

Abstract
The maternal embryonic leucine zipper kinase (MELK) has been implicated in the regulation of cancer cell proliferation. RNAi-mediated MELK depletion impairs growth and causes G2/M arrest in numerous cancers, but the mechanisms underlying these effects are poorly understood. Furthermore, the MELK inhibitor OTSSP167 has recently been shown to have poor selectivity for MELK, complicating the use of this inhibitor as a tool compound to investigate MELK function. Here, using a cell-based proteomics technique called multiplexed kinase inhibitor beads/mass spectrometry (MIB/MS), we profiled the selectivity of two additional MELK inhibitors, NVS-MELK8a (8a) and HTH-01-091. Our results revealed that 8a is a highly selective MELK inhibitor, which we further used for functional studies. Resazurin and crystal violet assays indicated that 8a decreases triple-negative breast cancer cell viability, and immunoblotting revealed that impaired growth is due to perturbation of cell cycle progression rather than induction of apoptosis. Using double-thymidine synchronization and immunoblotting, we observed that MELK inhibition delays mitotic entry, which was associated with delayed activation of Aurora A, Aurora B, and cyclin-dependent kinase 1 (CDK1). Following this delay, cells entered and completed mitosis. Using live-cell microscopy of cells harboring fluorescent proliferating cell nuclear antigen, we confirmed that 8a significantly and dose-dependently lengthens G2 phase. Collectively, our results provide a rationale for using 8a as a tool compound for functional studies of MELK and indicate that MELK inhibition delays mitotic entry, likely via transient G2/M checkpoint activation.
Keywords: mass spectrometry (MS), cell cycle, protein kinase, inhibitor, cell proliferation, G2/M checkpoint, kinome profiling, maternal embryonic leucine zipper kinase (MELK), mitotic delay, multiplexed kinase inhibitor beads/mass spectrometry (MIB/MS), inhibitor selectivity, NVS-MELK8a, AMPK family
Introduction
The maternal embryonic leucine zipper kinase (MELK),2 also known as MPK38 or pEg3, is a highly conserved member of the AMPK family of kinases (1, 2). The proposed role of MELK in regulation of cell growth has been controversial. MELK expression is increased in cancer relative to normal tissue (3), and high levels of MELK are correlated with tumor grade, poor prognosis, radioresistance, and recurrence in multiple cancers (4–10). RNAi-mediated depletion of MELK has been shown to cause impaired proliferation in a variety of cancers, including basal-like breast (4), glioma (5), acute myeloid leukemia (11), high-risk neuroblastoma (10), and hepatocellular carcinoma (9). In triple-negative breast cancer (TNBC), knockdown of MELK caused shrinking and radiosensitization of xenografts (4, 7). Significantly, numerous studies have shown that RNAi effects on growth are due specifically to MELK depletion by demonstrating rescue with ectopic MELK expression (4, 5, 8, 9, 12, 13). Complicating the interpretation of these findings, the requirement of MELK for cancer cell proliferation has been challenged by studies showing that genomic deletion of MELK with CRISPR/Cas9 caused no growth effects (14–16). Other studies contend that genomic MELK deletion does cause impaired proliferation of cancer cells (17), although this effect may only manifest under certain growth conditions (18).
The discordant results between acute MELK knockdown and longer-term genetic inactivation is reminiscent of other key cell cycle proteins whose essential roles are evident in experiments following acute inactivation by RNAi or with small molecules, but less clear in knockout experiments. Two examples in particular illustrate this phenomenon. First, the cyclin D-CDK4/6 complex is nonessential for proliferation in knockout cell lines and virtually all mouse tissue (19, 20), but chemical inhibitors halt progression into S phase in cell lines with a functional retinoblastoma protein (21). This example is particularly relevant, given the growing clinical use of CDK4/6 inhibitors for the treatment of cancer (22). Second, recent results using CRISPR/Cas9 gene editing suggest that Bub1 is nonessential for mitotic spindle checkpoint function in humans (23, 24), but it was later shown that these “knockout” cells express low levels of a truncated Bub1 protein, accounting for the negative results and lack of a phenotype (25–27). Together, these results highlight the importance of examining protein function using multiple, orthogonal approaches and of using caution in interpreting negative results from gene knockout experiments.
The recent controversy concerning the requirement of MELK in cancer has underscored the need for a more comprehensive understanding of MELK function. MELK has been reported to function in a multitude of processes, including cell cycle progression (3, 4, 28), maintenance of cancer cell stemness (5, 29, 30), protein synthesis (31), and apoptosis (8, 32–34). Many of the functional studies of MELK have focused on the role of this kinase in the cell cycle. MELK mRNA is cell cycle–regulated, and accordingly, protein levels of MELK oscillate throughout the cell cycle, with expression reaching a maximum in G2/mitosis (M) phase, before declining upon mitotic exit (4, 35, 36). Depletion of MELK has been shown to cause a G2/M arrest in cells (3, 4, 17, 37). Some studies have attributed this effect to a role for MELK in the G2/M DNA damage checkpoint, potentially mediated through interaction with CDC25B, a phosphatase that regulates CDK1 activity at the G2/M checkpoint (38). Additionally, loss of MELK activity may cause activation of the ATM/Chk2 DNA damage response (39). Other studies indicate that MELK knockdown specifically impairs mitotic progression. A circular relationship between MELK and FoxM1 has been described, whereby MELK expression is both regulated by this mitotic transcription factor, and phosphorylation by MELK regulates the activity and expression of FoxM1 (28, 39). MELK may also regulate synthesis of the anti-apoptotic protein MCL1 in M via interaction with eIF4B (31). Overall, whereas a number of studies have uncovered putative functions of MELK at G2/M, a comprehensive understanding of the role of MELK at these phases of the cell cycle remains elusive.
One of the key challenges in studying the biological functions of MELK has been the lack of a selective inhibitor. The most commonly used MELK inhibitor, OTSSP167 (OTS), also referred to as OTS167, has shown potent antiproliferative and apoptotic effects against multiple cancer types, including TNBC, acute myeloid leukemia, and high-risk neuroblastoma (4, 10, 11, 40, 41). Consequently, OTS is currently in multiple phase I clinical trials for the treatment of patients with advanced TNBC and refractory or relapsed leukemia (NCT01910545, NCT02795520, NCT02768519, and NCT02926690). While an effective inhibitor of cancer cell viability, OTS is poorly selective for MELK. Recent publications have shown that OTS is a broad-spectrum inhibitor that inhibits numerous kinases that are vital for proliferation, including multiple mitotic kinases (42, 43). A number of studies have relied upon OTS as a means to investigate the biological function of MELK; however, these results must be interpreted with caution due to the poor selectivity of this inhibitor. More recently, additional MELK inhibitors have been developed, including MELK-T1 (39), NVS-MELK8a (8a) (44), HTH-01-091 (HTH) (15), IN17 (45), and others (8, 46). 8a and HTH in particular appear to have more favorable selectivity profiles than OTS based upon kinase activity profiling assays (15, 44).
The key objectives of this study were to evaluate the selectivity profiles of three MELK inhibitors, 8a, HTH, and OTS, using a cell-based assay, to identify a highly selective inhibitor to subsequently investigate MELK function. To this end, we utilized a chemical proteomics approach called multiplexed kinase inhibitor beads/mass spectrometry (MIB/MS) to characterize the selectivity of these MELK inhibitors in TNBC cells (47, 48). OTS was observed to be highly nonselective, in agreement with previous studies, and HTH did not effectively inhibit MELK in cells. In contrast, we found the inhibitor 8a to be highly selective for MELK, providing the first reliable alternative to OTS for functional MELK studies. Using 8a as a tool compound to probe the function of MELK, we observed that MELK inhibition perturbed cell cycle progression by delaying entry into M, without an induction of apoptosis. Collectively, our results provide a rationale to utilize 8a for functional MELK studies as an orthogonal approach to knockdown and knockout techniques and provide insight into the role of MELK in cell cycle progression, specifically at the G2/M checkpoint.
Results
Identification of a selective MELK inhibitor by MIB/MS kinome profiling
MIB/MS is a proteomics technology used for enriching kinases from complex samples (47, 49). This technique can be used for selectivity profiling of inhibitors in a cell-based fashion, an application termed competition MIB/MS (see Fig. 1 for a detailed description of the technology) (48). Selectivity can also be assessed using MIB/MS by adding inhibitors directly to lysates. A similar chemical proteomics approach, using kinobeads and MS, was recently employed by Klaeger et al. (42) to comprehensively define the selectivity of all clinical and Food and Drug Administration–approved kinase inhibitors, validating the use of this approach for measuring inhibitor selectivity in cells. We used the competition MIB/MS approach to profile the selectivity of 8a and HTH in an effort to identify a highly selective MELK inhibitor suitable for functional studies.
Figure 1.

Schematic of competition MIB/MS workflow and sample selectivity output data. MDA-MB-468 cells were treated with DMSO (negative control) or MELK inhibitor for 30 min. This time point allows sufficient time for inhibitors to penetrate cells and engage kinase targets, but not for significant expression-level changes. After harvest, cell lysates were flowed over columns containing kinase inhibitors immobilized on Sepharose® beads, which bind kinases in the cell lysates (top). Kinases bound to the added MELK inhibitor are unable to bind to the MIBs and thus are not captured. Following wash steps to remove nonspecific binders, kinases were subsequently eluted and quantified by MS using label-free quantification. Kinases detected in lower abundance in MELK inhibitor conditions relative to DMSO control (bottom) are prevented from binding to beads due to the inhibitor and thus are targets of that inhibitor.
