

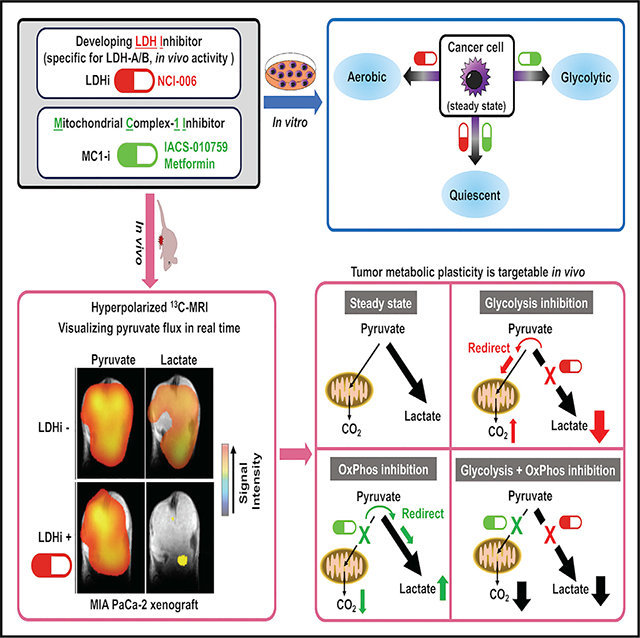
SUMMARY
The reliance of many cancers on aerobic glycolysis has stimulated efforts to develop lactate dehydrogenase (LDH) inhibitors. However, despite significant efforts, LDH inhibitors (LDHi) with sufficient specificity and in vivo activity to determine whether LDH is a feasible drug target are lacking. We describe an LDHi with potent, on-target, in vivo activity. Using hyperpolarized magnetic resonance spectroscopic imaging (HP-MRSI), we demonstrate in vivo LDH inhibition in two glycolytic cancer models, MIA PaCa-2 and HT29, and we correlate depth and duration of LDH inhibition with direct anti-tumor activity. HP-MRSI also reveals a metabolic rewiring that occurs in vivo within 30 min of LDH inhibition, wherein pyruvate in a tumor is redirected toward mitochondrial metabolism. Using HP-MRSI, we show that inhibition of mitochondrial complex 1 rapidly redirects tumor pyruvate toward lactate. Inhibition of both mitochondrial complex 1 and LDH suppresses metabolic plasticity, causing metabolic quiescence in vitro and tumor growth inhibition in vivo.
In Brief
Oshima et al. use hyperpolarized magnetic resonance spectroscopy to dynamically monitor tumor glycolysis and oxidative phosphorylation. LDH inhibition slows tumor growth but rapidly redirects pyruvate to support mitochondrial metabolism. Inhibiting both mitochondrial complex 1 and LDH suppresses metabolic plasticity of glycolytic tumors in vivo, significantly prolonging tumor growth inhibition.
Graphical Abstract
INTRODUCTION
Cancer metabolism has been an area of active interest ever since Warburg and Negelein (1924) proposed that cancer cells prefer glycolysis to oxidative phosphorylation, even when oxygen levels are not limiting (Warburg, 1956). In succeeding years, this perspective has evolved when it became clear that metabolic plasticity in cancer cells is an important contributor to tumorigenesis in vivo. Cancer cell metabolism is diverse, with varying reliance on glycolysis and mitochondrial oxidative phosphorylation working in concert to serve metabolic needs and to maintain redox homeostasis. Recent advances in metabolomics, medicinal chemistry, and imaging techniques now make it possible to test the hypothesis that glycolysis and oxidative phosphorylation are functionally integrated in vivo and that both are essential to maintaining metabolic plasticity and tumor growth (DeBerardinis and Chandel, 2016; Jia et al., 2019; Moreno-Sánchez et al., 2007; Porporato et al., 2018; Smolková et al., 2011; Weinberg et al., 2010; Weinberg and Chandel, 2015; Zu and Guppy, 2004). In the present study, we test that hypothesis using a specific in vivo-active LDH inhibitor (NCI-006), the specific mitochondrial complex 1 inhibitor IACS-010759 (Molina et al., 2018), and metabolic imaging techniques.
Lactate dehydrogenase (LDH) is the terminal enzyme in the glycolytic pathway and is responsible for conversion of pyruvate to lactate with concomitant regeneration of NAD+ from NADH. Studies demonstrating the negative effect of genetic ablation of lactate dehydrogenase A (LDHA, the predominately expressed isoform of LDH in most cancer cells) on tumor cell growth have provided support for the concept that glycolytic cancers may be effectively targeted at the molecular level by inhibitors of this key enzyme (Fantin et al., 2006; Le et al., 2010; Vander Heiden et al., 2009; Xie et al., 2014). Although this hypothesis has provided a valuable heuristic framework, specific and effective in vivo-active LDH inhibitors have not been available. Although the past 10 years have seen a significant effort by academic laboratories and biotechnology and pharmaceutical companies to develop LDHA inhibitors with cellular and in vivo activity (Billiard et al., 2013; Boudreau et al., 2016; Le et al., 2010), no highly specific inhibitors with durable in vivo activity have been reported. Importantly, although the natural product FX11 does display in vivo activity, it also has significant off-target effects and is not specific for LDH (Billiard et al., 2013; Fantin et al., 2006; Granchi et al., 2013; Le et al., 2010; Ward et al., 2012; Xie et al., 2014). Thus, the impact of a specific LDH inhibitor on tumor metabolism in vivo is, in fact, unknown.
An additional limitation with previous efforts has been a focus on only the LDHA isoform. A recent report demonstrated convincingly that double genetic disruption of both LDHA and LDHB may be necessary to fully suppress glycolysis in cancer cells (Ždralević et al., 2018), and LDHB has been shown to be an essential gene in triple-negative breast cancer (McCleland et al., 2012). In addition, LDHB is reported to regulate lysosome activity and autophagy in cancer (Brisson et al., 2016) and is essential for oncogenic transformation by mutant p53 and mutant Ras (Smith et al., 2016).
Here, we report a specific small-molecule LDHA/B (herein referred to as LDH) inhibitor with potent, on-target cellular and in vivo activity, derived by further structural refinement of a recently described series of LDH inhibitors (Rai et al., 2017; Yeung et al., 2019). This molecule provides a means for dynamic, noninvasive analysis of tumor pyruvate flux in a living subject and also displays both single-agent, in vivo activity and cooperativity with the specific mitochondrial complex 1 inhibitor IACS-010759. Of critical importance, we determined tumor pharmaco-dynamic response to these metabolic inhibitors using real-time, in vivo imaging of hyperpolarized [13C]pyruvate flux.
RESULTS
In Vitro and In Vivo Evaluation of On-Target Effects of the LDH Inhibitor NCI-006
To evaluate the in vitro activity of NCI-006 (Figure 1A), we examined in-gel redox activity of human LDH (hLDH) isozymes 2, 3, 4, and 5 obtained from normal kidney and the HEK293T kidney epithelial cell line, and five different LDH isozymes found in the mouse heart. The activities of hLDH (Figures 1B, left panel, and S1A, left panel) and mouse isozymes (Figure 1B, right panel) were dose-dependently inhibited by NCI-006, consistent with the similar biochemically determined potency of NCI-006 for LDHA and LDHB proteins (LDHA IC50 = 0.06 μM; LDHB IC50 = 0.03 μM). These data are in general agreement with results of a previous report that examined other members of this chemical series (Rai et al., 2017). In contrast to its effect on LDH, NCI-006 did not inhibit the activity of two unrelated mitochondrial dehydrogenases, malate dehydrogenase (MDH) and succinate dehydrogenase (SDH), isolated from human kidney (Figure S1A, right top and bottom panels). The cellular half-maximal response (EC50) of NCI-006, as determined by an in vitro lactate secretion assay using both mouse and human red blood cells (RBCs), was 1.6 and 2.1 μM, respectively (Figure 1C).
