

Abstract
Liquid-liquid phase separation (LLPS) of proteins and nucleic acids is a phenomenon that underlies membraneless compartmentalization of the cell. The underlying molecular interactions that underpin biomolecular LLPS have been of increased interest due to the importance of membraneless organelles in facilitating various biological processes and the disease association of several of the proteins that mediate LLPS. Proteins that are able to undergo LLPS often contain intrinsically disordered regions and remain dynamic in solution. Solution-state NMR spectroscopy has emerged as a leading structural technique to characterize protein LLPS due to the variety and specificity of information that can be obtained about intrinsically disordered sequences. This review discusses practical aspects of studying LLPS by NMR, summarizes recent work on the molecular aspects of LLPS of various protein systems, and discusses future opportunities for characterizing the molecular details of LLPS to modulate phase separation.
Keywords: intrinsically disordered protein, structural biology, heterogeneous nuclear ribonucleoprotein (hnRNP), nuclear magnetic resonance (NMR), protein-protein interaction, liquid-liquid phase separation, RNA-binding proteins
Introduction
Over the past decade, membraneless organelles have been characterized in cells as liquids (1–3). These dynamic assemblies are formed by the phenomenon of liquid-liquid phase separation (4). The underlying constituents of these assemblies are specific proteins and nucleic acids that are responsible for phase transitions. Many of the proteins that undergo physiologically relevant liquid-liquid phase separation (LLPS)3 contain intrinsically disordered domains with low sequence complexity (5). Because these sequences do not contain persistent secondary/tertiary structure, solution-state biomolecular NMR spectroscopy has become the leading biophysical technique to study intrinsically disordered proteins (IDPs) and regions (IDRs) associated with LLPS. In this review, we introduce protein LLPS and IDP NMR. We then describe the various types of samples used to probe protein LLPS by NMR, and finally, we highlight the NMR approaches for probing structure and motions of proteins that undergo LLPS and the information that each technique provides. We delve into practical aspects of studying protein LLPS using NMR. The opportunities and limitations presented by each NMR method are important for those familiar with LLPS, as NMR data become more common and applied in LLPS studies, and for NMR experts, because LLPS systems present unique constraints. Critically, we present background on the experimental observables to help both communities understand the capabilities of NMR in LLPS and critically evaluate data presented in the literature. Overall, we hope that the information presented herein will make the application of NMR spectroscopy to LLPS and its interpretation more accessible for a broader audience.
Protein liquid-liquid phase separation
In protein LLPS, proteins demix from the surrounding solvent to form a distinct, high-concentration phase in equilibrium with a dispersed phase, depleted in protein. Protein phase separation is common under the high-concentration conditions used for protein crystallization; however, proteins have only recently been shown to undergo phase separation at physiological concentrations and conditions (6). The sequence and interaction requirements for LLPS in a physiological context are still not well-understood, although several sequence motifs have been identified in structurally disordered, LLPS-prone proteins (Table 1) (7). A subset of disordered protein domains that facilitate LLPS are classified as prion-like, meaning that they have a sequence composition resembling yeast prion domains enriched in polar amino acids such as glutamine and asparagine (5). The RNA-binding proteins FUS, TDP-43, and hnRNPA2 contain prion-like low-complexity domains that mediate phase separation; however, even within this group, the amino acids and molecular interactions that contribute to phase separation are varied (8–10). Charge-patterned sequences are also able to facilitate phase separation via complex coacervation, the co-demixing of oppositely charged biopolymers. In particular, positively charged arginines in RGG motifs are able to interact with polyanions such as RNA to promote LLPS (11). Elastin-like peptides are enriched in hydrophobic amino acids and are also able to undergo LLPS (12). In addition, LLPS can be modulated by post-translational modifications that can change electrostatic, hydrophobic, and π-interactions (13).
Table 1.
Subset of proteins able to undergo LLPS
| Protein | MLO | Biological function | LLPS protein-protein interaction domain | Sequence motifs/structural features |
|---|---|---|---|---|
| TDP-43 | Stress granules, mRNA transport granules | RNA metabolism | 276–414 | Gly-rich polar transient α-helix (residues 320–343) |
| FUS | Stress granules, paraspeckles, DNA-damage foci, transcriptional granules | RNA metabolism | 1–163 | (S/G/X)Y(S/G/X) repeats |
| 164–267 | RG/RGG motifs | |||
| 372–422 | ||||
| 453–507 | ||||
| hnRNPA2 | Stress granules, mRNA transport granules | RNA metabolism | 190–341 | Glycine-rich RG/RGG motifs (G/X)(N/X)FG repeats |
| Elastin-like peptides | Extracellular matrix | Elastic matrix in vertebrate tissues | Hydrophobic domain | (GVPGV)7 |
| Ddx4 | Developmental granules | DEAD-box RNA helicase | 1–236 | Charge-patterning (F/R)G repeats |
| UBQLN2 | Stress granules | Proteasomal shuttle protein | 379–486 | Hydrophobic amino acids |
| Tau | Stress granules | Microtubule-binding protein | 1–441 | Charge-patterning KXGS repeats |
| FMRP | Stress granules, mRNA transport granules | RNA metabolism | 445–632 | Charge-patterning RG/RGG motifs |
NMR spectroscopy of IDPs
The phase separation of proteins into liquid and solid states is of interest because of the formation of functional liquid condensates and amyloid-like compartments in cells (13–15). Many of these proteins are involved in diseases characterized by the presence of protein aggregates. X-ray crystallography and cryo-EM have been utilized to study amyloid fibrils that are related to disease; however, these techniques generate static “snapshots” and are not able to give atomic-level information about regions that remain dynamic. NMR spectroscopy has emerged as a leading technique to measure transient formation of secondary structure, molecular motions and tumbling, and interactions of intrinsically disordered proteins.
Solution-state NMR spectroscopy can be used to provide detailed information on the structure and motions of individual components inside liquid-like assemblies. In brief, NMR spectroscopy relies on the interaction of atomic nuclei with a magnetic field to provide local information on the chemical environment of each nucleus. In an external magnetic field, nuclear spin magnetization resonates at a characteristic frequency, proportional to the strength of the magnetic field and gyromagnetic ratio of the nucleus. This resonant frequency is further influenced by atoms in close proximity either through bonds or space; the variation in the resonant frequency is often very small and measured in units of parts per million (called the chemical shift). In this way, NMR spectroscopy allows elucidation of the relationships through bonds and space between atoms within a molecule. Chemical shift perturbations of nuclei that correspond to the protein backbone can be used to monitor changes in secondary structure as well as intermolecular interactions. Interactions can be further probed by various techniques that allow for detection of short-range (<6 Å, NOE) and long-range (10–25 Å, paramagnetic relaxation enhancement (PRE)) interactions. NMR spectroscopy is also suited to characterizing molecular motions across different timescales and may be able to provide information on the phase transitions between liquid and solid states. It is important to note that for IDPs, these NMR observables represent a weighted average over the conformational ensemble.