We first determined the kinase target landscape of OTS, HTH, and 8a at a single concentration. MDA-MB-468 cells were selected for initial experiments, as many previous studies utilized this and other TNBC cell lines. MDA-MB-468 cells were treated with a 1 μm concentration of each putative MELK inhibitor or DMSO for 30 min. Lysates from DMSO- or inhibitor-treated cells were flowed over columns packed with MIBs. Kinases bound to MIBs in the absence or presence of inhibitors were eluted and quantified by MS using label-free quantification in MaxQuant with integrated Andromeda search engine. The specificity of each inhibitor was analyzed for a total of 235 protein kinases quantified in this assay (Fig. S1). Examining the 20 protein kinases detected in lowest abundance after OTS, HTH, or 8a treatment of cells (i.e. most prevented from binding to MIBs), relative to DMSO treatment, revealed stark differences in the selectivity and potency of these three compounds (Fig. 2A). As reported previously, OTS exhibited broad specificity, with 52 protein kinases including MELK captured by MIBs with at least 4-fold decreased abundance relative to DMSO (OTS/DMSO <0.25). Moreover, OTS decreased the binding of a number of kinases to MIBs more effectively than MELK. This finding is in agreement with previous studies, confirming that OTS has very poor selectivity, despite high potency for MELK (42, 43). HTH was found to have a much narrower selectivity profile, with only five protein kinases captured with at least 4-fold decreased abundance relative to DMSO, but surprisingly MELK binding was largely unaffected by this compound. These results suggest that HTH did not effectively inhibit MELK in this cell-based assay, inconsistent with in vitro enzyme assay data (15). By contrast, the target landscape of 8a was observed to be extremely narrow, with MELK being the only protein kinase captured with at least 4-fold decreased abundance relative to DMSO. These results indicated that 8a is the most selective of the three MELK inhibitors profiled using MIB/MS. Additional MS data, including number of peptides identified, sequence coverage, and abundance ratios can be found in Table S1, for this and all MS experiments.
Figure 2.

8a is a highly selective MELK inhibitor. A, MDA-MB-468 cells were treated for 30 min with either DMSO or 1 μm 8a, HTH, or OTS. Cells were harvested, and competition MIB/MS was completed, as described under “Experimental procedures,” to determine the selectivity profile of each MELK inhibitor. The 20 kinases detected in lowest abundance, relative to DMSO control, are displayed for each inhibitor. Arrows indicate the bar corresponding to MELK in each selectivity profile. Results shown are from one experiment. B, MDA-MB-468 cells were treated for 30 min with either DMSO, 1 μm 8a, or 1 μm HTH in three biological replicates (distinct from the replicate shown in A). Competition MIB/MS was used to determine selectivity as in A, and results are displayed as volcano plots to show both the -fold change and significance of the quantified changes in kinase abundance. Kinases with high significance and log2 -fold change <−1 are colored red and labeled. Kinases with only log2 -fold change <−1 are colored yellow. Kinases with log2 -fold change >−1 and nonsignificance are colored gray. t statistics were calculated by empirical Bayes moderation of S.E. values toward the S.E. estimated from all kinases (67). The Benjamini–Hochberg method was used for multiple-test correction with a 5% false discovery rate (68). C, MDA-MB-468 cells were treated with DMSO or 1, 3, or 10 μm 8a for 30 min. Cells were harvested, and lysates were subjected to competition MIB/MS as in A. Data were filtered for kinases that showed decreased binding at all three concentrations in a dose-dependent manner. The 32 kinases that met these criteria are displayed in a heat map, with a double-gradient color scheme ranging from dark blue (nearly total loss of binding to MIBs) to red (no loss of binding to MIBs). Results are indicative of one experiment.
Due to the striking specificity and potency differences between 8a and HTH, we sought to further validate these results in biological triplicate, again at a single concentration of 1 μm. Subsequent competition MIB/MS results are displayed as volcano plots to assess both kinase -fold change magnitude and significance (Fig. 2B). With 8a treatment, MELK was the only kinase that displayed statistically significant decreased binding to MIBs, confirming the high selectivity of this compound. As observed previously (Fig. 2A), HTH treatment did not significantly decrease MELK capture, whereas binding of GAK, CSNK2A2, CSNK2B, CSNK2A1, PIP4K2C, and RIPK2 were significantly decreased. These results further confirm that 8a is a highly selective MELK inhibitor in cells and underscore the importance of determining compound selectivity in a cellular context prior to biological studies.
Competition MIB/MS was next used to profile the selectivity of 8a at 10, 100, and 1000 nm. As expected, we observed a dose-dependent decrease in MELK binding to MIBs, with no major off-target inhibition over this concentration range (Fig. S2). Whereas high selectivity was maintained at 1 μm 8a, we did not observe greater than 90% loss of MELK binding to MIBs, suggesting incomplete inhibition. Because the percentage inhibition of MELK required to elicit a phenotype is not well-defined, we tested higher concentrations of 8a (3 and 10 μm), with the goal of assessing selectivity at concentrations required for nearly total inhibition of binding to MIBs. Following MIB/MS, 221 protein kinases were quantified by label-free quantification as above (Fig. S3), and the data were filtered to remove any protein kinases that did not exhibit reduced binding at all three concentrations in a dose-dependent manner. The 32 protein kinases that met these criteria are displayed in a heat map (Fig. 2C). At 1 μm, 8a was again observed to be highly selective for MELK, with only MELK, STK11, and MAP2K4 captured with 4-fold less abundance, relative to DMSO. Notably, competition MIB/MS results indicated that MELK was not detected at quantifiable levels following 3 μm 8a treatment, suggesting total loss of MELK binding to MIBs, while maintaining a high level of selectivity at this concentration. At 10 μm, however, the specificity of 8a was decreased, with robust off-target kinase inhibition observed.
Whereas STK11 and MAP2K4 appear to exhibit decreased binding in a somewhat dose-dependent manner, it should be noted that neither exhibited significantly reduced binding with 1 μm 8a treatment in biological triplicate (Fig. 2B). We further tested the binding affinity of 8a for these kinases using the Eurofins DiscoverX KINOMEscanTM assay, which quantitatively measures the ability of an inhibitor to compete with a kinase ligand. 8a was found to have no detectable binding affinity for STK11 (Kd > 30 μm) or MAP2K4 (Kd = 17 μm) at 3 μm or lower, whereas high affinity for MELK was observed (Kd = 14 nm) (Fig. S4). Taken together, results from this cell-based selectivity-profiling assay indicate that treatment of cells with 8a at 1–3 μm concentrations is sufficient for moderately strong levels of inhibition to nearly total inhibition of MELK, respectively, while maintaining high selectivity for this kinase.
Effects of MELK inhibition on TNBC cell viability
MELK has been reported to play a role in TNBC proliferation and radioresistance (4, 6, 7). In TNBC and other cancers, RNAi-mediated depletion of this kinase impairs growth, an effect that can be reversed with exogenous MELK rescue, indicating that MELK may be an attractive therapeutic target (4, 5, 7, 8, 12, 13). Recent results demonstrating that genetic knockout of MELK may cause no growth phenotype have called into question the approach of inhibiting this kinase as a cancer monotherapy (14, 15). In light of these disparate results, we sought to test whether selective MELK inhibition with 8a impaired the proliferation of two TNBC cell lines. Treatment of MDA-MB-231 and MDA-MB-468 cells with 8a for 72 h caused decreased cell viability, as measured by a resazurin assay, with an IC50 of 1.7 μm ± 0.4 and 2.3 μm ± 0.4, respectively (Fig. 3A). We further measured the viability effects of 8a using long-term crystal violet cell-staining assays. MDA-MB-231 and MDA-MB-468 cells were treated with 8a for 8 or 14 days, respectively, prior to staining with crystal violet. Similar IC50 values of between 1 and 3 μm were obtained for both cell lines (Fig. 3B), in agreement with shorter-term resazurin assay results. Notably, this inhibition of cell viability was observed at the same concentrations found to result in selective, moderately strong to nearly total inhibition of MELK (Fig. 2C).
Figure 3.

Treatment with 8a causes decreased viability and decreased phosphorylation of histone H3 (Ser-10) in TNBC cells. A, MDA-MB-231 (left) and MDA-MB-468 (right) cells were treated with DMSO or 8a at concentrations ranging from 1 nm to 30 μm. Cells were exposed to 8a for 72 h before viability was measured using a resazurin assay. Three biological replicates were completed for each cell line, and representative dose–response curves are displayed here. B, MDA-MB-231 and MDA-MB-468 cells were treated with DMSO or 8a at the indicated concentrations. The medium was changed, and fresh inhibitor was added every 2–3 days. Cells were monitored until nearly confluent in the DMSO condition (about 8 or 14 days, respectively), at which point they were stained with crystal violet and imaged. Three (MDA-MB-231) or two (MDA-MB-468) biological replicates were completed, and representative images are shown here. C, asynchronous MDA-MB-468 cells were treated with DMSO or 0.5, 1, or 3 μm 8a for 1, 6, or 24 h. Cells were harvested and immunoblotted with the indicated antibodies. Data displayed here are representative of three biological replicates.
We next examined whether reduced cell viability was mediated through the induction of apoptosis or perturbation of cell cycle progression. MDA-MB-468 cells were treated with 0.5, 1, or 3 μm 8a or with DMSO vehicle control, and cells were collected over a 24-h time course. Samples were immunoblotted for PARP/cleaved PARP; cyclins E1, A2, and B1; and phospho-histone H3 (p-H3) (Ser-10), to measure effects on apoptosis and cell cycle distribution (Fig. 3C). PARP cleavage was not observed at any concentration or time point, indicating that 8a did not induce apoptosis. 8a treatment also had no effect on the abundance of the measured cyclins, but there was a dose-dependent decrease in p-H3 (Ser-10), a marker of M that is induced in G2 and dephosphorylated upon mitotic exit. This decrease was observed as early as 1 h post-treatment and maintained through 24 h.