Figure 1. On-Target Inhibition of LDH In Vitro.

(A) Chemical structure of the LDH inhibitor NCI-006.
(B) Native gel assay demonstrating potency of LDHi toward all human (h, HEK293-T) and mouse (m, mouse heart) LDH isoforms. The bands indicate redox reactions mediated by active LDH in the gels. LDHi suppressed redox activity of all LDH isoforms in a dose-dependent manner. In contrast, the LDH inhibitor did not inhibit either human malate dehydrogenase (MDH) or succinate dehydrogenase (SDH) (see Figure S1A; proteins were obtained from normal human kidney); 10 μg of protein was applied in each lane. Superfluous lanes on either side of these bands were digitally eliminated, as indicated by the rectangular outlines.
(C) In vitro lactate secretion assay comparing sensitivity of mouse (m) and human (h) red blood cells (RBCs) to LDH inhibition. EC50 is 2.073 × 10−6 M and 1.628 × 10−6 M in hRBCs and mRBCs, respectively.
(D) The NAD/NADH ratio is affected by LDH inhibition in MIA PaCa-2 and HT29 cells. The NAD/NADH ratio was significantly decreased after 2 h exposure of cells to NCI-006 at the concentrations shown. The data are represented as means ± SEM (n = 4 in MIA PaCa-2 and n = 3 in HT29; **p < 0.01, t test).
(E) Effect of NCI-006 on LDH activity (as measured by lactate secretion) in MIA PaCa-2 and HT29 cells after 1 h of drug treatment (MIA PaCa-2 EC50 = 3.736 × 10−7 M; HT29 EC50 = 5.346 × 10−7 M).
(F) NCI-006 rapidly decreased glycolytic flux in MIA PaCa-2 cells. Extracellular acidification rate (ECAR) was monitored over time using the Seahorse bioanalyzer. After a stable baseline was established, LDHi was introduced to the medium, bathing the cells as shown. Oligomycin (O; 1 μg/mL), FCCP (F; 1 μM), and antimycin A + 2-deoxyglucose (AA+2-DG; 10 μM/50 mM) were sequentially introduced to the medium, bathing the cells as shown.
(G) Impact of NCI-006 on the oxygen consumption rate (OCR) in MIA PaCa-2 cells. Measurements were made simultaneously with the ECAR determinations described in (F).
(H) Increase in basal OCR after 1 μM LDHi. Data from (G) were extracted and plotted as mean basal OCR at 62 and 96 min after initiation of tracing (LDHi was injected at 27 minutes). See also Figure S1B.
The data are displayed as means ± SEM (n = 3–6/group; *p < 0.05).
A key role of LDH is to regenerate NAD+ from the NADH, which is generated during glycolysis. Accordingly, LDH inhibition should be associated with a decrease in the NAD+/NADH ratio (Chiarugi et al., 2012). This ratio was measured in MIA PaCa-2 and HT29 cells in vitro in the presence or absence of the LDH inhibitor (Figure 1D) and, as expected, was significantly decreased in both cell lines after 2 h of exposure to NCI-006. These data indicate that failure to regenerate the NAD+ pool is a major metabolic consequence of LDH inhibition.
Lactate secretion from MIA PaCa-2 and HT29 cells was similarly affected by LDH inhibitor (EC50 = 0.37 and 0.53 μM, respectively) (Figure 1E). Assessment of glycolytic flux in MIA PaCa-2 cells using a mitochondrial stress test showed that, after establishment of a stable baseline, addition of the LDH inhibitor resulted in a time-dependent decrease in basal extracellular acidification rate (ECAR) at a minimal concentration of 1 μM (Figure 1F). Injection of oligomycin, which inhibits mitochondrial ATP synthesis, resulted in the expected compensatory increase in glycolysis in vehicle-treated cells, which was also inhibited by NCI-006 at a concentration ≥1 μM. The final injection of antimycin and 2-deoxy-D-glucose resulted in a decreased ECAR, as expected. Oxygen consumption rate (OCR) was measured under the same conditions and is shown in Figure 1G. Importantly, we detected a stimulation of basal OCR upon injection of NCI-006 (1 μM, the lowest concentration to significantly inhibit ECAR under these conditions) (Figures 1H and S1B; also see Figure S4A). Although this may appear a small effect, it represents an approximately 19% increase in mitochondrial respiration dedicated to making ATP, which, in bioenergetic terms, is highly significant. Taken together, these data demonstrate that LDH inhibition results in suppression of glycolytic flux in MIA PaCa-2 cells in vitro concomitant with a small but significant increase in basal OCR, consistent with rapid metabolic rewiring upon inhibition of LDH.
To investigate the in vivo activity of NCI-006, an ex vivo lactate production assay was performed using mouse RBCs (mRBCs) prepared from blood sampled 2 and 24 h after drug administration (Figures 2A–2C). Mice were administered a single dose of LDH inhibitor either orally (PO, 10–200 mg/kg; Figure 2A) or intravenously (IV; 1 – 100 mg/kg; Figures 2B and 2C). LDH activity decreased dose dependently at 2 h but, in most cases, returned to baseline levels by 24 h. In mice dosed PO with 100 mg/kg NCI-006 or by IV at 50 mg/kg NCI-006, LDH activity returned to 75%–80% of baseline by 24 h. Recovery of LDH activity was confirmed by examining several time points after PO administration of 50 mg/kg NCI-006 (Figure 2D). LDH activity remained maximally inhibited at 8 h but recovered steadily thereafter, reaching pretreatment levels by 20 h.
Figure 2. On-Target Inhibition of RBC LDH Ex Vivo and Tumor LDH In Vivo.

(A–C) Ex vivo lactate secretion assay using mouse (m) RBCs. Mice were dosed orally (PO) (A) or intravenously (IV) (B and C) with NCI-006. (B) includes LDHi dosing between 1 and 50 μM; (C) includes 50 and 100 μM doses. At 2 h after LDH inhibitor administration, blood was drawn, and ex vivo lactate production in mRBCs was determined (see Method Details). Lactate production in RBCs obtained from mice treated with 50–200 mg/kg NCI-006 in the PO group and 10–50 mg/kg NCI-006 in the IV group was significantly reduced compared with lactate production in mice treated with PBS (n = 3 for each group, *p < 0.05, t test). By 24 h after LDH inhibitor administration, lactate production returned to normal levels, except in the PO group treated with 100 or 200 mg/kg NCI-006, and in the IV group treated with 50 or 100 mg/kg NCI-006. The data are displayed as means ± SEM (n = 3 for each group; *p < 0.05, t test). †50% reduction in RBC counts was seen in mice treated with 200 mg PO and 100 mg/kg IV of the LDH inhibitor.
(D) Ex vivo lactate recovery time measured using mRBCs. Mice were orally administered 50 mg/kg LDHi or PBS. Lactate production returned to baseline between 15 and 24 h after LDHi administration.
