

Abstract
Stimulant medication has long been effective in treating attention-deficit/hyperactivity disorder (ADHD) and is currently the first-line pharmacological treatment for children. Both methylphenidate and amphetamine modulate extracellular catecholamine levels through interaction with dopaminergic, adrenergic and serotonergic system components; it is therefore likely that catecholaminergic molecular components influence the effects of ADHD treatment. Using meta-analysis, we sought to identify predictors of pharmacotherapy to further the clinical implementation of personalized medicine. We identified 36 studies (3647 children) linking the effectiveness of methylphenidate treatment with DNA variants. Pooled-data revealed a statistically significant association between single nucleotide polymorphisms (SNPs) rs1800544 ADRA2A (odds ratio: 1.69; confidence interval: 1.12–2.55), rs4680 COMT (odds ratio (OR): 1.40; confidence interval: 1.04–1.87), rs5569 SLC6A2 (odds ratio: 1.73; confidence interval: 1.26–2.37) and rs28386840 SLC6A2 (odds ratio: 2.93; confidence interval: 1.76–4.90), and, repeat variants variable number tandem repeat (VNTR) 4 DRD4 (odds ratio: 1.66; confidence interval: 1.16–2.37) and VNTR 10 SLC6A3 (odds ratio: 0.74; confidence interval: 0.60–0.90), whereas the following variants were not statistically significant: rs1947274 LPHN3 (odds ratio: 0.95; confidence interval: 0.71–1.26), rs5661665 LPHN3 (odds ratio: 1.07; confidence interval: 0.84–1.37) and VNTR 7 DRD4 (odds ratio: 0.68; confidence interval: 0.47–1.00). Funnel plot asymmetry among SLC6A3 studies was identified and attributed largely to small study effects. Egger’s regression test and Duval and Tweedie’s ‘trim and fill’ were used to examine and correct for publication bias. These findings have major implications for advancing our therapeutic approach to childhood ADHD treatment.
INTRODUCTION
Attention-deficit/hyperactivity disorder (ADHD) is the most common neuropsychiatric disease treated in children today with a recent study reporting a pooled-prevalence of 5.3%.1 There are currently five FDA-approved medications for the treatment of childhood ADHD, each falling within one of two drug classes: stimulant (methylphenidate (MPH) or amphetamine (AMP)) or non-stimulant (atomoxetine (ATX), extended-release guanfacine or extended-release clonidine).2,3 The use of stimulant medication has long been considered safe and efficacious; prescription stimulant use continues to be the first-line psychopharmacological treatment for childhood ADHD4–9 and is exclusively indicated—in conjunction with behavioral therapy—for preschool-aged children (4–5 years of age).2 Many similarities exist between MPH and other psychostimulant preparations, both in terms of efficacy and side effect profile. Despite this, patient response varies greatly and some individuals preferentially respond to or tolerate one treatment over another.10–13
Over the past 20 years many independent studies have assessed the treatment of ADHD-diagnosed children and adolescents with MPH. Though MPH treatment proved beneficial by reducing hyperactive and inattentive behaviors,14,15 it is important to note that rare but serious adverse reactions occurred in about 3% of children.16,17 For example, the development of tic disorder, elongated QT durations or the onset of depression, psychosis and/or mania were identified in some MPH-treated individuals.11,18 Furthermore, a recent comprehensive meta-analysis including 185 studies of MPH use for ADHD treatment reported a high frequency of non-serious adverse events associated with treatment, including for example, insomnia and loss of appetite.19 Currently, no clinical guidelines exist to address these issues with a personalized approach for individualized medication.
Inter-individual variability in MPH response may be influenced by genetic factors.20 Classification of 60 thousand single nucleotide polymorphisms (SNPs), that fell within exon coding regions, were categorized through navigation of the 1.42 million SNPs within the human genome.21 As a means to direct ADHD treatment through use of clinical predictors, subsequent molecular genetic studies have facilitated the identification of relevant genetic markers likely responsible for interindividual drug response. The metabolic system influences plasma concentrations of MPH before it reaches the brain22 and studies in ADHD-children report significant inter-individual variability of MPH plasma concentrations following administration of standardized doses.23–25 Characterization and sequencing of the carboxylesterase 1A1 (CES1A1) gene led to the identification of variants that are associated with signficantly reduced enzyme activity.26–28 For example, C/T (rs71647871;Gly143Glu) heterozygotes required appreciably less MPH than C/C homozygotes, suggesting reduced CES1A1 enzyme activity.29 Furthermore, possession of the G allele (rs3815583; – 75T>G) was associated with worsening anorexia during MPH treatment in stimulant-naïve ADHD children.30
Moreover, polymorphisms within key monoaminergic genes have been associated with the response to stimulant medication, albeit through conflicting evidence. This is mechanistically intuitive as both MPH and AMP modulate extracellular catecholamine levels through interaction with dopaminergic, adrenergic and serotonergic system components. AMP increases synaptic dopamine (DA) levels through three major pathways: competitive inhibition of the DA transporter (DAT), enhanced release of vesicular DA and promotion of reverse DA transport into the synaptic cleft.31 AMP also disrupts vesicular DA storage and inhibits normal monoamine oxidase degradation.32 More recently, it was shown that at low doses, AMP cannot reach sufficient vesicular concentrations to facilitate emanation and this may account for phasic pharmacological effects.33 Similarly, MPH inhibits catecholamine reuptake and modulates DA and norepinephrine levels by binding to and blocking DA and norepinephrine transporters, thereby increasing extracellular concentrations.34 Therapeutic doses of MPH cause 80% blockage of DAT and significantly enhance extracellular DA in the basal ganglia.35 This effect is modulated by the rate of DA release, which can be influenced by age: a heightened effect can be seen in younger children.36 Thus, it is likely that catecholaminergic molecular components influence the effects of ADHD treatment.
Genetic variation may lead to differential symptom changes and side effect profiles. Often, discontinuation of treatment due to side effects or the administration of sub-optimal treatment regimens limits the effectiveness of stimulant treatment. This leaves children impaired and exposes them to undue consequences from untreated ADHD.14 There is currently no way to predict this preferential response in treatment naïve patients, nor to direct a personalized treatment regimen. Pharmacogenetics testing has the potential to reduce discontinuation due to adverse events and improve time to efficacy. Here we report a meta-analysis evaluating the aggregate body of literature in an effort to identify clinically significant predictors of MPH response in children. We report five genes—SLC6A2, SLC6A3, COMT, DRD4 and ADRA2A—that may cumulatively act as reliable predictors to MPH response.
METHODS
Search strategy
To identify eligible studies, a systematic review of the National Center for Biotechnology Information (NCBI) PubMed database (https://www.ncbi.nlm.nih.gov/pubmed/) was performed using the broad search terms ‘ADHD’, ‘Attention-Deficit/Hyperactivity Disorder’, ‘pharmacogenetics’, ‘stimulant medication’, ‘methylphenidate’, ‘medication response AND genetic’, ‘methylphenidate AND gene’, ‘methylphenidate AND genetic’. Further specification of literature relevant to clinical care and research was obtained through use of MeSH headings and filters, and by tailoring search terms using Boolean logic: methylphenidate OR MPH AND (Attention-Deficit/Hyperactivity Disorder OR ADHD) AND pharmacogenetic OR pharmacogenomic; amphetamine AND (Attention-Deficit/Hyperactivity Disorder OR ADHD) AND pharmacogenetic OR pharmacogenomic; stimulant AND (Attention-Deficit/Hyperactivity Disorder OR ADHD) AND pharmacogenetic OR pharmacogenomics. Searches were limited to articles available in English and included publications up to April 2017. Additionally, relevant citations were followed after initial searches. Of the available literature, only articles addressing pharmacogenetics, treatment efficacy and childhood ADHD were considered.
