SMPs
The electron transfer activities of OS-UQs were measured in the NADH-quinone oxidoreductase assay in SMPs in the presence of antimycin A and KCN to inhibit complex III and IV activities, respectively. Because the electron transfer activities of all OS-UQs were saturated in a range of 10–30 μm, apparent maximum activities measured in this range are shown in Fig. 3 along with activities of UQ2–UQ4 for reference. The electron transfer activities of OS-UQs varied like a bell shape against the spacer length with OS-UQ2 as the apex. Notably, the activity of OS-UQ2 was highest among the oversized UQs studied so far (17) and comparable with that of UQ2. Hydrophobic OS-UQ4 and OS-UQ5 no longer functioned as a practical electron acceptor. The variation in the electron transfer activities of OS-UQ2−OS-UQ5 may be explained as follows: the solubility of these UQs in water decreases with an increasing spacer length and, hence, partitioning of the UQs into SMPs decreases, as typically seen for UQ2–UQ4 (activity order of UQ2 > UQ3 > UQ4).
Figure 3.
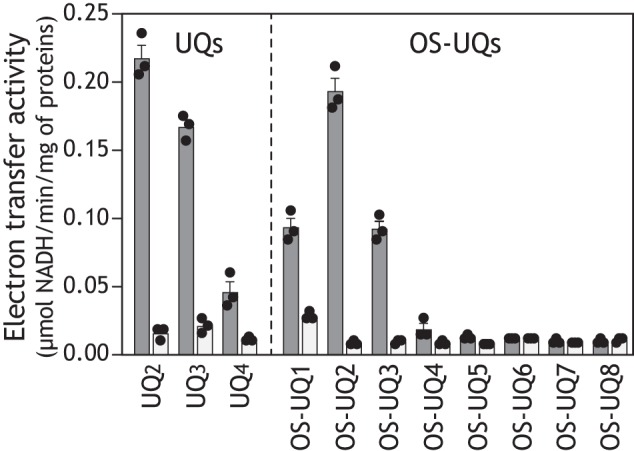
The electron transfer activity of OS-UQs in SMPs. The apparent maximum rates of NADH oxidation in the NADH-quinone oxidoreductase assay in the absence (black) or presence (gray) of bullatacin (50 nm) are shown. The apparent maximum activity of each UQ was determined in a concentration range of 10–30 μm. The rates for UQ2, UQ3, and UQ4 are shown as references. The concentrations of SMP proteins and NADH were set to 90 μg/ml and 100 μm, respectively. Values show means ± S.E. (error bars) (n = 3).
It is noteworthy that although the solubility of OS-UQ1 in water is superior to that of OS-UQ2, the electron transfer activity of the former was rather lower than that of the latter (Fig. 3). This reversal strongly suggests that access of the quinone ring of OS-UQ1 to the reaction site is markedly obstructed because its spacer is too short to avoid the steric hindrance arising from the block, as anticipated when we initially designed these OS-UQs. The electron transfer activities of OS-UQ1−OS-UQ5 were almost completely inhibited by different quinone-site inhibitors (piericidin A, rotenone, fenpyroximate, or bullatacin), and the residual activities were identical to that of UQ2, as shown in Fig. 3, taking bullatacin as an example (see Fig. S3 for other inhibitors).
Inhibition of the OS-UQ2 reduction by S1QEL2.3
Brand et al. (27) identified small chemicals S1QELs as specific suppressors of superoxide production from the quinone reaction site in mammalian complex I during reverse electron transfer, although the mechanism of their action in the enzyme was not disclosed. We recently demonstrated that S1QELs are inhibitors of complex I but do not occupy the binding pocket for ubiquinone or known quinone-site inhibitors; rather, they may indirectly modulate the ubiquinone redox reactions by inducing structural changes of the pocket through binding to the middle of the ND1 subunit (28). This indirect modulation of the ubiquinone redox reactions may be responsible for the direction-dependent (forward or reverse) and incomplete inhibition of electron transfer by S1QELs (28). Because the mode of action of S1QELs differs from other known inhibitors, they may be useful for characterizing the reaction manner of OS-UQs in complex I.
We examined the susceptibility of the catalytic reduction of OS-UQ2 to S1QEL2.3 (one of the S1QELs identified in Ref. 27; Fig. S1) in the NADH-quinone oxidoreduction assay, using UQ2 as a reference. The concentration of OS-UQ2 and UQ2 was set at 30 μm each. The dose-response curve for the inhibition of OS-UQ2 reduction was almost identical to that of UQ2 (Fig. 4). Substantial enzyme activity (∼60%) remained for both quinones even in the presence of excess S1QEL2.3. This incomplete inhibition is one of the unique inhibitory effects of S1QELs that is attributable to the indirect modulation of the quinone redox reactions (28). We initially considered that because a bulky OS-UQ2 is barely accommodated in the reaction pocket, the reduction of this quinone is more susceptibly disturbed by the structural changes induced via S1QEL2.3 binding to the ND1 subunit compared with UQ2; for example, the dose-response curve for OS-UQ2 may shift to a lower concentration range of S1QEL2.3 compared with that for UQ2, or the maximum inhibition of OS-UQ2 reduction may be greater than that of UQ2, or both. However, none of these was the case. The results strongly suggest that the reaction manners of the two quinones are not so different.
Figure 4.
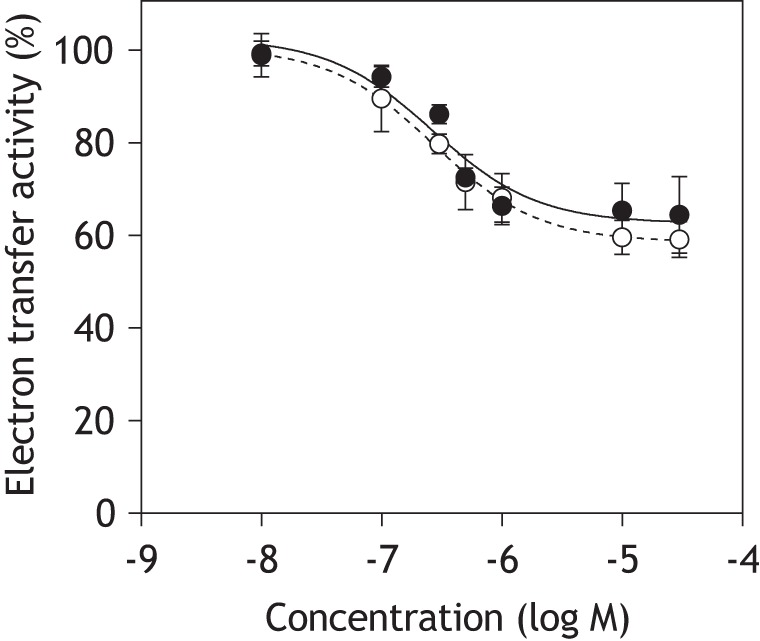
Dose-response curves for the inhibition of NADH-quinone oxidoreductase activity by S1QEL2.3. The reaction was initiated by adding NADH (100 μm) after incubation of SMPs with S1QEL2.3 and quinone (OS-UQ2 (open circles) or UQ2 (closed circles), 30 μm each) for 4 min at 30 °C. The final concentration of SMP protein was 90 μg/ml. Values show means ± S.D. (error bars) (n = 3).
Proton translocation coupled with the reduction of OS-UQs in SMPs
The marked susceptibility of the catalytic reduction of OS-UQ1−OS-UQ3 to quinone-site inhibitors (Fig. 3 and Fig. S3) suggests that their reduction takes place at the physiological reaction site for UQ. To verify this, we examined whether the reduction of OS-UQs generates a membrane potential in SMPs by monitoring changes in the absorbance of oxonol VI in the presence of nigericin (a K+/H+ exchanger). Although assessment of the proton translocation from a stoichiometric point of view is not the aim of this section, it should be noted that a considerable fraction of the protons pumped across the membrane are lost due to leakage in SMPs (29). As the proton leak significantly increases at a higher proton-motive force generated by respiratory enzymes (29–31), we determined membrane potential formation in a concentration range of UQs that led to relatively low electron transfer activities (i.e. low rates of NADH oxidation).