The insights garnered by NMR studies can provide information about the single-chain properties of proteins that undergo LLPS and the interactions that are important for assembly. A detailed molecular picture of how different protein sequences mediate LLPS affords an understanding of the function of membraneless organelles, engineering of LLPS systems for use as novel biomaterials, and development of small molecules that can modulate self-assembly.
Types of samples to study protein phase separation
Structural biologists often take a reductionist approach to studying the characteristics of biomolecules. The formation of membraneless organelles by LLPS is a complex phenomenon involving the presence of many different types of proteins, both those directly involved in LLPS and those that are clients for these assemblies, as well as nucleic acids. To gain insight into these assemblies, many in vitro studies have focused on the protein domains that directly mediate phase separation as minimal models of biomolecular condensates. In addition, some in vitro studies have included molecular crowding agents to mimic the intracellular environment and induce LLPS; however, most NMR studies have excluded these molecules as they may confound results due to protein-crowding agent interactions.
The phases present in NMR samples of LLPS proteins determine the type of information that can be observed. As for any biomolecular NMR experiment, high-quality sample preparation is integral to obtaining useful data. Protein assembly or aggregation typically presents problems in obtaining and interpreting solution NMR data. Because intermolecular interaction is a fundamental feature of LLPS proteins, care must be taken to gain reliable molecular insight into systems in which LLPS occurs. In this section, we review the classes of NMR sample preparations of LLPS proteins used thus far.
Dispersed phase
Insights into LLPS can be glimpsed through using samples in which the protein is at a concentration below that required for phase separation. Without prior knowledge of the boundaries of the phase diagram, it can be difficult to determine a concentration range appropriate for this type of sample. In addition, the critical concentration for some proteins to undergo LLPS can be below 1 μm, which can prevent acquisition of data within a reasonable experimental time due to low signal/noise ratio. If the protein system is amenable to this method, the dispersed phase (dilute phase) sample is easy to prepare and can be used to obtain information on the secondary structure of the protein, molecular motions, and interactions in the dispersed phase.
Chemical shift perturbations are sensitive probes of interactions with residue-by-residue resolution. Chemical shift perturbations as a function of increasing protein concentrations can therefore inform on which residues are important for self-association (Fig. 1A). One study on the phase separation–prone protein TDP-43 mapped a helical subregion (residues 321–343) important for LLPS by observing chemical shift perturbations, which was then confirmed by testing the effect of mutations in this region on phase separation (9, 16). In contrast, other LLPS systems display small chemical shifts with increasing protein concentrations across the entirety of the protein sequence, suggesting that the interactions that stabilize phase separation are not localized to a particular region (10). In samples near the saturation concentration where a small fraction of the protein undergoes LLPS, other studies have probed the onset of LLPS by measuring the signal intensities in standard one- and two-dimensional spectra of the protein remaining in the dispersed phase (17, 18). The fraction of protein in the condensed phase contributes very little to the total signal due to enhanced relaxation rates in the condensed phase, leading to extreme signal broadening and leaving only the signals from the dispersed phase (9, 17–22). Whereas dispersed phase samples are useful in characterizing certain aspects of protein systems that undergo LLPS, they are limited as conclusions about the condensed phase are indirect. Nonetheless, structural insights into proteins within the dispersed phase can provide quantitative information on the processes leading up to phase separation.
Figure 1.

Methods to study the condensed phase by NMR spectroscopy. A, the dispersed phase can be used to garner information about the condensed phase indirectly. A titration of the phase separation–prone C-terminal region of TDP-43 shows chemical shift perturbations of certain residues that are involved in LLPS. Adapted from Ref. 9). This research was originally published in Structure. Conicella, A. E., Zerze, G. H., Mittal, J., and Fawzi, N. L. Structure. 2016; 24:1537–1549. © Cell Press. B, a biphasic sample containing the dispersed and condensed phases can be used to study properties of both. Spectra of an elastin-like peptide recorded with an R2 relaxation rate filter or a pulsed-field gradient diffusion rate filter select for signals arising from either the dispersed or condensed phases, respectively. Adapted from Ref. 23). This research was originally published in Proceedings of the National Academy of Sciences of the United States of America. Reichheld, S. E., Muiznieks, L. D., Keeley, F. W., and Sharpe, S. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E4408–E4415. © United States National Academy of Sciences. C, the condensed phase can be studied directly by creating a macroscopic phase that fills the coil volume of the NMR spectrometer. Spectra of the condensed phase of the low-complexity domain of FUS produce one set of broad resonances. Adapted from Ref. 24). This research was originally published in Molecular Cell. Burke, K. A., Janke, A. M., Rhine, C. L., and Fawzi, N. L. Mol. Cell. 2015; 60:231–241. © Cell Press.
Biphasic sample
Several studies have been able to obtain structural information on protein LLPS systems using samples in which significant populations of both dispersed and condensed phases are present (i.e. a suspension of “droplets”) (8, 23). Due to the presence of two different protein populations, two sets of resonances with distinct chemical shifts are observed, reflecting the distinct chemical environments of the dispersed and condensed phases (Fig. 1B). It is possible to isolate signals from the different species because phase separation into micro-sized condensates influences both translational diffusion and rotational tumbling; therefore, diffusion or relaxation editing can be used to select for signals corresponding to either the dispersed or condensed phase. For diffusion editing, pulsed field gradients are used to create linear magnetic field variation across the sample. This results in a phase shift dependent on the position of the molecule. To rephase, magnetization is inverted with a 180° pulse, and another pulsed field gradient of the same duration and strength is applied. Efficient rephasing only works if no diffusion has occurred; otherwise, the intensity of the peak depends upon molecular diffusion and the strength and duration of the pulsed field gradient. In this way, fast-diffusing species like those in the dispersed phase can be removed, leaving only the signals in the condensed phase. Conversely, relaxation editing relies on the principle that large biomolecules or those in a viscous phase have long rotational correlation times, leading to enhanced R2 relaxation rates. Using standard experiments for the measurement of R2, if the relaxation delay is long (i.e. several hundreds of ms), then fast-decaying signals from large-molecular weight species like the condensed phase can be removed. In the study of an elastin-like peptide, the spectra of the dispersed and condensed phases were separated by using either a relaxation filter to select for fast tumbling (to remove signals with R2 > 5 s−1) molecules such as those in the dispersed phase or a diffusion filter to select for slow-diffusing (to remove signals with diffusion rates >10−7 cm2·s−1) molecules, such as those in the condensed phase (23).
There are several challenges associated with the use of a biphasic sample, namely that the sample is not stable over long periods of time and the signal intensity of the condensed phase is low. A biphasic sample type can be difficult to use for experiments that require long experiment times because the sample will change over time as the condensed phase settles due to gravity. In addition, broad line widths due to the increased viscosity and slowed motions within the condensed phase make resonances associated with the condensed phase difficult to detect above the noise, requiring high protein concentrations (>1 mm) and conditions that maximize the protein concentration in the condensed phase (i.e. recording experiments far above the saturation concentration for LLPS). Thus far, molecular crowding agents (high-molecular weight PEG and dextran) have not been used in NMR studies of LLPS systems; however, experiments conducted in the presence of these compounds may aid in driving more protein into the condensed phase and increasing signal intensity with appropriate controls.