Inclusion criteria
To minimize heterogeneity among studies we initially assessed literature for violation of inclusion criteria and included only studies that met set criteria in further analyses. Articles were included if the patient cohort comprised children and/or adolescents under 18 years of age. Review articles, meta-analyses and case studies with a sample size of nine or less were excluded; original research articles with a sample size of at least 10 subjects were required. Within studies ADHD diagnosis must have been determined by use of DSM criteria (third, fourth or fifth edition) or a comparable diagnostic method. Results from studies that were further stratified by comorbidity or ADHD subtype were not included in this analysis. Likewise, studies were excluded if their reported results did not include quantitative measures transformable into ORs (for example, Cohen’s d, Cohen’s f2, F-test, etc.). Upon completion of a robust search, further stratification of results was performed. Research articles correlating specific genetic variants to MPH efficacy in childhood ADHD were considered eligible. Few studies reported tolerability results and were therefore not analyzable. Furthermore, studies with overlapping patient samples were excluded to only include the study with the larger number of patients. Research articles were further categorized by gene and then genetic variant. Results from genetic variants reported in three or more independent research articles were combined and meta-analyzed.
Outcome measures
Pre- and post-treatment outcome measures and/or effect size changes were analyzed to evaluate differential treatment response. Publications were included regardless of measurement scale used to evaluate the treatment effect of MPH. Therefore, both categorical and dimensional measurements were extracted for analysis. For studies that employed a quantitative scale or instrument to gauge ADHD symptoms, only those studies using validated ADHD rating scales were used.
Study selection
Article abstracts were initially assessed to determine if they met overall inclusion criteria. Subsequently, full version articles were blindly reviewed by three independent researchers and evaluated for methodological soundness. Candidate genes were selected for review if evaluated by two or more studies and entered into meta-analysis if data for the same genetic variant was available from at least three studies.
Data extraction
Demographic information, including mean age, sex ratios and cohort ethnicity, were extracted. Relevant aspects of study design, including study type, sample size, drug choice and dosage, were compiled. Appropriate transformations were used to estimate effect sizes of MPH efficacy—expressed as odds ratios (ORs)—and those values were evaluated using Review Manager (RevMan) version 5.3, an open-source software publicly available from the Cochrane Collaboration (http://community.cochrane.org/tools/review-production-tools/revman-5). As mentioned previously, few studies reported tolerability data and we were therefore unable to obtain a MPH tolerability effect size. If more than one source of data was available in a publication (that is, CPT and CGI-I), we preferentially pooled data from a clinician-administered scale over those scales administered by a parent or teacher.
Statistical analysis
Individual OR and standard error (s.e.) values were calculated for each study. In several studies ORs for differential treatment response by genotype were reported. For cases in which ORs were not reported additional calculations were required. Any valid measure of differential treatment response among alleles was extracted or statistically derived. Transformations from these measures, including alternative metrics of effect size (r, Cohen’s d, η2, and Cohen’s f2) and measures of mean difference (ANOVA and χ2), were performed following the methods of Borenstein et al.37 Heterogeneity among studies was assessed in RevMan 5 with the χ2-test and calculation of I2. The overall effect for each SNP was calculated using a fixed effect model.
Publication bias analyses
To detect publication bias, funnel plot—ORs against s.e.(log[OR])—dispersion patterns were visually assessed for symmetry. Symmetry is based on a weighted linear regression of standard normal deviation of the OR (standardized effect) on the inverse of the s.e. of the OR (precision) (that is, the larger the deviation of each study from the funnel curve, the more pronounced the asymmetry). Publication bias was quantitatively measured using Egger’s Regression Tests, where a P-value of < 0.05 indicated statistically significant publication bias. For those genetic variants with significant publication bias Duval and Tweedie’s ‘Trim and Fill’ was used to generate an adjusted best estimate of the unbiased effect size.
Meta-regression
To assess the relationship between covariates (moderators)—mean age, gender ratio (% male), ethnicity (% white) or study quality—and ORs we performed individual meta-regressions for each genetic variant. Ethnicity regressions were incomplete as many studies did not include usable ethnicity data. For study quality analyses, studies were binned into ‘strong’, ‘moderate’ or ‘weak’ categories according to the Effective Public Health Practice Project (EPHPP) Quality Assessment Tool for Quantitative Studies. The regression coefficient was then assessed for statistical significance. A Welch’s unequal variances t-test comparing randomized controlled trials to all other study designs was also performed.
RESULTS
The study selection procedure is illustrated in Figure 1. A total of 161 eligible studies were identified both manually (n = 41 studies) and through systematic PubMed review (n = 120 studies). Several studies were excluded based on our a priori eligibility criteria: 15 included adult participants over the age of 18, 19 reported data that was not transformable into a binary effect size, 54 were not primary research articles (for example, review articles), 13 employed non-MPH drugs (that is, atomoxetine or amphetamine), 1 included a repeat patient cohort, 17 addressed unrelated topics (for example, autism disorder and MPH response), and 6 included genetic variants whose total study cohort did not exceed three. After filtering by these criteria, 36 total studies (n = 3647 children) remained and were used to assess MPH efficacy for childhood ADHD treatment.
Figure 1.
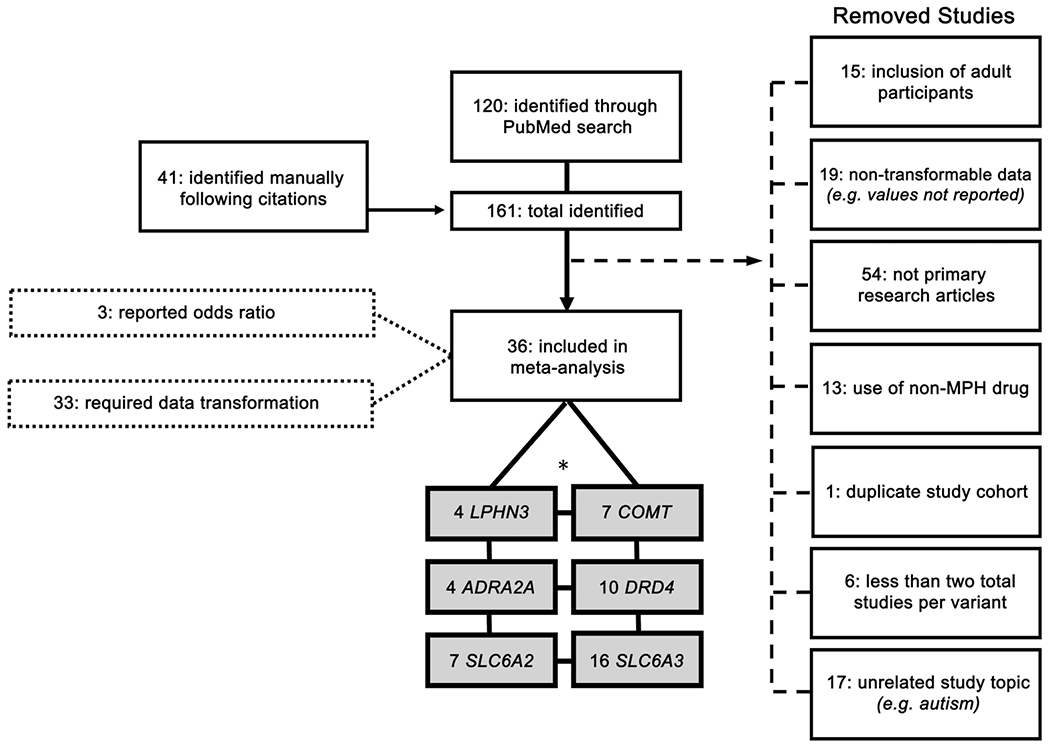
Flow chart of study inclusion. Diagram depicts the systematic search progression used in this study. Boxes attached to the body of the diagram by a dashed line represent removed studies. Boxes attached to the body of the diagram by dotted lines represent data-input methods. Grayed boxes indicate included studies. *The six genes chosen for further meta-analysis exceed 36 (represented in diagram as ‘36 included in further analysis’) because some studies included more than one gene and/or genetic variant.