A membrane potential was not generated without externally added UQ (Fig. 5A). We confirmed, as a reference, that reduction of UQ2 forms a membrane potential, but not in the presence of bullatacin (Fig. 5B). The extents of membrane potential formation coupled with reduction of OS-UQ2 or OS-UQ3 were comparable with those of UQ2 when determined at similar rates of NADH oxidation (Fig. 5, D and E). No membrane potential was formed in the presence of bullatacin for the two quinones. Notably, a membrane potential formed by OS-UQ1 reduction was negligibly small as compared with the three quinones (Fig. 5C). OS-UQ1 scarcely generated a membrane potential even at the concentration (20 μm) exhibiting maximum electron transfer activity, as seen by a comparison with UQ2 and OS-UQ2 (20 μm each) (Fig. 5F). These results indicate that the reduction of the quinone ring and subsequent proton translocation is less efficiently coupled for OS-UQ1 relative to the three quinones. This feature may be associated with the “cramped” reaction of OS-UQ1 in the binding pocket mentioned above, as further discussed below.
Figure 5.

Membrane potential formation coupled with reduction of OS-UQs in SMPs. The membrane potential generated by NADH:quinone oxidoreduction was monitored by following changes in the absorbance of oxonol VI (601–630 nm). A, without externally added UQ; B, UQ2 (2.0 and 5.0 μm); C, OS-UQ1 (2.0 and 5.0 μm); D, OS-UQ2 (2.0 and 5.0 μm); E, OS-UQ3 (5.0 and 10 μm); F, UQ2, OS-UQ1, and OS-UQ2 (20 μm each). The gray and black traces represent the absorbance at low and high concentrations, respectively, of each quinone. Membrane potential was not formed in the presence of bullatacin (50 nm). In A–E, the traces of NADH oxidation determined at 340 nm are also presented. The SMP protein concentration was set to 90 μg/ml. The arrows indicate the addition of NADH (100 μm). Data are representative of three independent experiments.
Electron transfer activity of OS-UQs in complex I–reconstituted PLs
Hirst and colleagues (25) established an excellent PL system suitable for measuring the electron transfer activity of extremely hydrophobic UQs, such as UQ8 and UQ10, in the catalytic reaction by complex I. Considering the marked hydrophobicity of our OS-UQs (particularly OS-UQ4 and OS-UQ5), we should make use of this assay system for examining electron transfer activities of these UQs. Their PLs contain a hydrophobic UQ, complex I isolated from bovine heart mitochondria, and alternative quinol oxidase (AOX) isolated from Trypanosoma brucei (16, 25). As the catalytic activity of isolated complex I is significantly dependent on the lipid composition, the liposomes are composed of bovine heart phosphatidylcholine, phosphatidylethanolamine, and cardiolipin (a mass ratio of 8:1:1). AOX, an interfacial membrane protein, is reconstituted into the PLs to recycle reduced UQ (UQH2) to UQ. Because AOX is present in excess in this system, the complex I reaction is rate-determining, and the UQ/UQH2 pool is kept predominantly oxidized under steady-state catalysis.
Using a phospholipid mixture extracted from bovine heart mitochondria, we prepared PLs that contain isolated bovine heart complex I (32), T. brucei AOX (33), and the OS-UQ of choice. It may be noted that this phospholipid composition is not necessarily ideal for the AOX activity. The experimental procedure for preparing liposomes was slightly modified from the original work (25) to produce as many unilamella vesicles as possible (see “Experimental Procedures”).
We confirmed that UQ9 efficiently functions as an electron mediator between the reconstituted complex I and AOX by generating a membrane potential (Fig. 6, A and B, left). The electron transfer activity and membrane potential formation were completely blocked by a specific inhibitor of either enzyme (bullatacin or ascofuranone (33)) (Fig. 6, A and B). The rate of NADH oxidation with 9.2 ± 0.5 mm UQ9 was 5.1 ± 0.4 μmol·min−1/mg complex I (note that the rate with ∼10 mm UQ8 was ∼17 μmol·min−1/mg complex I (16)). In this and the following experiments, the concentration of UQ reconstituted into the liposomal membranes was estimated assuming that 1 nmol of UQ/mg of phospholipids is equivalent to 1 mm (25). The molar ratio of complex I to AOX was set to ∼1:60.
Figure 6.

The electron transfer activity of UQ in complex I/AOX-reconstituted PLs. A, electron transfer activity of UQ9 (left), OS-UQ5−OS-UQ7 (middle), or OS-UQ8 (right) in the PLs was determined by monitoring NADH oxidation in the absence (black lines) or presence (gray lines) of ascofuranone (100 nm; a specific inhibitor of AOX). The concentrations of UQ (mm), complex I (μg proteins μl−1 lipids), and AOX (μg proteins μl−1 lipids) in the PLs were as follows: UQ9-reconstituted PLs, 9.2 ± 0.5, 3.5 ± 0.3, and 9.4 ± 0.3; OS-UQ5–reconstituted PLs, 11.0 ± 0.2, 3.7 ± 0.1, and 10.3 ± 0.1; OS-UQ6–reconstituted PLs, 8.9 ± 0.5, 3.3 ± 0.6, and 11.2 ± 0.6; OS-UQ7–reconstituted PLs, 6.2 ± 0.2, 3.8 ± 0.5, and 9.8 ± 0.6; and OS-UQ8–reconstituted PLs, 4.8 ± 0.2, 3.7 ± 0.2, and 8.3 ± 0.2. The arrows indicate the addition of NADH (100 μm) or ascofuranone (100 nm). B, membrane potential formation coupled with reduction of the UQ tested in the PLs was monitored by following absorbance changes of oxonol VI in the absence (black lines) or presence (gray lines) of bullatacin (100 nm). The experimental conditions were the same as those in A. The arrows indicate the addition of NADH (100 μm) or uncoupler SF6847 (0.20 μm). Data are representative of three or four independent experiments.
We next examined the electron transfer activity of OS-UQ1−OS-UQ5 using the PLs. Relatively hydrophilic OS-UQ1−OS-UQ3 was externally added to the complex I/AOX–reconstituted PLs, whereas OS-UQ4 and OS-UQ5 were previously reconstituted into the PLs (∼20 mm). Unexpectedly, none of these OS-UQs functioned as an electron mediator between complex I and AOX, as shown in Fig. 6, A and B (middle panel), taking OS-UQ5 as an example. Given that OS-UQ1−OS-UQ3 functioned as an electron acceptor from complex I in SMPs (Fig. 3), this result suggests that once reduced, OS-UQs cannot be reoxidized by AOX, such as in the case where AOX is completely inhibited by ascofuranone (Fig. 6A, left). To check this possibility, we examined whether reduced forms of OS-UQ1−OS-UQ5 serve as substrates of isolated AOX in the solution assay, not in the PLs. As a result, they did not serve as efficient substrates of AOX (Fig. 7).
Figure 7.
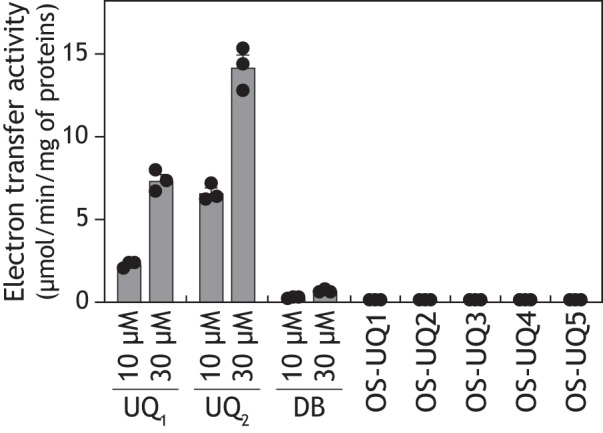
The electron-donating abilities of ubiquinols to isolated AOX. The electron-donating abilities of ubiquinols were examined using isolated T. brucei AOX by monitoring their oxidation (278 nm). The concentration of OS-UQ1−OS-UQ5 was set to 30 μm. The reaction conditions were as follows: 0.05% (w/v) C12E8, Tris-HCl (pH 7.4), and 2.5 μg of AOX/ml. Values in graphs are means ± S.E. (error bars) (n = 3).
Based on the structure of ubiquinol-bound AOX predicted from the inhibitor-bound state (33, 34), the ubiquinol oxidation site near a diiron active center is accessed from the membrane interior via a short cavity. Two (or three) isoprene units of ubiquinol may be accommodated in the cavity. In light of this model, the block in OS-UQ1−OS-UQ4 may hamper access of the quinol ring to the oxidation site. We are left, however, with the question of why OS-UQ5, which would be able to avoid the steric hindrance, did not serve as a substrate of AOX.