Histone H3 is phosphorylated at Ser-10 by Aurora B (50). To be certain that the observed decrease in p-H3 (Ser-10) was not due to direct inhibition of the Aurora kinases, we utilized the Thermo Fisher SelectScreen kinase assay to test the effects of 8a on Aurora A and B activity. Concentrations below 10 μm 8a did not inhibit Aurora A. The IC50 for inhibition of Aurora B was 24 μm, and only 10 and 21% inhibition was observed with 1.2 and 3.7 μm 8a treatment, respectively. The potency of 8a for the Aurora kinases was 1000-fold lower than that observed for MELK (IC50 = 17 nm) (Fig. S5), indicating that 8a does not significantly inhibit Aurora A and B. It should also be noted that these data were from a cell-free enzymatic assay, so inhibition in cells is likely to be considerably weaker at these concentrations. In agreement with this, we did not observe substantial decreases in Aurora A or B binding to MIBs with ≤3 μm 8a (Figs. S1 and S3). Collectively, these results show that MELK inhibition with 8a caused decreased viability through impaired cell cycle progression, specifically in G2/M phases.
MELK inhibition results in delayed mitotic entry mediated through delayed activation of Aurora A, Aurora B, and CDK1
Our results prompted further investigation into the effects of MELK inhibition on cell cycle progression. For subsequent experiments, HeLaS3 cells were used because of their robust and well-characterized response to cell cycle synchronization methods. We first confirmed that HeLaS3 cells exhibit similar sensitivity to 8a (IC50 of 1.9 μm ± 0.5) as TNBC cell lines (Fig. S6A). We further observed that 8a treatment caused the same dose-dependent decrease in p-H3 (Ser-10), with no effect on PARP cleavage or abundance of the measured cyclins, in HeLaS3 cells as in MDA-MB-468 cells (Fig. S6B). MELK expression levels were found to be similar between HeLaS3, U2OS, and TNBC cell lines as well (Fig. S6C).
To specifically examine effects on progression through G2 and M, HeLaS3 cells were synchronized with double-thymidine block and released into medium containing DMSO or 1 or 3 μm 8a and harvested every 2 h over a 12-h time course. Samples were immunoblotted for cell cycle markers to determine the effect of 8a treatment on progression through S phase and into M (Fig. 4A). No differences in cyclin E1 or A2 abundance were observed after 8a treatment from 0 to 8 h, indicating that S phase progression was unaffected by MELK inhibition. At 8 h after release, p-H3 (Ser-10) abundance was lower in 3 μm 8a-treated cells relative to DMSO- and 1 μm 8a-treated cells. Further, at 10 h after release, cyclins A2 and B1 and p-H3 (Ser-10) abundances were greater with 3 μm 8a treatment, indicating that entry into and progression through M was delayed (Fig. 4B). At the 12-h time point, cells appeared to have completed M, evident by major decreases in signal for cyclins E1, A2, and B1 and p-H3 (Ser-10). Cyclin E1 abundance was decreased by 3 μm 8a at 12 h, relative to other conditions, suggesting the effects of mitotic delay may persist after M. Treatment with 1 μm 8a had little effect on cell cycle progression as measured using this thymidine synchronization technique. Therefore, 3 μm 8a was used in subsequent synchronization experiments.
Figure 4.

MELK inhibition with 3 μm 8a causes delayed mitotic entry. A, HeLaS3 cells were synchronized at G1/S with double-thymidine block and released into DMSO or 1 or 3 μm 8a. Cells were collected every 2 h over a 12-h time course. Cells were lysed and immunoblotted with the indicated antibodies. The splits in the image indicate that the samples from DMSO and 1 and 3 μm 8a conditions were immunoblotted separately due to space constraints. Asynchronous HeLaS3 lysate was loaded in the lanes labeled A to normalize among the blots. The data shown here are representative of three independent experiments. B, the signal intensity of the cyclin E1, A2, and B1 and p-H3 (Ser-10) bands was quantified for the replicate shown in A using Bio-Rad Image LabTM software. The ratio of the quantified signal intensity at each time point relative to the 0-h time point was computed for each condition. The resulting ratios are plotted as bar charts for each protein, with 1.0 indicating no change relative to the 0-h time point.
To further investigate this apparent delay in mitotic entry, we completed another double-thymidine synchronization experiment with more frequent collections around M (8–12 h after release). Cells were not collected between 0 and 6 h after release, as no differences were observed with 8a over this time frame in the previous experiment (Fig. 4A). Additionally, we sought to investigate the mechanism underlying this mitotic delay further by expanding the number of cell cycle proteins that were measured by immunoblotting. Aurora A and B have critical roles in mitotic entry and progression, and their activity is regulated by autophosphorylation at Thr-288 and Thr-232, respectively (51, 52). CDK1, regarded as the key kinase for promoting M, is activated by cyclin binding and dephosphorylation of Tyr-15 and Thr-14. These regulatory sites are phosphorylated by Wee1 and Myt1 in interphase, respectively, and are dephosphorylated by CDC25B (53). We immunoblotted for Aurora A and B and their regulatory phosphorylation sites, CDK1 and its regulatory phosphorylation sites, Myt1, Wee1, CDC25B, and the cell cycle markers used previously (Fig. 5).
Figure 5.

Delayed mitotic entry caused by 8a is associated with delayed activation of Aurora A, Aurora B, and CDK1. HeLaS3 cells were synchronized at G1/S with double-thymidine block and released into DMSO or 3 μm 8a. Cells were collected starting at 6 h after release, with subsequent collections completed every 0.5 or 1 h, over a 12-h time course. Cells were lysed and immunoblotted with the indicated antibodies. The split in the image indicates that the samples from DMSO and 3 μm 8a conditions were blotted separately due to space constraints. Asynchronous HeLaS3 lysate was loaded in the lanes labeled A to normalize among the blots. The data shown here are representative of two independent experiments.
With more frequent time points, the delay in onset of M caused by 3 μm 8a was even more apparent than in the previous experiment. This was evident by delayed degradation of cyclins A2 and B1, delayed accumulation of the mitotic marker p-H3 (Ser-10), delayed phosphorylation and activation of Aurora A and B, and delayed activation of CDK1 by dephosphorylation of Tyr-15 and Thr-14. In control cells, cyclin A2 accumulation peaked at 7 h after release and persisted through 8.5 h, after which it was rapidly degraded. In 3 μm 8a-treated cells, cyclin A2 accumulated similarly, but expression persisted through 11 h. Cyclin B1 accumulation was also unaffected by 8a, but its degradation upon mitotic exit was delayed by ∼3 h, beginning at 9 h in control cells and 12 h in 8a-treated cells. Consistent with this, p-H3 (Ser-10) accumulation began at 8 h and decreased after 10 h in control cells, compared with accumulation beginning at 9 h, peaking at 11 h, and persisting at high levels through 12 h in 8a-treated cells. The activation of Aurora A and B by autophosphorylation was also delayed, correlating precisely with p-H3 (Ser-10) patterns. CDK1 activation by dephosphorylation of Tyr-15 and Thr-14 began at 8.5 h in control cells, compared with 11 h in 8a-treated cells, whereas CDK1 expression was unaffected. Interestingly, the dynamics of Wee1 expression appeared mostly unchanged between control and 8a conditions, whereas inactivation of Myt1 by hyperphosphorylation (slower migrating band on gel) mirrors the delayed dephosphorylation pattern observed with CDK1 Thr-14 and Tyr-15. Finally, the accumulation pattern of CDC25B may also be unaffected by MELK inhibition, although peak levels of CDC25B may be decreased relative to control. These results indicate that MELK inhibition results in delayed mitotic entry due to a delay in activation of multiple kinases essential for this process, namely Aurora A and B and CDK1.
MELK inhibition does not impair mitotic completion
After observing that MELK inhibition results in a delay in mitotic entry, we next asked whether it also had an effect on mitotic progression and completion. Following the previously described synchronization/release approach, cells were collected over a 20-h time course and immunoblotted for the same cell cycle markers (Fig. 6). Again, degradation of cyclins A2 and B1, and induction of p-H3 (Ser-10) were delayed by 3 μm 8a treatment, correlating with delayed activation of Aurora A and B and CDK1. Despite this delay, cells treated with 3 μm 8a appeared to progress through and complete M in a similar amount of time, once M began. This was evident by a return of p-H3 (Ser-10) to steady-state levels at 14 h, compared with 12 h for DMSO, and dephosphorylation of Myt1, resulting in a faster-migrating band relative to hyperphosphorylated mitotic Myt1. MELK expression also decreased rapidly immediately following completion of M, as described in other studies (4, 35). HeLaS3 cells treated with 3 μm 8a re-entered the subsequent cell cycle, as observed by an induction of cyclin E1 at 14 h, compared with 12 h for DMSO. Levels of this G1/S phase cyclin peak at ∼16–18 h with 3 μm 8a treatment, which is 2 h later than control cells, where cyclin E1 peaks at 14–16 h. These results indicate that the length of M appears to be largely unaffected by MELK inhibition, despite mitotic entry being delayed. These observations, along with delayed activation of Aurora A and B and CDK1, are consistent with activation of the G2/M checkpoint (54). Because M occurs quite rapidly (∼30 min), we sought to further examine these results using live-cell imaging for greater temporal resolution.