From the pooled childhood cohort (mean age = 9.5 years; age range = 4.3–12.8 years; % male = 83%), we report results from nine allelic variants distributed among six genes (Supplementary Table 1). Of these, we meta-analyzed data from three publications for rs28386840 (SLC6A2), seven for rs5569 (SLC6A2), four for rs1800544 (ADRA2A), seven for rs4680 (COMT), four for rs6551665 (LPHN3), three for rs1947274 (LPHN3), six for the 4-repeat VNTR of DRD4, five for the 7-repeat VNTR of DRD4, and 16 for the 10-repeat VNTR of SLC6A3. All but two studies used DSM-IV criteria to diagnose ADHD; DSM-III and ICD-10 codes were used by Winsberg et al38 and Seeger et al.,39 respectively. Most of the studies analyzed were of high quality—58.9% of studies were rated ‘strong’, 35.7% were rated ‘moderate’ and 3.6% were rated ‘weak’ using the Effective Public Health Practice Project (EPHPP) Quality Assessment Tool for Quantitative Studies (QA). All but two studies (Tharoor et al.,40 and Kirley et al,41) utilized a prospective study design.
Association of MPH and variants
Using a fixed-effect model for analysis, we identified a significant association between MPH efficacy and DNA variants tagging the genes. Five studies reported data on adverse events in addition to efficacy; however, there was not enough data to support statistical analysis. Several genes significantly affected the response to MPH: SLC6A2, ADRA2A, COMT, DRD4 and SLC6A3 (Figures 2, 3). Odds ratios were used as a measure of effect size and ranged from 0.95 to 2.9. When correlating MPH efficacy to polymorphisms, four SNPs reached statistical significance: rs28386840 (SLC6A2), rs5569 (SLC6A2), rs1800544 (ADRA2A) and rs4680 (COMT) (Figures 2a–d). For rs28386840 (SLC6A2), the T allele was associated with an improved response to MPH treatment when compared to the A/A genotype (OR: 2.93, CI: 1.76–4.90, P < 0.0001) (Figure 2a). The G/G genotype of rs5569 (SLC6A2) was associated with an improved MPH response compared to A allele carriers (OR: 1.73, CI: 1.26–2.37, P = 0.0007) (Figure 2b). Furthermore, the G allele of rs1800544 (ADRA2A) was associated with improved MPH response compared to C allele carriers (OR: 1.69, CI: 1.12–2.55, P = 0.01) (Figure 2c). Meta-analysis of the rs4680 variant in COMT revealed that the Val/Val genotype was associated with improved treatment response compared with Met allele carriers (OR: 1.40, CI: 1.04–1.87, P = 0.02) (Figure 2d). Two variants within the LPHN3 gene were not significantly associated with MPH response: rs5661665 (OR: 1.07, CI: 0.84–1.37, P = 0.59) and rs1947274 (OR: 0.95, CI: 0.71–1.26, P = 0.70) (Figures 2e and f).
Figure 2.
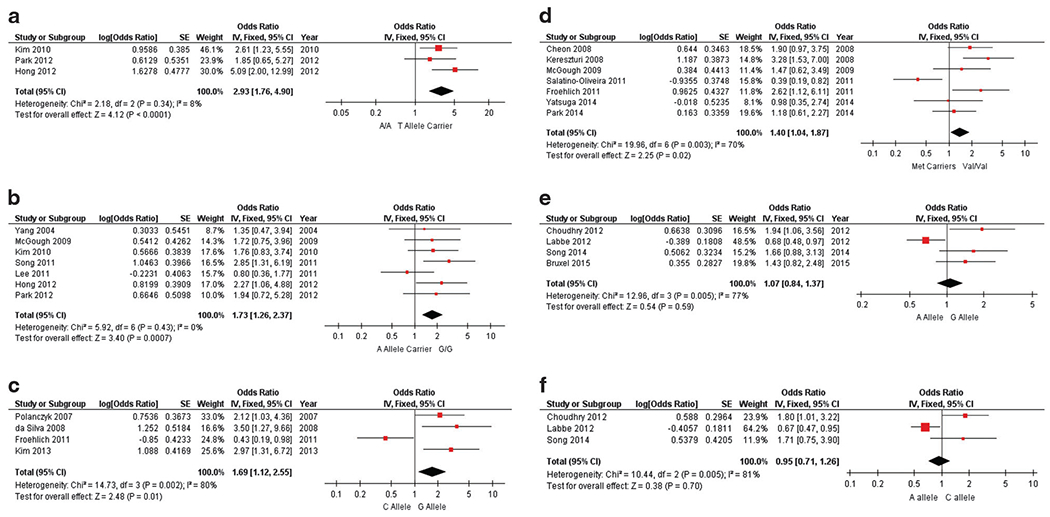
Pooled results correlating MPH efficacy to SNPs. Forest plots of association between MPH efficacy and the following SNPs: (a) SLC6A2 rs28386840; (b) SLC6A2 rs5569; (c) ADRA2A rs1800544; (d) COMT rs4680; (e) LPHN3 rs5661665; (f) LPHN3 rs1947274. Each forest plot represents one genetic variant and its computed effect size (odds ratio, measure of precision and P-value). Red squares are proportional to the studies’ weights and horizontal black bars represent the corresponding 95% confidence interval (precision measurement) for each study. The vertical line at 1 indicates the null effect line and represents no differences between groups. The location of the black box represents the direction and magnitude of the pooled odds ratio that was generated from a fixed-effect model; its width represents the upper and lower bounds of the 95% confidence interval. Assessments of heterogeneity by χ2 and I2 are located on the left of each forest plot. A P-value is given for a test of the null. MPH, methylphenidate; SNP, single nucleotide polymorphism.
Figure 3.