Electron transfer activity of isoprenyl derivatives (OS-UQ6-OS-UQ8) in complex I–reconstituted PLs
In connection with the question above, Čermáková et al. (35) reported that T. brucei AOX strictly recognizes the isoprene unit in the side chain of UQs; for example, the electron transfer activity of a reduced form of DB is much poorer than that of reduced UQ2 and UQ1 (activity order of UQ2H2 > UQ1H2 ≫ DBH2). We confirmed this tendency for the three quinols using our AOX preparation (Fig. 7). The electron-donating activities of these quinols were completely inhibited by ascofuranone (100 nm). Thus, to improve the ability of OS-UQs as a substrate of AOX, we synthesized isoprenyl derivatives OS-UQ6−OS-UQ8 (Fig. 1) and investigated their electron transfer activity in the PLs. Note that OS-UQ6−OS-UQ8 did not work as a practical electron acceptor of complex I in SMPs because of their extremely high hydrophobicities (Fig. 3). OS-UQ6 and OS-UQ7 did not function as an electron mediator between complex I and AOX (Fig. 6, A and B, middle panel), although they have a longer side chain than OS-UQ1−OS-UQ5. In contrast, OS-UQ8 worked as an efficient electron mediator by forming a membrane potential in an inhibitor-sensitive manner (Fig. 6, A and B, right). The rate of NADH oxidation with 4.8 ± 0.2 mm OS-UQ8 was 3.6 ± 0.2 μmol·min−1/mg complex I. It is, however, unclear solely from these results which enzyme (complex I or AOX (or both)) could not recognize OS-UQ6 and OS-UQ7 as quinone substrates.
Therefore, we adopted another approach to assess the electron-accepting ability of OS-UQ6 and OS-UQ7 from isolated complex I reconstituted into liposomes. Namely, we assessed the redox state of OS-UQ6 or OS-UQ7, which was reconstituted into the liposomal membrane with isolated complex I as above but without AOX, by monitoring its reduction by reverse-phase HPLC 5 min after the addition of NADH (1.3 mm) (36). If OS-UQ6 or OS-UQ7 is catalytically reduced by reconstituted complex I, a reduced form of it should be accumulated in the liposomal membrane. We confirmed, as a control, that a reduced form of UQ9 is produced under the experimental conditions, but not in the presence of bullatacin (Fig. 8A). Notably, no reduced form was detected for OS-UQ6 and OS-UQ7 (Fig. 8, B and C), whereas OS-UQ8 was catalytically reduced in an inhibitor-sensitive manner (Fig. 8D), consistent with the above results in the PLs (Fig. 6, A and B). We note that OS-UQ1−OS-UQ5 was not reduced under the same experimental conditions. Computational simulations of UQ10-bound bovine complex I predicted that seven or eight isoprene units of UQ10 enter the quinone-access channel (16). If so, the side chain of solely OS-UQ8 is long enough to avoid the steric hindrance because the block may be outside the channel cavity (Fig. 1). Taken together, all OS-UQs tested, except OS-UQ8, could not be catalytically reduced by the isolated complex I reconstituted into the liposomal membrane. These results contradict those obtained in SMPs.
Figure 8.

Catalytic reduction of UQs by complex I in the PLs. Catalytic reduction of different UQs (A, UQ9; B, OS-UQ6; C, OS-UQ7; D, OS-UQ8) by complex I reconstituted into liposomes was monitored by determining a reduced form of each UQ accumulated in the liposomal membrane by reverse-phase HPLC (see “Experimental procedures”). The concentrations of UQ (mm) and complex I (μg proteins μl−1 lipids) in the liposomal membrane were as follows: UQ9-reconstituted PLs, 9.7 ± 0.2 and 5.0 ± 0.1; OS-UQ6–reconstituted PLs, 8.9 ± 0.6 and 5.4 ± 0.3; OS-UQ7–reconstituted PLs, 9.9 ± 0.4 and 5.0 ± 0.5; OS-UQ8–reconstituted PLs, 9.0 ± 0.2 and 6.2 ± 0.3. The reaction was initiated by the addition of NADH (1.3 mm) and quenched by adding ethanol to the liposomal suspension at 5 min after the initiation. The top chromatogram in each panel represents the retention times of authentic reduced and oxidized forms of UQ tested. The middle and bottom chromatograms represent the analytical result in the absence and presence of bullatacin (5.0 μm), respectively. A mobile phase of the HPLC analysis for UQ9 and OS-UQ8 was a mixture of ethanol/methanol (3:2) containing 0.1% TFA. For OS-UQ6 and OS-UQ7, methanol containing 0.1% TFA was used as a mobile phase. Data are representative of two or three independent experiments.
Effects of UQs on the alkynylation of Asp160 in the 49-kDa subunit by AL1
To obtain a clue to the cause for the contradictory results between SMPs (native complex I) and PLs (isolated complex I), it may be useful to determine whether OS-UQs retain binding affinity to complex I in the PLs. Therefore, we examined the effects of OS-UQ2 and UQ2 (as a reference) on the pinpoint alkynylation of Asp160 in the 49-kDa subunit (49 kDa-Asp160) by an acetogenin-type ligand, AL1 (Fig. S4). We previously demonstrated that 49 kDa-Asp160 located deep inside the proposed ubiquinone-access channel (7, 9, 11) can be specifically alkynylated (Asp160(COO)-(CH2)2-C CH) via the so-called ligand-directed tosyl chemistry technique using AL1 in SMPs (19, 22). The pinpoint alkynylation can be achieved by incubating SMPs with AL1 for several hours and visualized by conjugating a fluorescent tag TAMRA-N3 to Asp160(COO)-(CH2)2-CCH via a Cu+-catalyzed click chemistry after solubilizing SMPs (Fig. S4). Because AL1 is an acetogenin-type inhibitor (IC50 value ∼3 nm (19)), the alkynylation was completely blocked by excess other quinone-site inhibitors (e.g. piericidin A, aminoquinazoline, bullatacin, and fenpyroximate) (19, 22).
CH) via the so-called ligand-directed tosyl chemistry technique using AL1 in SMPs (19, 22). The pinpoint alkynylation can be achieved by incubating SMPs with AL1 for several hours and visualized by conjugating a fluorescent tag TAMRA-N3 to Asp160(COO)-(CH2)2-CCH via a Cu+-catalyzed click chemistry after solubilizing SMPs (Fig. S4). Because AL1 is an acetogenin-type inhibitor (IC50 value ∼3 nm (19)), the alkynylation was completely blocked by excess other quinone-site inhibitors (e.g. piericidin A, aminoquinazoline, bullatacin, and fenpyroximate) (19, 22).
Before investigating with the PLs, we compared the effects of OS-UQ2 and UQ2 on the alkynylation in SMPs. SMPs (2.0 mg of proteins/ml) were incubated with AL1 (0.50 μm) for 12 h at 24 °C in the presence of different concentrations of OS-UQ2. OS-UQ2 suppressed the alkynylation in a concentration-dependent manner but did not achieve complete suppression up to 100 μm (Fig. 9A). This is probably because the binding affinity of AL1 to complex I is much higher than that of OS-UQ2 or because binding positions of the two ligands do not completely overlap, or both. We did not use concentrations of OS-UQ2 over 100 μm because high concentrations of hydrophobic chemicals can nonspecifically alter the enzyme structure. UQ2 suppressed the alkynylation as efficiently as OS-UQ2 in the same concentration range (Fig. 9A). We confirmed, as a reference, that excess bullatacin (10 μm) completely blocked the alkynylation (Fig. 9A).
Figure 9.
