

Abstract
Since their discovery, the matrix metalloproteinase (MMP) family proteases have been considered as therapeutic targets in numerous diseases and disorders. Unfortunately, clinical trials with MMP inhibitors have failed to yield any clinical benefits of these inhibitors. These failures were largely due to a lack of MMP-selective agents; accordingly, it has become important to identify a platform with which high selectivity can be achieved. To this end, we propose using MMP-targeting antibodies that can achieve high specificity in interactions with their targets. Using a scaffold of single-domain antibodies, here we raised a panel of MMP10-selective antibodies through immunization of llamas, a member of the camelid family, whose members generate conventional heavy/light-chain antibodies and also smaller antibodies lacking light-chain and CH1 domains. We report the generation of a highly selective and tightly binding MMP10 inhibitor (Ki < 2 nm). Using bio-layer interferometry–based binding assays, we found that this antibody interacts with the MMP10 active site. Activity assays demonstrated that the antibody selectively inhibits MMP10 over its closest relative, MMP3. The ability of a single-domain antibody to discriminate between the most conserved MMP pair via an active site–directed mechanism of inhibition reported here supports the potential of this antibody as a broadly applicable scaffold for the development of selective, tightly binding MMP inhibitors.
Keywords: antibody engineering, antibody, metalloprotease, protease inhibitor, single-domain antibody, sdAb, nanobody, matrix metalloproteinase, protease, MMP10, MMP3, antibody selectivity, camelid, orthosteric inhibitor
Introduction
Early in MMP2 research, the general consensus within the MMP community was that the few known MMPs were all driving disease, a dogma that led to the development of broad-spectrum MMP inhibitors within the pipelines of most major pharmaceutical companies (1–5). Despite promising preclinical data supporting broad-spectrum MMP inhibition in oncology, results of over 50 clinical trials were consistently negative (6). MMP inhibition failed to reach its clinical end point in every clinical trial, with increased disease burden and musculoskeletal side effects observed in some treatment groups (7). Broad-spectrum MMP inhibition was unsuccessful (8).
As more MMPs were discovered, researchers were uncovering the complexity of MMP activity in normal physiology (9, 10). Some MMPs were being observed to have a protective effect in certain pathological situations while other MMPs were driving pathology (8, 11). Accordingly, the consensus in the field concerning the MMPs as therapeutic targets shifted from broad-spectrum inhibition to selective inhibition: inhibit the “bad” MMPs while sparing the catalytic activity of the “good” MMPs (8).
The design of selective MMP inhibitors continues to be hindered by the pronounced structural homology shared within the active site of all the MMPs (12). Small molecules can penetrate the substrate-binding clefts of the MMPs to achieve active site–directed (orthosteric) inhibition. However, their small size also limits the extent of their target interactions, oftentimes preventing sufficient contacts with regions of an MMP that differentiate it from other family members. Conversely, protein-based inhibitors, such as antibodies, possess the structural bulk that enables them to bind to their targets with multiple different contacts, conferred by multiple complementarity-determining regions (CDRs). However, conventional antibodies generally prefer planar epitopes, and as such, they typically target exosites, or regions of a protease outside of the active site (13). Whereas success has been achieved with exosite-targeting antibodies, the most successful protease inhibitors target the active site, and as such, conventional antibodies offer a limited platform for selective protease inhibition (14–16). Other classes of protein-based inhibitors, including engineered tissue inhibitor of metalloproteases (TIMPs), have also been explored as selective MMP inhibitors with varying success; however, high selectivity using these scaffolds is yet to be achieved (17–19).
Single-domain antibodies, a more recently discovered class of antibodies devoid of a light chain and CH1 region, are gaining momentum as potent inhibitors of enzymes (20–22). To compensate for the lack of a light chain, single-domain antibodies have evolved a substantially larger antigen recognition region, particularly in their CDR3 (23). These longer CDR regions are thought to form convex antigen-binding surfaces, ideal for inserting into the active-site clefts of enzymes and toward orthosteric inhibition (24). Furthermore, single-domain antibodies still possess three complementarity-determining regions that are capable of making multiple contacts with their antigens. With single-domain antibodies capable of achieving orthosteric targeting and possessing multicontact antigen recognition, we sought to determine their applicability toward selective, active site–directed, MMP inhibition. Considering the 86% sequence identity in their catalytic domains and nearly identical active sites, MMP10 and MMP3 represent the most homologous pair of MMPs (25).
Our efforts were focused on MMP10 due to its tumorigenic potential (26–28). MMP10 is up-regulated at the RNA level in multiple different types of cancers, and knockout and knockdown strategies have highlighted its potential as a therapeutic target in cancer. However, a lack of selective inhibitors has prevented researchers from understanding whether the benefit seen with genetic modulation of MMP10 can be recapitulated with pharmacological intervention. Accordingly, we sought to generate a robust and highly selective inhibitor of MMP10, based on a single-domain antibody raised in llamas. A major goal was to obtain an inhibitor that selectively inhibits MMP10 over its closest homologue, MMP3.
Results
Generation of active MMP10
Zymogen MMP10 harbors a propeptide in the active site, rendering the active site unavailable to substrates. Upon activation, the propeptide is removed, leading to a 9-kDa decrease in the molecular mass of MMP10 (29). We therefore hypothesized that to direct the immunization toward the active site, our best chance was to use active MMP10 as our immunogen. To do so, full-length human MMP10 was cloned into a modified pCEP vector as a llama Fc fusion protein, making pCEP-hMMP10-LFc. The llama IgG Fc fragment both increases circulation time in the host llama due to Fc/FcRn interactions and provides an efficient purification tag when via protein A–Sepharose (30, 31). pCEP-hMMP10-LFc was subsequently transfected and expressed in mammalian cells and purified via protein A chromatography, yielding MMP10-Fc (Fig. 1A), Activation of MMP10-Fc was achieved by treatment with a small molecule activator of MMPs, aminophenylmercuric acetate (APMA), showing both a dose-dependent increase in MMP10 activity and a corresponding decrease in molecular weight (Fig. 1B).