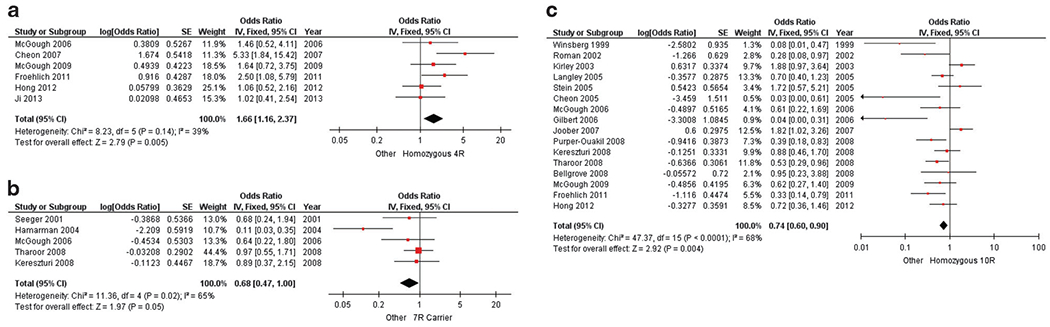
Pooled results correlating MPH efficacy to VNTRs. Forest plots of association between MPH efficacy and the following DNA variants: (a) DRD4 4-repeat; (b) DRD4 7-repeat; (c) SLC6A3 10-repeat. Each forest plot represents one genetic variant and its computed effect size (odds ratio, measure of precision and P-value). Red squares are proportional to the studies’ weights and horizontal black bars represent the corresponding 95% confidence interval (precision measurement) for each study. The vertical line at 1 indicates the null effect line and represents no differences between groups. The location of the black box represents the direction and magnitude of the pooled odds ratio that was generated from a fixed-effect model; its width represents the upper and lower bounds of the 95% confidence interval. Assessments of heterogeneity by χ2 and I2 are located on the left of each forest plot. A P-value is given for a test of the null. MPH, methylphenidate.
Two classifications of the DRD4 48bp VNTR polymorphism have been reported in the literature, the 4-repeat allele versus others and the 7-repeat allele versus others. The homozygous 4-repeat genotype demonstrated an association with improved MPH response when compared to other genotypes (OR: 1.66, CI: 1.16–2.37, P = 0.005) (Figure 3a). In contrast, meta-analysis of the 7-repeat allele versus others did not reach statistical significance (OR: 0.68, CI: 0.47–1.00, P = 0.05) (Figure 3b). Lastly, the homozygous 10-repeat genotype of the 40bp VNTR in the SLC6A3 gene was significantly associated with reduced efficacy (OR: 0.74, CI: 0.60–0.90, P = 0.004) (Figure 3c).
Three additional genetic variants—within the Glutamate Ionotropic Receptor NMDA Type Subunit 2B (GRIN2B), Brain-derived Neurotrophic Factor (BDNF) and Synaptosomal-associated Protein 25 (SNAP-25) genes—were found in our systematic search, but did not meet our inclusion criteria because the total study cohort for these variants did not exceed three.42–46 The C/C genotype of rs2284411 (GRIN2B) was associated with improved MPH treatment response (OR: 9.03, CI: 1.02–79.99, P = 0.05). The Val/Val genotype of rs6265 (BDNF) was not significantly associated with MPH response, despite having a substantial effect size (OR: 6.67, CI: 0.84–52.89, P = 0.07). Finally, the T/T genotype of rs3746544 (SNAP25) was not significantly associated with treatment response in two studies (OR:1.12, CI: 0.55–2.25, P = 0.86;OR: 1.66, CI: 0.51–5.39, P = 0.41).
Evidence for publication bias
We tested for publication bias by first assessing funnel plots for symmetry. Funnel plots present the relationship between effect size (odds ratio) and s.e. of log(OR) (Supplementary Figure 1). An asymmetrical distribution was visually identified in funnel plots for rs6551665 (LPHN3) and the 10-repeat VNTR (SLC6A3) (Supplementary Figures 1F and 1I, respectively). Egger’s regression tests detected significant publication bias (asymmetry) for both DNA variants (P = 0.01). To adjust for this publication bias, we recomputed the effect sizes for both DNA variants using Duval and Tweedie’s trim and fill method, which removes the most extreme small studies from the positive side of the funnel plot, provides an estimate of the number of studies that were not published, imputes the effect sizes of those missing studies, and calculates an adjusted overall effect.47,48 The adjusted effect size for the G allele of rs6551665 (LPHN3), as compared to the A allele, was not significant after imputing two studies (OR: 0.84, CI: 0.68–1.04, P < 0.1) (Supplementary Figure 2A). In contrast, the adjusted effect size for the 10-repeat homozygous VNTR (SLC6A3), compared with other repeat variants, remained significant after imputing four studies (OR: 0.82, CI: 0.67–1.00, P< 0.05) (Supplementary Figures 2A,B).
Accounting for heterogeneity
Heterogeneity and inconsistency statistics (χ2 and I2) were computed for all genetic variants meta-analyzed: rs28386840 (SLC6A2) (χ2 = 2.18, P = 0.34; I2 = 8%), rs5569 (SLC6A2) (χ2 = 5.92, P = 0.43; I2 = 0%), rs1800544 (ADRA2A) (χ2 = 14.73, P = 0.002; I2 = 80%), rs4680 (COMT) (χ2 = 19.96, P = 0.003; I2 = 70%), rs5661665 (LPHN3) (χ2 = 12.96, P = 0.005; I2 = 77%), rs1947274 (LPHN3) (χ2= 10.44, P = 0.005; I2 = 81%), 4-repeat VNTR (DRD4) (χ2 = 8.23, P = 0.14; I2 = 39%), 7-repeat VNTR (DRD4) (χ2 = 11.36, P = 0.02; I2 = 65%), 10-repeat VNTR (SLC6A3) (χ2 = 47.37, P < 0.0001 ; I2 = 68%). The ADRA2A, COMT, SLC6A3 and LPHN3 genes had high heterogeneity (> 65%) while DRD4 had moderate (30–64%) and SLC6A2 had no to low (< 29%) heterogeneity. As many of the meta-analyzed variants had moderate to high heterogeneity, we performed meta-regressions using a fixed-effect model to assess the relationship between covariates (mean age, gender and study quality) and effect size.
The majority of effect sizes were not significantly associated with the mean age of the study participants; however, the association between the G allele of rs1800544 (ADRA2A) and MPH response was stronger in studies of older cohorts ((k) = 4 (number of studies), b = 0.3662, P = 0.01). The opposite was true when examining the association between the G/G genotype of rs5569 (SLC6A2) and MPH efficacy—the association was stronger in studies with a relatively younger cohort (k = 7, b = −0.7139, P =0.04). Effect sizes of several genetic variants were moderated by study participant gender ratios. The association of MPH response and both the Val/Val genotype of rs4680 (COMT) and the 7-repeat VNTR (DRD4) was weaker in studies that included a higher proportion of male participants (k = 7, b = −0.0490, P =0.004 and k = 2, b = −0.1952, P = 0.005, respectively). The opposite was true for the associations of the G allele of rs1800544 (ADRA2A), the A allele of rs1947274 (LPHN3), and the G allele of rs6551665 (LPHN3); a larger proportion of male participants yielded a stronger association (k = 4, b = 0.2226, P = 0.002; k = 2, b = 0.0865, P = 0.004; k = 2, b = 12.4, P = 0.03, respectively). Study quality was significantly associated with the effect size of rs1800544 (ADRA2A) such that studies of higher quality revealed stronger associations between the G allele and MPH efficacy (k = 4, b = 1.0876, P = 0.01). Finally, genetic variants that reached statistical significance in the meta-analysis and showed no evidence of publication bias were combined in a meta-regression analysis to determine whether study quality predicted effect size. This analysis found no significant effect size associations with MPH response (k = 55, b = 0.0626, P = 0.4885) (Supplementary Figure 3). Finally, a Welch’s unequal variances t-test comparing randomized controlled trials to all other study designs was not significant (P = 0.35).