Macroscopic condensed-phase sample
To directly observe the condensed phase, several studies have taken advantage of sedimentation to fuse the dense droplets of the protein-rich condensed phase into macroscopic samples that fill the NMR coil observation volume (8, 10, 23–27) (Fig. 1C). To create macroscopic condensed-phase samples, some approaches involve preparing high-concentration samples that demix followed by allowing the condensed phase to settle due to gravity at the bottom of the NMR tube (25, 26), whereas others have expedited the process by centrifuging (<5000 × g) the sample and transferring it into an NMR tube or directly centrifuging into an NMR tube (8, 10, 24). A macroscopic condensed phase can be challenging to make as it requires large amounts of purified protein (>150 mg for a 5-mm diameter NMR tube with a sample volume of ∼400 μl). Using specialized NMR hardware, Sharpe and colleagues (23) were able to decrease the sample volume requirements by using a 1-mm MicroProbe that enables use of sample volumes as low as 20 μl.
Despite the challenges of making this type of sample, it is currently the best way to achieve direct information on the structure, molecular diffusion, and interactions within a stable condensed phase. To demonstrate the relevance of this sample to studies of in vitro droplets typically visualized by microscopy, chemical shifts have been used to compare the environments of the different types of samples for the low-complexity domain of RNA-binding protein FUS (FUS LC) (8). The chemical shifts of the macroscopic condensed phase overlaid well with the set of peaks in the biphasic sample arising from suspended droplets. Furthermore, the diffusion rate for FUS LC in the condensed phase by NMR (see below) matched that estimated from fluorescence recovery after photobleaching kinetic microscopy experiments on spontaneously formed droplets at the same conditions. Together, these observations suggest that the macroscopic condensed phase of FUS LC created for NMR retains all of the biophysical properties of microscopic droplets. Interestingly, another study on the low-complexity domain of the germ line granule protein Ddx4, which is enriched in FG/RG sequence motifs, compared the macroscopic condensed phase of Ddx4 with a high-concentration dispersed phase (∼400 mg/ml) made from a Ddx4 variant unable to phase-separate at tested conditions (25). This high concentration dispersed phase mimicked the enhanced viscosity effects of a macroscopic condensed phase but failed to recapitulate the extensive intermolecular interactions that are present within the condensed phase.
The macroscopic condensed phase may also be a good candidate for solid-state NMR (ssNMR) studies as not all systems remain liquid and stable for long periods of time at such high protein concentration. For example, the macroscopic condensed phase of the low-complexity domain of FUS remains liquid for months (8); however, the macroscopic condensed phase of hnRNPA2 low-complexity domain is solid at room temperature and requires heating to 65 °C to liquify (10). ssNMR has proven useful to study the interplay between folded and disordered regions of LLPS systems (28, 29). An ssNMR study has been conducted on the two-component condensed phase of the nucleolar protein nucleophosmin (NPM1) and p14ARF tumor suppressor (28). The folded domain of NPM1 that mediates oligomerization into pentamers was detected within the condensed phase by cross-polarization techniques (sensitive to solid regions) and found to form an immobilized scaffold, whereas the disordered regions remained mobile. In the future, combining both solution-state and ssNMR may provide further insights into the interactions that mediate phase separation of full-length proteins. Solid-state NMR has also been used to study the amyloid fibrillar states of proteins that undergo LLPS as well as the transition between the liquid and hydrogel phases (29–31).
These three categories of NMR samples provide distinct information about characteristics of LLPS systems. In the future, it will be important to begin to recapitulate multicomponent systems to understand the structural characteristics of biomolecular condensates. This may involve all of the different types of samples presented above and also the use of isotopic labeling techniques to differentiate multiple components.
Structure and contacts mediating LLPS
We now examine the NMR techniques appropriate for determining structure and contacts of phase-separated proteins using solution NMR spectroscopy.
Chemical shifts
One of the requisite approaches in biomolecular NMR is to obtain chemical shift information for the protein backbone for each amino acid in a protein sequence. The two-dimensional 1H,15N heteronuclear single quantum coherence (HSQC) experiment provides information for each covalently bonded 1H-15N pair corresponding to the peptide backbone and amide-containing side chains. Similarly, the 1H,13C HSQC provides information for each 1H-13C pair corresponding to the Cα, Cβ, and side chains of each amino acid. Because these experiments probe the peptide backbone and side chains, they are sensitive to secondary structure and perturbations that can result from ligand binding or conformational change. The 1H,15N HSQC of a disordered protein typically contains little 1HN signal dispersion, indicating that each residue is exposed to the solvent and experiences a similar chemical environment (32). Importantly, all of the 1H,15N HSQC spectra of the low-complexity domains of phase separation–prone proteins have characteristics of IDPs in both the dispersed and condensed phases, as seen by the narrow signal dispersion in the 1H dimension (9, 10, 23–25) (Fig. 1, B and C). The observed chemical shift represents the population-weighted average shift across the conformational ensemble (33) (assuming similar relaxation properties of the ensemble members) (34, 35). The repetitive sequence motifs found in many proteins that undergo LLPS also introduce spectral crowding and overlap of resonances, making spectral interpretation difficult. However, advances in isotopic labeling and resonance assignment have made studying IDPs easier (reviewed in Ref. 36).
Changes in the position or intensity of the resonances corresponding to each residue have been used to map self-interactions and interactions with protein-binding partners and ligands of LLPS systems (9, 17, 18, 20, 24, 27, 37, 38). The proteasomal shuttle protein, ubiquilin (UBQLN), phase-separates and localizes to stress granules (37). By observing chemical shift perturbations in the dispersed phase, Castañeda and co-workers (37) mapped the domains responsible for phase separation of UBQLN and found that oligomerization of the folded domains coupled with interactions of intrinsically disordered regions mediate UBQLN LLPS. Alternatively, titrations can also be performed with protein-binding partners to determine binding sites. For example, Burke et al. (24) mapped the nonspecific interactions between the low-complexity domain of FUS and RNA polymerase II C-terminal heptad tail in the dispersed phase. Finally, interactions between LLPS-prone proteins and small molecules can be monitored using chemical shifts (20, 39). For example, the interaction of the low-complexity domain of FUS and potential therapeutic small molecules was quantified—the chemotherapeutic mitoxantrone was found to interact with tyrosine residues present in the low-complexity domain of FUS in the dispersed phase (39).
In addition to the information on intrinsic disorder and binding sites derived from amide chemical shifts, quantification of stable and transient protein secondary structure can be obtained by comparing the chemical shifts of each α- and β-carbon with a true “random coil” reference. There are several libraries and methods available to aid in this analysis and take into account neighboring residue effects (32, 40, 41). Many of these methods have been used to evaluate the dispersed and condensed phases of LLPS-prone domains (9, 10, 24). We used secondary shift analysis to compare the different material states of the low-complexity domain of FUS (8). The dispersed and condensed phases of FUS LC show secondary shifts consistent with disorder, whereas the fibrillar state has values with large deviations from random coil, consistent with β-sheet structure (30).
