

Abstract
Multiple observations implicate T-cell dysregulation as a central event in the pathogenesis of rheumatoid arthritis. Here, we investigated mechanisms for suppressing T-cell activation via the inhibitory receptor leukocyte-associated immunoglobulin-like receptor 1 (LAIR-1). To determine how LAIR-1 affects T-cell receptor (TCR) signaling, we compared 1) T cells from LAIR-1–sufficient and –deficient mice, 2) Jurkat cells expressing either LAIR-1 mutants or C-terminal Src kinase (CSK) mutants, and 3) T cells from mice that contain a CSK transgene susceptible to chemical inhibition. Our results indicated that LAIR-1 engagement by collagen or by complement C1q (C1Q, which contains a collagen-like domain) inhibits TCR signaling by decreasing the phosphorylation of key components in the canonical T-cell signaling pathway, including LCK proto-oncogene SRC family tyrosine kinase (LCK), LYN proto-oncogene SRC family tyrosine kinase (LYN), ζ chain of T-cell receptor–associated protein kinase 70 (ZAP-70), and three mitogen-activated protein kinases (extracellular signal–regulated kinase, c-Jun N-terminal kinase 1/2, and p38). The intracellular region of LAIR-1 contains two immunoreceptor tyrosine-based inhibition motifs that are both phosphorylated by LAIR-1 activation, and immunoprecipitation experiments revealed that Tyr-251 in LAIR-1 binds CSK. Using CRISPR/Cas9-mediated genome editing, we demonstrate that CSK is essential for the LAIR-1–induced inhibition of the human TCR signal transduction. T cells from mice that expressed a PP1 analog–sensitive form of CSK (CskAS) corroborated these findings, and we also found that Tyr-251 is critical for LAIR-1's inhibitory function. We propose that LAIR-1 activation may be a strategy for controlling inflammation and may offer a potential therapeutic approach for managing autoimmune diseases.
Keywords: signal transduction, receptor, autoimmunity, collagen, inflammation, CD305, kinase signaling, leukocyte-associated immunoglobulin-like receptor 1, rheumatoid arthritis
Introduction
Inhibitory receptors serve as a critical counterbalance to hold the immune system in check and protect against tissue damage induced by hyperactive immune responses or autoimmune dysfunction. Although cells may express multiple inhibitory receptors, some individual receptors are important in the regulation of particular immune functions (1). One such receptor is leukocyte-associated Ig-like receptor 1 (LAIR-1)2 or CD305. Human LAIR-1 was first cloned in 1997; its structure was delineated, and its function as a regulator of NK cells was established (1). LAIR-1 contributes to the regulation of the immune system by binding to extracellular ligands and delivering a signal to immune cells. The ligands to which LAIR-1 binds are collagenous structures. Multiple collagens can serve as functional, high-affinity ligands for LAIR-1, including transmembrane collagens, extracellular matrix collagens, and the collagenous portion of the complement protein C1q. Structurally, LAIR-1 has an extracellular binding domain coupled to a cytoplasmic tail containing two immunoreceptor tyrosine-based inhibition motifs (ITIMs). When LAIR-1 is activated by binding to a collagenous ligand, both of the ITIMs are phosphorylated, and additional downstream events are triggered.
We have previously shown that activation of LAIR-1 can prevent collagen-induced autoimmune arthritis (CIA) (2). The precise mechanism of this activity is unknown, but immune regulation is critical. Although LAIR-1 is found on multiple immune cells, T cells were selected as the focus of this study because both Th1 and Th17 CD4+ T cells play a prominent role in the initiation of systemic immune responses in CIA. It has previously been shown that a subset of T cells, including naïve T cells, express high levels of LAIR-1 (3). It is also known that LAIR-1 down-regulates T-cell receptor (TCR) signal transduction, but there is no comprehensive understanding of the mechanism by which LAIR-1 exerts this activity. This study explores the mechanism by which LAIR-1 modifies T-cell function and prevents signaling through the TCR.
To accomplish these experiments, we utilized several transgenic mouse strains. The DR1 strain is a partially humanized strain developed to study experimental autoimmune arthritis (4). It has a C57Bl/6 background with a chimeric human/mouse immune response transgene and is highly susceptible to CIA. We have bred two variants of this strain, one of which is also deficient in LAIR-1 (2). We also used a mouse expressing a mutant form of C-terminal Src kinase (CskAS). Csk is the primary negative regulator of Src-family kinases (5). CskAS mice express a conditionally inhibitory CSK protein and lack endogenous CSK expression. CskAS catalytic activity is specifically and rapidly inhibited by 3-iodo-benzyl-PP1 (3-IB-PP1).
Collagen exists in tissue as a trimer that is extensively cross-linked. During inflammation, collagen is degraded releasing fragments of the component polypeptide chains. Both the collagen peptides and C1q are present in areas of inflammation including inflammation resulting from arthritis. These denatured fragments serve as the natural ligands for LAIR-1. In the experiments reported here, we used denatured α chains of type I collagen (α1(I)), type II collagen (α1(II)), and intact C1q to engage LAIR-1. We examined both the resulting surface expression of LAIR-1 and the deactivation of critical components of the canonical T-cell signaling pathway. Individual cytoplasmic tyrosine residues in the ITIMs were modified to determine which are necessary for LAIR-1 suppression of the TCR signaling. Based on the results of these experiments, we believe that therapies directed toward stimulating LAIR-1 or augmenting the intracellular pathways used by LAIR-1 hold promise for down-regulating unwanted inflammation (6) and that this natural inhibitory molecule can be exploited to suppress the tissue injury occurring in autoimmune arthritis.
Results
Inhibition of murine T cell signaling using α1(II)
Initial experiments used spleen cells isolated from DR1 mice. Naïve CD4+ T cells were isolated by negative selection from the spleens of DR1 LAIR-1–sufficient and –deficient mice. The purified T cells were pretreated with α1(II) to stimulate the LAIR-1 and were subsequently activated with antibodies to CD3 (α-CD3). Proteins were collected from whole-cell lysates to evaluate for phosphorylation of the Src family tyrosine kinases Lck and Lyn. Early activation of these kinases is an early step in the canonical T-cell signaling pathway. CD3 stimulation induced phosphorylation of both Lck and Lyn in WT cells, whereas LAIR-1 engagement with α1(II) inhibited their activation. On the other hand, CD3 stimulation led to phosphorylation of both Lck and Lyn in cells lacking LAIR-1, regardless of the presence or absence of α1(II) (Fig. 1). Downstream events, specifically the activation of Zap-70, ERK1/2, JNK-2, and p38, were evaluated in a similar manner. CD4+ T cells pretreated with α1(II) and stimulated with α-CD3 experienced a significant reduction in phosphorylation of Zap-70, ERK1/2, JNK-2, and p38 compared with cells not pretreated with α1(II). This inhibition depends upon the presence of LAIR-1 because the LAIR-1–deficient cells were not affected by treatment with α1(II).