Figure 6.

HeLaS3 cells treated with 8a complete mitosis and begin the subsequent cell cycle following delayed mitotic entry. HeLaS3 cells were synchronized at G1/S with double-thymidine block and released into DMSO or 3 μm 8a. Cells were collected starting at 6 h after release, with subsequent collections completed every 2 h over a 20-h time course. Cells were lysed and immunoblotted with the indicated antibodies. The split in the image indicates that the samples from DMSO and 3 μm 8a conditions were blotted separately due to space constraints. Asynchronous HeLaS3 lysate was loaded in the lanes labeled A to normalize among the blots. The data shown here are representative of at least two biological replicates.
Live-cell imaging quantifies the mitotic entry delay caused by MELK inhibition
To more precisely measure the lengths of G2 and M phases in response to 8a treatment, we conducted live-cell imaging of U2OS osteosarcoma cells stably expressing a fluorescent cell cycle reporter based on PCNA localization (55). U2OS cells are routinely used for cell cycle studies, particularly with imaging methods, and our results indicate that MELK expression is similar between U2OS and TNBC cells (Fig. S6C). This live-cell imaging approach employs tracking of individual cells from an unperturbed, asynchronous population. Fluorescent PCNA delineated the start and end of S phase, whereas nuclear envelope breakdown and cytokinesis delineated the start and end of M. Cells were treated with DMSO or 1 or 3 μm 8a, and G2 length was measured. We observed significant, dose-dependent G2 lengthening in cells treated with either 1 or 3 μm 8a, compared with DMSO-treated cells (Fig. 7A). In contrast to the lower-time resolution immunoblotting experiments with synchronized cells (Fig. 4), the increased sensitivity of live-cell imaging revealed that G2 was indeed significantly lengthened by 1 μm 8a treatment (∼30% increase, 81 min). Following 3 μm 8a treatment, median G2 length was markedly increased (>250% increase, 426 min) compared with untreated cells (Fig. 7C).
Figure 7.
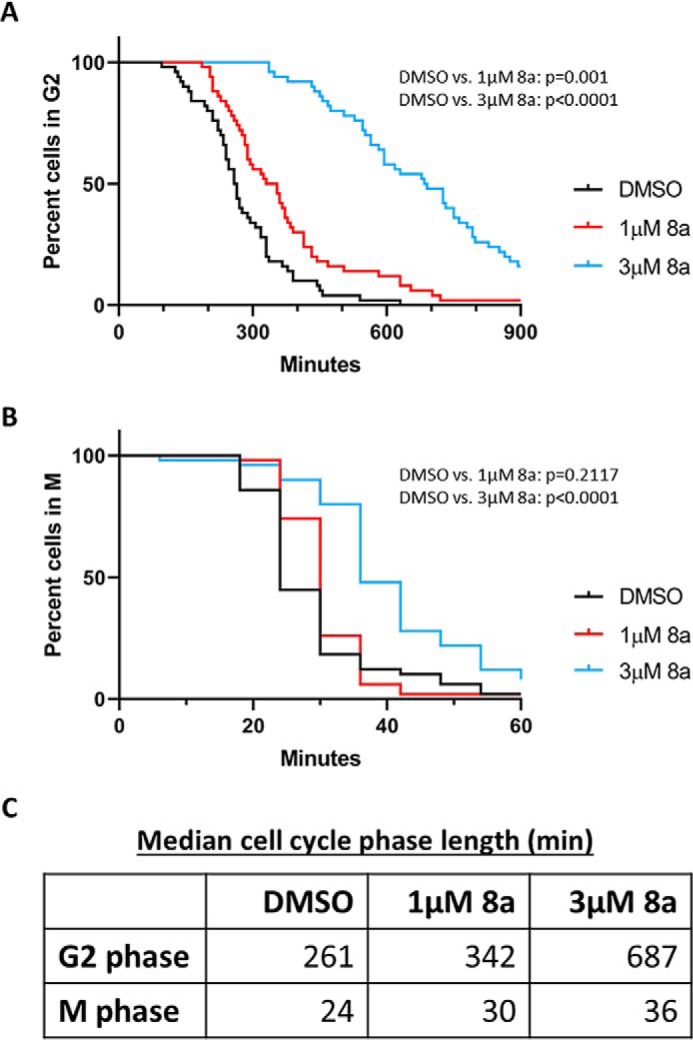
8a causes a major, dose-dependent increase in G2 length. A, asynchronous U2OS cells harboring fluorescent PCNA were treated with DMSO or 1 or 3 μm 8a. Microscopy images were acquired every 6 min over the course of 72 h. Cells were manually tracked, and the length of G2 was calculated by measuring the amount of time between disappearance of PCNA foci (end of S phase) and nuclear envelope breakdown (start of M). For each of three biological replicates, 50 cells were tracked per condition. Kaplan–Meier curves were generated, and significance was calculated in GraphPad Prism using the log-rank test for survival curves. Representative data are shown here, and results from additional replicates are shown in Fig. S7. B, the length of M was manually calculated in the cells in A by measuring the amount of time between nuclear envelope breakdown (start of mitosis) and cytokinesis (end of mitosis). For each of three biological replicates, 50 cells were tracked per condition. Curves were generated, and significance was calculated as in A. Data shown here, indicating M to be significantly lengthened by 3 μm 8a, are representative of two of three biological replicates. Additional experiments (Fig. S7) indicate that M was not significantly lengthened in one replicate. C, median length of G2 and M, in minutes. The displayed cell cycle phase lengths were calculated for the experiment shown in A and B.
Live-cell imaging was next used to quantify the length of M phase. Whereas no significant difference in time to complete M was observed between cells treated with DMSO and 1 μm 8a, 3 μm 8a caused detectable lengthening of M in two of three replicates (Fig. 7B and Fig. S7). This increase in mitotic length from 24 min in control cells to 36 min in 3 μm 8a-treated cells was much smaller in magnitude than the observed increase in G2 (Fig. 7C), which likely explains why it was not detected using synchronization methods. An increase in G2 length appears to be the predominant phenotype resulting from MELK inhibition. Taken together, our results indicate that selective MELK inhibition causes a delay in mitotic entry that is consistent with transient G2 arrest.
Discussion
Currently, one of the main questions in the MELK field is how to rectify the seemingly disparate observations that RNAi-mediated MELK depletion and pharmacological inhibition cause a strong growth defect in cancer cells (3–5, 9, 12, 28, 31, 37, 39, 44), whereas CRISPR/Cas9 knockout of MELK may have conditional or no effect on proliferation (14–18). It is vital to recognize that genomic knockout techniques are not identical to RNAi-mediated knockdown or inhibition, but rather that all are valuable orthogonal approaches. Thus, divergent results should be interpreted as evidence that the functional role of the protein being investigated is likely complex, rather than evidence that one set of results is incorrect in its entirety. It is quite possible that the total loss of protein expression and activity, via knockout, could have different effects than partial inhibition of activity with unperturbed expression, via pharmacological inhibition. Further, and because of the inherent differences between these techniques, it is also possible that genomic knockout could result in cellular reprogramming of signaling networks such that compensation for growth defects occurs, whereas partial inhibition or depletion of the same enzyme may not result in the same changes. As described in the Introduction, divergent phenotypes have been observed, but not yet fully explained, between cyclin D-CDK4/6 knockout and inhibition, demonstrating that this phenomenon is not constrained solely to MELK (19–21). At this point, these potential explanations are purely speculative, but this will undoubtedly be an important area of investigation for future MELK studies.
This controversy has underscored the fact that our understanding of the function of MELK in cancer is still lacking. As a further complication, functional studies of MELK have long relied upon OTS, an inhibitor with a remarkable lack of selectivity, to complement results obtained using knockdown and other techniques. Whereas RNAi-mediated knockdown and genomic knockout approaches obviously have their merits, the use of a pharmacological inhibitor has distinct advantages as well. First, pharmacological inhibition generally allows for much tighter temporal control than other techniques, such that effects can be measured on a short timescale before significant changes in signaling networks occur. Second, inhibition of kinase activity often does not directly affect expression of the inhibited kinase, which allows for differentiation between activity-dependent and -independent functions. Thus, a selective MELK inhibitor would clearly be a valuable addition to the toolkit available for studying the mechanistic underpinnings of this kinase in cancer.
In an effort to identify a selective MELK inhibitor, we used competition MIB/MS technology to evaluate the selectivity profiles of OTS, HTH, and 8a. Our results (Fig. 2A) are in agreement with other studies in demonstrating that OTS is highly nonselective and therefore is unsuitable for mechanistic studies of MELK biology (42, 43). In contrast to published enzyme assay data demonstrating HTH to be a potent MELK inhibitor, we found that this inhibitor did not significantly affect MELK binding to MIBs (Fig. 2, A and B) (15). This illustrates the disparate results that can be observed between enzyme- and cell-based selectivity-profiling methods, emphasizing the importance of evaluating compound selectivity in a cellular context. Our results indicating that 8a exhibits high selectivity for MELK (Fig. 2, A–C) are the first to characterize an exquisitely selective MELK inhibitor in cells and thus rationalize the use of 8a for future functional studies. Because thoroughly validated MELK substrates are lacking, one intriguing possibility is to use MS to compare the phosphoproteome of cells treated with 8a to control cells, in an effort to identify novel MELK substrates. Such studies could serve to establish a much-needed cellular readout for MELK activity and to further elucidate the role of MELK in the cell cycle and cancer. To date, MELK phosphoproteomics studies have only been completed using a mutant null allele of the MELK homolog in Caenorhabditis elegans (pig-1) (56) or with OTS treatment (13).