Because the condensed phase of many protein systems is highly viscous, the intrinsic line widths that result are broadened due to the decreased molecular motions in the condensed phase (10, 20, 24, 25). To retain residue-by-residue resolution obtained in the dispersed state, one may change the apodization function used to process the data to increase resolution. In essentially all biomolecular NMR experiments, the raw time domain data are multiplied by a decreasing exponential or cosine-bell function to improve signal/noise ratio and remove artifactual peak shapes that arise from Fourier transformation. In addition to typical line broadening, line sharpening was also used to process 1H,15N HSQC spectra of the macroscopic condensed phase of FUS LC; this choice of apodization function improved resolution but at the cost of decreased signal/noise ratio, which is tolerated in very high-concentration condensed samples (Fig. 1C) (24). Another source of line-broadening for amide 1H positions in samples of intrinsically disordered proteins (in dispersed and condensed phases) is hydrogen exchange with water, especially at physiological temperatures and pH (T > 25 °C and pH > 7.0). To circumvent the need to lower temperature or pH for optimal 1H,15N HSQC spectra, 13C-direct-detected experiments produce narrow, sharp peaks, as there is no contribution to line broadening from water exchange, while retaining a resolution similar to that of 1H,15N HSQC. 13C-Direct-detected experiments have been used to study LLPS-prone proteins in the dispersed and condensed phases (20, 27, 37). For example, Zweckstetter and colleagues (20) used 13CO/15N correlation spectra in the dispersed phase to improve spectral resolution to determine the regions of microtubule-binding protein tau K18 that are responsible for phase separation. In addition, Forman-Kay and co-workers (27) used 13CO/15N correlation spectra to observe stress/transport granule-associated FMRP/CAPRIN1 macroscopic condensed phases at physiological pH (7.4), which was essential for appropriate protonation of phosphorylated residues.
NOESY
Whereas chemical shift perturbations can detect transient population of secondary structural elements and intermolecular interaction sites, these observables do not provide direct information on intermolecular contacts. NOE is commonly used in solution NMR to measure internuclear distances (<6 Å) via through-space dipolar coupling of 1H positions for structure determination. Whereas NOEs cannot be used to directly quantitate distances in conformationally heterogenous IDPs due to the lack of inherent structure, they have been useful in LLPS systems to identify protein-protein interactions between residues that come in contact and hence are important to phase separation (8, 23, 25). In a basic NOE experiment, magnetization is transferred during the NOE mixing time via cross-relaxation between 1H positions that are within close proximity, identifying molecular contacts (Fig. 2A). This experiment reports on all of the 1H positions within close proximity to one another, meaning that it provides information on both the intramolecular and intermolecular interactions within a protein system.
Figure 2.
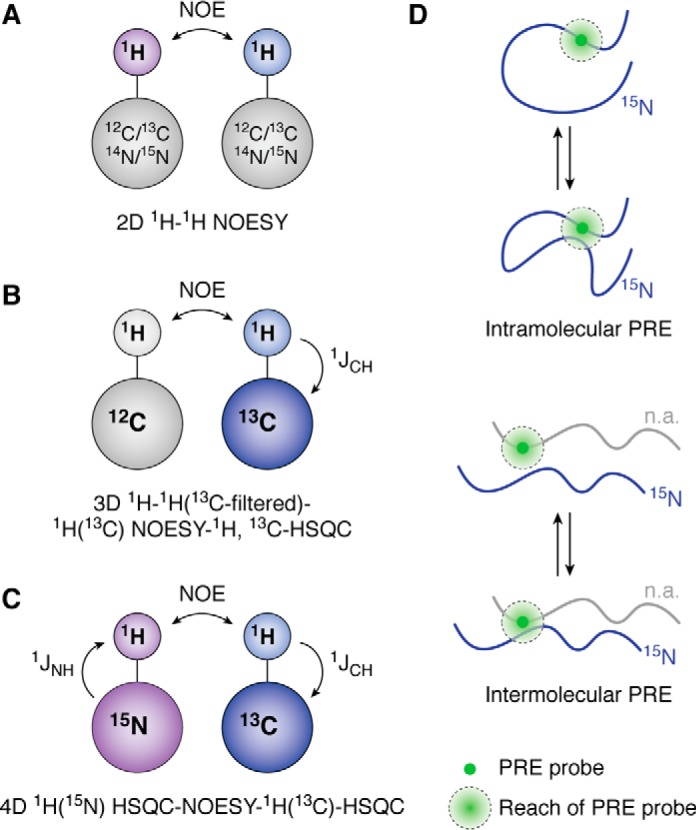
Structural analysis of LLPS systems using NOESY and PREs. A, basic 1H-1H NOESY experiment transfers magnetization between protons in close proximity through space. This is used to study both intra- and intermolecular contacts within protein systems. B, 13C/12C-filtered/edited NOESY experiments utilize the basic 1H-1H NOESY but select for differentially isotopic-labeled protein through the HSQC transfer. C, to increase selectivity, an HSQC-NOESY-HSQC experiment can be run on a sample containing both 15N-labeled protein and 13C-labeled protein. D, intra- and intermolecular paramagnetic relaxation enhancement experiments measure the frequency of contact within a single molecule to investigate collapse (intramolecular PRE) or between two molecules (one NMR visible by 15N isotopic labeling and the other at natural isotopic abundance (n.a.) and hence NMR invisible) to detect interactions (intermolecular PRE).
To differentiate between intra- and intermolecular contributions, differential isotopic labeling schemes are often combined with heteronuclear filtering/editing (“selecting”). This method allows for differentiation between signals arising from isotopically labeled nuclei and natural abundance nuclei. Studies on an elastin-like peptide and on nuage granule protein Ddx4 implemented 13C-filtered/edited experiments to isolate intermolecular interactions within the condensed phase (23, 25). In these experiments, magnetization of 1H positions attached to 13C and/or 15N heteronuclei was removed in the filtering step, leaving only signals from 1H positions attached to 12C and 14N heteronuclei (Fig. 2B). After the NOE mixing time, in the editing step magnetization of 1H positions attached to 13C and/or 15N heteronuclei is selected and read out in an INEPT transfer (HSQC). If this experiment is conducted on a sample that contains 50% 15N,13C-labeled protein and 50% (14N,12C) unlabeled protein (i.e. a 1:1 mixture), then only the intermolecular contacts (i.e. between unlabeled residues and 15N,13C-labeled residues) can be detected. Because of natural abundance of 13C present in the unlabeled sample and incomplete heavy isotope incorporation in the labeled sample (∼1 and ∼99%, respectively), artifacts due to incomplete isotopic labeling may arise. One strategy to avoid incomplete filtering artifacts is to perform doubly edited HSQC-NOESY-HSQC experiments on samples containing 50% 13C-labeled protein and 50% 15N-labeled protein within the condensed phase (8). In this experiment, two INEPT transfers between 1H positions and attached heteronuclei select for signals without relying on filtering. For example, in one version of the experiment, there are three steps: a heteronuclear editing step to select for magnetization starting on 15N-attached 1H nuclei, a conventional NOE step, and a second heteronuclear editing step to select for magnetization ending on 13C-attached 1H nuclei (Fig. 2C). Whereas this experiment does decrease filtering artifacts, it can be challenging due to the loss of signal because of the multiple INEPT transfers. To combat the decreased signal, one may extend the NOE mixing time at the risk of increasing artifacts from spin diffusion (i.e. observation of NOEs between positions that do not directly interact but rather are mutually close to another 1H position). However, the significant motions present in the condensed phases suggest that spin diffusion artifacts will not make major contributions to NOEs. Finally, it is important to note that because of the highly repetitive disordered sequence, the side-chain chemical shifts are overlapped, and hence these NOEs provide primarily residue-type information (e.g. tyrosine side-chain positions interact with glutamine side-chain amide positions).