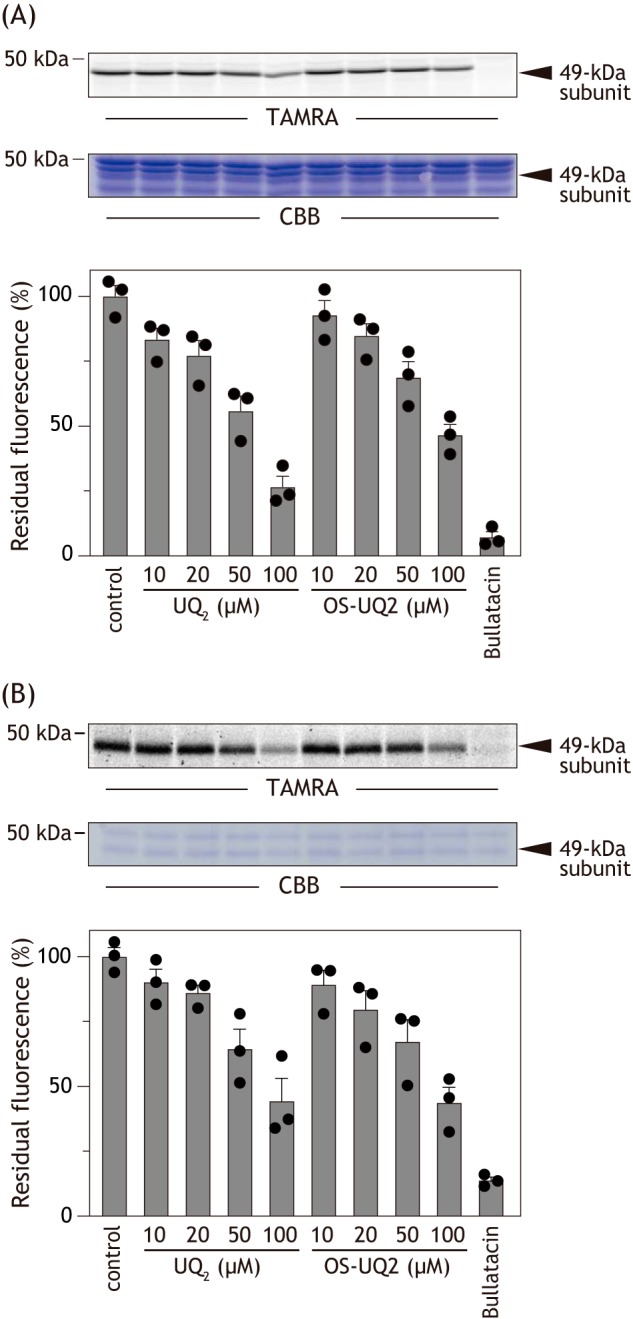
The effects of OS-UQ2 and UQ2 on the chemical modification of 49 kDa-Asp160 by AL1.
A, the effects of OS-UQ2 and UQ2 were examined in SMPs. SMPs (2.0 mg of proteins/ml) were incubated with AL1 (0.5 μm) for 12 h at 24 °C in the presence of different concentrations of OS-UQ2 or UQ2. The alkynylation was visualized by conjugating a fluorescent tag TAMRA-N3 to Asp160(COO)-(CH2)2-CCH via Cu+-catalyzed click chemistry. Bullatacin (10 μm) was used as a reference. Top, gel image of SDS-PAGE analysis used for the alkynylation. Bottom, the extents of suppression by UQs. B, the effects of OS-UQ2 and UQ2 on the alkynylation were examined in the PLs. The PLs (∼10 μg of proteins/mg of phospholipids (equivalent to ∼0.15 mg of proteins/ml)) were incubated with AL1 (2.0 μm) for 12 h at 24 °C in the presence of different concentrations of OS-UQ2 or UQ2. Visualization of the alkynylation was conducted according to the same procedure used for SMPs. Top, gel image of SDS-PAGE analysis used for the alkynylation. Bottom, the extents of suppression by UQs. Values in the graphs are means ± S.E. (error bars) (n = 3).
The alkynylation by AL1 also took place with complex I reconstituted into liposomes (∼10 μg of proteins/mg of phospholipids) (Fig. 9B). Although the suppressive effects cannot be directly compared between SMPs and PLs (because of, for example, different partitioning equilibriums of the quinones), the relative suppressive effects by OS-UQ2 and UQ2 were similar between the two experimental systems (Fig. 9B). These results strongly suggest that OS-UQ2 does not necessarily lose its binding affinity to complex I even in the PLs. Altogether, it is likely that OS-UQ2 can bind to complex I in the PLs, but its quinone head-ring cannot reach the deep reaction site near an Fe-S cluster N2 to be reduced.
Discussion
Structural biology studies have proposed the existence of a long and narrow quinone-access channel in complex I from different biological species (5–12). Because the quinone reduction is a key step in the energy conversion processes in the enzyme, it is essential to examine the physiological relevance of the channel models by different experimental approaches. Here, we investigated accessibility of the UQ reduction site in bovine complex I to a series of oversized UQs, which are highly unlikely to transit the predicted channel (Fig. 2). OS-UQ1−OS-UQ3 functioned as an electron acceptor from complex I in SMPs; particularly, the apparent electron transfer activity of OS-UQ2 was comparable with that of UQ2. Hydrophobic OS-UQ4 and OS-UQ5 did not work as a practical electron acceptor because of their extremely low solubility in water. Importantly, reduction of OS-UQ2 and OS-UQ3 and the membrane potential formation coupled with their reduction were both completely blocked by different inhibitors. The catalytic reduction of OS-UQ2 and UQ2 was similarly inhibited by the unique inhibitor S1QEL2.3. These results provide strong evidence that the reduction of OS-UQ2 and OS-UQ3 takes place at the physiological reaction site for UQ, not at the FMN site. On the other hand, although higher solubility of short-chain UQ in water is favorable for apparent electron transfer activity in SMPs (e.g. activity order of UQ2 > UQ3 > UQ4), the activity of OS-UQ1 was rather poorer than that of OS-UQ2. This reversal strongly suggests that the steric hindrance arising from the block becomes more severe for the catalytic reduction of OS-UQ1 compared with the longer analogues. Conversely, it is surprising that the short spacer of OS-UQ2 is just enough to allow its quinone ring to gain access to the reaction site, avoiding the steric hindrance (Fig. 1). Taking the position of the Fe-S cluster N2 into consideration (5–12), this finding suggests that the block of OS-UQ2 enters fairly deeply into the interior of the enzyme led by the quinone head-ring.
Although the monitoring of membrane potential formation (Fig. 5) is a semiquantitative assessment, it is clear that the proton-translocating efficiency coupled with reduction of OS-UQ1 is significantly lower than that of OS-UQ2 and OS-UQ3 (also UQ2). This finding strongly suggests that reduction of OS-UQ1 does not sufficiently induce the predicted structural changes of the UQ reaction pocket required for triggering proton translocation. In this context, we previously showed that catalytic reduction of “decoupling” UQs (e.g. QT1 in Fig. S1) by complex I in SMPs scarcely derives proton translocation, and the extent of this decoupling phenomenon varies with the side-chain structures (21, 37). It should be noted that QT1 can access the deep interior of the UQ reaction site because a sulfonate group attached to the side chain of this quinone reacts with nucleophilic Asp160 in the 49-kDa subunit to form a covalent bond (21). Thus, the proton-translocating efficiency coupled with reduction of the artificial UQs is significantly affected by their side chain structures. How can we explain this finding? In connection with this question, Brandt and colleagues (38) recently reported that blocking the movement of a loop connecting TMH1–2 in the ND3 subunit of Yarrowia lipolytica complex I by tethering the loop to the nearby subunit PSST via a disulfide bond significantly reduces the proton-translocating efficiency coupled with the reduction of DB. Their study strongly suggests that conformational rearrangements of this flexible loop induced by the DB reduction are critical for energy transmission from the site of the UQ reduction to remotely located proton pumps of complex I. Taking this into consideration, we cannot rule out the possibility that the extent and/or nature of conformational rearrangements of the loop induced by reduction of the artificial UQs may vary with their reaction manners, reflecting different side-chain structures, thereby resulting in different proton-translocating efficiencies. OS-UQ1 may be unable to induce the intrinsic conformational rearrangements required for triggering proton translocation because reduction of this quinone barely takes place due to the marked steric hindrance. Collectively, we conclude that the quinone rings of OS-UQ2 and OS-UQ3, but not OS-UQ1, can reach the physiological reaction site in the native complex I and can be catalytically reduced in a similar manner with ordinary short-chain UQs, such as UQ2.