As previously described, 8a treatment caused decreased viability of TNBC cells (Fig. 3, A and B) at concentration ranges that significantly prevented MELK binding to MIBs (Fig. 2C) (44). We did not observe an induction of apoptosis with 8a (Fig. 3C), despite some others describing this effect as a result of MELK knockdown (4, 9, 17, 31). It is possible that the longest time point used in our study (24 h) is not sufficient for induction of apoptosis with 8a and that the observed cell cycle perturbation precedes apoptosis. Both prosurvival and proapoptotic roles have been attributed to MELK, and the specific contexts that determine these opposing functions remain to be elucidated (34).
Although many studies describe a role for MELK in cell cycle progression, the precise details of this role are not well-understood. In response to MELK silencing with RNAi, numerous studies have reported an accumulation of cells in G2/M phases (3, 17, 37, 44, 57). It should be noted that due to the methods used (quantification of cellular DNA by flow cytometry), these particular studies were not able to differentiate between an accumulation in G2 versus M. Paradoxically, overexpression of MELK may induce a G2/M accumulation of cells as well (3, 38). Other studies have used more specific techniques to differentiate between G2 and M phase effects. MELK knockdown has been reported to impair mitotic progression by varied mechanisms, including by decreasing activity and expression of the oncogenic mitotic transcription factor FoxM1 (28, 39), by regulating MCL1 synthesis in M (31), by inducing mitotic catastrophe (8), and by causing cytokinetic defects (4, 58, 59). A role for MELK in G2, distinct from M, has been described as well. Davezac et al. (38) observed that ectopic expression of MELK (formerly known as pEg3) caused an accumulation of cells in G2, which could be rescued by co-expression of CDC25B. MELK was found to directly interact with CDC25B in Xenopus and phosphorylate this phosphatase in vitro, leading the authors to hypothesize that MELK negatively regulates the G2/M checkpoint through interaction with CDC25B (38).
As there is no clear consensus whether MELK plays a role in G2, M, or both, we used 8a, as an alternative to RNAi knockdown, to further investigate the cell cycle function of MELK. Time-lapse microscopy experiments (Fig. 7) indicated that MELK inhibition caused a major, significant lengthening of G2 phase. Of note, we observed many cells that delayed in G2 phase after exposure to 8a within that same cell cycle, suggesting that the G2 delay phenotype is proximal to MELK inhibition. An additional minor delay in mitotic progression was observed, warranting further investigation in light of previous studies indicating that MELK has mitotic functions. However, we focused on the more predominant delay in mitotic entry. Using thymidine synchronization techniques (Figs. 4–6), we observed that MELK inhibition caused delayed mitotic entry, likely mediated through delayed activation of CDK1, which was ultimately overcome to allow cells to complete M and begin the subsequent cell cycle. These observations are consistent with a transient activation of the G2/M checkpoint (54). Further studies are required to elucidate the underlying mechanism, although it is possible to speculate based upon the work of others.
One potential explanation for the observed G2 delay phenotype is that MELK positively regulates CDC25B in G2, such that inhibition of MELK causes CDC25B to be unable to dephosphorylate and activate CDK1, thereby delaying mitotic entry. MELK phosphorylates multiple sites on CDC25B to regulate both progression through the G2/M checkpoint and CDC25B localization to centrosomes during M (38, 60). Importantly, this putative interaction has never been examined in the context of cancer. A possible interaction with CDC25C, which has a critical role at the G2/M checkpoint and shares high sequence homology with CDC25B, has not been explored either. Additional studies are needed to more clearly define the interaction between MELK and the CDC25 phosphatases in cancer. An alternative hypothesis is that MELK inhibition causes DNA damage, which subsequently activates the G2/M checkpoint. A study by Beke et al. (39) found that MELK inhibition with MELK-T1 caused replication stress in MCF-7 cells, manifesting in replication fork stalling and DNA double-strand breaks. This resulted in activation of the ATM/Chk2 DNA damage repair pathway, which stimulates the G2/M checkpoint (39, 61). Although this pathway would certainly explain the putative checkpoint activation, one would also expect strong ATM activation to lengthen S phase, which was not observed in our study (Fig. 4).
In conclusion, we have validated 8a as a highly selective MELK inhibitor suitable for functional studies and have shown that MELK inhibition causes a mitotic delay consistent with activation of the G2/M checkpoint.
Experimental procedures
Antibodies and reagents
MELK-8a hydrochloride was purchased from MedChemExpress (HY-100368A). OTSSP167 was purchased from Selleck Chemicals (S7159). HTH-01-091 was a generous gift from Dr. Nathanael Gray (Harvard University). The following primary antibodies were used in this study. Aurora A (14475, 1:1000), Aurora B (3094, 1:1000), CDC25B (9525, 1:1000), cyclin A2 (4656, 1:2000), cyclin E1 (4129, 1:2500), MELK (2274, 1:1000), Myt1 (4282, 1:1000), PARP (9532, 1:1000), phospho-Aurora A/B/C (2914, 1:1000), phospho-CDK1 (Thr-14) (2543, 1:1000), phospho-CDK1 (Tyr-15) (4539, 1:1000), and p-H3 (Ser-10) (9701, 1:1000) were purchased from Cell Signaling Technology. β-Actin (SC47778, 1:500) was purchased from Santa Cruz Biotechnology, Inc. CDK1 (MAB8878, 1:2000) was purchased from Millipore. Cyclin B1 (ab32053, 1:10,000) was purchased from Abcam.
Cell culture and viability assays
MDA-MB-468 and MDA-MB-231 cells were kindly provided by Dr. Gary Johnson (University of North Carolina). These cell lines were cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS; Millipore) and 1% antibiotic-antimycotic (Sigma). HeLaS3 cells were kindly provided by Dr. Michael Emanuele (University of North Carolina) and were cultured in Dulbecco's modified Eagle's medium (DMEM), high-glucose (Gibco), supplemented with 10% FBS and 1% antibiotic-antimycotic.
Resazurin assays were completed as described previously (62). Briefly, MDA-MB-231 (1500 cells/well), MDA-MB-468 (4000 cells/well), or HeLaS3 (1000 cells/well) cells were plated on a 96-well plate. After 24 h, medium was aspirated and replaced with fresh medium containing 8a at concentrations ranging from 1 nm to 30 μm or DMSO (negative control). Each treatment condition was tested in technical quadruplicate. Cells were incubated in medium with 8a or DMSO for 72 h prior to the addition of resazurin dye (0.6 mm, Acros Organics 62758-13-8). After color change was observed (3 h for MDA-MB-231, 2 h for MDA-MB-468, 0.75 h for HeLaS3), signaling that measurable reduction of resazurin to resorufin had occurred, fluorescence intensity was measured using a PHERAstar (BMG Labtech) plate reader with fluorescence module (FI: 540-20, 590-20). Dose-response curves were created, and IC50 values were calculated using GraphPad Prism version 8.
For crystal violet assays, MDA-MB-231 and MDA-MB-468 cells were seeded at a density of 5000 and 10,000 cells/well, respectively, on each well of a cell culture-treated 6-well plate (Corning, Inc.). The following day, medium was aspirated from all wells and replaced with medium supplemented with either DMSO or 0.1, 0.3, 1, 3, or 10 μm 8a. Every 2 days throughout the course of the assay, medium was replaced on all wells with fresh medium containing 8a or DMSO. Cells were observed daily using a bright-field microscope, and confluence was noted. The assay was continued until cells in the DMSO well were observed to be nearly confluent (∼8 days for MDA-MB-231 and ∼14 days for MDA-MB-468). Medium was then aspirated from all wells, and cells were washed gently with ice-cold PBS (Gibco). Next, 2 ml of crystal violet stain (0.5% crystal violet (w/v) (Sigma), 20% methanol (Fisher)) was gently added to each well, and plates incubated for 10 min at room temperature. Wells were subsequently washed three times with water and allowed to dry, and plates were imaged with a Canon LiDE 110 document scanner.
Double-thymidine synchronization
HeLaS3 cells were seeded on 6-cm plates and allowed to adhere overnight. During this and all subsequent incubation steps, cells were maintained in a humidified chamber with 5% CO2 at 37 °C. DMEM was aspirated from all plates (except for asynchronous control plates, which were maintained in an incubator until the end of the assay) and replaced with medium supplemented with 2 mm thymidine (Sigma). 16 h after thymidine block, medium was aspirated, plates were washed with warm PBS (1×) and warm DMEM (2×), and fresh medium was added for release from block. Plates were incubated for 9 h, at which point medium was aspirated and again replaced with DMEM supplemented with 2 mm thymidine and incubated for 16 h. Medium was aspirated and cells were washed as above and released from thymidine block into DMEM supplemented with 8a or DMSO. 0-h time points were collected at the time of release, and subsequent collections were completed at the time points shown in the figures. Cells were collected by scraping in medium, and cell suspension was transferred to a 15-ml conical tube. Cells were pelleted at 3000 rcf in a centrifuge with swinging bucket rotor, washed with cold PBS, and stored at −80 °C. After all time points were collected, cell pellets were lysed and prepared for immunoblotting as described below.