Figure 1.

Inhibition of murine T-cell receptor signaling by α1(II) in CD4+ T cells isolated from WT or LAIR-1-deficient mice. Naive CD4+ T cells were isolated by negative selection as described under “Experimental procedures” and pretreated with α chains of type II collagen (α1(II)) (200 μg/ml) overnight prior to stimulation with α-CD3. LAIR-1 expression peaked after 18 h of culture with collagen. Cells lysates were collected and separated using SDS-PAGE gels. The separated proteins were then electrotransferred onto nitrocellulose membranes and analyzed for phosphorylation of the indicated proteins by Western blotting. The upper panel shows activation of the Src kinases Lck and Lyn. The lower panels show activation of ZAP-70 (pZAP-70–Tyr-493), as well as the MAP kinases JNK (pJNK-2), p38 (pp38), and ERK 1/2 (pERK). The membrane was stripped and reblotted with non–phospho-specific antibodies. This figure is a composite of several individual gels and is representative of three separate experiments.
LAIR-1 in human T cells
Having established the ability of LAIR-1 to attenuate murine T-cell receptor signaling, we next tested the effect of LAIR-1 stimulation using human T cells. T cells from Hut78, a human T-cell lymphoma cell line, were cultured overnight with a mAb recognizing human LAIR-1 or an isotype control and evaluated by flow cytometry for the surface expression of LAIR-1. The protein level of LAIR-1 on the surface of the cell was significantly up-regulated by culture with the stimulatory antibody to LAIR-1 (Fig. 2A). In a second set of experiments, Jurkat cells overexpressing human LAIR-1 were treated with either α1(II) or C1q for 5–30 min and then analyzed for the phosphorylation status of LAIR-1 as an indication of LAIR-1 activation. By 5 min after exposure of these cells to either ligand, there was significant activation of LAIR-1 (Fig. 2B). These results demonstrate that LAIR-1 can be up-regulated on the cell surface and triggered by engagement with collagenous proteins. To establish how LAIR-1 affects TCR signaling through the human canonical signaling pathway, Jurkat T cells overexpressing LAIR-1 were pretreated overnight with α1(II) and stimulated with a combination of α-CD3 and α-CD28. The data showed that engagement of LAIR-1 by α1(II) reduces phosphorylation of Zap-70 and the MAP kinases (ERK1/2, JNK 2, and p38) compared with controls stimulated by α-CD3 and α-CD28 without treatment with α1(II) (Fig. 3). These results are similar to those observed in murine CD4+ T cells and confirm that engagement of LAIR-1 leads to a significant suppression of T-cell signaling in both murine and human T cells.
Figure 2.
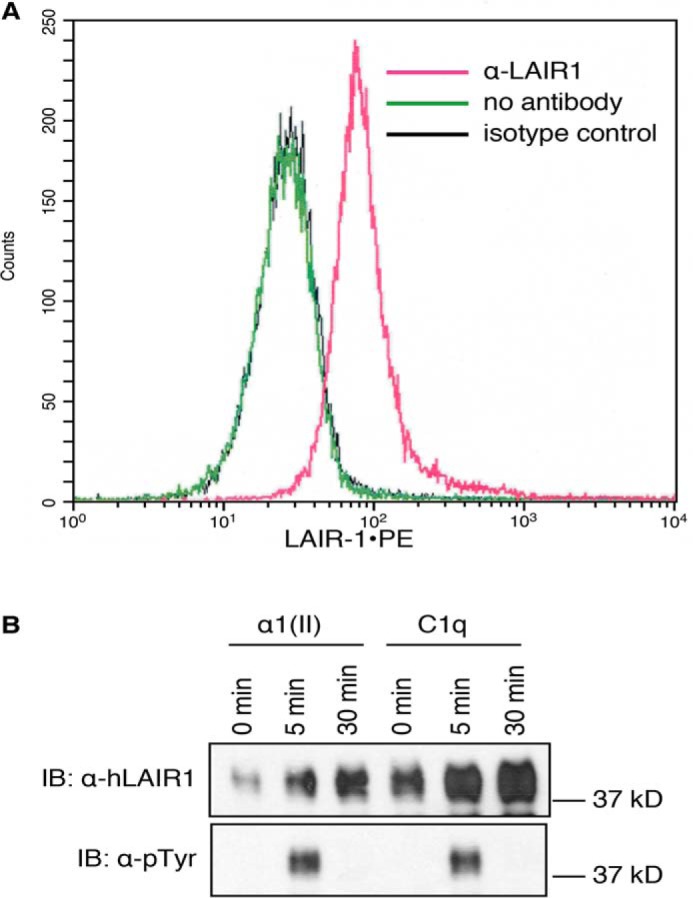
Activation of LAIR-1 in human T cells. A, Hut78 cells were stimulated with anti–LAIR-1 for 24 h, and expression of LAIR-1 was examined by flow cytometry. Mean fluorescence without stimulation was 789 ± 22; mean fluorescence with an isotype control was 779 ± 28; mean fluorescence with stimulation of anti–LAIR-1 was 2115 ± 53 (p ≤ 0.01). The data shown are representative of three separate analyses. B, Jurkat cells that express human LAIR-1 were activated by αI(II) or C1q for the indicated time periods. The LAIR-1 proteins in whole-cell lysates were immunoprecipitated using protein A/G beads conjugated with anti–LAIR-1. Phosphorylation status was examined by Western blotting analysis using anti-phosphotyrosine (α-pTyr) or anti–LAIR-1 (α-hLAIR-1) antibodies. Samples primed with an irrelevant protein (OVA) did not demonstrate any phosphorylation (data not shown). The data shown are representative of three separate analyses. IB, immunoblot.