The results obtained in SMPs are difficult to reconcile with the quinone-access channel models (5–9) unless the channel undergoes drastic structural changes under steady-state catalysis, which allow OS-UQs to move in and out of the channel. The channel was originally postulated to undergo slight structural rearrangement because the planar UQ ring is wider (∼6 Å across) than the diameter of the entry point (5, 11). Nevertheless, the structural change required here is more drastic than the originally postulated one because the block (∼13 Å across) is much wider than the channel entrance and body; therefore, we consider the likelihood of this to be very low. The results obtained by the MD simulations support this notion (Fig. 2).
Instead, it is conceivable that the access path for UQ is open to allow a wide range of ligand molecules access to the deep interior. In this regard, it should be noted that although the so-called quinone-site inhibitors have been considered to occupy the channel interior (5, 6, 13), we recently showed that the binding positions of different types of inhibitors are distributed around the channel (17, 28). Then, if the predicted channel is not the access path for OS-UQs, where is the open path in the native enzyme? We previously proposed that the putative path is located in the area where the regions that were photoaffinity-labeled by different types of inhibitors (fenpyroximate (39), quinazoline (40, 41), bullatacin (42), and various amilorides (17)) are in contact or close to one other (yellow circle in Fig. S5) because the binding of these inhibitors was competitive with one another, and short-chain UQs such as UQ2 and SF-UQ6 (Fig. S1) competitively suppressed the binding of amilorides (17). This area is not only closer to the cluster N2 than the predicted channel entrance but also contains or is in contact with the flexible TMH5–6ND1 and TMH1–2ND3 loops, respectively (see right panels in Fig. S5). Based on the static structural models (5–9), there is not enough space for UQ passage in this area because these loops enclose the reaction site of UQ, although a part of the loops is disordered to varying degrees. We propose that these flexible loops are dynamic “lids” that allow UQ (or some inhibitors) to enter the reaction site rather than rigid “walls” that encloses the site. Further studies with diversified approaches are needed to assess this.
With regard to the binding site of UQ, a recent cryo-EM structure of complex I from Y. lipolytica showed a tightly bound UQ9 molecule in the originally proposed UQ-access tunnel (43). Interestingly, the position of the head-ring of this bound UQ9 is at some distance from the Fe-S cluster N2 (∼27 Å) and in agreement with the position predicted by earlier MD simulations (44). Although the functional significance of the observed position of UQ9 in the catalytic cycle remains to be elucidated, this structural data would extend possible mechanistic models of the terminal electron transfer step in complex I (e.g. two-UQ molecule model (43, 45)).
It is noteworthy that the results obtained using the complex I–reconstituted PLs contradict those obtained using SMPs. Namely, not only OS-UQ1−OS-UQ5 but also isoprenyl derivatives OS-UQ6 and OS-UQ7 were not catalytically reduced by the isolated complex I in the PLs. OS-UQ8 solely functioned as an electron acceptor from the isolated enzyme (Figs. 6 and 8). We repeated the experiments shown in Fig. 8 using another complex I preparation, which was isolated by the procedure reported by Jones et al. (25), and obtained similar results. Considering that the side-chain length of solely OS-UQ8 is longer than that of the predicted channel cavity (therefore, its quinone ring may be able to reach the reaction site without steric hindrance from the block) (Fig. 1), the observations in the PLs appear to be consistent with the quinone-access channel model (5–12).
How can we explain the contradictory results between SMPs (native complex I) and PLs (isolated complex I)? Although we currently have no way of comparing structural differences, if any, in the channel cavity between the native and isolated enzymes, the results shown in Fig. 9 suggest that OS-UQ2 retains the binding affinity to complex I even in the PLs, but its quinone head-ring cannot reach to the reaction site near an Fe-S cluster N2. In light of this view, the most straightforward explanation for the contradictory results could be that the structure of the open access path for OS-UQs in the native enzyme is altered (or narrowed) by purification from the inner mitochondrial membrane; hence, the access of the ubiquinone ring of OS-UQs to the reaction site is obstructed. OS-UQ8, which has the longest side chain among the OS-UQs examined, would be able to avoid this obstruction. Alternatively, we cannot rule out the possibility that some specific positioning of the planar and polar terminal block, which contains multiple polar functional groups (-COO- and three -OCH3), in the highly ordered lipid bilayer membrane may restrict movement of the whole side chain in the membrane and, consequently, interfere with the quinone ring smoothly accessing the reaction site via the open path. This restricted movement could be considerably lessened in OS-UQ8 compared with other shorter OS-UQs. Structural models of OS-UQ–bound complex I with high resolution may provide the key to solving the current contradictions.
Experimental procedures
Materials
UQ1, UQ2, and UQ3 were kind gifts from Eisai (Tokyo, Japan). UQ4 and UQ9 were purchased from Sigma-Aldrich. S1QEL.2.3 was the same sample as used previously (28). All of other reagents were of analytical grade.
Syntheses of OS-UQs
The synthetic procedures of OS-UQ1−OS-UQ8 are described in the supporting information. A reduced form of OS-UQs was prepared by the method of Rieske (46).
Preparation of bovine heart SMPs and measurement of electron transfer activity of UQs in SMPs
Mitochondria were isolated from bovine heart. SMPs were prepared by the method of Matsuno-Yagi and Hatefi (47) and stored in buffer containing 0.25 m sucrose and 10 mm Tris/HCl (pH 7.4) at −80 °C until use. NADH-UQ oxidoreductase activity in SMPs was measured spectrophotometrically by following the oxidation of NADH with a Shimadzu UV-3000 instrument (340 nm, ϵ = 6.2 mm−1 cm−1) at 30 °C (17). The reaction medium (2.5 ml) contained 0.25 m sucrose, 1.0 mm MgCl2, and 50 mm phosphate buffer (pH 7.4). The final mitochondrial protein concentration was 90 μg of proteins/ml. The reaction was initiated by adding 100 μm NADH after the equilibration of SMPs with UQ for 4 min.
Measurement of membrane potential formation in SMPs
Membrane potential formation coupled with NADH-UQ oxidoreduction in SMPs was measured by following changes in the absorbance of oxonol VI (601–630 nm) with a Shimadzu UV-3000 instrument in the dual-wavelength mode in reaction medium (2.5 ml) containing 0.25 m sucrose, 1.0 mm MgCl2, 0.8 μm antimycin A, 4.0 mm KCN, 2.5 mm oligomycin, 0.10 μm nigericin, 1.0 μm oxonol VI, and 50 mm phosphate buffer (pH 7.4) at 30 °C (17). The final mitochondrial protein concentration was set to 90 μg of proteins/ml. The reaction was initiated by adding 100 μm NADH after the equilibration of SMP with UQ for 4 min. A protonophoric uncoupler, SF6847, was used at a final concentration of 0.10 μm to confirm the complete dissipation of the membrane potential.
Purification of complex I and AOX
Complex I was isolated from bovine heart mitochondria by solubilization with sodium deoxycholate and n-decyl-β-d-maltoside and purified by sucrose density gradient centrifugation and anion-exchange chromatography, as described previously (32). The recombinant AOX from T. brucei was expressed in Escherichia coli (48). After the E. coli membrane was solubilized by n-octylglucoside, AOX was purified by cobalt affinity chromatography according to the previous method (48). Electron transfer activities of UQ and UQH2 with isolated complex I and AOX, respectively, were measured according to the previous procedures (32, 33).
Extraction of phospholipid mixture from bovine heart mitochondria
A phospholipid mixture was extracted from bovine heart mitochondria (49) to be used for the preparation of liposomes. Bovine mitochondria were gently suspended in a mixture of chloroform/methanol/water (1:2:1) at room temperature. Then a mixture of chloroform/methanol (1:1) was added, and the mixture was shaken for 1 min, followed by removal of the organic (lower) layer. The aqueous layer was further extracted twice with chloroform. After the combined organic layer was evaporated under N2 atmosphere, phospholipids were purified by a silica gel column (Wako gel® C-200) (50). A total lipid sample was applied in chloroform, followed by 10 column volumes of chloroform to elute nonpolar lipids. Then phospholipids were eluted with 10 column volumes of methanol, evaporated under N2 atmosphere, and dissolved in chloroform/methanol (2:1) solution.
The phospholipid composition was assessed by TLC using a chloroform/methanol/water (65:25:4) mixture as an eluent. The purified phospholipids predominantly contained phosphatidylcholine (PC), phosphatidylethanolamine, and cardiolipin, as shown in Fig. S6. The PC content, which was determined using the choline assay kit LabAssayTM Phospholipids (Wako Pure Chemicals, Osaka, Japan), in the total phospholipid mixture was ∼40%. We confirmed that the phospholipid mixture did not contain UQ10.