Immunoblotting
HeLaS3 cells were plated in 6-well plates and treated with DMSO or 0.5, 1, or 3 μm 8a for 1, 2, 4, 8, 12, or 24 h. Cells were collected as described above for double-thymidine synchronization protocol. Pellets from asynchronous and synchronized experiments were lysed by the addition of radioimmune precipitation assay buffer (20 mm Tris, pH 7.4, 137 mm NaCl, 10% glycerol, 0.5% sodium deoxycholate, 1% Nonidet P-40 substitute (Fluka), 0.1% SDS, 2 mm EDTA) supplemented with protease and phosphatase inhibitors (2 mm Na(VO3)4, 10 mm NaF, 0.0125 μm calyculin A, and cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science). Benzonase was added to each lysate (42 units/sample), and samples were incubated on ice for 10 min. Lysates were clarified by centrifugation at 21,000 rcf, 4 °C for 15 min. Protein concentrations were normalized by Bradford assay (Bio-Rad) and prepared for SDS-PAGE by the addition of 4× Laemmli sample buffer (250 mm Tris, pH 6.8, 40% glycerol, 8% SDS, 8% β-mercaptoethanol, 0.4% bromphenol blue). Samples (10–30 μg) were applied to a 10% SDS-polyacrylamide gel for separation of proteins, after which proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad). Membranes were blocked for 1 h with 5% nonfat dry milk or 5% BSA (for phosphospecific antibodies) in TBS-T (20 mm Tris, pH 7.6, 137 mm NaCl, 0.05% Tween 20). Membranes were incubated in primary antibody dilutions (described above) in 5% BSA made in TBS-T at 4 °C with gentle shaking. Following overnight incubation, membranes were washed with TBS-T three times and incubated for 1 h at room temperature with anti-mouse or -rabbit IgG-HRP–conjugated secondary antibody (Promega) dilutions in 5% milk in TBS-T. Membranes were again washed three times in TBS-T, and bands were imaged using a Chemi-Doc Touch Imaging System (Bio-Rad) after the addition of Clarity ECL reagent (Bio-Rad).
MIB/MS selectivity-profiling sample preparation
The selectivity of MELK inhibitors for kinase targets was profiled using competition MIB/MS. MDA-MB-468 cells were treated with MELK inhibitor, at concentrations listed in the figure legends, or DMSO for 30 min. This time point is sufficient for kinase engagement and inhibition by compound, but insufficient for major expression-level changes. Cells were then washed twice with ice-cold PBS before being scrape-harvested in PBS. Cell lysis, sample preparation, and MIB kinase enrichment were completed as described previously (63), with the exception of the composition of the kinase inhibitor mix used for enrichment. To enrich for kinases in each sample, a 350-μl volume of the following kinase inhibitors, immobilized on beads, was applied to one Poly-Prep® chromatography column (Bio-Rad) per sample: CTx-0294885, PP58, Purvalanol B, UNC2147A, VI-16832, and UNC8088A.
LC-MS/MS analysis
Samples were analyzed by LC-MS/MS using a Thermo Easy nLC 1200 coupled to an Orbitrap Q Exactive HF mass spectrometer equipped with an EasySpray nano source. Samples were loaded onto an EasySpray C18 column (75-μm inner diameter × 25 cm, 2-μm particle size) and eluted over a 120-min method. The gradient for separation consisted of 5–40% B at a 250-nl/min flow rate, where mobile phase A was water, 0.1% formic acid, and mobile phase B was 80% acetonitrile, 0.1% formic acid. The Q Exactive HF was operated in data-dependent mode, where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 350–1700) was set to 120,000 with a target value of 3 × 106 ions, 100-ms maximum injection time. MS/MS scans resolution was set to 15,000 with a target value of 1 × 105 ions, 75-ms maximum injection time. The normalized collision energy was set to 27% for higher-energy collisional dissociation. Dynamic exclusion was set to 30 s, and precursors with unknown charge or a charge state of 1 and ≥8 were excluded.
MS data analysis
Data were processed using the MaxQuant software suite (version 1.6.1.0) (64, 65). The data were searched against a reviewed Uniprot human database (downloaded in February 2018) containing 20,245 sequences. Precursor mass tolerance was set to 4.5 ppm, and fragment mass tolerance was set to 20 ppm. A maximum of two missed tryptic cleavages were allowed. The fixed modification specified was carbamidomethylation of cysteine residues. The variable modification specified was oxidation of methionine. A default protein false discovery rate (FDR) of 1% was used to filter all data within MaxQuant. Proteins were quantified across all samples using MaxLFQ (66). Matching between runs was allowed with the default retention time window. Kinases were parsed from the data set, and those with >1 unique peptide were quantified using label-free quantification (LFQ). Kinases with >50% missing values were removed, and LFQ intensities for the missing values were imputed using a constant value (average lower 10% of all LFQ intensities). The ratio of the LFQ intensity for each kinase in MELK inhibitor conditions to that kinase in DMSO control was computed. GraphPad Prism version 8 was used to generate bar plots and heat maps. For data used to generate volcano plots, kinases that were not quantified in all three replicates in at least one condition within each analysis were removed. Missing LFQ values were then imputed by randomly sampling from a normal distribution with a mean of 1.8 S.D. values lower than the mean in the original data, and an S.D. value of 0.3 times the S.D. in the original data. Data were log2-transformed, and moderated t tests were computed using the Limma package (67). Briefly, a linear model predicting signal intensity given treatment condition was fit for each kinase, followed by t statistics calculated by empirical Bayes moderation of S.E. values toward the S.E. estimated from all kinases. Multiple-test correction was performed using the Benjamini–Hochberg method to control for a 5% FDR (68). The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (69) partner repository with the data set identifier PXD016022.
Live-cell imaging
U2OS cells were a gift from Dr. Michael Whitfield (Dartmouth College) and maintained in DMEM (Sigma) supplemented with 10% FBS (Sigma), 1× penicillin-streptomycin, and 4 mm l-glutamine and incubated at 5% CO2. Cells were transduced with fluorescent PCNA (pLenti-Hyg-turq2-PCNA) and histone H2B (pBabe-Puro-mCh-H2B) using standard techniques, as described previously (55). One day prior to imaging, cells were plated on #1.5 glass-bottom plates (Cellvis) in FluoroBrite DMEM (Gibco) supplemented with FBS, penicillin-streptomycin, and l-glutamine, as above. Imaging was performed on a Nikon Ti Eclipse inverted microscope using a 40× (numerical aperture 0.95) Plan Apochromat dry objective lens and the Nikon Perfect Focus System. Still images were captured using an Andor Zyla 4.2 sCMOS detector with 12-bit resolution. During imaging, cells were maintained in a humidified chamber with 5% CO2 at 37 °C. Filter sets were as follows: CFP, 436/20 nm; 455 nm; 480/40 nm (excitation; beam splitter; emission filter) and mCherry, 560/40 nm; 585 nm; 630/75 nm (Chroma). Images were obtained every 6 min using NIS-Elements AR software. No photobleaching or phototoxicity was observed in cells imaged with this protocol.
8a or DMSO (negative control) was added 24 h after the beginning of the imaging run as described in the figure legends. Cells were manually tracked and scored. The onset and end of S phase were determined visually using the DNA replication–associated patterns of PCNA localization in the nucleus. Mitosis length was defined visually by the length of time between nuclear envelope breakdown and cytokinesis. Three individual biological replicates were imaged with 50 cells counted per condition per replicate. Cells that traveled out of the field of view or did not complete a cell cycle (S phase to cytokinesis) were excluded from the analysis.
Author contributions
I. M. M., G. D. G., M. P. E., T. S. G., C. V., J. G. C., M. J. E., and L. M. G. conceptualization; I. M. M., G. D. G., M. P. E., E. M. W., D. G., J. B., L. E. H., and C. V. data curation; I. M. M., G. D. G., M. P. E., D. G., J. B., L. E. H., and C. V. formal analysis; I. M. M., J. G. C., and L. M. G. funding acquisition; I. M. M., L. E. H., and G. D. G. writing-original draft; I. M. M., M. P. E., T. S. G., J. G. C., M. J. E., and L. M. G. writing-review and editing; G. D. G., M. P. E., T. S. G., E. M. W., L. E. H., and C. V. methodology; J. G. C. resources.
Supplementary Material
Acknowledgments
We thank Thomas Bonacci for sharing protocols and reagents, as well as Emily Fennell and Lucas Aponte-Collazo for assistance with experimental troubleshooting. We also thank Dr. Nathanael Gray for the generous gift of the compound HTH-01-091. This research was based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
This work was supported by National Institutes of Health Grants R01 CA199064 (to L. M. G.), GM083024 and GM102413 (to J. G. C.), and T32 CA009156 (to G. D. G.). This work was also supported in part by a UNC Dissertation Completion Fellowship (to I. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (69) partner repository with the data set identifier PXD016022.
This article contains Table S1 and Figs. S1–S7.
- MELK
- maternal embryonic leucine zipper kinase
- MIB
- multiplexed kinase inhibitor beads
- CDK1
- cyclin-dependent kinase 1
- PCNA
- proliferating cell nuclear antigen
- TNBC
- triple-negative breast cancer
- M
- mitosis
- OTS
- OTSSP167
- 8a
- NVS-MELK8a
- HTH
- HTH-01-091
- FDR
- false discovery rate
- LFQ
- label-free quantification
- PARP
- poly(ADP-ribose) polymerase
- p-H3
- phospho-histone H3
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- rcf
- relative centrifugal force.