DISCUSSION
Here, we report the plausibility of SLC6A2, COMT, ADRA2A, SLC6A3 and DRD4 as genetic candidates to predict MPH efficacy in children. Several polymorphic variants tagging these genes significantly affected MPH treatment efficacy in ADHD-children: rs28386840 and rs5569 (SLC6A2), rs4680 (COMT), rs1800544 (ADRA2A) (Figure 2). We also report a significant association between MPH efficacy and DNA repeat variants, 10-repeat VNTR (SLC6A3) and 4-repeat VNTR (DRD4) (Figure 3). Both rs5661665 and rs1947274 (LPHN3) failed to reach statistical significance, as did the 7-repeat VNTR(DRD4); all three of these non-significant variants had moderate-to-high heterogeneity. Among the significant associations identified, SLC6A2 variants showed no to low heterogeneity, whereas DRD4, SLC6A3, COMT and ADRA2A variants showed moderate to high heterogeneity. Finally, an Egger’s Regression Test identified publication bias for the SLC6A3 DNA variant; however, the association remained significant after correction with Duval and Tweedie’s Trim and Fill (Supplementary Figure 2B).
Nineteen studies were not included in the meta-analyses because an odds ratio could not be calculated from the published data. In studies where an effect size could not be computed, relevant results were reviewed qualitatively. Three studies investigated the effect of rs1800544 (ADRA2A) and MPH response by measuring differential treatment outcomes in the GG genotype and C allele carriers. Each found no significant association between the SNP and an improved response to MPH.49–51 Sengupta et al.52 examined the association between COMT rs4680 and MPH response by rating task-oriented behavior, but found no significant genotype by treatment interaction effect. Two studies showed that there was no significant association between the 10-repeat VNTR in SLC6A3 and response to MPH.53,54 Another study that examined the SLC6A3 VNTR showed an association between the 10R/10R genotype and an improved neurocognitive response.55
SLC6A2 is a sensible biological candidate for predicting MPH efficacy. Encoding the norepinephrine transporter, it plays a vital role in the adrenergic system and is the main target of atomoxetine, a non-stimulant with ADHD efficacy. In addition, previous studies have shown that MPH is a potent inhibitor of the norepinephrine transporter, providing evidence of its mechanism of action. Here we report two SNPs, both of which have shown compelling associations with ADHD risk in previous studies.56–58 Firstly, rs28386840 is at position – 3081 upstream of the transcription initiation site. The T allele significantly reduces promoter function relative to the A allele and our analyses show it to be associated with a 193% increased risk of poor MPH efficacy59 (Figure 2a). Also, the A allele of rs5569, located at position 1287 in exon 9, was associated with a 73% increased risk for poor MPH efficacy (Figure 2b). Among the DNA variants examined in our meta-analysis, variants within the SLC6A2 gene showed the strongest correlation to MPH efficacy in ADHD-children. Statistical results from these analyses have little to no heterogeneity and both exceeded our a priori standards for significance.
A similarly pivotal component of the noradrenergic system is the alpha-2-adrenergic receptor, encoded by the ADRA2A gene. This receptor is the main target of guanfacine and clonidine, both of which are non-stimulants. Evidence supporting ADRA2A involvement in the etiology of ADHD comes from studies suggesting that prefrontal cortex alpha 2a-adrenoreceptors influence executive functions like attention and inhibitory control.60 Here, it is important to note that, generally speaking, the biology of treatment response and ADHD etiology are distinct. The two do not have to be linked and are often not (for example, in the case of narcolepsy). Further, our findings are consistent with prior evidence that MPH has been shown to increase stimulation of alpha-2A-adrenergic receptors.61 Owing to its implication in the mode of action of MPH, it is not surprising that our meta-analysis reports a significant association between possession of a G allele and enhanced MPH efficacy for the promoter region SNP that creates a Msp1 restriction site (rs1800544). The C allele was associated with a 69% increased risk for poor MPH response (Figure 2c). In addition to the Msp1 polymorphism, Hha1 (rs1800545) and Dra1 (rs553668) restriction polymorphisms have been studied in relation to ADHD, though, they were not meta-analyzed here as they did not meet our criteria for inclusion (that is, three or more independent research articles per SNP were not identified).
Catechol-o-methyltransferase is an enzyme responsible for the degradation of catecholamines and plays an especially important role in regulating extracellular DA levels.62 Encoded by the COMT gene, a SNP in exon 4 leads to a valine (Val) to methionine (Met) amino acid switch that ultimately causes reduced enzyme activity; homozygosity for the Val amino acid has been linked to 3–4 times reduced enzyme activity.63 Owing to its function in the dopaminergic pathway, this gene was initially implicated in ADHD risk and is likely highly relevant to stimulant medication response.56,64 We demonstrate here that Val/Val homozygotes demonstrate a 40% increased chance for efficacious MPH response (Figure 2d).
The 48-bp VNTR is the most widely studied DRD4 gene polymorphism and has been associated with ADHD susceptibility in a number of case-controlled studies and meta-analyses.56 The dopamine D4 receptor is a G-protein-coupled receptor that modulates signal transduction by altering intracellular cyclic AMP levels. Though there is ethnic variability, the most common alleles of this DNA variant have 2-, 4-, and 7-repeats.65 While non-significant results were obtained when comparing the 7R variant to other repeat variants, children with a 4R/4R genotype showed a 66% increased chance for efficacious MPH response (Figures 3a and b). This is consistent with reports that the 4-repeat VNTR leads to higher receptor expression and increased sensitivity to dopamine, as compared to the 7-repeat variant.66,67 From this and our results, we postulate that 7-repeat variant carriers have reduced dopamine 4 receptor expression and hence comparatively poor MPH efficacy, whereas 4-repeat carriers likely have ‘baseline’ dopamine 4 receptor expression.
SLC6A3 encodes the transmembrane DAT. Much research on this gene has focused on the 40-base pair VNTR polymorphism located in the 3′-untranslated region (3′UTR), which plays a regulatory role during transcription. The two common alleles, 9- and 10-repeats (9R and 10R), have shown mixed associations with stimulant efficacy during ADHD treatment.64 In a recent meta-analysis, the 40-bp VNTR polymorphism of DAT was shown to regulate striatal DA levels and the amount of DAT expression in the striatum.68 While this gene has been implicated in the underlying pathophysiology of ADHD,14,58 it is also a key pharmacological target of stimulant ADHD medications approved for use in treating childhood ADHD.69 As stimulant medications block the DAT, thereby increasing the concentration of DA in the synaptic cleft, it is intuitive to associate polymorphisms within SLC6A3 with variable stimulant efficacy. Despite having identified publication bias among the body of SLC6A3 studies meta-analyzed, after correction, the association between SLC6A3 and MPH efficacy in children was significant with 10R/10R homozygotes demonstrating a 26% decreased risk for improved MPH efficacy (Figure 3c).
With regard to clinical implications, our results suggest that interventions accounting for individual genetic variability may improve outcomes of childhood-ADHD treatment. Although not all children will experience a poor response to MPH, it is unclear how to identify those who will respond and what alternative clinical interventions would remediate the liability in this ‘poor response’ subgroup. While there are not enough pharmacogenetic studies of MPH response, there are even fewer examining genetic variability and AMP, atomoxetine, guanfacine or clonidine response in children. Additionally, of the 36 studies meta-analyzed here, only five reported tolerability data. Linking genetic profiles to serious tolerability concerns (for example, tic disorder) that plague some MPH recipients would truly yield clinically valuable data. Even predicting minor adverse events could be useful when choosing an initial medication. Future research should focus on replicating these pooled-findings, assessing their specificity under various environmental conditions and combining multiple variants.