Figure 1.
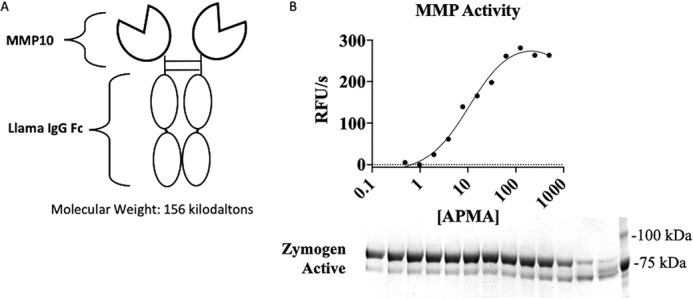
Active recombinant MMP10. A, schematic representing MMP10 immunogen as a bivalent Fc fusion protein; B, evidence for activation of proMMP10 by APMA. Top, cleavage of quenched fluorescent substrate by MMP10; bottom, SDS-PAGE showing generation of active MMP10 evidenced by loss of the propeptide. RFU, relative fluorescence units.
Immunization
Members of the camelid family, including camels, llamas, and alpacas, generate conventional heavy/light chain antibodies. They also generate smaller antibodies lacking light chain and CH1 domains (Fig. 2A) (32). Three llamas were immunized with MMP10-Fc and monitored for responses against MMP10 over time. All three immunized llamas responded to MMP10, and the Vhh repertoires of each of the llamas were cloned into a yeast display platform to allow for enrichment of monoclonal antibodies targeting MMP10.
Figure 2.

Generation of yeast display libraries. A, top, schematic of conventional antibody structure versus heavy chain–only antibodies. Bottom, representative DNA strands with priming regions highlighted. B, agarose gel showing two bands from the round 1 PCR. The top band (∼900 bp) represents Vh from conventional antibodies, and the bottom (∼600 bp) represents the Vh of heavy chain–only antibodies.
Antibody library construction
cDNA to build the yeast libraries was generated as explained under “Experimental procedures” (33). The PCR output using this method generates two bands, one corresponding to heavy chains from conventional antibodies and a smaller band corresponding to heavy chains from heavy chain–only antibodies (Fig. 3B). The smaller DNA band was used as the template for the generation of a yeast library. Once separate yeast display libraries were built for each llama, the libraries were enriched using a combination of magnetic bead separation and fluorescence-activated cell sorting. In total, 23 unique mAb sequences were isolated. All 23 monoclonal sequences were subcloned and transfected into a mammalian expression system and expressed as bivalent hIgG1 Fc fusion proteins. 20 antibodies expressed at appreciable levels (>100 μg/ml) and were further evaluated in an MMP10 binding and inhibition assay.
Figure 3.

Screening of MMP10-Fc binders and inhibitors. A, antibody clones were tested for affinity to MMP10 using biolayer interferometry. B, four of the binders inhibited MMP10 activity as measured by a decrease in cleavage of a quenched fluorescence substrate of MMP10. Antibodies were also measured for inhibition of MMP3 to screen for selectivity. RFU, relative fluorescence units.
Binding and inhibition of MMP10
All 20 antibodies were subject to a binding assay using bio-layer interferometry (BLI) to check for specific binding to MMP10-Fc. 14 of the 20 expressed antibodies showed specific binding to MMP10-Fc with no association detected against an irrelevant coated antigen. The MMP10-reactive antibodies displayed a range of affinities from subnanomolar to nearly 50 nm (Fig. 3A).
To measure MMP10 inhibition, each of the 14 binding antibodies were titrated onto a constant concentration of active MMP10-Fc and subjected to an activity assay. Inhibitors were characterized as antibodies that decreased the activity of MMP10. In a parallel screen, the antibodies were titrated onto a constant concentration of active MMP3-Fc, also generated in-house, to determine selectivity. Of the 14 binders, four inhibited MMP10-Fc, with two monoclonal antibodies, E5 and H3, inhibiting MMP10-Fc more strongly than the others. Of the two strong inhibitors, H3 displayed an inhibition profile similar to TIMP2, an endogenous inhibitor of MMPs, while showing only slight reactivity toward MMP3-Fc (Fig. 3B). On the basis of this screen, we considered H3 as our lead inhibitory antibody of MMP10.
H3 selectivity
To further probe the selectivity of H3, its inhibitory activity was tested against a panel of MMPs generated for this study: MMP1, MMP2, MMP3, MMP9, and MMP13. In this experiment, TIMP1, an endogenous inhibitor of most MMPs, served as a positive control. Whereas TIMP1 inhibited all of the MMPs tested, H3 treatment had no effect on MMP1, MMP2, MMP9, and MMP13 activity; minimal effect on MMP3; and complete inhibition of MMP10 (Fig. 4).
Figure 4.