PRE
Because of the transient, weak nature of many of the contacts that are involved in LLPS, it may be difficult to detect self-interactions or interactions between binding partners using NMR chemical shifts or NOEs. Furthermore, because NOE-based experiments do not provide much sequence-position information (see above), additional techniques are needed. Alternatively, transient short-range interactions or persistent long-range interactions up to 25 Å can be probed using PRE NMR. PRE experiments require conjugation of a paramagnetic probe that contains unpaired electrons (e.g. functionalized stabilized nitroxide radical or EDTA-Mn2+) site-specifically, often to one endogenous or engineered cysteine residue. A reduced form (diamagnetic) of the spin label is used as a control. The PREs arise from dipolar interactions between a nucleus (often 1H) and the unpaired electron(s) in the paramagnetic probe and are measured as the difference in the transverse relaxation rates between otherwise identical samples made with the paramagnetic or diamagnetic PRE probes (Fig. 2D). Because the addition of the spin label requires protein engineering, it is important to ensure that the addition of a cysteine site and conjugation of a label does not dramatically alter LLPS behavior and structure of the protein. In addition, nonspecific interactions between the protein and spin label can occur. Hence, an experiment where free spin label is added to the protein serves as a useful control.
Several studies have used PREs to investigate transient intermolecular contacts and intramolecular collapse in the dispersed and condensed phases (8–10, 42). The intra- and intermolecular contacts disrupted by phosphorylation and phosphomimetic substitution in FUS LC were monitored by intra- and intermolecular PRE experiments, respectively (42). The interaction between the low-complexity domains of hnRNPA2 and TDP-43 in the dispersed phase was characterized using intermolecular PREs (10). TDP-43 formed dynamic interactions across the entire LC domain of hnRNPA2, whereas the helical segment in TDP-43 seemed to participate more in contact formation. Importantly, studying the transient interactions of proteins within the dispersed phase may provide information on which contacts exist within condensed phases. For example, we measured PREs within the condensed phase of FUS LC and found that the contacts formed within the phase did not favor a particular subregion of the sequence and are distributed throughout the domain (8). Interestingly, the PREs in the condensed phase followed a similar trend as PREs measured in the dispersed phase, with the N-terminal region of FUS LC exhibiting higher PREs than other regions (42). It is important to note that, in our experience, PREs within a condensed phase can be challenging to interpret due to the variability in partitioning of the PRE labeled protein and the extent of labeling, and hence the data are best interpreted qualitatively.
Protein motions and conformational changes in the dispersed and condensed phase
Solution-state NMR spectroscopy has the advantage of being able to characterize motions of protein systems with atomistic resolution. A variety of techniques discussed below have been used to probe the timescale of molecular motions within the dispersed and condensed phases (Fig. 3). These NMR methods also provide information on transiently populated structure (as seen by experiments probing picosecond-to-nanosecond timescale motions) and exchange between conformational states (microsecond-to-second timescale motions).
Figure 3.
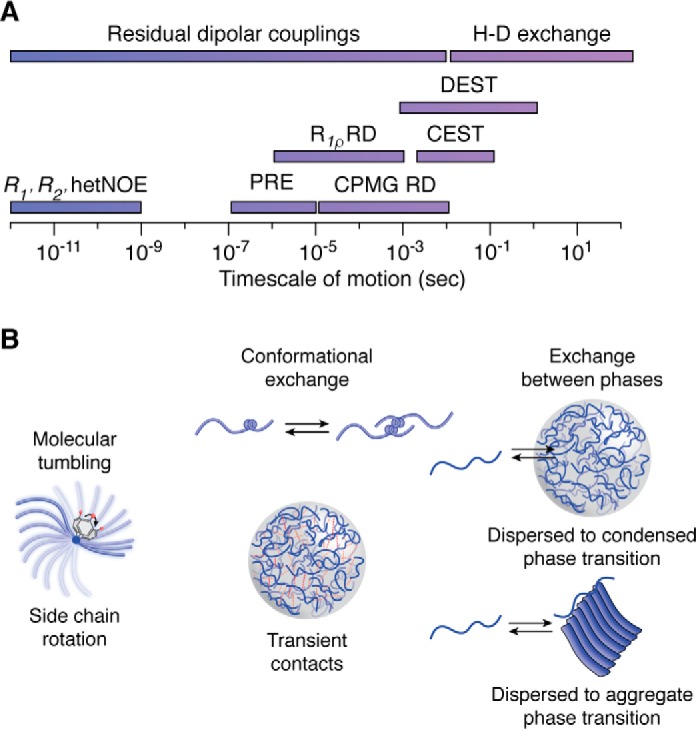
NMR timescale of motion for studying the dynamics of LLPS systems. A, various types of NMR experiments can probe for processes from the picosecond to second timescale. B, fast motions (picosecond-to-nanosecond) that involve overall molecular tumbling and fluctuations of the peptide backbone and side-chain rotations can be measured using R1, R2, and heteronuclear NOE experiments. Intermediate motion processes (microsecond-to-millisecond) that involve conformational exchange and transient contacts can be measured by a variety of experiments, such as paramagnetic relaxation enhancement, CPMG relaxation dispersion, and R1ρ. Slower processes (millisecond-to-second), such as the exchange between liquid and solid phases can be probed using saturation transfer techniques as well as hydrogen-deuterium exchange.
Picosecond-to-nanosecond motions
NMR relaxation of the protein backbone is most easily probed using 15N relaxation experiments that probe the reorientational motion of the amide bond vector. Together, R1, R2, and heteronuclear {1H}-15N NOE experiments probe the flexibility of each nonproline amino acid position on the picosecond-to-nanosecond timescale to gain information about structure and motions. These observables can also be combined to examine the conformational exchange contributions to R2 (see below). The spin relaxation parameters for proteins in the dilute and condensed phase remain predominantly uniform across the sequence, consistent with predominant, uniform disorder for these protein systems. In contrast to protein systems that remain intrinsically disordered across the entire domain, the low-complexity domain of TDP-43 contains a short helical segment (∼20 residues) that displays higher R2 and heteronuclear NOE values in the dispersed phase (9). The transient formation of structure within the helical region rigidifies the amide bond vector and induces slower motions. In general, slowed reorientational motions are a feature of condensed phases due to increased viscosity and high protein concentrations, with an increase in R2 and heteronuclear NOE values and alterations in R1 (8, 10, 23–25). In summary, fast motions in the dispersed and condensed phases can be probed and give insights into transiently populated structural features.