Figure 3.

Inhibition of the TCR in human cells by stimulation of LAIR-1. Jurkat cells transduced with MSCV retroviruses containing an expression vector encoding either an empty control or FLAG-tagged (human LAIR-1 FLAG) or untagged (human LAIR-1-stop) human LAIR-1 were pretreated with αI(II)) and stimulated with α-CD3 + α-CD28 for the indicated time period (A). The phosphorylation of ZAP-70 and MAPKs was examined with Western blotting analysis using anti–phospho-ZAP-70 (Tyr-493), anti–phospho-JNK, anti–phospho-p38, or anti–phospho-ERK1/2 antibodies. The membrane was stripped and reblotted with non–phospho-specific antibodies. None of the cells were activated using isotype controls (data not shown). In B, the indicated cells were pretreated with αI(II), and the LAIR-1 proteins in whole-cell lysates were immunoprecipitated (IP) using protein A/G beads conjugated with anti–LAIR-1. Western blotting analysis confirmed the presence of LAIR-1 in the FLAG-tagged and LAIR-1 stop cell lines. The data shown are representative of three separate analyses.
Tyrosine phosphorylation of ITIMs in LAIR-1 is required for LAIR-1–mediated inhibition of TCR signaling in T cells
The human LAIR-1 receptor contains two ITIM modules in the cytoplasmic tail (7). The presence of these ITIM receptor subunits suggests that they are critical for the regulation of signal transduction pathways in T cells. ITIMs typically contain conserved tyrosine residues that regulate the receptor-linked signal transduction pathways in a negative fashion. To investigate whether tyrosine residues in ITIMs are critical for LAIR-1 function and to clarify which tyrosine is required for the inhibition of the T cell, we developed mutant LAIR-1 constructs in which individual tyrosine residues were replaced by phenylalanine. Jurkat cells were stably transfected with either WT LAIR-1 or LAIR-1 with mutations to convert the tyrosine at amino acid residue 251 (in ITIM 1) or 281 (in ITIM 2) or both to phenylalanine. Jurkat cells that express LAIR-1 empty control or deletion mutants of human LAIR-1 were activated by α1(II) and C1q prior to immunoprecipitation with anti–LAIR-1 antibody. When phosphorylation was examined by Western blotting analysis using an anti-phosphotyrosine antibody, the data revealed the deletion of each individual ITIM resulted in decreased phosphorylation of the LAIR-1, whereas deletion of both ITIMs abrogated this activation. Mutations replacing both tyrosines led to a complete abrogation of LAIR-1 phosphorylation (Fig. 4). When the cells expressing tyrosine mutants of LAIR-1 were pretreated with α1(II) to stimulate LAIR-1 prior to activation of the TCR with α-CD3 and α-CD28, it was clear that LAIR-1 stimulation effectively reduced phosphorylation of ZAP-70 in WT cells and those containing the Y281F mutations, whereas cells expressing LAIR-1 with the Tyr-251 mutation were ineffective in inhibiting TCR-mediated ZAP-70 activation (Fig. 4C). Taken together, our results demonstrate that cognate ligand binding to LAIR-1 leads to tyrosine phosphorylation at Tyr-251 and Tyr-281 residues in ITIMs of LAIR-1 and that tyrosine phosphorylation at Tyr-251 of LAIR-1 is essential for its function of suppressing TCR signal transduction.
Figure 4.

Induction of tyrosine phosphorylation in ITIMs of LAIR-1 by α1(II) and its impact on TCR signaling. A, Jurkat cells expressing either an empty control, WT human LAIR-1, or ITIM-deletion mutants of LAIR-1 were activated by α1(II) or C1q for the indicated time period. LAIR-1 was immunoprecipitated (IP) from each whole-cell lysate with protein A/G beads conjugated with anti–LAIR-1 antibody. Phosphorylation of LAIR-1 in the immunoprecipitants was examined by Western blotting analysis using anti–LAIR-1 and anti-phosphotyrosine antibodies, respectively. The big ranges of hLAIR-1 on the Western blotting with anti–hLAIR-1 antibody may be explained by other posttranslational modifications, such as ubiquitination or SUMOylation. B, Jurkat cells expressing either an empty control, WT human LAIR-1, or Tyr-to-Phe point mutation at aa 251, aa 281, or both of LAIR-1 were activated by α1(II) or C1q for 5 min. LAIR-1 was immunoprecipitated from each whole-cell lysate with protein A/G beads conjugated with anti–LAIR-1 antibody. The phosphorylation of LAIR-1 in the immunoprecipitants was examined by Western blotting analysis using anti–LAIR-1 and anti-phosphotyrosine antibodies, respectively. C, Jurkat cells expressing either an empty control, WT human LAIR-1, or a Tyr-to-Phe point mutation at aa 251, aa 281, or both of LAIR-1 were pretreated media (left panel) or α1(II) (right panel) and then activated with α-CD3 and α-CD28 for 5 min. Whole-cell lysates were collected, and the phosphorylation of ZAP-70 was detected by Western blotting assay using anti-pZAP70 Tyr-493 as an indication of ZAP-70 activation. The membrane was stripped and reblotted with anti-ZAP 70 antibody. The data shown are representative of three separate analyses. IB, immunoblot.
C-terminal Src kinase binds to the phosphorylated Tyr-251 in LAIR-1
LAIR-1 engages other molecules that in turn intersect with the TCR signaling pathway. Csk is a molecule implicated in the regulation of immune cells bearing ITIM-containing receptors (8). Therefore, we determined whether activation of LAIR-1 initiated recruitment of Csk to the LAIR-1 ITIMS. To that end, Jurkat cells expressing various LAIR-1 mutants were activated with collagen, and immunoprecipitation of LAIR-1 was performed to verify the presence of Csk in the LAIR-1 receptor signaling complex. Csk co-precipitated with activated LAIR-1, whereas replacement of the Tyr-251 residue of LAIR-1 completely abrogated Csk binding. Replacement of the Tyr-281 residue had no effect (Fig. 5). These data establish that Csk binds to the phosphorylated tyrosine residue 251 in LAIR-1 and suggest that Csk is involved in the regulation of the immune system by the ITIM bearing LAIR-1.
Figure 5.