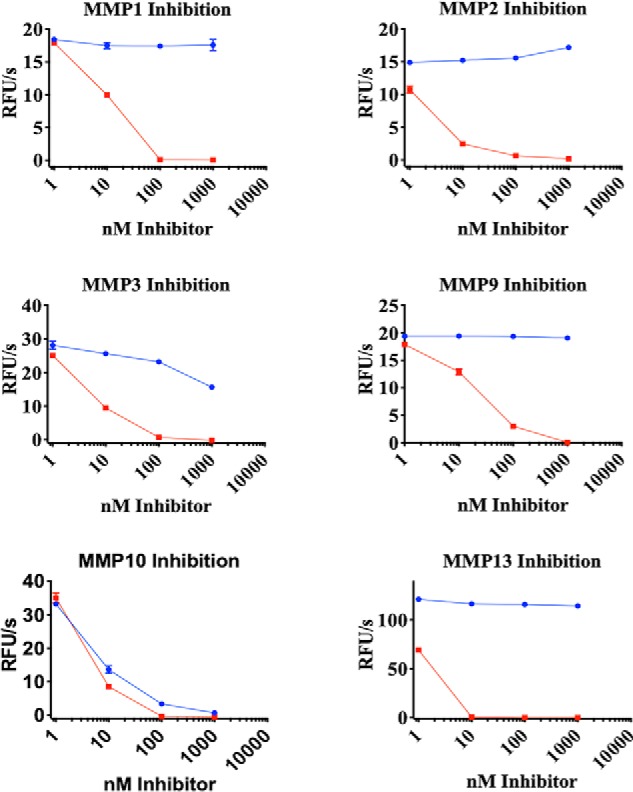
H3 selectivity toward MMP10. H3 (blue) and TIMP1 (red) were titrated against the indicated MMPs. Activity was monitored using a fluorescence plate reader. Error bars, S.D.; n = 3. RFU, relative fluorescence units.
In-depth profiling of H3-mediated inhibition of MMP10
In an attempt to attain precise kinetic information, H3 was assayed for its ability to inhibit the minimum concentration of MMP10 capable of eliciting reliable activity in the system used. TIMP1 was titrated as a positive control, as was the negative control antibody C10, which is a binder, noninhibitor, of MMP10 also acquired from llama immunization (Fig. 3A). H3 inhibited MMP10 with an IC50 of 657 pm, whereas TIMP1 inhibited MMP10 with an IC50 of 296 pm (Fig. 5). The inhibition kinetics imply that the interaction is extremely tight and mechanistically cannot be distinguished from an irreversible mechanism of inhibition. Accordingly, we conclude that H3 is an extremely tight and selective inhibitor of MMP10.
Figure 5.
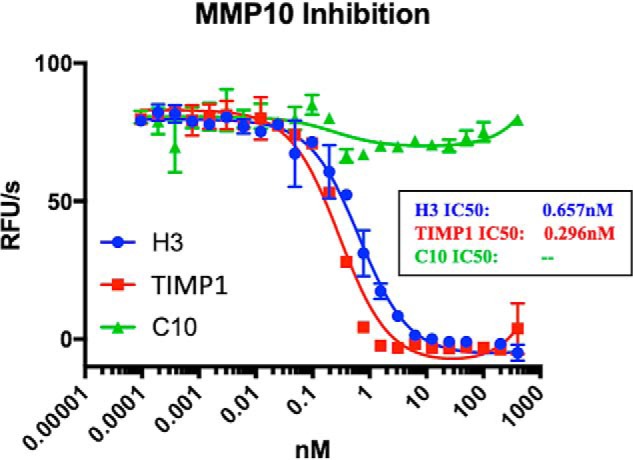
In-depth kinetics of H3. H3, TIMP1, and C10 were titrated with MMP10 as described under “Experimental procedures” to determine kinetics of inhibition. Error bars, S.D.; n = 3. RFU, relative fluorescence units.
H3 inhibition of MMP10-mediated cleavage of α1-antitrypsin
H3 potently inhibited MMP10-mediated cleavage of a peptide substrate, but we also wanted to determine whether it would inhibit cleavage of a protein substrate. The serpin α1-antitrypsin (AAT) has been reported as a substrate for numerous MMPs (34, 35). To determine whether AAT was also an MMP10 substrate, active MMP10-Fc was titrated and incubated with a constant concentration of AAT overnight at 37 °C, with cleavage observed by SDS-PAGE. Having demonstrated that AAT is a substrate for MMP10, we aimed to determine whether H3 inhibited this activity. H3 was titrated in the presence of a constant concentration of active MMP10-Fc. Following a short incubation, AAT was added, and mixtures were incubated overnight. Cleavage of AAT was analyzed by SDS-PAGE, which demonstrated that H3 prevented MMP10-Fc from cleaving AAT (Fig. 6A). Cleavage of AAT by MMPs abrogates the ability of this serpin to inhibit its target enzyme, neutrophil elastase (NE) (34, 35). Samples from Fig. 6A were incubated with NE to determine whether H3 was able recover AAT activity in regard to NE inhibition. Incubation of MMP10 with H3 prevented MMP10's ability to inactivate AAT, as indicated by a decrease in NE activity with increasing levels of H3 (Fig. 6B). Both of these experiments demonstrate that H3 is able to inhibit the proteolytic activity of MMP10. Moreover, they indicate AAT may be a physiologic substrate of MMP10, much as it is for MMP9 (34).
Figure 6.

Inhibition of MMP10 cleavage of endogenous substrates. A, H3 was serially diluted into wells containing a constant concentration of MMP10. Samples were then incubated with AAT to observe whether H3 prevents MMP10-mediated cleavage of AAT. Samples were analyzed by reducing SDS-PAGE. Data are representative of three separate experiments. B, NE activity was measured after the addition of H3/MMP10/AAT samples. Error bars. S.D.; n = 3. RFU, relative fluorescence units.
Mechanism of H3 inhibition
H3's strong interaction with MMP10 coupled to a lack of highly concentrated quenched fluorescent substrates of MMP10 made determining the mechanism of binding, orthosteric (active site–directed) versus allosteric (exosite-directed), impossible to determine using traditional competitive enzyme kinetics. Furthermore, an attempt to co-crystallize MMP10 with H3 failed to produce crystals. Therefore, a series of competition assays were performed to validate the mechanism of H3 inhibition of MMP10. These assays involved observing the binding of MMP10 derivatives to H3 by biolayer interferometry (Fig. 7). H3 bound to active MMP10 but was unable to bind to proMMP10 or MMP10 inhibited by a small molecule (marimastat). Moreover, MMP10 in complex with H3 did not bind by either TIMP1 or TIMP2. Consequently, H3 did not tolerate the presence of a propeptide, small molecule, or protein-based inhibitor within the MMP10 active site. These characteristics support a model where H3 binds to MMP10's active site as its mode of inhibition.