(E and F) Correlation between intra-tumor concentration of LDH inhibitor and LDH activity in MIA PaCa-2 xenografts, comparing oral (E) and IV (F) drug administration. Tumors were excised 1 h after NCI-006 administration, and the LDH activity in the tumor and the NCI-006 concentration in the plasma and tumor were measured.
Next, we examined how intratumoral NCI-006 drug levels affected LDH activity in MIA PaCa-2 xenografts. Tumors were excised between 1 and 25 h after either PO or IV administration of 50 mg/kg NCI-006 (Figures 2E and 2F). PO administration resulted in maximal LDH inhibition of 20%–40%, measured by biochemical assay, between 3 and 6 h after dosing, returning to baseline by 24 h (Figure 2E). Peak plasma drug concentration (between 1 and 6 h) was 3–4 μM, whereas peak intratumoral drug concentration (reached at 6 h) was between 2 and 3 μM. In contrast, IV administration of an equal dose of LDH inhibitor resulted in a peak intratumoral concentration (measured at 1 h and persisting to 6 h) of 20 μM, whereas peak plasma concentration of the LDH inhibitor approached 50 μM at 1 h. Under these conditions, intratumoral LDH activity was inhibited by nearly 80% between 1 and 6 h after IV dosing. At 24 h, LDH activity approached 60% of baseline (Figure 2F). These data suggest that intratumoral drug level reflects the plasma Cmax (peak drug concentration) more than the AUC (total area under the curve). The slow decline in tumor drug level after both PO and IV administration, in contrast to the change in plasma concentration of NCI-006, suggests the significant tumor retention of the drug, possibly because of the long target residence time that is a characteristic of this LDH inhibitor series (Rai et al., 2017). Importantly, the results show that approximately 20 μM intratumoral drug level is necessary to inhibit LDH by 80%.
Non-invasive Monitoring of Tumor LDH Inhibition In Vivo Using HP-MRSI
Several studies have reported that 13C-MRSI (magnetic resonance spectroscopic imaging) with hyperpolarized (HP) 13C-labeled pyruvate ([1-13C]Pyr) can be used for real-time monitoring of LDH activity in vivo (Albers et al., 2008; Ardenkjaer-Larsen et al., 2003; Kurhanewicz et al., 2008; Scroggins et al., 2018; Zierhut et al., 2010), with one study using a non-specific LDH inhibitor (Dutta et al., 2013). Consequently, we used 13C-MRSI to non-invasively explore the in vivo efficacy of NCI-006 in mice bearing MIA PaCa-2 or HT29 xenografts. Individual mice were imaged before and after LDH inhibitor administration (see Figure 3A). Imageable products of [1-13C]pyruvate metabolism are shown in Figure S1C. Figures 3B and 3C show representative 13C spectra, obtained from MIA PaCa-2 tumors, and signal intensity curves of [13C]Pyr, [13C]Lac, [13C]alanine(Ala), and H13CO3-([13C]Bic) after HP [13C]Pyr administration. [13C] Lac to [13C]Pyr (Lac/Pyr) ratio was calculated from the AUCs using time-intensity data as described in previous reports (Hill et al., 2013; Saito et al., 2015).
Figure 3. Monitoring LDH Inhibitor On-Target Activity in Tumor Xenografts Using 13C MR Spectroscopy with Hyperpolarized (HP) [1-13C] Pyruvate (Pyr).

(A) A schematic representation of the HP [1-13C]Pyr MR study. The 13C MR studies were performed with mice bearing MIA PaCa-2 tumors. Except for (E), each mouse was imaged 24 h before and 30 min after LDHi administration, thus serving as its own control.
(B and C) Representative 13C-MR spectra (B) and signal intensity curves (C) of [1-13C]pyruvate (Pyr), [1-13C]lactate (Lac), [1-13C]alanine (Ala), and H13CO3- (Bic) detected in a MIA PaCa-2 xenograft after HP [1-13C]Pyr injection.
(D) 13C MR spectroscopy showed dose-dependent LDH inhibitor suppression of the tumor Lac/Pyr ratio.
(E) Lactate recovery time following a single dose (IV) of 50 mg/kg NCI-006 was determined by 13C MR spectroscopy. The [1-13C]Lac/Pyr ratio returned to baseline within 12–16 h after IV dosing.
(F) Representative 13C-MR spectra (top) and signal intensity curves (bottom) were obtained from before (left) and after (right) evaluations, as described above. The scale bar of the MR image insert (white bar, left side of image) is 8 mm. For a magnified MR image and scale bar (the same for all MR images), please refer to Figure S1E. Conversion of [1-13C]Pyr to [1-13C]Lac was clearly suppressed by IV NCI-006 administration.
(G) [1-13C]Lac/Pyr ratio is significantly decreased after in vivo NCI-006 administration (IV, 50 mg/kg) to mice bearing MIA PaCa-2 (n = 3) or HT29 (n = 3) xenografts.
(H) Average inhibition of tumor LDH activity ± standard deviation (vehicle group, n = 3; treated group, n = 4), determined by biochemical assay of tumors removed from mice 1 h after an IV dose of 50 mg/kg NCI-006 is 79.3% ± 7.4% (**p < 0.01, t test).
See also Figure S1.
Drug-dose response and recovery time experiments using 13C-MR spectroscopic measurements were performed by IV dosing of MIA PaCa-2 tumor xenografts (Figures 3D and 3E). 13C-MRSI revealed that the tumor [13C]lactate/[13C]pyruvate ratio (providing a snapshot of real-time LDH activity) was dose dependently suppressed by NCI-006, with 50 mg/kg being most effective (approximately 80% inhibition at 30 min; Figure 3D). In addition, 13C-MRSI also revealed that the [13C]Lac/[13C]Pyr ratio returned to baseline by 12 h after IV injection (Figure 3E). In contrast, 13C-MRSI showed that oral administration of 50 mg/kg LDH inhibitor did not affect the [13C]lactate/[13C] pyruvate ratio in MIA PaCa-2 tumors (up to 7 h after drug administration) (Figure S1D). Together, these findings are consistent with the cellular and ex vivo data presented in Figures 2D–2F.
Next, we investigated the effect of IV administration of NCI-006 (50 mg/kg) on metabolic flux in HT29 compared with MiaPaCa-2 tumor xenografts (Figures 3F and S1E). 13C-MRSI revealed that the [13C]Lac/[13C]Pyr ratio decreased by 83.3% ± 4.4% in MIA PaCa-2 and 74.7% ± 8.4% in HT29 tumors 30 min after a single IV dose of the LDH inhibitor (Figure 3G). These data are consistent with the results obtained by biochemical assay of LDH activity in MIA PaCa-2 tumors excised 1 h after IV dosing (50 mg/kg; Figure 3H). Taken together, these results show that dynamic MR imaging of intratumoral hyperpolarized [13C]pyruvate flux provides a reliable snapshot of tumor LDH activity in vivo.
[13C]MRSI of the same region of the leg of non-tumor bearing mice showed that the reduction in the ratio of [13C]Lac/[13C]Pyr was appreciably smaller than that seen in either MIA PaCa-2 or HT29 xenografts (compare Figure S1F to Figure 3G). Although these findings are consistent with the possibility that these glycolytic tumors maintain a greater glycolytic flux compared with adjacent non-tumor tissue under the same metabolic conditions, these differences could also be explained by reduced LDH expression or activity or by more efficient lactate export in the healthy tissue.