References
- 1. Heyer B. S., Warsowe J., Solter D., Knowles B. B., and Ackerman S. L. (1997) New member of the Snf1/AMPK kinase family, Melk, is expressed in the mouse egg and preimplantation embryo. Mol. Reprod. Dev. 47, 148–156 [DOI] [PubMed] [Google Scholar]
- 2. Ganguly R., Mohyeldin A., Thiel J., Kornblum H. I., Beullens M., and Nakano I. (2015) MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin. Transl. Med. 4, 11 10.1186/s40169-014-0045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gray D., Jubb A. M., Hogue D., Dowd P., Kljavin N., Yi S., Bai W., Frantz G., Zhang Z., Koeppen H., de Sauvage F. J., and Davis D. P. (2005) Maternal embryonic leucine zipper kinase/murine protein serine-threonine kinase 38 is a promising therapeutic target for multiple cancers. Cancer Res. 65, 9751–9761 10.1158/0008-5472.CAN-04-4531 [DOI] [PubMed] [Google Scholar]
- 4. Wang Y., Lee Y.-M., Baitsch L., Huang A., Xiang Y., Tong H., Lako A., Von T., Choi C., Lim E., Min J., Li L., Stegmeier F., Schlegel R., Eck M. J., et al. (2014) MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife 3, e01763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakano I., Masterman-Smith M., Saigusa K., Paucar A. A., Horvath S., Shoemaker L., Watanabe M., Negro A., Bajpai R., Howes A., Lelievre V., Waschek J. A., Lazareff J. A., Freije W. A., Liau L. M., et al. (2008) Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J. Neurosci. Res. 86, 48–60 10.1002/jnr.21471 [DOI] [PubMed] [Google Scholar]
- 6. Pickard M. R., Green A. R., Ellis I. O., Caldas C., Hedge V. L., Mourtada-Maarabouni M., and Williams G. T. (2009) Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 11, R60 10.1186/bcr2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Speers C., Zhao S. G., Kothari V., Santola A., Liu M., Wilder-Romans K., Evans J., Batra N., Bartelink H., Hayes D. F., Lawrence T. S., Brown P. H., Pierce L. J., and Feng F. Y. (2016) Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin. Cancer Res. 22, 5864–5875 10.1158/1078-0432.CCR-15-2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Minata M., Gu C., Joshi K., Nakano-Okuno M., Hong C., Nguyen C.-H., Kornblum H. I., Molla A., and Nakano I. (2014) Multi-kinase inhibitor C1 triggers mitotic catastrophe of glioma stem cells mainly through MELK kinase inhibition. PLoS ONE 9, e92546 10.1371/journal.pone.0092546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia H., Kong S. N., Chen J., Shi M., Sekar K., Seshachalam V. P., Rajasekaran M., Goh B. K. P., Ooi L. L., and Hui K. M. (2016) MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence. Cancer Lett. 383, 85–93 10.1016/j.canlet.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 10. Guan S., Lu J., Zhao Y., Yu Y., Li H., Chen Z., Shi Z., Liang H., Wang M., Guo K., Chen X., Sun W., Bieerkehazhi S., Xu X., Sun S., et al. (2018) MELK is a novel therapeutic target in high-risk neuroblastoma. Oncotarget 9, 2591–2602 10.18632/oncotarget.23515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alachkar H., Mutonga M. B., Metzeler K. H., Fulton N., Malnassy G., Herold T., Spiekermann K., Bohlander S. K., Hiddemann W., Matsuo Y., Stock W., and Nakamura Y. (2014) Preclinical efficacy of maternal embryonic leucine-zipper kinase (MELK) inhibition in acute myeloid leukemia. Oncotarget 5, 12371–12382 10.18632/oncotarget.2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hebbard L. W., Maurer J., Miller A., Lesperance J., Hassell J., Oshima R. G., and Terskikh A. V. (2010) Maternal embryonic leucine zipper kinase is upregulated and required in mammary tumor-initiating cells in vivo. Cancer Res. 70, 8863–8873 10.1158/0008-5472.CAN-10-1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janostiak R., Rauniyar N., Lam T. T., Ou J., Zhu L. J., Green M. R., and Wajapeyee N. (2017) MELK promotes melanoma growth by stimulating the NF-κB pathway. Cell Rep. 21, 2829–2841 10.1016/j.celrep.2017.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin A., Giuliano C. J., Sayles N. M., and Sheltzer J. M. (2017) CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife 6, e24179 10.7554/eLife.24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang H.-T., Seo H.-S., Zhang T., Wang Y., Jiang B., Li Q., Buckley D. L., Nabet B., Roberts J. M., Paulk J., Dastjerdi S., Winter G. E., McLauchlan H., Moran J., Bradner J. E., et al. (2017) MELK is not necessary for the proliferation of basal-like breast cancer cells. Elife 6, e26693 10.7554/eLife.26693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giuliano C. J., Lin A., Smith J. C., Palladino A. C., and Sheltzer J. M. (2018) MELK expression correlates with tumor mitotic activity but is not required for cancer growth. Elife 7, e32838 10.7554/eLife.32838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y., Zhou X., Li Y., Xu Y., Lu K., Li P., and Wang X. (2018) Inhibition of maternal embryonic leucine zipper kinase with OTSSP167 displays potent anti-leukemic effects in chronic lymphocytic leukemia. Oncogene 37, 5520–5533 10.1038/s41388-018-0333-x [DOI] [PubMed] [Google Scholar]
- 18. Wang Y., Li B. B., Li J., Roberts T. M., and Zhao J. J. (2018) A conditional dependency on MELK for the proliferation of triple-negative breast cancer cells. iScience 9, 149–160 10.1016/j.isci.2018.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malumbres M., Sotillo R., Santamaría D., Galán J., Cerezo A., Ortega S., Dubus P., and Barbacid M. (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504 10.1016/j.cell.2004.08.002 [DOI] [PubMed] [Google Scholar]
- 20. Malumbres M., and Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 10.1038/nrc2602 [DOI] [PubMed] [Google Scholar]
- 21. Choi Y. J., Li X., Hydbring P., Sanda T., Stefano J., Christie A. L., Signoretti S., Look A. T., Kung A. L., von Boehmer H., and Sicinski P. (2012) The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451 10.1016/j.ccr.2012.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otto T., and Sicinski P. (2017) Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115 10.1038/nrc.2016.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Currie C. E., Mora-Santos M., Smith C. A., McAinsh A. D., and Millar J. B. A. (2018) Bub1 is not essential for the checkpoint response to unattached kinetochores in diploid human cells. Curr. Biol. 28, R929–R930 10.1016/j.cub.2018.07.040 [DOI] [PubMed] [Google Scholar]
- 24. Raaijmakers J. A., van Heesbeen R. G. H. P., Blomen V. A., Janssen L. M. E., van Diemen F., Brummelkamp T. R., and Medema R. H. (2018) BUB1 is essential for the viability of human cells in which the spindle assembly checkpoint is compromised. Cell Rep. 22, 1424–1438 10.1016/j.celrep.2018.01.034 [DOI] [PubMed] [Google Scholar]
- 25. Rodriguez-Rodriguez J. A., Lewis C., McKinley K. L., Sikirzhytski V., Corona J., Maciejowski J., Khodjakov A., Cheeseman I. M., and Jallepalli P. V. (2018) Distinct roles of RZZ and Bub1-KNL1 in mitotic checkpoint signaling and kinetochore expansion. Curr. Biol. 28, 3422–3429.e5 10.1016/j.cub.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang G., Kruse T., Guasch Boldú C., Garvanska D. H., Coscia F., Mann M., Barisic M., and Nilsson J. (2019) Efficient mitotic checkpoint signaling depends on integrated activities of Bub1 and the RZZ complex. EMBO J. 38, e100977 10.15252/embj.2018100977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meraldi P. (2019) Bub1—the zombie protein that CRISPR cannot kill. EMBO J. 38, e101912 10.15252/embj.2019101912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joshi K., Banasavadi-Siddegowda Y., Mo X., Kim S. H., Mao P., Kig C., Nardini D., Sobol R. W., Chow L. M. L., Kornblum H. I., Waclaw R., Beullens M., and Nakano I. (2013) MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 31, 1051–1063 10.1002/stem.1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakano I., Paucar A. A., Bajpai R., Dougherty J. D., Zewail A., Kelly T. K., Kim K. J., Ou J., Groszer M., Imura T., Freije W. A., Nelson S. F., Sofroniew M. V., Wu H., Liu X., et al. (2005) Maternal embryonic leucine zipper kinase (MELK) regulates multipotent neural progenitor proliferation. J. Cell Biol. 170, 413–427 10.1083/jcb.200412115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gu C., Banasavadi-Siddegowda Y. K., Joshi K., Nakamura Y., Kurt H., Gupta S., and Nakano I. (2013) Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells 31, 870–881 10.1002/stem.1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y., Begley M., Li Q., Huang H.-T., Lako A., Eck M. J., Gray N. S., Mitchison T. J., Cantley L. C., and Zhao J. J. (2016) Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc. Natl. Acad. Sci. U.S.A. 113, 9810–9815 10.1073/pnas.1606862113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin M.-L., Park J.-H., Nishidate T., Nakamura Y., and Katagiri T. (2007) Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 9, R17 10.1186/bcr1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jung H., Seong H.-A., and Ha H. (2008) Murine protein serine/threonine kinase 38 activates apoptosis signal-regulating kinase 1 via Thr838 phosphorylation. J. Biol. Chem. 283, 34541–34553 10.1074/jbc.M807219200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pitner M. K., Taliaferro J. M., Dalby K. N., and Bartholomeusz C. (2017) MELK: a potential novel therapeutic target for TNBC and other aggressive malignancies. Expert Opin. Ther. Targets 21, 849–859 10.1080/14728222.2017.1363183 [DOI] [PubMed] [Google Scholar]