Figure 7.

Interaction of MMP10 derivatives with H3 as measured by BLI. A biotinylated protein (biotinylated MMP10-Fc in A and B, biotinylated H3 in C and D) was loaded onto a streptavidin sensor. Once the baseline response (BL) was established, the sensor was introduced into solutions containing the proteins indicated above the sensorgrams to measure the association phase. A, H3 binding to activated MMP10-Fc or proMMP10-Fc. B, H3 binding to active MMP10 or marimastat-inhibited MMP10-Fc. C, binding of TIMP1 to the H3/MMP10 complex. D, binding of TIMP2 to the H3/MMP10 complex.
Discussion
Whereas broad-spectrum MMP inhibitors failed in the clinic, selectively targeting the MMPs is still considered to be of clinical value (36). Some MMPs show an mRNA expression bias toward cancer, and there exists a large body of evidence implicating MMP activity in various diseases (8). Accordingly, it has become important to generate a panel of selective MMP inhibitors to validate individual MMPs as therapeutic targets.
The primary question addressed in this study was whether single-domain antibodies, in addition to achieving robust MMP inhibition, could offer an adequate level of selectivity. This question was addressed by focusing our efforts toward the inhibition of MMP10 over MMP3, the two most closely related MMPs, sharing 86% identity within their catalytic domains and nearly identical active sites. Whereas low-level inhibition of MMP3 was observed with H3, the window of inhibition between MMP3 and MMP10 was vast, with full inhibition of MMP3 not observed even with high concentrations of H3. The ability of a single-domain antibody to differentiate between the most identical MMPs provides a rationale for single-domain antibodies as a broadly applicable scaffold for selective MMP inhibition.
Conventional antibodies targeting proteases generally target regions outside of the active site. Indeed, the several reported conventional antibodies targeting the catalytic activity of proteases have invariably failed to target the active site, with inhibition achieved through allosteric targeting (14). Conversely, heavy chain–only antibodies produced by camelid family members are hypothesized to prefer concave epitopes, such as the active site clefts of enzymes (23). To determine whether H3 orthosterically targets MMP10 via traditional kinetics, it would be necessary to utilize better substrates than commercially available. Therefore, to define the mechanism of inhibition, we developed a series of competition assays focusing on the interaction of H3 with various forms of MMP10. In this series of experiments, H3 did not tolerate the presence of a propeptide, marimastat, or TIMPs within the active site of MMP10, indicating its preference to target a free active site. Whereas further validation of the mechanism of inhibition of H3 would require a crystal structure, the data presented in this study strongly support H3's status as an orthosteric inhibitor.
In addition to probing the ability of single-domain antibodies to selectively inhibit MMP10 over its closest homologue, MMP3, we also sought to target MMP10 because it represents a potential cancer target. Based on mRNA expression data from the GENT website, MMP10 shows a strong cancer bias with virtually undetectable mRNA levels in healthy tissue (37). Furthermore, a large body of research suggesting MMP10's role as a cancer driver, both in vitro and in vivo, has been published (26, 28, 29, 38). Combined, these data provide a strong rationale for the potential of MMP10 as a therapeutic target. Developing a selective inhibitor of MMP10 may be challenging with classical MMP inhibitor scaffolds; however, we demonstrate that single-domain antibodies are up to this task. As such, this antibody could be evaluated further as a therapeutic candidate.
Experimental procedures
MMP10 expression
The nucleotide sequence encoding the ORF of human MMP10, corresponding to Uniprot limits 1–476, was cloned into a modified pCEP vector in frame with a C-terminal llama IgG Fc, encoding proMMP10-Fc. 293 freestyle cells were transfected with the MMP10-Fc construct using polyethyleneimine. Following a 7-day incubation in cell culture conditions, supernatants were harvested, and proMMP10-Fc was purified with Thermo Scientific Gentle Ag/Ab Elution Buffer (product no. 21013). Expression was measured via absorbance at 280 nm and visualized by SDS-PAGE. The eluted protein was subjected to buffer exchange via a PD-10 column (GE Healthcare) into 50 mm Tris, 10 mm CaCl2, 150 mm NaCl, 0.05% (w/v) Brij-35, pH 7.5 (TCNB). Following buffer exchange, hMMP10-FL-LFc was activated with a titration of 4-aminophenylmercuric acid (Sigma, catalogue no. A-9563) starting at 1 mm with a 1:4 dilution in TCNB. Samples were run on SDS-PAGE using an Invitrogen 4–12% bis-tris gel to visualize loss of the MMP10 propeptide. To check for activity, each sample from the APMA titration was incubated with a 10 μm final concentration of quenched fluorescent MMP10 substrate, MCA-RPKPVE-NVal-WRK(DNP)-NH2 (R&D Systems, catalog no. ES002). Fluorescence was measured on the BMG Labtech Clariostar plate reader at 320-nm excitation and 405-nm emission. Rates were defined as the slope of fluorescence generation (y axis) over time (x axis).