Although the [13C]Ala/[13C]Pyr ratio in MIA PaCa-2 xenografts remained relatively unchanged in the presence of NCI-006, the [13C]Bic/[13C]Pyr ratio was significantly increased (Figure S1G). Taken together, these data suggest that tumor LDH inhibition in two xenograft models resulted in reduced conversion of pyruvate to lactate concomitant with enhanced flux of pyruvate to bicarbonate and mitochondrial oxidation without a detectable increase in transaminase-mediated flux of pyruvate to alanine. This hypothesis is consistent with the increased OCR seen in vitro after inhibition of ECAR by LDHi (Figures 1H, S1B, and S4A).
Chemical Shift Imaging Using 13C-MRSI
Next, we sought to monitor metabolic flux in different tumor regions divided into 2-mm × 2-mm × 8-mm voxels using chemical shift imaging (CSI) with 13C-MRSI. The distribution of [13C]Pyr and [13C]Lac in MIA PaCa-2 and HT29 tumors after HP [13C]Pyr administration was monitored using 13C-MRSI with the same scheme as shown in Figure 3A. The data document the intratumoral heterogeneity of pyruvate to lactate conversion (Figures 4A–4D). However, tumor LDH activity was dramatically and uniformly suppressed by NCI-006 in each voxel within the tumor region in both MIA PaCa-2 and HT29 xenografts (Figures 4A and 4C). This information is displayed as false-color intensity images (Figures 4B and 4D). The total [13C]Lac/[13C]Pyr ratio in the tumor is integrated from the [13C]Lac/[13C]Pyr ratio in each voxel and is displayed as a mean value (bars), with the data from individual animals superimposed as either filled ([13C] lactate/[13C]pyruvate ratio before treatment with LDH inhibitor) or open (ratio determined 30 min after treatment with LDH inhibitor) circles (Figure 4E).
Figure 4. Chemical Shift Imaging (CSI).

(A–D) CSI of a MIA PaCa-2 (A) and an HT29 (C) xenograft obtained before and after treatment, as described in Figure 3A. Images were acquired 25 s after hyperpolarized [1-13C]Pyr injection. The left peak in each voxel (small square) is [1-13C]Lac, and the right peak is [1-13C]Pyr. [1-13C]Lac signal decreased in each 2-mm × 2-mm × 8-mm voxel in the tumor region after LDH inhibitor injection (IV, 50 mg/kg). Most of the tissue volume inside the coil is tumor (approximately 75% from analysis of the fast low-angle shot magnetic [FLASH] anatomical MRI images). False-color images of [1-13C]Pyr (left), [1-13C]Lac (middle), and the [1-13C]Lac/Pyr ratio (right), in before (top) and after (bottom) treatment evaluations of a MIA PaCa-2 (B) and an HT29 (D) xenograft. False-color images (B and D) are generated from the chemical shift data (A and C).
(E) The [1-13C]Lac/Pyr ratio calculated from these data was significantly decreased 30 min after LDH inhibitor injection in both MIA PaCa-2 (n = 4) and HT29 (n = 3) xenografts. The data are displayed as means ± SEM (**p < 0.01, t test).
(F) Representative CSIs of a MIA PaCa-2 xenograft before and 30 min after LDH inhibitor administration (left), with equivalent false-color images of [1-13C]pyr (middle) and [1-13C]lac (right). Top panels display pre-LDH inhibitor images, and bottom panels display images collected 30 min after LDH inhibitor. Data were obtained using a clinical grade imager (see Method Details).
Finally, imaging using a clinical grade HP-MRSI system (Nelson et al., 2013; see Method Details) agreed with our previous data and showed a significant decrease in the [13C]Lac/[13C]Pyr ratio upon LDH inhibition in mice bearing MIA PaCa-2 xenografts (Figure 4F), confirming the reproducibility of the findings presented in Figures 4A–4D. These results support the utility of this technology for repeated, non-invasive, dynamic monitoring of tumor pyruvate metabolism in patients receiving metabolically directed therapy.
Anti-Tumor Activity of NCI-006
In vitro cell proliferation assays showed that MIA PaCa-2 and HT29 cell growth was dose dependently suppressed by NCI-006 (Figures S2A and S2B). Based on these data, we assessed the in vivo anti-tumor activity of the compound in mice bearing MIA PaCa-2 xenografts. NCI-006 (50 mg/kg) was intravenously administered every other day for 1 week (three injections, Monday, Wednesday, and Friday), or for 2 weeks (six injections) with a break of 1 week between cycles. Tumor growth was significantly impeded by LDH inhibition, and 2 weeks of treatment (two cycles) was significantly more effective than was 1 week of treatment (Figures 5A and 5B). Further, in mice treated for two cycles, tumor volumes in four of eight animals measured nearly 3 weeks after treatment cessation were not markedly different from their volume at initiation of treatment (Figure 5B). Mouse body weights were monitored throughout the treatment course as a means to assess drug toxicity. The difficulty in maintaining intravenous treatment for longer periods is a technical one, and in our hands, six tail-vein injections was the maximum that was possible. Mean body weights of the drug-treated mice did not vary significantly from that of the vehicle controls (Figure S2C). We repeated this experiment using an alternative treatment schedule (IV injection of 50 mg/kg NCI-006, Monday, Wednesday, and Friday for a consecutive 2 weeks) (Figures S2D and S2E). Similarly, LDH inhibition significantly slowed the growth of MIA PaCa-2 xenografts without a marked effect on mouse body weight (Figure S2F).
Figure 5. In Vivo Anti-Tumor Activity of NCI-006.

(A) MIA PaCa-2 cells (1 × 106) were subcutaneously injected into the right leg of athymic mice. LDH inhibitor treatment (IV, 50 mg/kg) commenced 3 weeks after cell inoculation. NCI-006 or an equivalent volume of vehicle was administered three times per week (every other day). Mice in the Veh-3 and LDHi-3 groups were treated for one cycle (treatment days identified by white arrowheads), whereas mice in the Veh-6 and LDHi-6 groups were treated for two cycles (during the third and fifth weeks after tumor cell inoculation; treatment days identified by black arrowheads). Tumor growth rates in LDHi-3 and LDHi-6 groups were clearly slower than those in the Veh-3 and Veh-6 groups, and tumor growth was slower in mice receiving two cycles of LDHi compared with one cycle.
(B) Individual tumor volumes at day 53 after cell inoculation. Mean tumor volume in mice receiving two cycles of NCI-006 was significantly less than that in all other groups (*p < 0.05; **p < 0.01, one-way ANOVA with uncorrected Fisher’s least significant difference). Comparing LDHi-3 to LDH-6 groups by t test yields a p value of 0.017.
See also Figure S2.