A number of genetic variants associated with MPH efficacy displayed high heterogeneity (I2 values). This heterogeneity was not accounted for by study quality (Supplementary Figure 2) and other covariates were not available for analysis. Where publication bias was identified, effect sizes were adjusted using Duval and Tweedie’s trim and fill method. Simulation studies have found that the trim and fill methods detects missing studies even in the absence of bias. This method may therefore give conservative estimates of the true effect size.70
In addition to methodological limitations, results are further confounded by small sample sizes and a low number of included studies. Specifically, LPHN3 variants rs5661665 and rs1947274 show some potential for predicting MPH response, although we obtained non-significant results (Figures 2e and f). Such results should be viewed as intriguing pilot data. LPHN3 encodes a member of the latrophilin subfamily of G-protein-coupled receptors in GABA-ergic neurotransmission and has been associated with ADHD susceptibility.71 Furthermore, genetic—as well as cultural—diversity is correlated to ethnicity. Patients from different ethnic groups likely possess unique risk/protective factors that influence ADHD treatment efficacy.
Individual response to stimulant medication is complex and heterogeneous. In gene-by-environment analyses, prenatal and perinatal risk factors like prenatal smoking—in conjunction with functional polymorphisms—have been identified as potential predictors of MPH efficacy and tolerability.72 Furthermore, ancestral informative markers are missing from our analysis. Comorbid disorders and ADHD sub-type were not accounted for in this analysis and may contribute additional heterogeneity to pooled-studies.
Currently, there is no reliable biological predictor for pharmacological ADHD treatment choice. Leveraging individual genetic variants within SLC6A2, COMT, ADRA2A, SLC6A3 and DRD4 present a plausible multivariate to assess risk for poor MPH efficacy. Although the odd ratios for each variant are low, it is possible that a multivariate predictor would be sufficiently accurate for clinical use. Clinically controlling ADHD symptomology is especially important in preschool- and adolescent-aged children as they face adversities and an increased assumption of risk from uncontrolled ADHD. ADHD-diagnosed preschool-aged children are more likely to become socially or academically deficient whereas adolescents are at greater risk for substance abuse and more likely to engage in promiscuous behavior.73 Only half of children with ADHD followed pharmacological treatment regimens consistently over the course of a 5-year prospective study, and many reported adverse effects.74 Perceived tolerability may also be an impediment to adherence to pharmacological treatment regimens.75 Thus, it is not surprising that patients receiving personalized treatment were found to be more medication adherent.76
While much work has been done to unravel the genetic predictors of ADHD, there is a call in the literature for ‘a more practical clinical application surrounding prediction of side effect risk and medication tolerability’ and to ‘compile a portfolio of biomarker [genetic] information that would guide pharmacological treatment in order to quickly adopt an efficient regimen’.77 This would likely prove helpful in treatment resistant or treatment intolerant patients. Collectively evaluating genetic variability among plausible biological markers for treatment success would eliminate trial-and-error treatment used today.64 This approach has proven to be practical during a pharmacogenetics trial linking genetic predictors of irritability, social withdrawal and abnormal movements to treatment with MPH in preschool-aged children.42 Through multidisciplinary research, future work should aim to actualize accurate environmental and genetic predictors that confer perilous or protective factors in order to provide a comprehensive understanding of individualized risks during ADHD treatment with MPH.
Supplementary Material
ACKNOWLEDGMENTS
SVF is supported by the K.G. Jebsen Centre for Research on Neuropsychiatric Disorders, University of Bergen, Bergen, Norway, the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 602805, the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 667302 and NIMH grants 5R01MH101519 and U01 MH109536-01.
Footnotes
CONFLICT OF INTEREST
NMM and JRB were employed by Genomind during the research and authorship of this manuscript. In the past year, SVF received income, potential income, travel expenses, continuing education support and/or research support from Lundbeck, KenPharm, Rhodes, Arbor, Ironshore, Shire, Akili Interactive Labs, CogCubed, Alcobra, VAYA, Sunovion, Genomind and NeuroLifeSciences. With his institution, he has US patent US20130217707 A1 for the use of sodium-hydrogen exchange inhibitors in the treatment of ADHD.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
REFERENCES
- 1.Polanczyk GV, Willcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol 2014; 43: 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Academy of Pediatrics. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics 2011; 128: 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchison SL, Ghuman JK, Ghuman HS, Karpov I, Schuster JM. Efficacy of atomoxetine in the treatment of attention-deficit hyperactivity disorder in patients with common comorbidities in children, adolescents and adults: a review. Ther Adv Psychopharmacol 2016; 6: 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Safer DJ. Recent trends in stimulant usage. J Atten Disord 2015; 20: 471–477. [DOI] [PubMed] [Google Scholar]
- 5.Olfson M, Druss BG, Marcus SC. Trends in mental health care among children and adolescents. N Engl J Med 2015; 372: 2029–2038. [DOI] [PubMed] [Google Scholar]
- 6.Jensen PS, Arnold LE, Swanson JM, Vitiello B. 3-year follow-up of the NIMH MTA study. J Am Acad Child Adolesc Psychiatry 2007; 46: 989–1002. [DOI] [PubMed] [Google Scholar]
- 7.Jensen PS, Hinshaw SP, Swanson JM. Findings from the NIMH Multimodal Treatment Study of ADHD (MTA): implications and applications for primary care providers. J Dev Behav Pediatr 2001; 22: 60–73. [DOI] [PubMed] [Google Scholar]
- 8.Faraone SV, Biederman J, Spencer TJ, Aleardi M. Comparing the efficacy of medications for ADHD using meta-analysis. MedGenMed 2006; 8: 4. [PMC free article] [PubMed] [Google Scholar]
- 9.Katzman MA, Sternat T. A review of OROS methylphenidate (Concerta®) in the treatment of attention-deficit/hyperactivity disorder. CNS Drugs 2014; 28: 1005–1033. [DOI] [PubMed] [Google Scholar]
- 10.Faraone SV, Buitelaar J. Comparing the efficacy of stimulants for ADHD in children and adolescents using meta-analysis. Eur Child Adolesc Psychiatry 2010; 19: 353–364. [DOI] [PubMed] [Google Scholar]
- 11.Efron D, Jarman F, Barker M. Side effects of methylphenidate and dexamphetamine in children with attention deficit hyperactivity disorder: a double-blind, crossover trial. Pediatrics 1997; 100: 662–666. [DOI] [PubMed] [Google Scholar]
- 12.Cortese S, Faraone SV, Konofal E. Sleep in children with attention-deficit/hyperactivity disorder: meta-analysis of subjective and objective studies. J Am Acad Child Adolesc Psychiatry 2009; 48: 894–908. [DOI] [PubMed] [Google Scholar]