Microsecond-to-millisecond motions
To probe transient formation of structured conformations, the contribution of conformational exchange (Rex) to R2 can be determined by reduced spectral density mapping, where R1, R2, and heteronuclear {1H}-15N NOE observables are analyzed together (43). For example, the contribution of conformational exchange within the condensed phase of FUS LC was evaluated using reduced spectral density mapping, and the Rex term was determined to be effectively zero, suggesting that significant minor populations of structured states were not populated with microsecond-to-millisecond exchange rates (8). This is critical information, as the presence of transient β-sheet structure has been hypothesized to underlie phase separation, although these direct experiments failed to provide support for this hypothesis.
Probing motions on an intermediate timescale (τ ∼100 μs to 10 ms), including exchange between two conformational states, relaxation dispersion experiments provide information on the kinetics of assembly, chemical shift information of the minor state, and the relative distribution of the two populations. This family of techniques is particularly applicable to systems where the minor state is transiently populated and invisible to other NMR techniques (45). CPMG and R1ρ experiments enable quantification of the effect of conformational exchange on R2, Rex, by varying the repetition rate of 180° (π) refocusing pulses or the strength of a spin-lock radio frequency field, respectively (46). These techniques have been used to probe assembly and interactions in the dispersed and condensed states of TDP-43 and Ddx4 (9, 47). For the low-complexity domain of TDP-43, the exchange between the monomeric state and the helix-mediated oligomer important for LLPS was quantified in dilute solution using CPMG relaxation dispersion. In this system, large differences in chemical shifts (up to 1 ppm) between the monomer and assembled state and the transverse relaxation rates of the assembled state are extracted from that relaxation dispersion analysis, consistent with enhancement and extension of helical structure (9). Kay and colleagues (47) probed conformational exchange within a condensed phase of Ddx4. They found that the R2 rates are increased in a high-concentration control as well as in the condensed phase, reflecting the increased concentration and viscosity. Interestingly, relaxation dispersion (<1 s−1) was found, and off-resonance R1ρ experiments were used to elucidate the exchange within the condensed phase. This technique is useful for elucidating exchange with larger rates between the two states than CPMG experiments but where ΔR2 is much smaller than required for dark-state exchange saturation transfer (DEST) between the two states (see below). In the condensed phase, the R1ρ data are consistent with a model where Ddx4 residues populate a minor state with higher transverse relaxation rates but small chemical shift differences with the major state with an exchange rate of ∼18 s−1, suggesting that these interactions are weak and yet relatively long-lived. Crucially, the structural nature and significance of this minor state remain unknown, but perhaps it represents probing of formation and breaking of the weak interactions that lead to LLPS. It is important to note that extraction of chemical shifts and exchange rate constants requires selection and application of an equilibrium chemical kinetic model for the exchange process. The simplest, two-state models (i.e. bound and unbound) have been able to describe the observables thus far, but the true nature of the exchange processes present in condensed phases remains poorly understood (47).
Slow motions (millisecond-to-second)
Observing and quantifying slow conformational or phase-exchange processes that may occur in LLPS necessitates a different set of solution NMR approaches. Possible slow-exchange processes may include the formation and breaking of contacts in the phase or formation of, or interaction with, hypothesized large, structured conformations including amyloid fibrils. DEST probes the slow exchange between large assemblies and monomeric species that may have utility in probing interaction or exchange between different conformations within or between phases (48). High-molecular-weight assemblies have slow molecular tumbling, which results in large increases in the transverse relaxation rate R2 and therefore extreme line-broadening, making these species invisible in most NMR experiments. Thus, DEST takes advantage of the chemical exchange between an NMR-visible (i.e. low-molecular weight or monomeric) population and a high-molecular weight species (e.g. a peptide bound to a large aggregate) to obtain dynamic information about the assembly. In this experiment, a weak saturating B1 field is applied off-resonance from the monomeric signal, resulting in selective attenuation of only the invisible high-molecular weight species. Importantly, the attenuation is proportional to the site-specific R2 in the invisible species, providing residue-by-residue information on the invisible state. This signal attenuation is then transferred to the monomeric state by chemical exchange (i.e. unbinding) and read out as intensity changes in two-dimensional spectra. DEST can be useful in characterizing the regions mediating, and kinetics of, binding/unbinding of disordered domains in the dispersed phase to a solid and/or hydrogel phase also present in equilibrium/pseudo-equilibrium (48). DEST has been used to probe the interaction of the C-terminal heptad-repeat tail of RNA polymerase II with TAF15 hydrogels (49), a simple model of transcriptional activation assemblies (50). Due to the aggregation-prone nature of many of the protein systems that undergo LLPS, DEST may be useful in characterizing the interactions between the dispersed phase and aggregate/fibrillar phase. However, no transient interaction between the disordered major species and potential oligomeric or fibrillar species in the liquid condensed phase of FUS LC was observed using DEST, providing no evidence for the population of large stable structures (e.g. amyloids or gels) within these LLPS samples (8). This observation is important because these experiments directly tested a previous hypothesis that amyloid fibril conformations are important for mediating LLPS.
The presence of two sets of chemical shifts, corresponding to the dispersed and condensed phases, in biphasic samples indicates that these states are in slow exchange. In other words, the time for a given protein to transit from one phase to another is large on the chemical shift timescale, (i.e. approaching 1 s). ZZ-exchange NMR, which can directly measure exchange rates between species in slow exchange, may be appropriate to characterize the kinetics of exchange between the two phases. In addition, the timescale of proline isomerization in condensed phases, which may be significantly slowed compared with the dispersed phase, may be an interesting future target for measurement by ZZ-exchange (51).
Future directions
Solution-state NMR spectroscopy has emerged as a leading technique for characterizing intrinsically disordered systems, transient interactions, and conformational exchange. Probing all of these structural and motional features is essential for understanding the molecular details of systems that undergo liquid-liquid phase separation. Through various NMR experiments, common features of condensed phases are beginning to emerge: the maintenance of protein disorder, restricted motions due to high viscosities and protein concentrations, and transient, “fuzzy” interactions (52). In the future, it will be interesting to see how various mechanisms of phase separation between different systems can be characterized.
Currently, there remains a disconnect in the field between in vitro and in vivo studies of LLPS. Experimental techniques in cells typically measure bulk properties of membraneless organelles and often do not provide quantitative information (53). As a result, it has been difficult to connect the atomic-level observations of NMR spectroscopy to biomolecular condensates present in cells; however, extending current NMR techniques and taking a multidisciplinary approach can allow the field to reconcile structural information about biomolecular condensates. Many questions remain open: What are the driving forces for LLPS? How do complex mixtures of biomolecules consisting of nucleic acids and proteins undergo LLPS? What confers specificity for components within distinct types of biomolecular condensates? How does biochemistry occur within biomolecular condensates? How are biomolecular condensates assembled and disassembled?