Combined Treatment with the Mitochondrial Complex 1 Inhibitor IACS-010759 and LDH Inhibitor Leads to Energetic Quiescence In Vitro and Tumor-Growth Inhibition In Vivo
Our data showing that NCI-006 decreased pyruvate to lactate flux while enhancing pyruvate to bicarbonate flux in vivo and causing a small, but reproducible, increase in OCR in vitro, suggest the possibility of tumor adaptation to NCI-006 through metabolic rewiring that leads to increased mitochondrial oxidative metabolism via redirection of pyruvate metabolism, as previously proposed based on in vitro observations (Boudreau et al., 2016). This metabolic plasticity might also contribute to the variable, long-term response of these glycolytic xenografts to LDH inhibition alone. Therefore, we sought to determine whether NCI-006 treatment exposes tumor vulnerability to combination therapy with inhibitors of mitochondrial metabolism. We first performed a synergy screen in vitro using the recently reported specific mitochondrial complex 1 inhibitor IACS-010759, currently undergoing clinical evaluation (Molina et al., 2018). The data demonstrated robust synergy of both agents (Figure S3). We confirmed those data by monitoring MIA PaCa-2 cell viability 48 h after exposure to either IACS-010759 or NCI-006 alone (each at 1 μM) or in combination. Although single-agent treatment with either inhibitor significantly reduced cell viability compared with vehicle control, combined treatment at the equivalent dose for each inhibitor caused a nearly 100% loss in viability (Figure 6A). To confirm the cooperative metabolic effect of these agents, we used the Seahorse XF bioanalyzer to generate an energy phenotype of MIA PaCa-2 cells exposed to either agent separately or together (each inhibitor at 1 μM concentration, treatment for 100 min) (Figures 6B and S4A). As expected, inhibition of mitochondrial complex 1 with IACS-010759 reduced mitochondrial oxygen consumption by more than 80% and increased glycolysis 2-fold. Conversely, LDH inhibition made the cells more aerobic, reducing glycolysis by nearly 70% and increasing mitochondrial oxygen consumption by nearly 25%. However, combined treatment with each inhibitor led to a 67% reduction in oxygen consumption and a 75% reduction in glycolysis, placing the cells in an energetically adverse state.
Figure 6. Impact of the Combination of LDH Inhibitor NCI-006 and Mitochondrial Complex 1 Inhibitor IACS-010759 in MIA PaCa-2 Cells and Xenografts.

(A) Cell viability after 48 h of exposure to 1 μM of NCI-006 combined with 1 μM of IACS-010759 in MIA PaCa-2 cells was significantly reduced compared with cells treated with either vehicle or single agent treatment. The data are displayed as means ± SEM (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA with Tukey-Kramer method). Viability was assessed with CellTiter-Glo, according to the manufacturer’s instructions.
(B) The levels of OCR/ECAR in MIA PaCa-2 cells with or without treatments were evaluated (see Figure S4A) and used to generate cell-energy phenotype profiles. The baseline activity (0 min; open symbols) and the metabolic activity in response to treatments (100 min; closed symbols) are connected by dashed lines; 1 μM of IACS-010759 and/or 1 μM of NCI-006 were used as shown.
(C) The [13C]Lac/Pyr ratio obtained from 13C MR spectroscopy data (see Figure S4B) was significantly increased and [13C]Bic/Pyr ratio was significantly decreased after IACS-010759 administration to mice bearing MIA PaCa-2 xenografts (n = 4, *p < 0.05, **p < 0.01; paired t test). IACS-010759 (2.5 mg/kg) was orally administered 3.5 h before HP 13C MR spectroscopy.
(D) The [13C]Lac/Pyr ratio and the [13C]Bic/Pyr ratio were both significantly decreased in MIA PaCa-2 xenografts after combination treatment (n = 5, *p < 0.05, **p < 0.01; paired t test). IACS-010759 (2.5 mg/kg) was orally administered 3 h before NCI-006 administration, and NCI-006 (50 mg/kg) was intravenously administered 30 min before HP 13C MR spectroscopy. Representative 13C-MR spectra are shown in Figure S4C.
(E and F) MIA PaCa-2 cells (2 × 106) were subcutaneously injected into the right leg of athymic mice. NCI-006 (50 mg/kg) was administered intravenously every Monday, Wednesday, and Friday for 2 weeks. IACS-010759 (20 mg/kg) was orally administered from Monday to Friday for 2 weeks. Tumor volumes at day 37 in MIA PaCa-2 (E) were significantly reduced in mice receiving the combination treatment of NCI-006 and IACS-010759 when compared with treatment with vehicle or either monotherapy (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; one-way ANOVA with Tukey-Kramer method; data represents means ± SEM). Tumor growth curves are shown in Figure S4D. Kaplan-Meier analysis (time on study) (F) showed that combination therapy caused the greatest increase in overall survival (median survival: vehicle, 44 days; NCI-006: 50 days; IACS-010759: 49 days; combination: 63 days) compared with any other treatment (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; Mantel-Cox log-rank test). Mice were taken off study when tumors reached a volume of 1,800 mm3.
See also Figures S3, S4, S5, and S6.
Using HP 13C-MRSI with [13C]pyruvate in mice bearing MIA PaCa-2 xenografts, we found that 3.5 h after treatment with 2.5 mg/kg IACS-010759, conversion of [13C]pyruvate to [13C] lactate increased significantly, whereas conversion of [13C]pyruvate to [13C]bicarbonate decreased significantly (Figures 6C and S4B). Using the same in vivo model but treating with the combination of IACS-010759 (2.5 mg/kg) followed 3 h later with NCI-006 (50 mg/kg) and performing HP-MRSI 30 min later, we observed that conversion of [13C]pyruvate to both [13C]lactate and [13C]bicarbonate was significantly reduced (Figures 6D and S4C). Interestingly, these in vivo observations are fully consistent with the effect of these agents in vitro on the energy profile of MIA PaCa-2 cells (see Figure 6B).
With these data in hand, we compared single-agent to combination therapy using MIA PaCa-2 xenografts. Two million cells were subcutaneously injected into the right leg of athymic mice, and drug treatments commenced 21 days later when tumors reached an average volume of 300 mm3. Both drugs were administered over 2 weeks (IACS-010759: 20 mg/kg PO, daily for 5 days with weekends off; NCI-006: 50 mg/kg IV, Monday, Wednesday, and Friday with weekends off). Tumor volumes at day 37 were significantly reduced by combination treatment compared with vehicle and either monotherapy (Figure 6E). Full tumor-growth curves are shown in Figure S4D. During the treatment period, tumor growth was arrested in the combination treatment group, notably with very little variation in tumor volume within this group (n = 10). We followed the mice after cessation of treatment to determine “overall survival” (i.e., when mice had to be taken off the study because of excessive tumor size; Figure 6F). Median time on the study was 63 days for the combination group versus 44 days for the vehicle group, 50 days for the NCI-006 group, and 49 days for the IACS-010759 group. Although body weight initially decreased (maximum decrease <10%) during combination treatment, it rapidly returned to normal after treatment cessation (Figure S4E). The stronger impact of combination treatment and the small variation in tumor volume between animals in this group compared with either single-agent treatment group, suggests that tumor escape is minimized by combination treatment with complex 1 and LDH inhibitors, even in a highly glycolytic tumor model. The significantly prolonged effect of combination treatment after treatment cessation is consistent with this interpretation.
Metformin has also been reported to inhibit mitochondrial respiratory chain complex 1 (Birsoy et al., 2014; Pollak, 2014) and to suppress oxidative phosphorylation in cancer cells (Xie et al., 2014). To confirm that mitochondrial complex 1 inhibition is at least additive with LDH inhibition in vivo, we investigated the effect of metformin on the [13C]pyruvate flux in both MIA PaCa-2 and HT29 xenografts using HP 13C-MRSI. Based on previous reports (Foretz et al., 2010; Madiraju et al., 2014), we administered metformin (50 mg/kg) orally to tumor-bearing mice. HP 13C-MRSI revealed a significant increase in tumor [13C]Lac/[13C]Pyr ratio by 3 h after dosing for both MIA PaCa-2 and HT29 xenografts (Figures S5, S6A, and S6B). These results are consistent with inhibition of mitochondrial pyruvate oxidation. Lower doses of metformin were ineffective (data not shown). Next, we explored whether this drug combination was reflected in greater growth inhibition of HT29 xenografts. Metformin and NCI-006 were administered either as single agents or in combination following the 2-week schedule described above (metformin was administered daily per os as was IACS-010759). As with the IACS-010759/NCI-006 combination, we found that combination treatment caused significantly greater growth inhibition of HT29 xenografts (with much less variation in tumor volume from animal to animal) compared with either treatment alone (Figures S6C and S6C). Average body weights of mice in all groups varied by ≤10% (Figure S6E).