C-terminal Src kinase interacts with activated LAIR-1 via a phosphorylated Tyr-251 in LAIR-1. Jurkat cells expressing either an empty control, WT human LAIR-1, or tyrosine mutants of LAIR-1 Jurkat cells expressing either an empty control, WT human LAIR-1, or a Tyr-to-Phe point mutation at aa 251, aa 281, or both of LAIR-1 were activated by α1(II) for 5 min. The presence of LAIR-1 and Csk in the resulting immunoprecipitates (IP) were examined by Western blotting analysis using anti–LAIR-1 antibody and anti-Csk antibody, respectively. The phosphorylation patterns of LAIR-1 activation of these mutants is demonstrated in Fig. 4B. The data shown are representative of three separate analyses. IB, immunoblot.
LAIR-1–mediated inhibition of TCR signal transduction requires Csk
It is known that Csk can be a negative regulator of basal and inducible receptor signaling (9), and from our previous experiments it was clear that Csk binds to phosphorylated LAIR-1. To address the necessity of Csk for LAIR-1 signaling, we used the CRISPR–Cas9 genome editing system to delete the Csk gene in Jurkat T cells. Several Csk haploid mutant cell lines were developed, and Western blotting and PCR analyses were used to detect protein or RNA for Csk in the resulting clones. Two Jurkat mutants, Csk PO-61 and Csk PO-101, had decreased amounts of Csk, compared with the control cell (Control CO-2) (Fig. 6A). These cells were selected for functional testing to determine whether TCR activation with anti-CD3 and anti-CD28 in the presence or absence of the LAIR-stimulator α1(II) would alter the production of IL-2. As shown in Table 1, when LAIR-1 was stimulated by α1(II), there was a significant decrease in cytokine production (IL-2) in the control cells, whereas both cells with the knockdown of Csk had no change in the secretion of IL-2. Similarly, overnight culture with α1(II) followed by TCR activation with anti-CD3 and anti-CD28 significantly decreased the phosphorylation of ZAP-70 in control cells, whereas the two cell lines with the knockdown of CSK showed no change in activation of ZAP-70 following TCR activation in the presence of α1(II) (Fig. 6B). These data demonstrate that Csk not only binds to the phosphorylated Tyr-251 residue of LAIR-1 but also is a critical molecule for the attenuation of the TCR signaling induced by LAIR-1 engagement.
Figure 6.

Csk is essential for the suppression of TCR-induced ZAP-70 activation by LAIR-1. A, Jurkat cells, a control CRISPR (clone CO-2), or Csk CRISPR (clones PO-61 and PO-101) cells were examined for Csk protein expression by Western blotting analysis (left panel) or for Csk mRNA by RT-PCR (right panel). Actin was used as control. The data presented are representative of three separate analyses. B, Jurkat cells, a control CRISPR (clone CO-2), or Csk CRISPR (clones PO-61 and PO-101) cells were pretreated with medium (no) or type II collagen (α1(II)) overnight and then stimulated with anti-CD3 and anti-CD28 antibodies for the indicated time period. Whole-cell lysates were collected and analyzed for activation of ZAP-70 by Western blotting analysis using anti–phospho-ZAP70 Tyr-493 (α-pZAP70) antibody. The membrane was stripped and reblotted with non–phospho-specific antibody for ZAP-70 (α-ZAP70). These data are representative of three separate analyses. C, CD4+ T cells from CskAS mice were pretreated with either vehicle (no), PP1* (3-IB-PP1, 10 μm), αI(I) (200 μg/ml), or PP1* + αI(I) overnight and stimulated with anti-CD3 antibody (10 μg/ml) for 5 min. Whole-cell lysates were prepared and analyzed for activation of ZAP-70 by Western blotting analysis using an anti–pZAP-70 Tyr-493 antibody. The membrane was stripped and reblotted with a non–phospho-specific antibody ZAP-70 (α-ZAP70). In the absence of α-CD3, none of these agents were able to elicit T-cell activation (data not shown). The data shown are representative of three separate analyses.
Table 1.
IL-2 concentrations
Either Csk haploid–sufficient (POCO2 control) or Csk haploid–deficient (PO61 and PO101) Jurkat cells were treated with α-CD3 either in the presence or absence of type I collagen (BI, 100 μg/ml). After 72 h of culture, the supernatants were tested for quantities of IL-2 by ELISA. The numbers shown represent the means and standard deviations of three separate experiments (in pg/ml).
| Cell line | +α-CD3 | +α-CD3 +BI | Media |
|---|---|---|---|
| POCO2 control | 296 ± 52 | 133 ± 43a | <10 |
| PO61 Csk KO | 375 ± 22 | 412 ± 35 | <10 |
| PO101 Csk KO | 280 ± 15 | 285 ± 25 | <10 |
a p ≤ 0.05 when stimulation with CD3 +BI is compared with stimulation with α-CD3 alone.
LAIR-1–mediated suppression the phosphorylation of ZAP-70 can be abolished by 3-IB-PP1 treatment of T cells from CskAS mice
Our data using human Jurkat cells expressing mutant forms of LAIR-1 established a crucial role for Csk in LAIR-1 regulation of TCR signaling in this cell line. To confirm this result in natural unimmortalized cells, we utilized the CskAS mouse, which expresses a PP1 analog (3-IB-PP1)–sensitive form of Csk (10). Deletion of Csk is lethal in mice; however, the phenotype can be rescued by replacement of the deleted endogenous WT Csk by a transgene that has only 25% of the activity of WT Csk. The catalytic activity of this particular transgene can be specifically and rapidly inhibited by a small molecule (3-IB-PP1). The dose required for inhibition is sufficiently low that it will not inhibit WT Csk. Murine CD4+ T cells from the CskAS mice were collected and stimulated with α-CD3 and collagen in the presence or absence of the 3–1B-PP1. In the presence of 3–1B-PP1, transgenic Csk completely abrogated the suppressive effect of LAIR-1 on TCR signaling as indicated by phosphorylation of ZAP-70. The phosphorylation of ZAP-70 was equivalent to that observed in cells activated with α-CD3 and treated with either the vehicle control or inhibitor alone. As expected, cells activated with α-CD3 in the presence of collagen showed substantially decreased levels of ZAP-70 phosphorylation. On the other hand, levels of phosphorylated ZAP-70 in cells activated with α-CD3 in the presence of collagen with the transgenic Csk inhibitor were comparable with those in cells activated by α-CD3 in the absence of collagen. These data confirm that Csk is critical for LAIR-1–induced suppression of TCR signaling in both human and murine T cells.