Llama immunization
Three llamas were each immunized (outsourced to Abcore, Inc., Ramona, CA) with 0.5 mg of hMMP10-LFc in complete Freund's adjuvant for the first immunization and were immunized every 2 weeks with 0.5 mg of hMMP10-LFc with incomplete Freund's adjuvant. Test bleeds were received every 2 weeks and were tested by ELISA. For the ELISA, each well was coated with 50 ng of human MMP10 from R&D Systems (910-MP-010), received as a zymogen and activated as recommended by R&D Systems and validated in an activity assay on a 96-well Medisorp plate (Thermo Scientific, catalog no. 467320). Commercial MMP10 was used due to the presence of a LFc on our immunogen, and an anti-llama IgG antibody was used for detection in the ELISA. Llama serum from test bleeds was diluted 1:100 in Dulbecco's PBS and titrated 1:5 onto coated MMP10. Plates were then incubated with horseradish peroxidase–conjugated anti-llama IgG H+L (Bethyl, catalog no. A160-100P) at the manufacturer's recommended concentration and developed using the horseradish peroxidase substrate TMB (Seracare KPL SureBlue TMB Microwell Peroxidase Substrate, catalog no. 5120-0077). Plates were read for absorbance at 650 nm using a Molecular Devices Emax plate reader. Llama immunizations were performed under their institutional animal care and use committee–approved protocols and United States Department of Agriculture guidelines via USDA 93-R-0574.
Library construction
Peripheral blood mononuclear cells enriched from three llamas were acquired by layering 400 ml of fresh blood onto a Ficoll gradient. Total RNA was isolated using a Qiagen RNeasy Maxi Kit, which was used as a template to generate cDNA with an Invitrogen SuperScript IV First-Strand Synthesis System with random hexamer and oligo(dT) as primers. Primers corresponding to llama Vh germ line genes were used in a first-round PCR as described previously (33). Two bands were visible on an agarose gel from the first round PCR, with the larger band corresponding to the Vh from a conventional antibody and a smaller band corresponding to Vhhs from heavy chain–only antibodies. The smaller band was excised and used as a template in a second round PCR to incorporate NheI/EcoNI restriction sites amenable for cloning into a proprietary yeast display plasmid, pAR5, placing the Vhhs N-terminal to the AGA2 protein classically utilized in yeast display (39). 1.5 μg of Vhh insert was ligated into 5 μg of pAR5 plasmid and transformed into a lithium acetate–treated proprietary yeast strain designed for use in yeast surface display. Libraries were grown in SD−CAA medium, and transformation efficiency was measured via serial dilution onto SD−CAA plates. Yeast surface display was induced using SG-CAA medium and incubated overnight at room temperature with shaking. Three separate libraries were generated, representing each immunized llama.
Enrichment of MMP10-binding Vhhs
For magnetic bead enrichments, activated hMMP10-LFc was biotinylated using Thermo Scientific EZ-LinkTM NHS-Biotin. 200 nm biotinylated hMMP10-LFc was then incubated with the input library generated above and incubated on ice for 1 h. After washing with TCNB-B buffer (TCNB + 1.5% BSA), libraries were incubated with 200 μl of New England Biolabs streptavidin magnetic beads in 3 ml of TCNB-B for 15 min on ice. Yeast bound to MMP10 were then magnetically separated on an EasySepTM magnetic tube holder from Stemcell Technologies. The output libraries were then grown up in SD−CAA and were subsequently induced in SG-CAA for a second enrichment by FACS. The FACS output libraries were incubated with 100 nm biotinylated hMMP10-LFc for 1 h, washed with TCNB-B, and labeled with an Alexa Fluor 647 streptavidin. The yeast were washed in TCNB-B and sorted on a Sony SH800 cell sorter for Alexa Fluor 647 positivity, which varied among the three llamas immunized. Output libraries were then plated and grown up as colonies, with each colony representing one Vhh sequence. Colonies were picked into a 96-well culture plate in SD−CAA medium and were induced in a 96-well format with SG−CAA. Induced colonies were then labeled with 50 nm biotinylated hMMP10-LFc for 1 h at 4 °C, labeled with Alexa Fluor 647–conjugated streptavidin as above, and run on an Accuri C6 flow cytometer. Colonies showing reactivity to MMP10 were then boiled in 0.1% SDS to lyse the cells, and lysates were used as a template to amplify their Vhh sequences using a forward and reverse primer specific to the backbone of pAR5. PCR products were sequenced using the same forward primer they were generated with.
Generation of recombinant antibodies
Unique sequences were subcloned into a modified pCEP vector, placing them in frame with a C-terminal hIgG1-Fc fragment. The antibody-Fc constructs were then transfected into 293 freestyle cells and grown at 37 °C for 3 days at 5% CO2. Supernatants were harvested, and antibodies were incubated with protein A (GE Healthcare) and eluted with Thermo Scientific IgG elution buffer (catalog no. 21009). Eluted proteins were subsequently buffered with the addition of 10% final 1 m Tris, pH 8. Recombinant proteins were quantified via absorbance at 280 nm and visualized with SDS-PAGE.
Binding of monoclonal antibodies
Recombinant antibodies were buffer-exchanged into TCNB buffer using a GE Healthcare PD-10 column to eliminate buffer effects in a BLI assay. Biotinylated MMP10-LFc (as generated above) was coated onto the surface of a ForteBio streptavidin dip-and-read sensor (part no. 18-5020). MMP10 was then measured for its affinity toward each of the antibodies separately using the ForteBio Octet Red 96 biolayer interferometry device. Sensorgrams were generated, and Kd values were calculated for each antibody with MMP10 using ForteBio data analysis software. For a negative control, a biotinylated llama IgG1 Fc fragment was coated onto ForteBio streptavidin sensors and measured for its affinity to each antibody.
Expression of TIMPs
Full-length TIMP1 and TIMP2 (corresponding to Uniprot limits 1–217 and 1–220, respectively) were cloned into a modified pCEP-mFc vector, placing them in frame and N-terminal to a mIgG2a Fc fragment. Ligations, transformations, plasmid prepping, HEK 293 freestyle transfections, and protein A purifications were the same as highlighted above for the recombinant antibody expressions. Once purified, proteins were quantitated by A280 and visualized by SDS-PAGE.