Finally, hemolytic anemia is an on-target consequence of a chemically induced, inactivating mutation within the Ldh-1 locus of the murine lactate dehydrogenase A gene, leading to systemic LDHA deficiency in homozygous mutants (Pretsch et al., 1993; Xie et al., 2014). Mice with homozygous, inactivating mutations in the Ldh-1 locus display a marked decrease in erythrocyte and hematocrit numbers (Kremer et al., 1987). Although murine RBCs express only LDHA, human RBCs express both LDHA and LDHB. Thus, hemolysis and hemolytic anemia may represent a significant on-target systemic toxicity of this class of LDH inhibitor in humans. With this in mind, we explored the effect on murine hematocrit levels of the intermittent IV dosing schedule for NCI-006, which we selected for the in vivo studies described above. After two consecutive treatment cycles of NCI-006 (separated by a 2-day break), either alone (50 mg/kg, IV) or in combination (30 mg/kg, IV) with metformin (50 mg/kg, PO), hematocrit level was reduced by <10% when measured 3 days after the final treatment, and it returned to normal levels within an additional 6–10 days (Figure S6F). However, treating mice with higher doses (200 mg/kg, PO; or 100 mg/kg, IV) of NCI-006 alone caused a 50% reduction in RBCs at 24 h and repeated dosing led to splenomegaly and extramedullary hematopoiesis (not shown). Taken together, these data confirm that it is possible to minimize on-target systemic toxicity (e.g., hemolysis) of this in vivo-active LDH inhibitor with appropriate dosing and scheduling while maintaining significant anti-tumor activity, particularly when combined with a mitochondrial complex 1 inhibitor.
DISCUSSION
In the present study, we describe (1) a small molecule with specific and durable in vivo LDH inhibitory activity, (2) a bioprobe that allows for the study of tumor metabolic response to specific LDH inhibition in vivo, (3) the ability to non-invasively visualize in real time the rapid onset of metabolic rewiring in glycolytic tumors exposed to either LDH inhibitor or mitochondrial complex 1 inhibitor, and (4) the mechanistic basis of this metabolic rewiring and its susceptibility to exploitation with combination therapy.
Although much emphasis has been placed on LDHA as the key mediator of metabolic plasticity in cancer, a recent study has shown that double knockout of both LDHA and LDHB is necessary to fully abrogate cancer cell dependence on aerobic glycolysis (Ždralević et al., 2018). In addition, both LDHA and LDHB have been identified as essential for oncogenic p53 and mutant Ras-dependent oncogenic transformation (Smith et al., 2016). Thus, we consider the dual inhibition of both LDHA and LDHB to be a positive feature of NCI-006. All specific small-molecule LDH inhibitors reported to date have displayed no or poor in vivo activity (Billiard et al., 2013; Boudreau et al., 2016; Le et al., 2010). Although the natural-product derivative FX11 has shown in vivo activity, it is not specific for LDH and has several off-target effects (Billiard et al., 2013; Fantin et al., 2006; Granchi et al., 2013; Le et al., 2010; Ward et al., 2012; Xie et al., 2014). In contrast, we have demonstrated robust, dose-dependent, in vivo activity of NCI-006 by examining ex vivo lactate secretion from RBCs isolated from mice receiving the LDH inhibitor by either oral or intravenous administration. Using this model, we were able to establish an effective and tolerable dose range for PO and IV administration of NCI-006. These data revealed the rapid onset of LDH inhibition, as well as the persistence of inhibition in RBCs for several hours after a single dose, although LDH activity generally returned to normal within 24 h. Because hemolytic anemia from hemolysis is an on-target consequence of an inactivating LDH mutation in mice, we suspected that hemolysis would likely be a major contributor to the dose-limiting toxicity of NCI-006 in vivo. Our data, however, suggest that although a small decrease in hematocrit does occur in mice treated over 2 weeks on an intermittent IV dosing schedule, full recovery occurs within 6–10 days after cessation of drug treatment.
Although both PO and IV administration of NCI-006 effectively inhibited RBC LDH, the same is not the case for LDH in tumor xenografts. Using the glycolytic MIA PaCa-2 xenograft model, we found that a single PO dose (50 mg/kg) of NCI-006, which is efficacious for RBC LDH inhibition, resulted in a peak intratumoral drug concentration of <4 μM, which failed to translate to significant inhibition of tumor LDH (not surprising given the 10–20 μM LDH concentration in cancer cells). In contrast, IV administration of an equivalent dose of NCI-006 resulted in a maximum intratumoral concentration of 15–20 μM that persisted from 30 min to 5 h after dosing and correlated with approximately 80% inhibition of LDH over that time. The divergent effect of PO versus IV dosing of NCI-006 on xenograft LDH activity was confirmed by metabolic imaging of hyperpolarized [13C]pyruvate flux. The existence of diffusion barriers within the solid tumor, which interfere with drug penetration, likely contributes to the differential efficacy between PO and IV administration routes when comparing RBC and intratumoral LDH inhibition (Jain, 2012). For tumor xenografts, the large difference in maximum intratumoral drug level achieved after PO or IV dosing corresponds well with the plasma Cmax. Intermittent intravenous treatment with LDH inhibitor alone caused significant retardation of MIA PaCa-2 xenograft growth, even several weeks after cessation of treatment.
Because we observed an increased flux of pyruvate to bicarbonate in MIA PaCa-2 xenografts treated with NCI-006, even these highly glycolytic cells (Daemen et al., 2015) possess the capability to redirect pyruvate to the tricarboxylic acid (TCA) cycle when challenged with an LDH inhibitor. Our in vitro data identified synergistic effects of the mitochondrial complex I inhibitor IACS-010759 and NCI-006 on the viability of MIA PaCa-2 cells. Using HP-MRSI with [13C]pyruvate to assess alterations in pyruvate flux in vivo, we found that mitochondrial complex 1 inhibition with IACS-010759 increased pyruvate metabolism to lactate in tumor while decreasing pyruvate entry into the TCA cycle, consistent with redirection of pyruvate to metabolism by LDH. However, the combination of IACS-010759 and NCI-006 resulted in reduced pyruvate flux to both lactate and bicarbonate (i.e., the TCA cycle). This was accompanied by prolonged inhibition of MIA PaCa-2 xenograft growth even after cessation of treatment. Importantly, metformin, also reported to inhibit mitochondrial complex 1, phenocopied IACS-010759 when administered together with NCI-006 in mice harboring HT29 xenografts. Taken together, these data support our hypothesis that rational combination of metabolic inhibitors can achieve maximal effect on tumor growth.