Discussion
LAIR-1, also known as CD305, is an immune inhibitory receptor that regulates immune system balance and protects against tissue damage and autoimmune dysfunction (11). In this study, LAIR-1 engagement by α chains of collagen or C1q led to inhibition of TCR signaling and decreased activation levels of key components of the canonical T cell signaling pathway, including Lck, Lyn, Zap-70, and the three MAP kinase (ERK1/2, JNK1/2, and p38). Although both ITIMS of LAIR-1 can be activated, point mutants of LAIR-1 showed that only the first ITIM with Tyr-251 is essential for the LAIR-1 inhibitory function. Moreover, CRISPR–Cas9 genome editing demonstrated that the nonreceptor tyrosine protein kinase, Csk, bound Tyr-251 of LAIR-1 and was required for the LAIR-1–induced inhibition of the human TCR signal transduction. This finding was corroborated using CD4+ cells from CskAS transgenic mice in which inhibition of the Csk transgene abrogated the LAIR-1–mediated suppression.
Although LAIR-1 is found on almost all cells of the immune system and its inhibitory functions have been described in a variety of cellular systems, this study demonstrates that LAIR-1 is a major player in down-regulating TCR signaling in both human and murine T cells (6, 7, 11–14). These results are especially interesting because CD4+ T cells, both Th1 and Th17, play a prominent role in the pathogenesis of autoimmune responses in autoimmune arthritis, because levels of LAIR-1 can be up-regulated on the surface of T cells. These data suggest that this inhibitory receptor can be used as a critical counterbalance to hold the immune system in check, when T cells become dysregulated (15).
The discovery that collagens are functional ligands for LAIR-1 (15, 16) disclosed a novel role for collagen in the regulation of immune function. α chains of type I or type II collagen and C1q were used to engage LAIR-1, and each could activate LAIR-1 in the absence of the other chains. Collagen is the most abundant protein in animals and is a key component of extracellular matrices, providing essential structural support for connective tissues. All collagens possess triple-helical regions, consisting of three polypeptide chains, that are characterized by repeating Gly-Xaa-Yaa sequences. Amino acids in positions Xaa and Yaa are often proline and 4-hydroxyproline, respectively (17). It has been found that GPHyp, the relatively common sequence in the triple-helical collagen domain, is recognized by LAIR-1, whereas GPP is not (18–20). Because each of the 18 polypeptide chains of C1q contains an N-terminal collagen-like Gly-Pro-Hyp repeat region, this molecule has the ability to bind and activate LAIR-1.
Activation of T cells through the TCR is essential for effector T-cell function. The TCR complex has no intrinsic kinase activity but instead has two spatially separated tyrosine residues in the ITAMs located in the cytoplasmic tails of its non–ligand-binding CD3 and ζ-chain subunits. Phosphorylation of those ITAMs is mediated by the T-cell Src-family kinases, for example Lck and Lyn, and thereby creates docking sites for recruitment of the cytoplasmic kinase Zap70 via its tandem SH2 domains. The autoinhibited conformation of Zap70 is relieved by its ITAM binding as well as by phosphorylation. Because activation of Zap70 is critical for downstream signaling events that lead to cellular responses, the phosphorylation of CD3ζ, poises Zap70 for activation upon interaction of the TCR with agonist peptide–MHC complexes. The activation of the TCR is tightly regulated. One critical negative regulator is the Csk, which can act both basally and during inducible signaling and primarily acts by down-regulating the Src family kinases (5, 21–26).
Our data showing that LAIR-1 binds and utilizes Csk to down-regulate TCR signaling are similar to observations made with the B-cell receptor (27). A yeast trihybrid system and chick B cells were used to show that phosphorylated LAIR-1 bound Csk through its SH2 domain. Following activation of the N-terminal tyrosine in LAIR-1, B-cell receptor signaling was attenuated. Because chick DT40 B cells lack both SHP-1 and SHP-2, these data show that LAIR-1 can utilize Csk to suppress receptors without requiring any other SH2 domain–containing phosphatases (27), although in some circumstances LAIR-1 does bind SHP-1 (13). In the case of T cells, LAIR-1 receptor engagement activates its ITIMs, leading to the recruitment of Csk to the receptor site. Csk then delivers a negative signal to the ITAM-bearing TCR.
The general assumption is that lack of inhibition predisposes for autoimmune diseases. When testing was done in synovial fluids of RA patients, LAIR-1 expression was found to be decreased in circulating CD4+ T cells in RA patients compared with both OA patients and healthy individuals. Upon stimulation with TNFα, LAIR-1 expression decreased in Th1 and Th2 CD4+ T cells from healthy donors. Although some studies indicate that LAIR-1 can be elevated in CD14+ monocytes and local CD68+ macrophages in synovial tissues from RA patients, more work must be done to determine whether LAIR-1 may exert different functions on T cells and monocytes/macrophages (11). When antibodies that stimulate LAIR-1 were used therapeutically to treat autoimmunity in a murine model of arthritis (collagen-induced arthritis), the antibodies significantly suppressed arthritis if administered in vivo (2). Mice genetically deficient in the LAIR-1 receptor develop more severe arthritis than WT controls (2). These data suggest that LAIR-1 may be a potential therapeutic target for suppressing RA.
Manipulating LAIR-1 may be a strategy for regulating inflammation and disease activity in systemic lupus erythematosus as well. Son and Diamond (13) have demonstrated that C1q can engage LAIR-1 and suppresses the secretion of interferons in human monocytes. Moreover, an absence of C1q has been shown to lead to enhanced IFN production by both human and mouse pDCs. These findings suggest that C1q/LAIR-1 signaling mediates a major inhibitory pathway for the innate immune response and that the C1q collagen tail, which engages LAIR-1, inhibits TLR signaling. A recent report (28) showed that collagenous domains of C-type lectin surfactant protein D bind to LAIR-1 and regulate FcαR-mediated reactive oxygen production in neutrophils. These results suggest that LAIR-1 engagement by collagen-like domains may be a therapeutic strategy for controlling inflammation in autoimmune diseases such as RA, systemic lupus erythematosus, and many other inflammatory conditions (28).