Whereas most of these studies have been isolated to minimal models of protein LLPS (i.e. using disordered domains of larger proteins), it may be possible to look at full-length proteins with specific and segmental isotopic labeling, as well as with ssNMR, to begin to understand the interplay between oligomerization, association of IDRs, and LLPS (44, 54). In addition, to understand the behavior and interactions in multicomponent proteinaceous phases, individual components can also be studied via NMR spectroscopy using differential labeling schemes. It is also possible to study LLPS systems in their endogenous environment using in-cell NMR techniques.
Finally, many of the methods discussed above can be used to characterize how small molecules interact with LLPS-prone proteins as well as identify how they disrupt the weak, multivalent interactions that are important for phase separation to develop treatments for neurodegenerative disease where phase separation may play a role.
Acknowledgment
We thank George Lisi for helpful comments.
This work was supported by NIGMS, National Institutes of Health, Grant R01GM118530 (to N. L. F.), Human Frontier Science Program Grant RGP0045/2018 (to N. L. F.), and National Science Foundation Grant 1845734 (to N. L. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LLPS
- liquid-liquid phase separation
- IDP
- intrinsically disordered protein
- IDR
- intrinsically disordered region
- PRE
- paramagnetic relaxation enhancement
- FUS LC
- FUS low-complexity domain
- ssNMR
- solid-state NMR
- HSQC
- heteronuclear single quantum coherence
- UBQLN
- ubiquilin
- CPMG
- Carr-Purcell-Meiboom-Gill
- DEST
- dark-state exchange saturation transfer.
References
- 1. Brangwynne C. P., Mitchison T. J., and Hyman A. A. (2011) Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. U.S.A. 108, 4334–4339 10.1073/pnas.1017150108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brangwynne C. P., Eckmann C. R., Courson D. S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., and Hyman A. A. (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 10.1126/science.1172046 [DOI] [PubMed] [Google Scholar]
- 3. Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., Mittag T., and Taylor J. P. (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shorter J. (2019) Phase separation of RNA-binding proteins in physiology and disease: an introduction to the JBC Reviews thematic series. J. Biol. Chem. 294, 7113–7114 10.1074/jbc.REV119.007944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King O. D., Gitler A. D., and Shorter J. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dumetz A. C., Chockla A. M., Kaler E. W., and Lenhoff A. M. (2008) Protein phase behavior in aqueous solutions: crystallization, liquid-liquid phase separation, gels, and aggregates. Biophys. J. 94, 570–583 10.1529/biophysj.107.116152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vernon R. M., and Forman-Kay J. D. (2019) First-generation predictors of biological protein phase separation. Curr. Opin. Struct. Biol. 58, 88–96 10.1016/j.sbi.2019.05.016 [DOI] [PubMed] [Google Scholar]
- 8. Murthy A. C., Dignon G. L., Kan Y., Zerze G. H., Parekh S. H., Mittal J., and Fawzi N. L. (2019) Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 26, 637–648 10.1038/s41594-019-0250-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conicella A. E., Zerze G. H., Mittal J., and Fawzi N. L. (2016) ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549 10.1016/j.str.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryan V. H., Dignon G. L., Zerze G. H., Chabata C. V., Silva R., Conicella A. E., Amaya J., Burke K. A., Mittal J., and Fawzi N. L. (2018) Mechanistic view of hnRNPA2 low-complexity domain structure, interactions, and phase separation altered by mutation and arginine methylation. Mol. Cell. 69, 465–479.e7 10.1016/j.molcel.2017.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Banerjee P. R., Milin A. N., Moosa M. M., Onuchic P. L., and Deniz A. A. (2017) Reentrant phase transition drives dynamic substructure formation in ribonucleoprotein droplets. Angew. Chem. Int. Ed. Engl. 56, 11354–11359 10.1002/anie.201703191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muiznieks L. D., Sharpe S., Pomès R., and Keeley F. W. (2018) Role of liquid–liquid phase separation in assembly of elastin and other extracellular matrix proteins. J. Mol. Biol. 430, 4741–4753 10.1016/j.jmb.2018.06.010 [DOI] [PubMed] [Google Scholar]
- 13. Ryan V. H., and Fawzi N. L. (2019) Physiological, pathological, and targetable membraneless organelles in neurons. Trends Neurosci. 42, 693–708 10.1016/j.tins.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J., Grishin N. V., Frantz D. E., Schneider J. W., Chen S., Li L., et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boke E., Ruer M., Wühr M., Coughlin M., Lemaitre R., Gygi S. P., Alberti S., Drechsel D., Hyman A. A., and Mitchison T. J. (2016) Amyloid-like self-assembly of a cellular compartment. Cell 166, 637–650 10.1016/j.cell.2016.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Conicella A. E., Dignon G. L., Zerze G. H., Schmidt H. B., D'Ordine A. M., Kim Y. C., Rohatgi R., Ayala Y. M., Mittal J., and Fawzi N. L. (2019) TDP-43 α-helical structure tunes liquid-liquid phase separation and function. bioRxiv 10.1101/640615 [DOI] [PMC free article] [PubMed]
- 17. Amaya J., Ryan V. H., and Fawzi N. L. (2018) The SH3 domain of Fyn kinase interacts with and induces liquid-liquid phase separation of the low-complexity domain of hnRNPA2. J. Biol. Chem. 293, 19522–19531 10.1074/jbc.RA118.005120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ukmar-Godec T., Hutten S., Grieshop M. P., Rezaei-Ghaleh N., Cima-Omori M.-S., Biernat J., Mandelkow E., Söding J., Dormann D., and Zweckstetter M. (2019) Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 10, 2909 10.1038/s41467-019-10792-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nott T. J., Petsalaki E., Farber P., Jervis D., Fussner E., Plochowietz A., Craggs T. D., Bazett-Jones D. P., Pawson T., Forman-Kay J. D., and Baldwin A. J. (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 57, 936–947 10.1016/j.molcel.2015.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ambadipudi S., Reddy J. G., Biernat J., Mandelkow E., and Zweckstetter M. (2019) Residue-specific identification of liquid phase separation hot spots of the Alzheimer's disease-related protein Tau. Chem. Sci. 10, 6503–6507 10.1039/C9SC00531E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitrea D. M., Cika J. A., Guy C. S., Ban D., Banerjee P. R., Stanley C. B., Nourse A., Deniz A. A., and Kriwacki R. W. (2016) Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 5, e13571 10.7554/eLife.13571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turner A. L., Watson M., Wilkins O. G., Cato L., Travers A., Thomas J. O., and Stott K. (2018) Highly disordered histone H1-DNA model complexes and their condensates. Proc. Natl. Acad. Sci. U.S.A. 115, 11964–11969 10.1073/pnas.1805943115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reichheld S. E., Muiznieks L. D., Keeley F. W., and Sharpe S. (2017) Direct observation of structure and dynamics during phase separation of an elastomeric protein. Proc. Natl. Acad. Sci. U.S.A. 114, E4408–E4415 10.1073/pnas.1701877114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burke K. A., Janke A. M., Rhine C. L., and Fawzi N. L. (2015) Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 60, 231–241 10.1016/j.molcel.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brady J. P., Farber P. J., Sekhar A., Lin Y.-H., Huang R., Bah A., Nott T. J., Chan H. S., Baldwin A. J., Forman-Kay J. D., and Kay L. E. (2017) Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. U.S.A. 114, E8194–E8203 10.1073/pnas.1706197114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsang B., Arsenault J., Vernon R. M., Lin H., Sonenberg N., Wang L.-Y., Bah A., and Forman-Kay J. D. (2019) Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. U.S.A. 116, 4218–4227 10.1073/pnas.1814385116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim T. H., Tsang B., Vernon R. M., Sonenberg N., Kay L. E., and Forman-Kay J. D. (2019) Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829 10.1126/science.aax4240 [DOI] [PubMed] [Google Scholar]