- 13.Owens JA. A clinical overview of sleep and attention-deficit/hyperactivity disorder in children and adolescents. J Can Acad Child Adolesc Psychiatry 2009; 18: 92–102. [PMC free article] [PubMed] [Google Scholar]
- 14.Faraone SV, Asherson P, Banaschewski T, Biederman J, Buitelaar JK, Ramos-Quiroga JA et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers 2015; 1: 15020–15023. [DOI] [PubMed] [Google Scholar]
- 15.van Wyk GW, Hazell PL, Kohn MR, Granger RE, Walton RJ. How oppositionality, inattention, and hyperactivity affect response to atomoxetine versus methylphenidate: a pooled meta-analysis. J Atten Disord 2012; 16: 314–324. [DOI] [PubMed] [Google Scholar]
- 16.Aron AR, Dowson JH, Sahakian BJ, Robbins TW. Methylphenidate improves response inhibition in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 2003; 54: 1465–1468. [DOI] [PubMed] [Google Scholar]
- 17.Pliszka SR. The use of psychostimulants in the pediatric patient. Pediatr Clin North Am 1998; 45: 1085–1098. [DOI] [PubMed] [Google Scholar]
- 18.Ari ME, Cetin II, Ekici F, Kocabas A, Eminoglu S, Guney E et al. Assessment of cardiovascular risks due to methylphenidate in six months of treatment in children with Attention Deficit and Hyperactivity Disorder. Bull Clin Psychopharmacol 2014; 24: 248–252. [Google Scholar]
- 19.Storebø OJ, Ramstad E, Krogh HB, Nilausen TD, Skoog M, Holmskov M et al. Methylphenidate for children and adolescents with attention deficit hyperactivity disorder (ADHD). Cochrane Database Syst Rev 2015; 11: CD009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polanczyk G, Caspi A, Houts R, Kollins SH, Rohde LA, Moffitt TE. Implications of extending the ADHD age-of-onset criterion to age 12: results from a prospectively studied birth cohort. J Am Acad Child Adolesc Psychiatry 2010; 49: 210–216. [PubMed] [Google Scholar]
- 21.Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001; 409: 928–933. [DOI] [PubMed] [Google Scholar]
- 22.Sun Z, Murry DJ, Sanghani SP, Davis WI, Kedishvili NY, Zou Q et al. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J Pharmacol Exp Ther 2004; 310: 469–476. [DOI] [PubMed] [Google Scholar]
- 23.Hungund BL, Perel JM, Hurwic MJ, Sverd J, Winsberg BG. Pharmacokinetics of methylphenidate in hyperkinetic children. Br J Clin Pharmac 1979; 8: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivas NR, Hubbard JW, Quinn D, Korchinski ED, Midha KK. Extensive and enantioselective presystemic metabolism of dl-threo-methylphenidate in humans. Prog Neuropsychopharmacol Biol Psychiatry 1991; 15: 213–220. [DOI] [PubMed] [Google Scholar]
- 25.Shader RL, Harmatz JS, Oesterheld JR, Parmelee DX, Sallee FR, Greenblatt DJ. Population pharmacokinetics of methylphenidate in children with attention-deficit hyperactivity disorder. Pharmacology 1999; 39: 775–785. [DOI] [PubMed] [Google Scholar]
- 26.Hosokawa M, Endo T, Fujisawa M, Hara S, Iwata N, Sato Y et al. Interindividual variation in carboxylesterase levels in human liver microsomes. Drug Metab Dispos 1995; 23: 1022–1027. [PubMed] [Google Scholar]
- 27.Zhu H-J, Patrick KS, Yuan H-J, Wang J-S, Donovan JL, DeVane CL et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am J Hum Genet 2008; 82: 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter Soria N, Belaus A, Galván C, Ana Pasquali M, Velez P, del Carmen Montes C et al. A simple allele-specific polymerase chain reaction method to detect the Gly143Glu polymorphism in the human carboxylesterase 1 gene: importance of genotyping for pharmacogenetic treatment. Genet Test Mol Biomarkers 2010; 14: 749–751. [DOI] [PubMed] [Google Scholar]
- 29.Nemoda Z, Angyal N, Tarnok Z, Gadoros J, Sasvari-Szekely M. Carboxylesterase 1 gene polymorphism and methylphenidate response in ADHD. Neuropharmacology 2009; 57: 731–733. [DOI] [PubMed] [Google Scholar]
- 30.Bruxel EM, Salatino-Oliveira A, Genro JP, Zeni CP, Polanczyk GV, Chazan R et al. Association of a carboxylesterase 1 polymorphism with appetite reduction in children and adolescents with attention-deficit/hyperactivity disorder treated with methylphenidate. Pharmacogenomics J 2013; 13: 476–480. [DOI] [PubMed] [Google Scholar]
- 31.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol 2007; 47: 681–698. [DOI] [PubMed] [Google Scholar]
- 32.Berman SM, Kuczenski R, McCracken JT, London ED. Potential adverse effects of amphetamine treatment on brain and behavior: a review. Mol Psychiatry 2009; 14: 123–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daberkow DP, Brown HD, Bunner KD, Kraniotis SA, Doellman MA, Ragozzino ME et al. Amphetamine paradoxically augments exocytotic dopamine release and phasic dopamine signals. J Pharmacol Exp Ther 2013; 33: 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L et al. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci 2001; 59: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swanson JM, Volkow ND. Pharmacokinetic and pharmacodynamic properties of stimulants: implications for the design of new treatments for ADHD. Behav Brain Res 2002; 130: 73–78. [DOI] [PubMed] [Google Scholar]
- 36.Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord 2002; 6: S31–S43. [DOI] [PubMed] [Google Scholar]
- 37.Borenstein M, Hedges LV, Higgins JPT, Rothstein HR. Criticisms of meta-analysis In: Sharples K (ed). Introduction to Meta-Analysis. John Wiley & Sons, Ltd: Chichester, UK, 2009, pp 377–387. [Google Scholar]
- 38.Winsberg BG, Comings DE. Association of the dopamine transporter gene (DAT1) with poor methylphenidate response. J Am Acad Child Adolesc Psychiatry 1999; 38: 1474–1477. [DOI] [PubMed] [Google Scholar]
- 39.Seeger G, Schloss P, Schmidt MH. Marker gene polymorphisms in hyperkinetic disorder-predictors of clinical response to treatment with methylphenidate?. Neurosci Lett 2001; 313: 45–48. [DOI] [PubMed] [Google Scholar]
- 40.Tharoor H, Lobos EA, Todd RD, Reiersen AM. Association of dopamine, serotonin, and nicotinic gene polymorphisms with methylphenidate response in ADHD. Am J Med Genet B Neuropsychiatr Genet 2008; 147: 527–530. [DOI] [PubMed] [Google Scholar]
- 41.Kirley A, Lowe N, Hawi Z, Mullins C, Daly G, Waldman I et al. Association of the 480 bp DAT1 allele with methylphenidate response in a sample of Irish children with ADHD. Am J Med Genet B Neuropsychiatr Genet 2008; 121: 50–54. [DOI] [PubMed] [Google Scholar]
- 42.McGough J, McCracken J, Swanson J, Riddle M, Kollins S, Greenhill L et al. Pharmacogenetics of methylphenidate response in preschoolers with ADHD. J Am Acad Child Adolesc Psychiatry 2006; 45: 1314–1322. [DOI] [PubMed] [Google Scholar]
- 43.Song J, Kim SW, Hong HJ, Lee MG, Lee BW, Choi TK et al. Association of SNAP-25, SLC6A2, and LPHN3 with OROS methylphenidate treatment response in attention-deficit/hyperactivity disorder. Clin Neuropharmacol 2014; 37: 136–141. [DOI] [PubMed] [Google Scholar]
- 44.Kim BN, Cummins TD, Kim JW, Bellgrove MA, Hong SB, Song SH et al. Val/Val genotype of brain-derived neurotrophic factor (BDNF) Val66Met polymorphism is associated with a better response to OROS-MPH in Korean ADHD children. Int J Neuropsychopharmacol 2011; 14: 1399–1410. [DOI] [PubMed] [Google Scholar]
- 45.Kim JI, Kim J-W, Park J-E, Park S, Hong S-B, Han D-H et al. Association of the GRIN2B rs2284411 polymorphism with methylphenidate response in attention-deficit/hyperactivity disorder. J Psychopharmacol 2017; 31: 1070–1077. [DOI] [PubMed] [Google Scholar]