Preparation of complex I/AOX-reconstituted PLs
The procedures for the preparation of liposomes were slightly modified from the original work (25) regarding the following two points. First, we used the phospholipid mixture extracted from bovine heart mitochondria in our laboratory (as described above) in place of commercially available phospholipids. Second, the lipid suspension was subjected to seven rounds of freeze-thawing before the extrusion operation using a LiposoFast device (equipped with a 100-nm pore polycarbonate filter) to produce as many unilamella vesicles as possible (51, 52).
PLs were prepared from the unilamella vesicles (675 μl) containing the phospholipid mixture (10 mg) and UQ (160 nmol) of choice. The vesicles were partially solubilized by the addition of 160 μl of n-octyl-d-glucoside from an aqueous 10% (w/v) solution. Then the purified complex I (100 μg) and AOX (100 μg) were added to the solubilized vesicle suspension (800 μl). The detergent was gradually removed from the mixture by the successive addition of SM Biobeads (Bio-Rad, 40 mg, four times) to afford PLs containing complex I, AOX, and UQ. Outward orientation of the reconstituted complex I, which was evaluated from acceleration of the electron transfer activity by the addition of the pore-forming antibiotic alamethicin (25), was ∼45% of the total enzyme.
The PC content in the PLs was quantified by the choline assay. The total phospholipid content in the PLs was estimated using the PC content, which was quantified by the choline assay, and its ratio in the phospholipid mixture (∼40%, as determined above) on the assumption that each phospholipid was equally reconstituted into the liposomal membrane.
The UQ content in the PLs was quantified by HPLC (Shimadzu LC-20AD system) using a standard calibration curve of the authentic sample (16). The suspension of PLs (10 μl) was mixed with a 10-fold volume of ethanol (90 μl), followed by sonication (1 min) and centrifugation (15,000 rpm, 5 min). The supernatant was separated on a reverse-phase column (COSMOSIL 5C18MS-II, 4.6 × 15 mm, Nacalai-Tesque, Kyoto, Japan) using a mixture of ethanol/methanol (3:2) containing 0.1% TFA as a mobile phase with a flow rate of 0.80 ml/min. The elution profiles were monitored at 280 nm.
The total protein content was determined by an Amido Black assay (53). The complex I content in the PLs was estimated by the recovery of NADH-ferricyanide oxidoreduction activity after preparation of the PLs. The AOX content was defined as the difference between the total protein and complex I content (16).
The concentrations of UQ and complex I in the liposomal membrane were estimated assuming that 1 mg of phospholipids occupies ∼1 μl; therefore, 1 nmol of UQ or complex I per mg of phospholipids is equivalent to 1 mm (25).
Electron transfer activity of UQs and membrane potential formation in complex I/AOX-reconstituted PLs
Electron transfer activity of UQ in the complex I/AOX-reconstituted PLs was determined by following the oxidation of NADH with a Shimadzu UV-3000 instrument (340 nm, ϵ = 6.2 mm−1 cm−1) at 30 °C (16, 25). The reaction medium (2.0 ml) contained 50 mm KCl and 10 mm Tris/SO4 (pH 7.5). The reaction was started by adding NADH (100 μm). Bullatacin (100 nm) and ascofuranone (100 nm) were used as a specific inhibitor of complex I and AOX, respectively, to achieve complete inhibition.
The membrane potential formation coupled with the NADH-UQ oxidoreduction in the PLs was determined by following absorbance changes of oxonol VI (601–630 nm). The assay conditions were identical to those for the electron transfer activity except that the reaction medium contained oxonol VI (1.0 μm) and nigericin (0.10 μm).
Determination of the redox state of UQs in the PLs
The complex I-reconstituted PLs (∼5.0 μg of complex I/mg of phospholipids) containing ∼10 mm UQ were incubated with NADH (1.3 mm) in reaction medium (50 mm KCl and 10 mm Tris/SO4 (pH 7.5), 20 μl) at 30 °C on a heat block for 5 min. The electron transfer reaction was stopped by adding argon-purged ethanol (80 μl), followed by gentle homogenization and centrifugation (16,000 × g at 4 °C for 5 min) (36). The supernatant was immediately analyzed by HPLC with the same experimental conditions as those for the quantification of the total UQ content above. UQ and UQH2 were identified by retention times of the authentic samples. The elution profiles were monitored at 292.5 nm, an isosbestic point for UQ and UQH2.
Alkynylation of 49 kDa-Asp160 of complex I by AL-1 in SMPs and PLs
SMPs (2.0 mg of proteins/ml, 50 μl), suspended in a buffer containing 250 mm sucrose, 1.0 mm MgCl2, and 50 mm KPi (pH 7.5), were incubated with AL1 (0.50 μm) in the absence or presence of UQ2 (or OS-UQ2) for 12 h at 24 °C (19). SMPs were then collected by ultracentrifugation (200,000 × g, 20 min, 4 °C) and denatured in 1.0% (w/v) SDS. For visualization of the 49-kDa subunit alkynylated by AL1, the subunit was conjugated with a fluorescent TAMRA-N3 tag via Cu+-catalyzed click chemistry using the Click-iT reaction buffer kit (Life Technologies, Inc.) according to the manufacturer's protocols. Proteins were recovered by precipitation with methanol/chloroform and subjected to Laemmli-type 12.5% SDS-PAGE, followed by the fluorescent gel imaging and Coomassie Brilliant Blue stain (19).
The alkynylation of complex I reconstituted into liposomes (∼15 mg of phospholipids/ml, 50 μl) by AL1 (2.0 μm) was carried out by the same procedure for SMPs. The concentration of complex I in the PLs was set to ∼10 μg of proteins/mg of phospholipids. The reaction medium contained 50 mm KCl and 10 mm Tris/SO4 (pH 7.5). Visualization was also conducted by the same procedure for SMPs.
Computational methods
Atomistic MD simulations were performed on mammalian complex I from Bos taurus (Protein Data Bank entry 5LC5) (7), with the system restricted to core subunits around the UQ channel (ND3, ND1, 49-kDa, 30-kDa, PSST, and TYKY subunits) in a 3-palmitoyl-2-oleoyl-d-glycero-1-phosphatidylcholine bilayer, as described previously (26). OS-UQ2 and OS-UQ3 were placed at the reaction site near the Fe-S cluster N2 based on the alignment with previous setups of natural UQ10. Systems were initially minimized using the steepest-descent algorithm in NAMD (54) to eliminate steric clashes, followed by solvation with TIP3 water molecules and 0.1 m NaCl. 100-ps NVT and 1-ns NPT equilibration steps were then performed with constraints on the protein backbone, followed by 100-ps NVT and 10-ns NPT without constraints. Simulations were performed using the CHARMM force field for lipids, protein, water, and ions (55, 56). The parameters for the bulky tails of OS-UQ2 and OS-UQ3 were obtained from the CHARMM-GUI Ligand Reader and Modeler tool (57), whereas parameters for the quinone headgroup were used from previous studies (58) (see also Ref. 13). GROMACS 2018 (59) was used to perform the simulations at 310 K and 1 atmosphere, with the temperature and pressure being controlled by the Nose–Hoover thermostat (60, 61) and Parrinello–Rahman barostat (62), respectively. The electrostatic interactions were computed using the PME method (63), and the LINCS algorithm (64) was used to achieve a step size of 2 fs. The production runs (n = 3) were performed, totaling ∼1.2 μs each for OS-UQ2 and OS-UQ3. Steered molecular dynamics simulations of bound OS-UQ3 at the site near the entrance to the UQ reaction site near the N2 cluster were also performed. A constant pulling velocity (7 Å ns−1) was applied to the UQ headgroup with a force constant of 1000 kJ mol−1 nm−2 in the direction toward Tyr108 (49 kDa) of the UQ reaction site. All trajectory analysis was performed with VMD (65).
Author contributions
S. U., T. M., M. M., and H. M. conceptualization; S. U., T. M., K. S.-I., J. L., O. H., T. S., D. I., V. S., M. M., and H. M. data curation; S. U., T. M., K. S.-I., J. L., O. H., T. S., D. I., V. S., M. M., and H. M. formal analysis; S. U., T. M., M. M., and H. M. investigation; S. U., T. M., V. S., M. M., and H. M. writing-original draft; K. S.-I., T. S., and D. I. resources; V. S., M. M., and H. M. supervision; V. S., M. M., and H. M. funding acquisition.