- 28. Gibbs E., Perrone B., Hassan A., Kümmerle R., and Kriwacki R. (2020) NPM1 Exhibits structural and dynamic heterogeneity upon phase separation with the p14ARF tumor suppressor. J. Magn. Reson. 310, 106646 10.1016/j.jmr.2019.106646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Damman R., Schütz S., Luo Y., Weingarth M., Sprangers R., and Baldus M. (2019) Atomic-level insight into mRNA processing bodies by combining solid and solution-state NMR spectroscopy. Nat. Commun. 10, 4536 10.1038/s41467-019-12402-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murray D. T., Kato M., Lin Y., Thurber K. R., Hung I., McKnight S. L., and Tycko R. (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615–627.e16 10.1016/j.cell.2017.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ackermann B. E., and Debelouchina G. T. (2019) Heterochromatin protein HP1α gelation dynamics revealed by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 58, 6300–6305 10.1002/anie.201901141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wishart D. S., Sykes B. D., and Richards F. M. (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651 10.1021/bi00121a010 [DOI] [PubMed] [Google Scholar]
- 33. Jensen M. R., Ruigrok R. W., and Blackledge M. (2013) Describing intrinsically disordered proteins at atomic resolution by NMR. Curr. Opin. Struct. Biol. 23, 426–435 10.1016/j.sbi.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 34. Libich D. S., Fawzi N. L., Ying J., and Clore G. M. (2013) Probing the transient dark state of substrate binding to GroEL by relaxation-based solution NMR. Proc. Natl. Acad. Sci. U.S.A. 110, 11361–11366 10.1073/pnas.1305715110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skrynnikov N. R., Dahlquist F. W., and Kay L. E. (2002) Reconstructing NMR spectra of “invisible” excited protein states using HSQC and HMQC experiments. J. Am. Chem. Soc. 124, 12352–12360 10.1021/ja0207089 [DOI] [PubMed] [Google Scholar]
- 36. Gibbs E. B., Cook E. C., and Showalter S. A. (2017) Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys. 628, 57–70 10.1016/j.abb.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 37. Dao T. P., Kolaitis R. M., Kim H. J., O'Donovan K., Martyniak B., Colicino E., Hehnly H., Taylor J. P., and Castañeda C. A. (2018) Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol. Cell. 69, 965–978.e6 10.1016/j.molcel.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoshizawa T., Ali R., Jiou J., Fung H. Y. J., Burke K. A., Kim S. J., Lin Y., Peeples W. B., Saltzberg D., Soniat M., Baumhardt J. M., Oldenbourg R., Sali A., Fawzi N. L., Rosen M. K., and Chook Y. M. (2018) Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22 10.1016/j.cell.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wheeler R. J., Lee H. O., Poser I., Pal A., Doeleman T., Kishigami S., Kour S., Anderson E. N., Marrone L., Murthy A. C., Jahnel M., Zhang X., Boczek E., Fritsch A., Fawzi N. L., et al. (2019) Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. bioRxiv 10.1101/721001 [DOI]
- 40. Camilloni C., De Simone A., Vranken W. F., and Vendruscolo M. (2012) Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 51, 2224–2231 10.1021/bi3001825 [DOI] [PubMed] [Google Scholar]
- 41. Krzeminski M., Marsh J. A., Neale C., Choy W. Y., and Forman-Kay J. D. (2013) Characterization of disordered proteins with ENSEMBLE. Bioinformatics 29, 398–399 10.1093/bioinformatics/bts701 [DOI] [PubMed] [Google Scholar]
- 42. Monahan Z., Ryan V. H., Janke A. M., Burke K. A., Rhoads S. N., Zerze G. H., O'Meally R., Dignon G. L., Conicella A. E., Zheng W., Best R. B., Cole R. N., Mittal J., Shewmaker F., and Fawzi N. L. (2017) Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967 10.15252/embj.201696394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farrow N. A., Zhang O., Szabo A., Torchia D. A., and Kay L. E. (1995) Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR 6, 153–162 10.1007/BF00211779 [DOI] [PubMed] [Google Scholar]
- 44. Debelouchina G. T., and Muir T. W. (2017) A molecular engineering toolbox for the structural biologist. Q. Rev. Biophys. 50, e7 10.1017/S0033583517000051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anthis N. J., and Clore G. M. (2015) Visualizing transient dark states by NMR spectroscopy. Q. Rev. Biophys. 48, 35–116 10.1017/S0033583514000122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Massi F., Johnson E., Wang C., Rance M., and Palmer A. G. 3rd (2004) NMR R1ρ rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 126, 2247–2256 10.1021/ja038721w [DOI] [PubMed] [Google Scholar]
- 47. Yuwen T., Brady J. P., and Kay L. E. (2018) Probing conformational exchange in weakly interacting, slowly exchanging protein systems via off-resonance R 1ρ experiments: application to studies of protein phase separation. J. Am. Chem. Soc. 140, 2115–2126 10.1021/jacs.7b09576 [DOI] [PubMed] [Google Scholar]
- 48. Fawzi N. L., Ying J., Ghirlando R., Torchia D. A., and Clore G. M. (2011) Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 480, 268–272 10.1038/nature10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Janke A. M., Seo D. H., Rahmanian V., Conicella A. E., Mathews K. L., Burke K. A., Mittal J., and Fawzi N. L. (2018) Lysines in the RNA polymerase II C-terminal domain contribute to TAF15 fibril recruitment. Biochemistry 57, 2549–2563 10.1021/acs.biochem.7b00310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kwon I., Kato M., Xiang S., Wu L., Theodoropoulos P., Mirzaei H., Han T., Xie S., Corden J. L., and McKnight S. L. (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 10.1016/j.cell.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sarkar P., Reichman C., Saleh T., Birge R. B., and Kalodimos C. G. (2007) Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell 25, 413–426 10.1016/j.molcel.2007.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fuxreiter M., and Tompa P. (2012) Fuzzy complexes: a more stochastic view of protein function. Adv. Exp. Med. Biol. 725, 1–14 10.1007/978-1-4614-0659-4_1 [DOI] [PubMed] [Google Scholar]
- 53. McSwiggen D. T., Mir M., Darzacq X., and Tjian R. (2019) Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 33, 1619–1634 10.1101/gad.331520.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tugarinov V., and Kay L. E. (2004) An isotope labeling strategy for methyl TROSY spectroscopy. J. Biomol. NMR 28, 165–172 10.1023/B:JNMR.0000013824.93994.1f [DOI] [PubMed] [Google Scholar]