- 46.McGough JJ, McCracken JT, Loo SK, Manganiello M, Leung MC, Tietjens JR et al. A candidate gene analysis of methylphenidate response in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2009; 48: 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sutton AJ, Duval SJ, Tweedie RL, Abrams KR, Jones DR. Empirical assessment of effect of publication bias on meta-analyses. BMJ 2000; 320: 1574–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duval S, Tweedie R. Trim and fill: A simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics 2000; 56: 455–463. [DOI] [PubMed] [Google Scholar]
- 49.Hong S-B, Kim J-W, Cho S-C, Shin M-S, Kim B-N, Yoo H-J. Dopaminergic and noradrenergic gene polymorphisms and response to methylphenidate in Korean children with attention-deficit/hyperactivity disorder: is there an interaction? J Child Adolesc Psychopharmacol 2012; 22: 343–352. [DOI] [PubMed] [Google Scholar]
- 50.Kim J-W, Sharma V, Ryan ND. Predicting methylphenidate response in ADHD using machine learning approaches. Int J Neuropsychopharmacol 2015; 18: pyv052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park S, Kim JW, Kim BN, Hong SB, Shin MS, Yoo HJ et al. No significant association between the alpha-2A-adrenergic receptor gene and treatment response in combined or inattentive subtypes of attention-deficit hyperactivity disorder. Pharmacopsychiatry 2013; 46: 169–174. [DOI] [PubMed] [Google Scholar]
- 52.Sengupta S, Grizenko N, Schmitz N, Schwartz G, Bellingham J, Polotskaia A et al. COMT Val108/158Met polymorphism and the modulation of task-oriented behavior in children with ADHD. Neuropsychopharmacology 2008; 33: 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Meulen EM, Bakker SC, Pauls DL, Oteman N, Kruitwagen CLJJ, Pearson PL et al. High sibling correlation on methylphenidate response but no association with DAT1-10R homozygosity in Dutch sibpairs with ADHD. J Child Psychol Psychiatry 2005; 46: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 54.Zeni CP, Guimarães AP, Polanczyk GV, Genro JP, Roman T, Hutz MH et al. No significant association between response to methylphenidate and genes of the dopaminergic and serotonergic systems in a sample of Brazilian children with attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2007; 144B: 391–394. [DOI] [PubMed] [Google Scholar]
- 55.Pasini A, Sinibaldi L, Paloscia C, Douzgou S, Pitzianti MB, Romeo E et al. Neurocognitive effects of methylphenidate on ADHD children with different DAT genotypes: a longitudinal open label trial. Eur J Paediatr Neurol 2013; 17:407–414. [DOI] [PubMed] [Google Scholar]
- 56.Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet 2009; 126: 51–90. [DOI] [PubMed] [Google Scholar]
- 57.Hawi Z, Cummins TDR, Tong J, Johnson B, Lau R, Samarrai W et al. The molecular genetic architecture of attention deficit hyperactivity disorder. Mol Psychiatry 2015; 20: 289–297. [DOI] [PubMed] [Google Scholar]
- 58.Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 2005; 57: 1313–1323. [DOI] [PubMed] [Google Scholar]
- 59.Kim C-H, Hahn MK, Joung Y, Anderson SL, Steele AH, Mazei-Robinson MS et al. A polymorphism in the norepinephrine transporter gene alters promoter activity and is associated with attention-deficit hyperactivity disorder. Proc Natl Acad Sci USA 2006; 103: 19164–19169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnsten AFT, Li BM. Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry 2005; 57: 1377–1384. [DOI] [PubMed] [Google Scholar]
- 61.Andrews GD, Lavin A. Methylphenidate increases cortical excitability via activation of alpha-2 noradrenergic receptors. Neuropsychopharmacology 2006; 31: 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schacht JP. COMT val158met moderation of dopaminergic drug effects on cognitive function: a critical review. Pharmacogenomics J 2016; 16: 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I et al. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry 1995; 34: 4202–210. [DOI] [PubMed] [Google Scholar]
- 64.Stein MA, McGough JJ. The pharmacogenomic era: promise for personalizing attention deficit hyperactivity disorder therapy. Child Adolesc Psychiatr Clin N Am 2008; 17: 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kidd KK, Pakstis AJ, Yun L. An historical perspective on ‘The world-wide distribution of allele frequencies at the human dopamine D4 receptor locus’. Hum Genet 2014; 133: 431–433. [DOI] [PubMed] [Google Scholar]
- 66.Schoots O, Van Tol HHM. The human dopamine D4 receptor repeat sequences modulate expression. Pharmacogenomics J 2003; 3: 343–348. [DOI] [PubMed] [Google Scholar]
- 67.Opmeer EM, Kortekaas R, Aleman A. Depression and the role of genes involved in dopamine metabolism and signalling. Prog Neurobiol 2010; 92: 112–133. [DOI] [PubMed] [Google Scholar]
- 68.Faraone SV, Spencer TJ, Madras BK, Zhang-James Y, Biederman J. Functional effects of dopamine transporter gene genotypes on in vivo dopamine transporter functioning: a meta-analysis. Mol Psychiatry 2014; 19: 880–889. [DOI] [PubMed] [Google Scholar]
- 69.Coghill DR, Banaschewski T, Lecendreux M, Soutullo C, Zuddas A, Adeyi B et al. Post hoc analyses of the impact of previous medication on the efficacy of lisdexamfetamine dimesylate in the treatment of attention-deficit/hyperactivity disorder in a randomized, controlled trial. Neuropsychiatr Dis Treat 2014; 10: 2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sterne JA, Egger M, Smith GD. Systematic reviews in health care: investigating and dealing with publication and other biases in meta-analysis. BMJ 2001; 323: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akutagava-Martins GC, Salatino-Oliveira A, Kieling CC, Rohde LA, Hutz MH. Genetics of attention-deficit/hyperactivity disorder: current findings and future directions. Expert Rev Neurother 2013; 13: 435–445. [DOI] [PubMed] [Google Scholar]
- 72.Pagerols M, Richarte V, Sánchez-Mora C, Garcia-Martínez I, Corrales M, Corominas M et al. Pharmacogenetics of methylphenidate response and tolerability in attention-deficit/hyperactivity disorder. Pharmacogenomics J 2017; 17: 98–104. [DOI] [PubMed] [Google Scholar]
- 73.Wolraich ML, Wibbelsman CJ, Brown TE, Evans SW, Gotlieb EM, Knight JR. Attention-deficit/hyperactivity disorder among adolescents: a review of the diagnosis, treatment, and clinical implications. Pediatrics 2005; 115: 1734–1746. [DOI] [PubMed] [Google Scholar]
- 74.Charach A, Ickowicz A, Schachar R. Stimulant treatment over five years: adherence, effectiveness, and adverse effects. J Am Acad Child Adolesc Psychiatry 2004; 43: 559–567. [DOI] [PubMed] [Google Scholar]
- 75.Wigal T, Greenhill L, Chuang S, McGough J, Vitiello B, Skrobala A et al. Safety and tolerability of methylphenidate in preschool children with ADHD. J Am Acad Child Adolesc Psychiatry 2006; 45: 1294–1303. [DOI] [PubMed] [Google Scholar]
- 76.Fagerness J, Fonseca E, Hess GP, Scott R, Gardner KR, Koffler M et al. Pharmacogenetic-guided psychiatric intervention associated with increased adherence and cost savings. Am J Manag Care 2014; 20: e146–e156. [PubMed] [Google Scholar]
- 77.Stahl SM. Psychiatric pharmacogenomics: how to integrate into clinical practice. CNS Spectr 2017; 22: 1–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.