Conclusion
Our study has elucidated the regulation and function of LAIR-1 on T-cell receptor activation. The data show that once LAIR-1 is triggered by its ligand, either collagen or C1q, both ITIMS are phosphorylated, leading to the binding of CSK to Tyr-251 of LAIR-1. This CSK recruitment then delivers a negative signal to the ITAMs of the T-cell receptor, causing a down-regulation of the canonical T-cell receptor signaling pathway. These data suggest that LAIR-1 could be a potential therapeutic target in autoimmune diseases, as well as other diseases with an immune component.
Experimental procedures
Preparation of cartilage-derived type II collagen (CII) and skin-derived type I collagen
Native type II collagen was solubilized from fetal calf articular cartilage and native type I collagen from bovine hides by limited pepsin digestion and purification as described earlier (29). The purified collagen was dissolved in cold 10 mm acetic acid at 4 mg/ml and stored frozen at −70 °C until used. In some experiments, type I collagen from Advanced Biomatrix (Carlsbad, CA) was used. α1(II) and α1(I) represent the constituent protein chains of bovine CII and CI, respectively, isolated by carboxymethyl-cellulose chromatography. C1q, which contains Gly-Xaa-Yaa sequences resembling collagen was purchased from Sigma–Aldrich.
Animals
C57BL/6 mice expressing the chimeric (human/mouse) DRB1*0101 construct were obtained from Taconic Biosciences, (Hudson, NY) and bred in the animal core facility of the Rheumatic Diseases Research Core Center, University of Tennessee. The chimeric DRB1*0101 construct has been previously described, as has the production of transgenic mice expressing this construct (4). Mice transgenic for a CII-specific TCR in the context of DR1 were developed and bred in our animal core facility as described previously (30). LAIR-1 KO (knockout) mice (16) were cross-bred to B6.DR1 transgenic mice with a B6 background for 12 generations. Genomic DNA was obtained from blood samples, and PCR was used to identify mice homozygous for either the LAIR-1−/− or LAIR+/+ and expressing the DR1 transgene. CskAS mice were purchased from the Jackson Laboratory (Bar Harbor, ME). A description of their generation has been previously reported (9). Briefly mice that contained a modified Csk transgene were crossed to Csk+/− mice and intercrossed. The mice were genotyped with PCR, and pups were selected that contained both a Csk−/− deletion and the Csk transgene.
All mice were fed standard rodent chow (Ralston Purina Co., St. Louis, MO) and water ad libitum. Sentinel mice were routinely tested for a panel of mouse pathogens. All animals were kept until the age of 7–10 weeks before being used for experiments. All animal care and housing requirements set forth by the National Institutes of Health Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources were followed, and animal protocols were reviewed and approved by the University of Tennessee and Veterinary Affairs Animal Care and Use Committees.
Flow cytometery
Human Hut78 cells or modified Jurkat cells were collected, and the phenotype was determined by multiparameter flow cytometry using an LSRII flow cytometer (Becton Dickinson). The Cells were labeled with fluorochrome antibodies including FITC-conjugated anti-CD4 and phycoerythrin-conjugated LAIR-1. All were used according to the manufacturer's recommendations. A minimum of 10,000 cells was analyzed from each sample, and the final analysis was performed using FlowJo software (Tree Star, Ashland, OR).
CD4+ T-cell isolation and activation
Spleens were collected from either LAIR-1–sufficient or –insufficient DR1 mice, and single-cell suspensions were prepared by mechanical disruption in complete Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 IU/ml of penicillin, 100 μg/ml streptomycin, 2.5 μm β-mercaptoethanol, and 2 mm l-glutamine. CD4+ naive T cells were isolated using a CD4+ T-cell isolation kit (Miltenyi Biotec, Auburn, CA) based on a negative selection protocol. The purity of the recovered CD4+ T cells was determined by flow cytometry after staining with anti-CD4+ mAb, and they were >95% pure. In some experiments the cells were cultured for varying lengths of time (30 s to 60 min) with α1(II) or α1(I) (100 μg/ml). After an overnight culture at 37 °C, the cells were collected and lysed, and insoluble materials were removed by centrifugation at 10,000 × g at 4 °C for 15 min.
Analysis of protein phosphorylation
To study the proximal signaling events following TCR stimulation, CD4+ T cells were isolated from mice. The lysates of whole cells following stimulation with CD3/CD28 or collagen were separated using SDS-PAGE gels and electrotransferred onto nitrocellulose membranes. After transfer, the membrane was blocked in TBS with Tween 20 buffer (TBST) containing 5% BSA for 1 h and incubated for 2 h with phospho-specific antibodies. The membrane was then incubated with a secondary antibody (Bio-Rad) for 1 h and subjected to ECL detection (ECL2 Western blotting kit; Thermo Scientific/Pierce) according to the manufacturer's protocol. To detect protein levels, the membranes were stripped and reblotted with antibodies pan-specific for the proteins of interest.
Generation of LAIR-1–overexpressing Jurkat cells
Retroviral vectors expressing WT or various mutant forms of human LAIR-1 were developed as previously described (31). Briefly, the coding region of each was amplified by PCR using human cDNA as a template and then inserted into a MSCV-IRES-GFP retroviral vector. 1 × 107/10 ml of HEK293T cells in D10 (Dulbecco's modified Eagle's medium + l-Glu + sodium pyruvate + 10% fetal bovine serum) medium were seeded in the morning. After 9 h, the cells were transfected with the MSCV construct encoding empty control, WT, or various mutant forms of human LAIR-1 and helper DNAs using the calcium method (10 μg of MSCV + 3 μg of 3240 helper DNA + 3 μg of pSR αG (VSV-G envelop) + 450 μl of endo-free H2O + 50 μl of 2.5 m CaCl2 (in 10 mm HEPES buffer) + 500 μl of 2× HEPES buffer). The next morning, the medium was changed with fresh D10 medium. Virus particles in D10 medium were harvested after 1 and 2 days. The virus particles were 20× concentrated by ultracentrifugation (25,000 rpm for 90 min at 4 °C). 5 × 104/ml of Jurkat cells were infected with the MSCV-IRES-GFP retrovirus that expressed empty control, WT, or mutant form of human LAIR-1 in the presence of 8 mg/ml of Polybrene. After 1 day, the medium was changed with fresh D10 medium. Further selection was done by GFP-positive sorting. Six days later, the levels of LAIR-1 in these cells were analyzed by SDS-PAGE followed by Western blotting analysis using anti-human LAIR-1 antibodies (1:1000 dilution factor).