MMP10 inhibition screen
Each antibody was titrated 1:10 across four points starting at 100 μg/ml in the presence of 20 μg/ml MMP10-LFc or 20 μg/ml MMP3-LFc and incubated for 15 min at room temperature. MMP3-Fc was generated as with MMP10. In short, the entire sequence of MMP3 as dictated by Uniprot was cloned into a modified pCEP vector, placing it N-terminal to an IgG llama Fc fragment. Purification, activation, and validation of MMP3-Fc was performed as described previously for MMP10. Samples were then transferred into wells containing a 20 μm concentration of an MMP10 quenched fluorescent substrate at a 1:1 volumetric ratio. Plates were then mixed and read for fluorescence on a BMG Clariostar at wavelengths of excitation 320 nm and emission 405 nm. Rates are calculated as the slope of fluorescence generation (y axis) over time (x axis). Curves were generated using GraphPad Prism 7 for Mac OS X.
Expression of MMP1, -2, -9, and -13
MMP1, -2, -9, and -13 were all cloned as MMP10 and MMP3 were. In short, gBlock gene synthesis fragments were ordered for MMP1, -2, -9, and -13, corresponding to Uniprot limits 1–469, 1–660, 1–707, and 1–471, respectively. Each gene synthesis had flanking sequences that were cut with the restriction enzymes NheI/AgeI and cloned into pPL1-LFc as with MMP10 and MMP3. All subsequent steps up to the activation of the protease were the same as for MMP10; however, treatments of MMPs with APMA varied in terms of length of incubation. MMP1, -2, and -13 were incubated in 1 mm APMA after being buffer-exchanged into TCNB for 2 h, whereas MMP9 was activated in 1 mm APMA for 24 h at 37 °C. Activity was measured by titrating enzyme in TCNB and transferring to a black 96-well NUNC plate containing 2 μm MCA-RPKPVE-NvalWRK-dpa-NH2 for MMP3 and -10 and 2 μm Mca-PLGL-Dpa-AR-NH2 for MMP1, -2, -9, and -13. Activity was checked on a BMG Clariostar plate reader with 320-nm excitation/405-nm emission. Slopes were analyzed in GraphPad Prism software.
Cross-reactivity of H3
TIMP1-mFc and H3-hFc were titrated onto each protease starting at a concentration of 1 μm and titrated 1:10 with constant MMP concentration (MMP1, 30 nm; MMP2, 30 nm; MMP3, 30 nm; MMP9, 0.5 nm; MMP13, 5 nm) as determined by A280, and titrations were left to complex for 15 min at room temperature. Titrations were then transferred to 96-well black NUNC plates containing a 2 μm concentration of the respective substrates for each of the MMPs, and activity was checked by measurement on the BMG Labtech Clariostar plate reader as highlighted in the experiments above. Slopes were analyzed in GraphPad Prism software.
Characterization of H3-mediated MMP10 inhibition
mAb H3, mAb C10, and TIMP1 were titrated 1:2 starting at 400 nm across 23 points in the presence of 2 nm human MMP10 (R&D Systems; see above) and left to complex for 15 min at room temperature. Titrations were then transferred to a quenched fluorescent substrate of MMP10 (R&D Systems, catalog no. ES-002) at a 1:1 volumetric ratio and read for fluorescence generation on a BMG Clariostar. Rates are calculated as the slope of fluorescence generation (y axis) over time (x axis). Curves representing inhibition with the antibodies or TIMPs on MMP10 were generated using GraphPad Prism 7 software.
Inhibition of MMP10-mediated cleavage of AAT with H3
To test inhibition of MMP10-mediated cleavage of AAT (provided as a gift from Inhibrx), a serial dilution of H3 was placed onto 80 nm MMP10-Fc and complexed for 15 min at room temperature. The titrations were then transferred to wells containing 1.5 μm AAT and incubated at 37 °C for 16 h and analyzed by SDS-PAGE. Titrations were then diluted 1:100 and incubated with 20 nm neutrophil elastase (Athens Bio) for 15 min. H3/MMP10/AAT/NE complexes were then transferred to wells containing 100 μm AAPV-AMC (Enzo), and fluorescence (NE activity) was monitored on a BMG Clariostar microplate reader.
H3 binding active versus zymogen MMP10
To test the mechanism of inhibition, a series of binding and competition assays were performed to determine the binding region of H3. First, H3 was checked to determine binding against active and zymogen MMP10 using BLI. H3 was biotinylated using Thermo Scientific EZ-LinkTM NHS-Biotin. H3 was then coated onto a streptavidin sensor and was introduced to 2.4 μg/ml of either APMA-treated or non-APMA-treated hMMP10-LFc with the entire assay being performed in TCNB buffer.
H3 binding marimastat inhibited MMP10
H3 was tested for its ability to bind small-molecule inhibited MMP10 using BLI. For this assay, 7 μg/ml hMMP10-LFc was treated with 10 μm marimastat (Sigma, catalog no. M2699-5MG) and incubated for 30 min. Biotinylated H3 (see above) was then coated onto a streptavidin sensor and introduced to noninhibited active hMMP10-LFc or inhibited hMMP10-LFc.
H3 competition with TIMPs for MMP10 binding
TIMP1 and TIMP2 were tested for their ability to bind an H3/MMP10 complex using BLI. First, biotinylated H3 was coated onto the surface of a streptavidin sensor and introduced to active hMMP10-LFc. The sensor was then transferred to a well containing either TIMP1 or TIMP2 and measured for binding.