Finally, we visualized intratumoral LDH activity dynamically and in real time using non-invasive MR imaging of hyperpolarized [13C]pyruvate, and we showed clearly that LDH is druggable in vivo. By correlating the degree of change in the tumor [13C] lactate/[13C]pyruvate ratio with the LDH inhibitor dose and the intratumoral LDH inhibitor concentration, we have established the degree and durability of tumor LDH inhibition necessary for causing significant growth inhibition of tumor xenografts with acceptable on-target systemic toxicity. Although different cancers and individual patient tumors are very likely to show differences in LDH activity in vivo, we show, in this study, that we are also able to image the on-target activity of the clinically evaluated mitochondrial complex 1 inhibitor IACS-010759. Thus, by allowing for metabolic characterization of a patient’s tumor, HP-MRSI can assist in selecting patients whose cancer is likely to respond to a targeted therapy, and by visualizing both the degree and duration of effect on pyruvate flux in real time, this imaging approach can correlate target inhibition with tumor response.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Len Neckers (neckersl@mail.nih.gov). Currently, there are restrictions to the availability of NCI-006 due to a licensing agreement with Chinook Therapeutics (Vancouver, Canada). Requests for NCI-006 will be considered on a case-by-case basis and compound may be supplied upon receipt of an approved MTA.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and tumor models
All animal studies were performed in compliance with the Guide for the Care and Use of Laboratory Animal Resources (National Research Council, 2011) and were approved by the NCI Animal Care and Use Committee. Female athymic nude mice (20 – 25 g) were supplied by the Frederick Cancer Research Center Animal Production Facility. A total of 1, 2, or 3 million tumor cells (depending on the experiment; MIA PaCa-2 or HT29) in 100 mL of serum-free PBS were injected subcutaneously into the right hind leg. The tumor volume was calculated by the formula 0.5 × L × W2 (L: length, W: width). Tumors were allowed to reach 100 – 300 mm3 in volume, depending on the experiment, before treatment was initiated. Prior to beginning treatment, mice were randomized into each treatment (or vehicle control) group. Mice were fed with a defined gel meal (Nutra-Gel, NCI, Sterile, # S4950) for the entire course of all animal experiments. During magnetic resonance spectroscopic imaging (MRSI) measurements, the breathing rate of the mouse was monitored with a pressure transducer (SA Instruments Inc., Stony Brook, NY, USA) and was maintained at 80 ± 10 breaths/min. Core body temperature was monitored using a nonmagnetic rectal temperature probe (FISO, Quebec, QC, Canada) and was maintained at 36 ± 1°C with a flow of warm water (Saito et al., 2015). All efforts were made to minimize suffering in each animal experiment. The LDH inhibitor (doses described in text) was administered either intravenously (IV) via tail vein injection or orally (PO) by gavage. Metformin (50 mg/kg) and IACS-010759 (20 mg/kg for efficacy studies, 2.5 mg/kg for HP-MRSI studies) were administered orally. Hematocrit was determined using a microhematocrit reader disk by calculating a ratio of the packed red cell volume to total blood volume in a 75 mm capillary tube which was centrifuged at 12,000 rpm for 5 min in a hematocrit centrifuge (LW Scientific, Hampton, NH, USA).
Human tissue
Renal cortex tissue was obtained from a nephrectomy specimen from a fifty-three year old male who underwent surgery at the Urologic Oncology Branch (UOB) of the National Cancer Institute (NCI), National Institutes of Health (NIH). The study was approved by the Institutional Review Board of the National Cancer Institute and the patient provided written informed consent.
Cell lines
Human pancreatic and colon cancer cell lines [MIA PaCa-2 (male) and HT29 (female), respectively], and HEK293-T (human embryonic cell line) were purchased from ATCC (Manassas, VA, USA). The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Corning, Manassas, VA, USA) containing 10% fetal bovine serum (FBS, Life Technologies, Carlsbad CA, USA), penicillin (100 Units/ml) and streptomycin (100 μg/ml) (Life Technologies, Carlsbad, CA, USA). Cells were not authenticated; however, early passage cells direct from purchase were used for the experiments described in this study.
METHOD DETAILS
Cell viability
Cell viability was assessed using Cell Titer Glo (Promega, Madison, WI) and results were verified by trypan blue exclusion.
LDH inhibitor and other drugs
The LDH inhibitor used in these studies (NCI-006) was generated and validated by a consortium of investigators sponsored by the National Cancer Institute (NCI) Experimental Therapeutics Program (NExT) and included investigators from NCI, the University of Pennsylvania, National Center for Advancing Translational Sciences (NCATS), Vanderbilt University, the University of New Mexico and the University of Alabama at Birmingham. NCI-006 is a result of further optimization of the pyrazole series of recently described LDH inhibitors7. The IUPAC name of this compound is 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-((5-methylthiophen-2-yl)ethynyl) phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid, and its chemical formula is C31H24F2N4O4S (molecular weight = 650.73) (see Figure 1A). Chemical synthesis route of NCI-006 and determination of product purity are shown in Supplemental Information (Method S1). For in vitro use, NCI-006 was dissolved in dimethylsulfoxide (DMSO) to create a 10 mM stock solution and final concentration was achieved by dilution in phosphate-buffered saline (PBS). For in vivo use, NCI-006 was dissolved in 0.1 N NaOH/PBS and pH was adjusted to 7.4 – 7.8 by dropwise addition of 1N HCl. Final concentration of LDH inhibitor in this formulation was 10 mg/mL. This solution is stable when kept at 4°C for up to two weeks. The solution was warmed to 37°C prior to administration. Metformin hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO, USA) and adjusted to its final concentration with PBS. The mitochondrial complex 1 inhibitor IACS-010759 was previously described by Molina et al. (2018) and was synthesized following the published procedure. For in vitro experiments, IACS-010759 was dissolved in DMSO as a 10 mM stock solution and further diluted in culture medium. For in vivo use, the inhibitor was dissolved in 0.5% methylcellulose in water and administered by oral gavage. Metformin was obtained from the veterinary pharmacy, NIH.
Native Gel assay
LDH isozymes were separated by native gel electrophoresis at 125V for 1.25 hours using pre-cast 1 mm 6% Tris-glycine gel electrophoresis (Life Technologies, Carlsbad, CA, USA). Running buffer consisted of 25 mM Tris, 192 mM glycine, pH 8.3. After electrophoresis, the gel was soaked in 10 mL of 50mM Tris buffer, pH 8.0, containing LDH inhibitor at the specified concentrations for 30 min, followed by addition of reaction mixture [1 mg/mL iodonitrotetrazolium bromide (INT), 1 mM NAD+, 0.184 mg/mL phenazine methosulfate (PMS), and 5mM L-lactate], and gels were incubated on a rocker in the dark at 42°C for 7–14 minutes. After development of LDH activity bands, gels were destained with several rinses of de-ionized water in the dark until the background was clear. Finally, gels were scanned using an Epson V700 photo scanner in transmitted light mode to generate the images shown.
NAD/NADH assay
3×105 cells were seeded in 6 well plates and treated with LDH inhibitor for 2 h on the following day. The concentrations of NCI-006 used were 0.2 μM and 5 μM for MIA PaCa-2 cells and HT29 cells, respectively. NAD/NADH ratio was measured using NAD/NADH Quantitation Kit (Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer’s instructions. Reaction time was set to 2h.