Generation of Csk haploid deficient Jurkat T cells using the CRISPR/Cas9 genome editing method
Control CRISPR/Cas9 plasmids (sc-418922) and Csk/Cas9 CRISPR plasmid, (sc-400992) were purchased from Santa Cruz Biotechnology and used to develop Csk haploid–deficient Jurkat cells. Briefly, plasmids consisting of Csk-specific 20-nt guide RNA sequences derived from the GeCKO (v2) library were selected and used to transfect human Jurkat cells using electroporation (Nucleofector II, Lonza). gRNA sequences directed the Cas9 protein to induce a site-specific double-stranded break in the genomic DNA. The resulting Jurkat clones were examined by flow cytometry to detect the presence of the plasmid. GFP-positive cells were selected and further screened for the presence of Csk by Western blotting and PCR. Two haploid-deficient clones together with an empty plasmid control were selected for further analysis.
Measurement of cytokines
To measure cytokines, Jurkat T cells were stimulated with anti-CD3 in the presence or absence of collagen. Supernatants were collected at 72 h and analyzed for the presence of IL-2 using a Bio-plex cytokine assay (Bio-Rad) according to the manufacturer's protocol.
Statistical analysis
Cytokine levels and mean fluorescence were compared using the Mann–Whitney test.
Reagents
Antibodies
The following antibodies were used for these studies: anti-phospho- and non–phospho-specific antibodies for murine Lck, Lyn, ZAP-70 (pZAP-70–Tyr-493), JNK (pSAPK/JNK-Thr-183/185), p38 (pp38 MAP kinase–Thr-180/Tyr-182), or ERK (pp44/42 MAP kinase–Thr-202/204), as well as their anti-non–phospho-specific counterparts were acquired from Cell Signaling Technology, Inc. (Beverly, MA). The anti-human LAIR-1 and their anti-phosphotyrosine-specific antibodies were purchased from R&D Systems, Inc. (Minneapolis, MN). An antibody against the non–phospho-specific Lyn was purchased from Santa Cruz Biotechnology. Antibodies against murine CD3 and CD4 and human CD3 and CD28 were purchased from BD Biosciences, and an antibody against Csk (Csk Antibody (E-3), sc-166560) was purchased from Santa Cruz Biotechnology.
Chemicals
The 3-IB-PP1 inhibitor has been described (10) and was used at a concentration of 10 μm (Sigma–Aldrich).
Cell lines
Jurkat cells (clone E6–1) and Hut78 cells, both immortalized human T-cell lines, were purchased from ATCC.
Author contributions
J.-E. P. and L. K. M. data curation; J.-E. P., D. D. B., and E. F. R. investigation; D. D. B., A. H. K., and L. K. M. conceptualization; D. D. B., E. F. R., A.-K. Y., J. M. S., and A. H. K. writing-review and editing; E. F. R. and A.-K. Y. methodology; A.-K. Y., J. M. S., and A. H. K. supervision; J. M. S., A. H. K., and L. K. M. project administration; L. K. M. funding acquisition.
This work was supported, in part, by U.S. Public Health Service Grants AR064825, AR069010, and AR064723 and program-directed funds from the Department of Veterans Affairs and the Arthritis Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LAIR-1
- leukocyte-associated immunoglobulin-like receptor 1
- IL
- interleukin
- TCR
- T-cell receptor
- CSK
- C-terminal Src kinase
- ERK
- extracellular signal–regulated kinase
- MAP
- mitogen-activated protein
- JNK
- c-Jun N-terminal kinase
- ITIM
- immunoreceptor tyrosine-based inhibition motif
- CIA
- collagen-induced autoimmune arthritis
- 3-IB-PP1
- 3-iodo-benzyl-PP1
- α1(I)
- α chain of type I collagen
- α1(II)
- α chain of type II collagen
- Th
- T-helper
- RA
- rheumatoid arthritis.
References
- 1. Pritchard N. R., and Smith K. G. (2003) B cell inhibitory receptors and autoimmunity. Immunology 108, 263–273 10.1046/j.1365-2567.2003.01592.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim S., Easterling E. R., Price L. C., Smith S. L., Coligan J. E., Park J. E., Brand D. D., Rosloniec E. F., Stuart J. M., Kang A. H., and Myers L. K. (2017) The role of leukocyte-associated Ig-like receptor-1 in suppressing collagen-induced arthritis. J. Immunol. 199, 2692–2700 10.4049/jimmunol.1700271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maasho K., Masilamani M., Valas R., Basu S., Coligan J. E., and Borrego F. (2005) The inhibitory leukocyte-associated Ig-like receptor-1 (LAIR-1) is expressed at high levels by human naive T cells and inhibits TCR mediated activation. Mol. Immunol. 42, 1521–1530 10.1016/j.molimm.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 4. Rosloniec E. F., Brand D. D., Myers L. K., Whittington K. B., Gumanovskaya M., Zaller D. M., Woods A., Altmann D. M., Stuart J. M., and Kang A. H. (1997) An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 185, 1113–1122 10.1084/jem.185.6.1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith-Garvin J. E., Koretzky G. A., and Jordan M. S. (2009) T cell activation. Annu. Rev. Immunol. 27, 591–619 10.1146/annurev.immunol.021908.132706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyaard L. (2008) The inhibitory collagen receptor LAIR-1 (CD305). J. Leukocyte Biol. 83, 799–803 10.1189/jlb.0907609 [DOI] [PubMed] [Google Scholar]
- 7. Meyaard L., Adema G. J., Chang C., Woollatt E., Sutherland G. R., Lanier L. L., and Phillips J. H. (1997) LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 7, 283–290 10.1016/S1074-7613(00)80530-0 [DOI] [PubMed] [Google Scholar]
- 8. Otáhal P., Pata S., Angelisová P., Horejsi V., and Brdicka T. (2011) The effects of membrane compartmentalization of csk on TCR signaling. Biochim. Biophys. Acta 1813, 367–376 10.1016/j.bbamcr.2010.12.003 [DOI] [PubMed] [Google Scholar]