Author contributions
A. S. R., B. P. E., and G. S. S. conceptualization; A. S. R. data curation; A. S. R. and G. S. S. formal analysis; A. S. R. validation; A. S. R. investigation; A. S. R. and B. P. E. methodology; A. S. R. writing-original draft; A. S. R. and G. S. S. writing-review and editing; B. P. E. and G. S. S. resources; B. P. E. and G. S. S. funding acquisition; G. S. S. supervision; G. S. S. project administration.
Acknowledgments
We thank Inhibrx for material contributions to this study. We also thank Gillian Murphy for intellectual contributions to this study.
This work was supported by NIGMS, National Institutes of Health, Grant GM-099040 (to G. S. S.) and an Academic/Industry Fellowship from Inhibrx (to A. S. R.). The authors declare the following financial relationships. Amir S. Razai and Guy S. Salvesen consult for Inhibrx, and Brendan P. Eckelman is employed by Inhibrx. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MMP
- matrix metalloproteinase
- CDR
- complementarity-determining region
- bis-tris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- TIMP
- tissue inhibitor of metalloprotease
- APMA
- aminophenylmercuric acetate
- BLI
- bio-layer interferometry
- AAT
- α1-antitrypsin
- NE
- neutrophil elastase.
References
- 1. Peterson J. T. (2004) Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 9, 63–79 10.1023/B:HREV.0000011395.11179.af [DOI] [PubMed] [Google Scholar]
- 2. Gatto C., Rieppi M., Borsotti P., Innocenti S., Ceruti R., Drudis T., Scanziani E., Casazza A. M., Taraboletti G., and Giavazzi R. (1999) BAY 12-9566, a novel inhibitor of matrix metalloproteinases with antiangiogenic activity. Clin. Cancer Res. 5, 3603–3607 [PubMed] [Google Scholar]
- 3. Brown P. D. (1999) Clinical studies with matrix metalloproteinase inhibitors. APMIS 107, 174–180 10.1111/j.1699-0463.1999.tb01541.x [DOI] [PubMed] [Google Scholar]
- 4. Liotta L. A., Tryggvason K., Garbisa S., Hart I., Foltz C. M., and Shafie S. (1980) Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 284, 67–68 10.1038/284067a0 [DOI] [PubMed] [Google Scholar]
- 5. Liotta L. A., Thorgeirsson U. P., and Garbisa S. (1982) Role of collagenases in tumor cell invasion. Cancer Metastasis Rev. 1, 277–288 10.1007/BF00124213 [DOI] [PubMed] [Google Scholar]
- 6. Fingleton B. (2008) MMP inhibitor clinical trials: the past, present, and future. in The Cancer Degradome: Proteases and Cancer Biology (Sloane B. F., Blasi F., Edwards E., and Hoyer-Hansen G., eds) pp. 759–785, Springer, Berlin [Google Scholar]
- 7. Mazurek S. G. N., Li J., Nabozny G. H., Reinhart G. A., Muthukumarana A. C., Harrison P. C., and Fryer R. M. (2011) Functional biomarkers of musculoskeletal syndrome (MSS) for early in vivo screening of selective MMP-13 inhibitors. J. Pharmacol. Toxicol. Methods 64, 89–96 10.1016/j.vascn.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 8. Overall C. M., and Kleifeld O. (2006) Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 6, 227–239 10.1038/nrc1821 [DOI] [PubMed] [Google Scholar]
- 9. Rodríguez D., Morrison C. J., and Overall C. M. (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54 10.1016/j.bbamcr.2009.09.015 [DOI] [PubMed] [Google Scholar]
- 10. Overall C. M., and Dean R. A. (2006) Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 25, 69–75 10.1007/s10555-006-7890-0 [DOI] [PubMed] [Google Scholar]
- 11. Fingleton B. (2008) MMPs as therapeutic targets-Still a viable option? Semin. Cell Dev. Biol. 19, 61–68 10.1016/j.semcdb.2007.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sela-Passwell N., Rosenblum G., Shoham T., and Sagi I. (2010) Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 1803, 29–38 10.1016/j.bbamcr.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 13. Marshall D. C., Lyman S. K., McCauley S., Kovalenko M., Spangler R., Liu C., Lee M., O'Sullivan C., Barry-Hamilton V., Ghermazien H., Mikels-Vigdal A., Garcia C. A., Jorgensen B., Velayo A. C., Wang R., et al. (2015) Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS ONE 10, e0127063 10.1371/journal.pone.0127063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santamaria S., and de Groot R. (2019) Monoclonal antibodies against metzincin targets. Br. J. Pharmacol. 176, 52–66 10.1111/bph.14186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., and Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 10.1073/pnas.0505379103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drag M., and Salvesen G. S. (2010) Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701 10.1038/nrd3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iyer S., Wei S., Brew K., and Acharya K. R. (2007) Crystal structure of the catalytic domain of matrix metalloproteinase-1 in complex with the inhibitory domain of tissue inhibitor of metalloproteinase-1. J. Biol. Chem. 282, 364–371 10.1074/jbc.M607625200 [DOI] [PubMed] [Google Scholar]
- 18. Nagase H., Meng Q., Malinovskii V., Huang W., Chung L., Bode W., Maskos K., and Brew K. (1999) Engineering of selective TIMPs. Ann. N.Y. Acad. Sci. 878, 1–11 10.1111/j.1749-6632.1999.tb07670.x [DOI] [PubMed] [Google Scholar]
- 19. Nagase H., and Brew K. (2002) Engineering of tissue inhibitor of metalloproteinases mutants as potential therapeutics. Arthritis Res. 4, S51–S61 10.1186/ar573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buelens K., Hassanzadeh-Ghassabeh G., Muyldermans S., Gils A., and Declerck P. J. (2010) Generation and characterization of inhibitory nanobodies towards thrombin activatable fibrinolysis inhibitor. J. Thromb. Haemost. 8, 1302–1312 10.1111/j.1538-7836.2010.03816.x [DOI] [PubMed] [Google Scholar]