- 35. Badouel C., Chartrain I., Blot J., and Tassan J.-P. (2010) Maternal embryonic leucine zipper kinase is stabilized in mitosis by phosphorylation and is partially degraded upon mitotic exit. Exp. Cell Res. 316, 2166–2173 10.1016/j.yexcr.2010.04.019 [DOI] [PubMed] [Google Scholar]
- 36. Grant G. D., Brooks L. 3rd, Zhang X., Mahoney J. M., Martyanov V., Wood T. A., Sherlock G., Cheng C., and Whitfield M. L. (2013) Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell. 24, 3634–3650 10.1091/mbc.e13-05-0264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li S., Li Z., Guo T., Xing X.-F., Cheng X., Du H., Wen X.-Z., and Ji J.-F. (2016) Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer. Oncotarget 7, 6266–6280 10.18632/oncotarget.6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davezac N., Baldin V., Blot J., Ducommun B., and Tassan J.-P. (2002) Human pEg3 kinase associates with and phosphorylates CDC25B phosphatase: a potential role for pEg3 in cell cycle regulation. Oncogene 21, 7630–7641 10.1038/sj.onc.1205870 [DOI] [PubMed] [Google Scholar]
- 39. Beke L., Kig C., Linders J. T. M., Boens S., Boeckx A., van Heerde E., Parade M., De Bondt A., Van den Wyngaert I., Bashir T., Ogata S., Meerpoel L., Van Eynde A., Johnson C. N., Beullens M., et al. (2015) MELK-T1, a small-molecule inhibitor of protein kinase MELK, decreases DNA-damage tolerance in proliferating cancer cells. Biosci. Rep. 35, e00267 10.1042/BSR20150194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung S., Suzuki H., Miyamoto T., Takamatsu N., Tatsuguchi A., Ueda K., Kijima K., Nakamura Y., and Matsuo Y. (2012) Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 3, 1629–1640 10.18632/oncotarget.790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chlenski A., Park C., Dobratic M., Salwen H. R., Budke B., Park J.-H., Miller R., Applebaum M. A., Wilkinson E., Nakamura Y., Connell P. P., and Cohn S. L. (2019) Maternal embryonic leucine zipper kinase (MELK), a potential therapeutic target for neuroblastoma. Mol. Cancer Ther. 18, 507–516 10.1158/1535-7163.MCT-18-0819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J., Reiter K., Qiao H., Helm D., Koch H., et al. (2017) The target landscape of clinical kinase drugs. Science 358, eaan4368 10.1126/science.aan4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ji W., Arnst C., Tipton A. R., Bekier M. E. 2nd, Taylor W. R., Yen T. J., and Liu S.-T. (2016) OTSSP167 abrogates mitotic checkpoint through inhibiting multiple mitotic kinases. PLoS ONE 11, e0153518 10.1371/journal.pone.0153518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Touré B. B., Giraldes J., Smith T., Sprague E. R., Wang Y., Mathieu S., Chen Z., Mishina Y., Feng Y., Yan-Neale Y., Shakya S., Chen D., Meyer M., Puleo D., Brazell J. T., et al. (2016) Toward the validation of maternal embryonic leucine zipper kinase: discovery, optimization of highly potent and selective inhibitors, and preliminary biology insight. J. Med. Chem. 59, 4711–4723 10.1021/acs.jmedchem.6b00052 [DOI] [PubMed] [Google Scholar]
- 45. Edupuganti R., Taliaferro J. M., Wang Q., Xie X., Cho E. J., Vidhu F., Ren P., Anslyn E. V., Bartholomeusz C., and Dalby K. N. (2017) Discovery of a potent inhibitor of MELK that inhibits expression of the anti-apoptotic protein Mcl-1 and TNBC cell growth. Bioorg. Med. Chem. 25, 2609–2616 10.1016/j.bmc.2017.03.018 [DOI] [PubMed] [Google Scholar]
- 46. Boutard N., Sabiniarz A., Czerwińska K., Jarosz M., Cierpich A., Kolasińska E., Wiklik K., Gluza K., Commandeur C., Buda A., Stasiowska A., Bobowska A., Galek M., Fabritius C. H., Bugaj M., et al. (2019) 5-Keto-3-cyano-2,4-diaminothiophenes as selective maternal embryonic leucine zipper kinase inhibitors. Bioorg. Med. Chem. Lett. 29, 607–613 10.1016/j.bmcl.2018.12.051 [DOI] [PubMed] [Google Scholar]
- 47. Duncan J. S., Whittle M. C., Nakamura K., Abell A. N., Midland A. A., Zawistowski J. S., Johnson N. L., Granger D. A., Jordan N. V., Darr D. B., Usary J., Kuan P.-F., Smalley D. M., Major B., He X., et al. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 10.1016/j.cell.2012.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krulikas L. J., McDonald I. M., Lee B., Okumu D. O., East M. P., Gilbert T. S. K., Herring L. E., Golitz B. T., Wells C. I., Axtman A. D., Zuercher W. J., Willson T. M., Kireev D., Yeh J. J., Johnson G. L., et al. (2018) Application of integrated drug screening/kinome analysis to identify inhibitors of gemcitabine-resistant pancreatic cancer cell growth. SLAS Discov. 23, 850–861 10.1177/2472555218773045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stuhlmiller T. J., Miller S. M., Zawistowski J. S., Nakamura K., Beltran A. S., Duncan J. S., Angus S. P., Collins K. A. L., Granger D. A., Reuther R. A., Graves L. M., Gomez S. M., Kuan P.-F., Parker J. S., Chen X., et al. (2015) Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404 10.1016/j.celrep.2015.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Crosio C., Fimia G. M., Loury R., Kimura M., Okano Y., Zhou H., Sen S., Allis C. D., and Sassone-Corsi P. (2002) Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian aurora kinases. Mol. Cell. Biol. 22, 874–885 10.1128/MCB.22.3.874-885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirota T., Kunitoku N., Sasayama T., Marumoto T., Zhang D., Nitta M., Hatakeyama K., and Saya H. (2003) Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 114, 585–598 10.1016/S0092-8674(03)00642-1 [DOI] [PubMed] [Google Scholar]
- 52. Yasui Y., Urano T., Kawajiri A., Nagata K., Tatsuka M., Saya H., Furukawa K., Takahashi T., Izawa I., and Inagaki M. (2004) Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J. Biol. Chem. 279, 12997–13003 10.1074/jbc.M311128200 [DOI] [PubMed] [Google Scholar]
- 53. Lindqvist A., Rodríguez-Bravo V., and Medema R. H. (2009) The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 185, 193–202 10.1083/jcb.200812045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaltiel I. A., Krenning L., Bruinsma W., and Medema R. H. (2015) The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 128, 607–620 10.1242/jcs.163766 [DOI] [PubMed] [Google Scholar]
- 55. Grant G. D., Kedziora K. M., Limas J. C., Cook J. G., and Purvis J. E. (2018) Accurate delineation of cell cycle phase transitions in living cells with PIP-FUCCI. Cell Cycle 17, 2496–2516 10.1080/15384101.2018.1547001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Offenburger S.-L., Bensaddek D., Murillo A. B., Lamond A. I., and Gartner A. (2017) Comparative genetic, proteomic and phosphoproteomic analysis of C. elegans embryos with a focus on ham-1/STOX and pig-1/MELK in dopaminergic neuron development. Sci. Rep. 7, 4314 10.1038/s41598-017-04375-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Du T., Qu Y., Li J., Li H., Su L., Zhou Q., Yan M., Li C., Zhu Z., and Liu B. (2014) Maternal embryonic leucine zipper kinase enhances gastric cancer progression via the FAK/paxillin pathway. Mol. Cancer 13, 100 10.1186/1476-4598-13-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Le Page Y., Chartrain I., Badouel C., and Tassan J. P. (2011) A functional analysis of MELK in cell division reveals a transition in the mode of cytokinesis during Xenopus development. J. Cell Sci. 124, 958–968 10.1242/jcs.069567 [DOI] [PubMed] [Google Scholar]
- 59. Chartrain I., Le Page Y., Hatte G., Körner R., Kubiak J. Z., and Tassan J. P. (2013) Cell-cycle dependent localization of MELK and its new partner RACK1 in epithelial versus mesenchyme-like cells in Xenopus embryo. Biol. Open 2, 1037–1048 10.1242/bio.20136080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mirey G., Chartrain I., Froment C., Quaranta M., Bouché J.-P., Monsarrat B., Tassan J.-P., and Ducommun B. (2005) CDC25B phosphorylated by pEg3 localizes to the centrosome and the spindle poles at mitosis. Cell Cycle 4, 806–811 10.4161/cc.4.6.1716 [DOI] [PubMed] [Google Scholar]
- 61. Guleria A., and Chandna S. (2016) ATM kinase: much more than a DNA damage responsive protein. DNA Repair (Amst.) 39, 1–20 10.1016/j.dnarep.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 62. Graves P. R., Aponte-Collazo L. J., Fennell E. M. J., Graves A. C., Hale A. E., Dicheva N., Herring L. E., Gilbert T. S. K., East M. P., McDonald I. M., Lockett M. R., Ashamalla H., Moorman N. J., Karanewsky D. S., Iwanowicz E. J., et al. (2019) Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem. Biol. 14, 1020–1029 10.1021/acschembio.9b00222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arend K. C., Lenarcic E. M., Vincent H. A., Rashid N., Lazear E., McDonald I. M., Gilbert T. S. K., East M. P., Herring L. E., Johnson G. L., Graves L. M., and Moorman N. J. (2017) Kinome profiling identifies druggable targets for novel human cytomegalovirus (HCMV) antivirals. Mol. Cell. Proteomics 16, S263–S276 10.1074/mcp.M116.065375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 65. Cox J., Michalski A., and Mann M. (2011) Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectrom. 22, 1373–1380 10.1007/s13361-011-0142-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 10.1074/mcp.M113.031591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 1–25 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- 68. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 [Google Scholar]
- 69. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yılmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.