- 9. Tan Y. X., Manz B. N., Freedman T. S., Zhang C., Shokat K. M., and Weiss A. (2014) Inhibition of the kinase Csk in thymocytes reveals a requirement for actin remodeling in the initiation of full TCR signaling. Nat. Immunol. 15, 186–194 10.1038/ni.2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schoenborn J. R., Tan Y. X., Zhang C., Shokat K. M., and Weiss A. (2011) Feedback circuits monitor and adjust basal Lck-dependent events in T cell receptor signaling. Sci. Signal. 4, ra59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Lv K., Zhang C. M., Jin B. Q., Zhuang R., and Ding Y. (2014) The role of LAIR-1 (CD305) in T cells and monocytes/macrophages in patients with rheumatoid arthritis. Cell Immunol. 287, 46–52 10.1016/j.cellimm.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 12. Boraschi-Diaz I., Mort J. S., Brömme D., Senis Y. A., Mazharian A., and Komarova S. V. (2018) Collagen type I degradation fragments act through the collagen receptor LAIR-1 to provide a negative feedback for osteoclast formation. Bone 117, 23–30 10.1016/j.bone.2018.09.006 [DOI] [PubMed] [Google Scholar]
- 13. Son M., and Diamond B. (2015) C1q-mediated repression of human monocytes is regulated by leukocyte-associated Ig-like receptor 1 (LAIR-1). Mol. Med. 20, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agashe V. V., Jankowska-Gan E., Keller M., Sullivan J. A., Haynes L. D., Kernien J. F., Torrealba J. R., Roenneburg D., Dart M., Colonna M., Wilkes D. S., and Burlingham W. J. (2018) Leukocyte-associated Ig-like receptor 1 inhibits Th1 responses but is required for natural and induced monocyte-dependent Th17 responses. J. Immunol. 201, 772–781 10.4049/jimmunol.1701753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lebbink R. J., Raynal N., de Ruiter T., Bihan D. G., Farndale R. W., and Meyaard L. (2009) Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 28, 202–210 10.1016/j.matbio.2009.03.005 [DOI] [PubMed] [Google Scholar]
- 16. Tang X., Tian L., Esteso G., Choi S. C., Barrow A. D., Colonna M., Borrego F., and Coligan J. E. (2012) Leukocyte-associated Ig-like receptor-1-deficient mice have an altered immune cell phenotype. J. Immunol. 188, 548–558 10.4049/jimmunol.1102044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brodsky B., and Persikov A. V. (2005) Molecular structure of the collagen triple helix. Adv. Protein Chem. 70, 301–339 10.1016/S0065-3233(05)70009-7 [DOI] [PubMed] [Google Scholar]
- 18. Myllyharju J., and Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 10.1016/j.tig.2003.11.004 [DOI] [PubMed] [Google Scholar]
- 19. Shoulders M. D., and Raines R. T. (2009) Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 10.1146/annurev.biochem.77.032207.120833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lebbink R. J., de Ruiter T., Adelmeijer J., Brenkman A. B., van Helvoort J. M., Koch M., Farndale R. W., Lisman T., Sonnenberg A., Lenting P. J., and Meyaard L. (2006) Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J. Exp. Med. 203, 1419–1425 10.1084/jem.20052554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chakraborty A. K., and Weiss A. (2014) Insights into the initiation of TCR signaling. Nat. Immunol. 15, 798–807 10.1038/ni.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stepanek O., Prabhakar A. S., Osswald C., King C. G., Bulek A., Naeher D., Beaufils-Hugot M., Abanto M. L., Galati V., Hausmann B., Lang R., Cole D. K., Huseby E. S., Sewell A. K., Chakraborty A. K., et al. (2014) Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell 159, 333–345 10.1016/j.cell.2014.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan A. C., Desai D. M., and Weiss A. (1994) The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annu. Rev. Immunol. 12, 555–592 10.1146/annurev.iy.12.040194.003011 [DOI] [PubMed] [Google Scholar]
- 24. Heyeck S. D., Wilcox H. M., Bunnell S. C., and Berg L. J. (1997) Lck phosphorylates the activation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J. Biol. Chem. 272, 25401–25408 10.1074/jbc.272.40.25401 [DOI] [PubMed] [Google Scholar]
- 25. Lowell C. A. (2004) Src-family kinases: rheostats of immune cell signaling. Mol. Immunol. 41, 631–643 10.1016/j.molimm.2004.04.010 [DOI] [PubMed] [Google Scholar]
- 26. Nika K., Soldani C., Salek M., Paster W., Gray A., Etzensperger R., Fugger L., Polzella P., Cerundolo V., Dushek O., Höfer T., Viola A., and Acuto O. (2010) Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777 10.1016/j.immuni.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verbrugge A., Rijkers E. S., de Ruiter T., and Meyaard L. (2006) Leukocyte-associated Ig-like receptor-1 has SH2 domain-containing phosphatase-independent function and recruits C-terminal Src kinase. Eur. J. Immunol. 36, 190–198 10.1002/eji.200535226 [DOI] [PubMed] [Google Scholar]
- 28. Jin J., Wang Y., Ma Q., Wang N., Guo W., Jin B., Fang L., and Chen L. (2018) LAIR-1 activation inhibits inflammatory macrophage phenotype in vitro. Cell Immunol. 331, 78–84 10.1016/j.cellimm.2018.05.011 [DOI] [PubMed] [Google Scholar]
- 29. Rosloniec E. F., Cremer M., Kang A. H., Myers L. K., and Brand D. D. (2010) Collagen-induced arthritis. Curr. Protoc. Immunol. Chapter 15, Unit 15.15.1–25 10.1002/0471142735.im1505s89 [DOI] [PubMed] [Google Scholar]
- 30. Tang B., Kim S., Hammond S., Cullins D. L., Brand D. D., Rosloniec E. F., Stuart J. M., Postlethwaite A. E., Kang A. H., and Myers L. K. (2014) Characterization of T cell phenotype and function in a double transgenic (collagen-specific TCR/HLA-DR1) humanized model of arthritis. Arthritis Res. Ther. 16, R7 10.1186/ar4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim Y. I., Park J. E., Martinez-Hernandez A., and Yi A. K. (2008) CpG DNA prevents liver injury and shock-mediated death by modulating expression of interleukin-1 receptor-associated kinases. J. Biol. Chem. 283, 15258–15270 10.1074/jbc.M709549200 [DOI] [PMC free article] [PubMed] [Google Scholar]