Supplementary Material
Acknowledgments
We acknowledge the CSC–IT Center for Science, Finland, for computational resources, including the Pilot Grand Challenge Project. We acknowledge PRACE for awarding us access to MareNostrum at the Barcelona Supercomputing Center (BSC), Spain.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants JP18H02147 and JP19K22278 (to H. M.) and JP18K05458 (to M. M.), by the Academy of Finland (294652), the University of Helsinki and the Sigrid Jusélius Foundation grants (to V. S.), and by the doctoral program in chemistry and molecular sciences (CHEMS) of the University of Helsinki funding (to O.H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6 and Schemes S1–S5.
- complex I
- proton-translocating NADH-quinone oxidoreductase
- AOX
- alternative quinol oxidase
- DB
- n-decyl benzoquinone
- MD
- molecular dynamics
- OS-UQ
- oversized ubiquinone
- PC
- phosphatidylcholine
- PL
- proteoliposome
- TMH
- transmembrane helix
- SMP
- submitochondrial particle
- UQ
- ubiquinone
- UQH2
- ubiquinol
- UQn
- ubiquinone-n
- TAMRA
- 6-carboxy-N,N,N′,N′-tetramethylrhodamine.
References
- 1. Hirst J. (2013) Mitochondrial complex I. Annu. Rev. Biochem. 82, 551–575 10.1146/annurev-biochem-070511-103700 [DOI] [PubMed] [Google Scholar]
- 2. Sazanov L. A. (2015) A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell. Biol. 16, 375–388 10.1038/nrm3997 [DOI] [PubMed] [Google Scholar]
- 3. Wirth C., Brandt U., Hunte C., and Zickermann V. (2016) Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 1857, 902–914 10.1016/j.bbabio.2016.02.013 [DOI] [PubMed] [Google Scholar]
- 4. Wong H.-S., Dighe P. A., Mezera V., Monternier P.-A., and Brand M. D. (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baradaran R., Berrisford J. M., Minhas G. S., and Sazanov L. A. (2013) Crystal structure of the entire respiratory complex I. Nature 494, 443–448 10.1038/nature11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zickermann V., Wirth C., Nasiri H., Siegmund K., Schwalbe H., Hunte C., and Brandt U. (2015) Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 10.1126/science.1259859 [DOI] [PubMed] [Google Scholar]
- 7. Zhu J., Vinothkumar K. R., and Hirst J. (2016) Structure of mammalian respiratory complex I. Nature 536, 354–358 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blaza J. N., Vinothkumar K. R., and Hirst J. (2018) Structure of the deactive state of mammalian respiratory complex I. Structure 26, 312–319.e3 10.1016/j.str.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fiedorczuk K., Letts J. A., Degliesposti G., Kaszuba K., Skehel M., and Sazanov L. A. (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 10.1038/nature19794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu M., Gu J., Guo R., Huang Y., and Yang M. (2016) Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167, 1598–1609.e10 10.1016/j.cell.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 11. Agip A.-N. A., Blaza J. N., Bridges H. R., Viscomi C., Rawson S., Muench S. P., and Hirst J. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 10.1038/s41594-018-0073-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo R., Zong S., Wu M., Gu J., and Yang M. (2017) Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 170, 1247–1257.e12 10.1016/j.cell.2017.07.050 [DOI] [PubMed] [Google Scholar]
- 13. Sharma V., Belevich G., Gamiz-Hernandez A. P., Róg T., Vattulainen I., Verkhovskaya M. L., Wikström M., Hummer G., and Kaila V. R. I. (2015) Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 112, 11571–11576 10.1073/pnas.1503761112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Di Luca A., Gamiz-Hernandez A. P., and Kaila V. R. I. (2017) Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, E6314–E6321 10.1073/pnas.1706278114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gamiz-Hernandez A. P., Jussupow A., Johansson M. P., and Kaila V. R. I. (2017) Terminal electron-proton transfer dynamics in the quinone reduction of respiratory complex I. J. Am. Chem. Soc. 139, 16282–16288 10.1021/jacs.7b08486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fedor J. G., Jones A. J. Y., Di Luca A., Kaila V. R. I., and Hirst J. (2017) Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, 12737–12742 10.1073/pnas.1714074114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uno S., Kimura H., Murai M., and Miyoshi H. (2019) Exploring the quinone/inhibitor-binding pocket in mitochondrial respiratory complex I by chemical biology approaches. J. Biol. Chem. 294, 679–696 10.1074/jbc.RA118.006056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. James A. M., Sharpley M. S., Manas A. R., Frerman F. E., Hirst J., Smith R. A. J., and Murphy M. P. (2007) Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 282, 14708–14718 10.1074/jbc.M611463200 [DOI] [PubMed] [Google Scholar]
- 19. Masuya T., Murai M., Morisaka H., and Miyoshi H. (2014) Pinpoint chemical modification of Asp160 in the 49 kDa subunit of bovine mitochondrial complex I via a combination of ligand-directed tosyl chemistry and click chemistry. Biochemistry 53, 7816–7823 10.1021/bi501342w [DOI] [PubMed] [Google Scholar]
- 20. Murai M., Murakami S., Ito T., and Miyoshi H. (2015) Amilorides bind to the quinone binding pocket of bovine mitochondrial complex I. Biochemistry 54, 2739–2746 10.1021/acs.biochem.5b00187 [DOI] [PubMed] [Google Scholar]
- 21. Okuda K., Murai M., Aburaya S., Aoki W., and Miyoshi H. (2016) Reduction of synthetic ubiquinone QT catalyzed by bovine mitochondrial complex I is decoupled from proton translocation. Biochemistry 55, 470–481 10.1021/acs.biochem.5b01090 [DOI] [PubMed] [Google Scholar]
- 22. Masuya T., Murai M., Ito T., Aburaya S., Aoki W., and Miyoshi H. (2017) Pinpoint chemical modification of the quinone-access channel of mitochondrial complex I via a two-step conjugation reaction. Biochemistry 56, 4279–4287 10.1021/acs.biochem.7b00612 [DOI] [PubMed] [Google Scholar]
- 23. Estornell E., Fato R., Pallotti F., and Lenaz G. (1993) Assay conditions for the mitochondrial NADH:coenzyme Q oxidoreductase. FEBS Lett. 332, 127–131 10.1016/0014-5793(93)80498-J [DOI] [PubMed] [Google Scholar]
- 24. Fato R., Estornell E., Di Bernardo S., Pallotti F., Parenti Castelli G., and Lenaz G. (1996) Steady-state kinetics of the reduction of coenzyme Q analogs by complex I (NADH:ubiquinone oxidoreductase) in bovine heart mitochondria and submitochondrial particles. Biochemistry 35, 2705–2716 10.1021/bi9516034 [DOI] [PubMed] [Google Scholar]
- 25. Jones A. J. Y., Blaza J. N., Bridges H. R., May B., Moore A. L., and Hirst J. (2016) A self-assembled respiratory chain that catalyzes NADH oxidation by ubiquinone-10 cycling between complex I and the alternative oxidase. Angew. Chem. Int. Ed. Engl. 55, 728–731 10.1002/anie.201507332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haapanen O., Djurabekova A., and Sharma V. (2019) Role of second quinone binding site in proton pumping by respiratory complex I. Front. Chem. 7, 221 10.3389/fchem.2019.00221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brand M. D., Goncalves R. L. S., Orr A. L., Vargas L., Gerencser A. A., Borch Jensen M., Wang Y. T., Melov S., Turk C. N., Matzen J. T., Dardov V. J., Petrassi H. M., Meeusen S. L., Perevoshchikova I. V., Jasper H., et al. (2016) Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 24, 582–592 10.1016/j.cmet.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banba A., Tsuji A., Kimura H., Murai M., and Miyoshi H. (2019) Defining the mechanism of action of S1QELs, specific suppressors of superoxide production in the quinone-reaction site in mitochondrial complex I. J. Biol. Chem. 294, 6550–6561 10.1074/jbc.RA119.007687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones A. J. Y., Blaza J. N., Varghese F., and Hirst J. (2017) Respiratory complex I in Bos taurus and Paracoccus denitricans pumps four protons across the membrane for every NADH oxidized. J. Biol. Chem. 292, 4987–4995 10.1074/jbc.M116.771899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nicholls D. G. (1974) The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur. J. Biochem. 50, 305–315 10.1111/j.1432-1033.1974.tb03899.x [DOI] [PubMed] [Google Scholar]