- 21. Muyldermans S. (2001) Single domain camel antibodies: current status. J. Biotechnol. 74, 277–302 10.1016/s1389-0352(01)00021-6 [DOI] [PubMed] [Google Scholar]
- 22. Lauwereys M., Arbabi Ghahroudi M., Desmyter A., Kinne J., Hölzer W., De Genst E., Wyns L., and Muyldermans S. (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17, 3512–3520 10.1093/emboj/17.13.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nam D. H., Rodriguez C., Remacle A. G., Strongin A. Y., and Ge X. (2016) Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc. Natl. Acad. Sci. U.S.A. 113, 14970–14975 10.1073/pnas.1609375114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Revets H., De Baetselier P., and Muyldermans S. (2005) Nanobodies as novel agents for cancer therapy. Expert Opin. Biol. Ther. 5, 111–124 10.1517/14712598.5.1.111 [DOI] [PubMed] [Google Scholar]
- 25. Cuniasse P., Devel L., Makaritis A., Beau F., Georgiadis D., Matziari M., Yiotakis A., and Dive V. (2005) Future challenges facing the development of specific active-site-directed synthetic inhibitors of MMPs. Biochimie 87, 393–402 10.1016/j.biochi.2004.09.025 [DOI] [PubMed] [Google Scholar]
- 26. Mariya T., Hirohashi Y., Torigoe T., Tabuchi Y., Asano T., Saijo H., Kuroda T., Yasuda K., Mizuuchi M., Saito T., and Sato N. (2016) Matrix metalloproteinase-10 regulates stemness of ovarian cancer stem-like cells by activation of canonical Wnt signaling and can be a target of chemotherapy-resistant ovarian cancer. Oncotarget 7, 26806–26822 10.18632/oncotarget.8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Regala R. P., Justilien V., Walsh M. P., Weems C., Khoor A., Murray N. R., and Fields A. P. (2011) Matrix metalloproteinase-10 promotes Kras-mediated bronchio-alveolar stem cell expansion and lung cancer formation. PLoS ONE 6, e26439 10.1371/journal.pone.0026439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Justilien V., Regala R. P., Tseng I.-C., Walsh M. P., Batra J., Radisky E. S., Murray N. R., and Fields A. P. (2012) Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS ONE 7, e35040 10.1371/journal.pone.0035040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gill J. H., Kirwan I. G., Seargent J. M., Martin S. W., Tijani S., Anikin V. A., Mearns A. J., Bibby M. C., Anthoney A., and Loadman P. M. (2004) MMP-10 is overexpressed, proteolytically active, and a potential target for therapeutic intervention in human lung carcinomas. Neoplasia 6, 777–785 10.1593/neo.04283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dickinson B. L., Badizadegan K., Wu Z., Ahouse J. C., Zhu X., Simister N. E., Blumberg R. S., and Lencer W. I. (1999) Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J. Clin. Invest. 104, 903–911 10.1172/JCI6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jendeberg L., Persson B., Andersson R., Karlsson R., Uhlén M., and Nilsson B. (1995) Kinetic analysis of the interaction between protein a domain variants and human Fc using plasmon resonance detection. J. Mol. Recognit. 8, 270–278 10.1002/jmr.300080405 [DOI] [PubMed] [Google Scholar]
- 32. Vu K. B., Ghahroudi M. A., Wyns L., and Muyldermans S. (1997) Comparison of llama V(H) sequences from conventional and heavy chain antibodies. Mol. Immunol. 34, 1121–1131 10.1016/S0161-5890(97)00146-6 [DOI] [PubMed] [Google Scholar]
- 33. Dong J., Thompson A. A., Fan Y., Lou J., Conrad F., Ho M., Pires-Alves M., Wilson B. A., Stevens R. C., and Marks J. D. (2010) A single-domain llama antibody potently inhibits the enzymatic activity of botulinum neurotoxin by binding to the non-catalytic α-exosite binding region. J. Mol. Biol. 397, 1106–1118 10.1016/j.jmb.2010.01.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Z., Zhou X., Shapiro S. D., Shipley J. M., Twining S. S., Diaz L. A., Senior R. M., and Werb Z. (2000) The serpin α1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell 102, 647–655 10.1016/S0092-8674(00)00087-8 [DOI] [PubMed] [Google Scholar]
- 35. Mast A. E., Enghild J. J., Nagase H., Suzuki K., Pizzo S. V., and Salvesen G. (1991) Kinetics and physiologic relevance of the inactivation of α1-proteinase inhibitor, α1-antichymotrypsin, and antithrombin III by matrix metalloproteinases-1 (tissue collagenase), -2 (72-kDa gelatinase/type IV collagenase), and -3 (stromelysin). J. Biol. Chem. 266, 15810–15816 [PubMed] [Google Scholar]
- 36. Yang Y., and Rosenberg G. A. (2015) Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 1623, 30–38 10.1016/j.brainres.2015.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shin G., Kang T. W., Yang S., Baek S. J., Jeong Y. S., and Kim S. Y. (2011) GENT: gene expression database of normal and tumor tissues. Cancer Inform. 10, 149–157 10.4137/CIN.S7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deraz E. M., Kudo Y., Yoshida M., Obayashi M., Tsunematsu T., Tani H., Siriwardena S. B. S. M., Keikhaee M. R., Qi G., Iizuka S., Ogawa I., Campisi G., Muzio Lo L., Abiko Y., et al. (2011) MMP-10/stromelysin-2 promotes invasion of head and neck cancer. PLoS ONE 6, e25438 10.1371/journal.pone.0025438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boder E. T., and Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 10.1038/nbt0697-553 [DOI] [PubMed] [Google Scholar]