- 31. Garlid K. D., Beavis A. D., and Ratkje S. K. (1989) On the nature of ion leaks in energy-transducing membranes. Biochim. Biophys. Acta 976, 109–120 10.1016/S0005-2728(89)80219-1 [DOI] [PubMed] [Google Scholar]
- 32. Shimada S., Maeda S., Hikita M., Mieda-Higa K., Uene S., Nariai Y., and Shinzawa-Itoh K. (2018) Solubilization conditions for bovine heart mitochondrial membranes allow selective purification of large quantities of respiratory complexes I, III, and V. Protein Expr. Purif. 150, 33–43 10.1016/j.pep.2018.04.015 [DOI] [PubMed] [Google Scholar]
- 33. Shiba T., Kido Y., Sakamoto K., Inaoka D. K., Tsuge C., Ryoko Tatsumi R., Takahashi G., Balogun E. O., Nara T., Aoki T., Honma T., Tanaka A., Inoue M., Matsuoka S., Saimoto H., et al. (2013) Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. U.S.A. 110, 4580–4585 10.1073/pnas.1218386110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shiba T., Inaoka D. K., Takahashi G., Tsuge C., Kido Y., Young L., Ueda S., Balogun E. O., Nara T., Honma T., Tanaka A., Inoue M., Saimoto H., Harada S., Moore A. L., and Kita K. (2019) Insights into the ubiquinol/dioxygen binding and proton relay pathways of the alternative oxidase. Biochim. Biophys. Acta Bioenerg. 1860, 375–382 10.1016/j.bbabio.2019.03.008 [DOI] [PubMed] [Google Scholar]
- 35. Čermáková P., Kovalinka T., Ferenczyová K., and Horváth A. (2019) Coenzyme Q2 is a universal substrate for the measurement of respiratory chain enzyme activities in trypanosomatids. Parasite 26, 17 10.1051/parasite/2019017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robb E. L., Hall A. R., Prime T. A., Eaton S., Szibor M., Viscomi C., James A. M., and Murphy M. P. (2018) Control of mitochondrial superoxide production by reverse electron transfer at complex I. J. Biol. Chem. 293, 9869–9879 10.1074/jbc.RA118.003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masuya T., Okuda K., Murai M., and Miyoshi H. (2016) Characterization of the reaction of decoupling ubiquinone with bovine mitochondrial respiratory complex I. Biosci. Biotechnol. Biochem. 80, 1464–1469 10.1080/09168451.2016.1179095 [DOI] [PubMed] [Google Scholar]
- 38. Cabrera-Orefice A., Yoga E. G., Wirth C., Siegmund K., Zwicker K., Guerrero-Castillo S., Zickermann V., Hunte C., and Brandt U. (2018) Locking loop movement in the ubiquinone pocket of complex I disengages the proton pumps. Nat. Commun. 9, 4500 10.1038/s41467-018-06955-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shiraishi Y., Murai M., Sakiyama N., Ifuku K., and Miyoshi H. (2012) Fenpyroximate binds to the interface between PSST and 49 kDa subunits in mitochondrial NADH-ubiquinone oxidoreductase. Biochemistry 51, 1953–1963 10.1021/bi300047h [DOI] [PubMed] [Google Scholar]
- 40. Murai M., Sekiguchi K., Nishioka T., and Miyoshi H. (2009) Characterization of the inhibitor binding site in mitochondrial NADH-ubiquinone oxidoreductase by photoaffinity labeling using a quinazoline-type inhibitor. Biochemistry 48, 688–698 10.1021/bi8019977 [DOI] [PubMed] [Google Scholar]
- 41. Murai M., Mashimo Y., Hirst J., and Miyoshi H. (2011) Exploring interactions between the 49 kDa and ND1 subunits in mitochondrial NADH-ubiquinone oxidoreductase (complex I) by photoaffinity labeling. Biochemistry 50, 6901–6908 10.1021/bi200883c [DOI] [PubMed] [Google Scholar]
- 42. Nakanishi S., Abe M., Yamamoto S., Murai M., and Miyoshi H. (2011) Bis-THF motif of acetogenin binds to the third matrix-side loop of ND1 subunit in mitochondrial NADH-ubiquinone oxidoreductase. Biochim. Biophys. Acta 1807, 1170–1176 10.1016/j.bbabio.2011.05.012 [DOI] [PubMed] [Google Scholar]
- 43. Parey K., Haapanen O., Sharma V., Köfeler H., Züllig T., Prinz S., Siegmund K., Wittig I., Mills D. J., Vonck J., Kühlbrandt W., and Zickermann V. (2019) High-resolution cryo-EM structures of respiratory complex I: mechanism, assembly, and disease. Sci. Adv. 5, eaax9484 10.1126/sciadv.aax9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Warnau J., Sharma V., Gamiz-Hernandez A. P., Di Luca A., Haapanen O., Vattulainen I., Wikström M., Hummer G., and Kaila V. R. I. (2018) Redox-coupled quinone dynamics in respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 115, E8413–E8420 10.1073/pnas.1805468115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wikström M., Sharma V., Kaila V. R., Hosler J. P., and Hummer G. (2015) New perspectives on proton pumping in cellular respiration. Chem. Rev. 115, 2196–2221 10.1021/cr500448t [DOI] [PubMed] [Google Scholar]
- 46. Rieske J. S. (1967) Preparation and properties of reduced coenzyme Q-cytochrome c reductase (complex III of the respiratory chain). Methods Enzymol. 10, 239–245 10.1016/0076-6879(67)10047-5 [DOI] [Google Scholar]
- 47. Matsuno-Yagi A., and Hatefi Y. (1985) Studies on the mechanism of oxidative phosphorylation. J. Biol. Chem. 260, 11424–11427 [PubMed] [Google Scholar]
- 48. Kido Y., Sakamoto K., Nakamura K., Harada M., Suzuki T., Yabu Y., Saimoto H., Yamakura F., Ohmori D., Moore A., Harada S., and Kita K. (2010) Purification and kinetic characterization of recombinant alternative oxidase from Trypanosoma brucei brucei. Biochim. Biophys. Acta 1797, 443–450 10.1016/j.bbabio.2009.12.021 [DOI] [PubMed] [Google Scholar]
- 49. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 50. Brookes P. S., Rolfe D. F. S., and Brand M. D. (1997) The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: comparison with isolated mitochondria. J. Membr. Biol. 155, 167–174 10.1007/s002329900168 [DOI] [PubMed] [Google Scholar]
- 51. Shintou K., Nakano M., Kamo T., Kuroda Y., and Handa T. (2007) Interaction of an amphipathic peptide with phosphatidylcholine/phosphatidylethanolamine mixed membrane. Biophys. J. 93, 3900–3906 10.1529/biophysj.107.108399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abe M., Hasegawa Y., Oku M., Sawada Y., Tanaka E., Sakai Y., and Miyoshi H. (2016) Mechanism for remodeling of the acyl chain composition of cardiolipin catalyzed by Saccharomyces cerevisiae tafazzin. J. Biol. Chem. 291, 15491–15502 10.1074/jbc.M116.718510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaplan R. S., and Pedersen P. L. (1985) Determination of microgram quantities of protein in the presence of milligram levels of lipid with amido black 10B. Anal. Biochem. 150, 97–104 10.1016/0003-2697(85)90445-2 [DOI] [PubMed] [Google Scholar]
- 54. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu X., Lopes P. E., and Mackerell A. D. Jr. (2012) Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 167–185 10.1002/wcms.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 58. Galassi V. V., and Arantes G. M. (2015) Partition, orientation and mobility of ubiquinones in a lipid bilayer. Biochim. Biophys. Acta 1847, 1560–1573 10.1016/j.bbabio.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 59. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 60. Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 10.1063/1.447334 [DOI] [Google Scholar]
- 61. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 31, 1695–1697 10.1103/PhysRevA.31.1695 [DOI] [PubMed] [Google Scholar]
- 62. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
- 63. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 64. Hess B. (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 10.1021/ct700200b [DOI] [PubMed] [Google Scholar]
- 65. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.