Ex vivo Lactate production
Mouse blood samples were collected at several time points after in vivo administration of LDH inhibitor. Red blood cells (RBCs) were collected from 50 μL whole blood by centrifugation and washed three times in Dulbecco’s Modified Eagle’s medium (DMEM, containing 4.5 g/L glucose, Life Technologies, Carlsbad, CA, USA). After washing, RBCs were diluted (~15 × 106/mL) and incubated in DMEM at 37°C for 1.5 h. After incubation, samples were centrifuged at 500 × g for 10 min and supernatant was collected for lactate assay, performed using a Lactate Colorimetric/Fluorometric Assay Kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s instructions. MIA PaCa-2 cells were cultured as described above, and plated in 1536-well black clear bottom tissue culture plates using a Multidrop Combi peristaltic dispenser (ThermoFisher, Waltham, MA, USA) at a density of 500 cells/well in 4 μL of DMEM (Invitrogen 31053036, Carlsbad, CA, USA) culture medium. A 1536-well pin tool dispenser outfitted with 20 nL pins (Wako Automation, San Diego, CA, USA) was used to transfer 23 nL of compound in DMSO to the 1536-well assay plates. After 2 hours incubation at 37°C, 2 μL of reconstituted Lactate Reaction Mix (BioVision K607–100, Milpitas, CA, USA) was dispensed into each well using a BioRAPTR FRD dispenser (Beckman Coulter, Brea, CA, USA). Plates were incubated at room temperature for 30 minutes, transferred to a ViewLux microplate imager (PerkinElmer, Waltham, MA, USA) and the fluorescence (Ex/Em 525/598 nm) and absorbance (573 nm) were measured accordingly. For each cell line, data were normalized to DMSO control-treated cells.
Measurement of drug concentration and LDH activity in tumor xenografts
Tumors were harvested and snap frozen between 1 and 3 h following the final dose of NCI-006. Measurement of tumor and plasma concentrations of NCI-006 was by mass spectrometric analysis with internal standards (pure NCI-006) and was performed by Quintara Discovery (Hayward, CA, USA). For measurement of LDH activity, frozen tissues (10–50 mg) were pulverized using a liquid nitrogen-cooled steel mortar and transferred to pre-cooled Eppendorf tubes. PBS, pH 7.4 (10 volumes) containing 0.1% Triton X-100 was added to each tissue sample prior to incubation on ice for 60 min. Samples were mixed by vortexing every 15 min. The insoluble fraction was removed by centrifugation at 13,000 rpm for 15 min at 4°C. Cleared homogenates were transferred to fresh tubes and protein content was determined by DC Lowry assay (Bio-Rad, Hercules, CA, USA). LDH activity was measured spectrophotometrically in tissue homogenates by monitoring the rate of oxidation of NADH at 340 nm over time in the presence of 10 mM pyruvate. Reactions were performed at 37°C in PBS, pH 7.4 containing 0.1% Triton x-100.
Extracellular flux analysis
OXidative PHOSphorylation (OXPHOS) and glycolysis in MIA PaCa-2 cells were monitored in real time using the extracellular flux (XF) bioanalyzer XFe96 platform (Agilent, Santa Clara, CA, USA). Cells were cultured overnight in custom XF microplates with DMEM complete medium. Prior to measurements, cells were washed and incubated in unbuffered assay medium (Sigma-Aldrich, St. Louis, MO, USA) in the absence of CO2 for 1 h at 37°C. The rates of mitochondrial respiration and glycolysis were measured using the mitochondrial stress test (Agilent, Santa Clara, CA, USA) and displayed as the Oxygen Consumption Rate (OCR) and the Extracellular Acidification Rate (ECAR), respectively (Dranka et al., 2011). All measurements commenced after establishment of a stable baseline, followed by sequential addition of the LDH inhibitor NCI-006, oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and finally antimycin A and 2-deoxyglucose.
Multiplexed in vitro synergy screen
An in vitro synergy screen was conducted in 1536-well white flat bottom plates (Corning, Corning, NY, USA). MIA PaCa-2 cells were dispensed at a density of 500 cells in 5 μL of media (MultiDrop Combi, Thermofisher Scientific, Waltham, MA, USA) and seeded into plates pre-spotted with 20 nL of compounds (Echo 555, Labcyte, San Jose, CA, USA). Compounds IACS-010759 and NCI-006 were dispensed (10 nL each compound) in a 10-concentration point dose window (including a zero concentration), 2-fold dilution for each compound, as a ‘10×10’ block. After 72 hours of incubation, 3 μL of CellTiter-Glo reagent (Promega, Madison, WI, USA) was added to each well. Following 10 min incubation, luminescence was read using the Viewlux microplate reader (PerkinElmer, Waltham, MA, USA). Plate data were normalized to in-plate controls (bortezomib as positive control for cell death, DMSO as negative control) and the normalized data was deconvoluted to individual dose combination matrices using in-house software. We employed the Bliss model of additivity (Bliss, 1956) to characterize the presence or absence of synergy for each combination. Each 10×10 synergy block was run in triplicate. The synergistic activity of the in vitro combinations was assessed based on the Delta Bliss Sum Negative (DBSumNeg) score with the conservative synergy cutoff DBSumNeg < −3 (Mott et al., 2015). The calculated DBSumNeg value for synergy between NCI-006 and IACS-010759 was −15.11.
Hyperpolarized (HP) 13C-MRSI
Hyperpolarized 13C-MRSI was performed as described previously (Saito et al., 2015). [1-13C]pyruvate (30 μL) containing 15 mmol/L OXO63 and 2.5 mmol/L gadolinium chelate was hyperpolarized using the Hypersense DNP polarizer (Oxford Instruments, Abingdon, UK). The hyperpolarized sample was rapidly dissolved in 4.5 mL of a superheated alkaline buffer and intravenously injected (12 μL/g body weight). Hyperpolarized 13C MRSI was performed on a 3T scanner (MRSolutions, Guildford, Surrey, UK) using a 17 mm homebuilt 13C solenoid coil placed inside a saddle coil for detection of 1H (hydrogen atom). Anatomical images were taken with a fast spin-echo imaging sequence with a repetition time (TR) of 2,500 msec, an effective TE of 68 msec, one signal, an echo-train length of 8 echoes, a field of view of 28 mm × 28 mm with a 256 × 256 matrix, and a 2-mm section thickness with a 2-mm intersection gap. The 13C MR spectra were acquired every second with a spectral width of 3330 Hz, a repetition time of 1000 ms and a flip angle of 10°. 13C two-dimensional spectroscopic images were acquired 25 s after the start of the pyruvate injection, with a 28 × 28 mm field of view in an 8 mm axial slice, yielding a matrix size of 14 × 14 mm. The chemical shift values for each signal used in this study were - [1-13C] Pyruvate: 170.6 ppm, [1-13C] Lactate: 182.7 ppm, [1-13C] Alanine: 176 ppm and [1-13C] Bicarbonate: 163.5 ppm, respectively. Each mouse harboring xenografted tumors was imaged 24 h before (“pre-study”) and at various times (30 min – 24 h) after (“post-study”) drug treatment, thus serving as its own control. We are confident that data obtained from imaging tumor xenografts reflect tumor-specific metabolism for three reasons. 1) Most of the tissue volume inside the coil is tumor (approximately 75% from analysis of the FLASH anatomical MRI images). 2) In analyzing the non-localized 13C experiments with MIA PaCa-2 xenografts versus the control leg, we found that the signal intensity for the upstream metabolite for labeled 13C-bicarbonate (13C-pyruvate), is 6 times stronger in scans of the leg bearing the tumor compared to the control leg. 3) A bicarbonate signal is not detectable in any of the scans of the control leg.
Hyperpolarized 13C-MRSI using a clinical grade imager
13C-MRSI was also performed using an MRI approved for clinical use (Achieva 3.0T-X series, Philips, Andover, MA, USA) and a SPINlab MRI Hyperpolarizer (GE Healthcare, Waukesha, WI, USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
MRSI data were analyzed using MATLAB (MathWorks, Natick, MA, USA) and ImageJ 1.49v (NIH, Bethesda, MD, USA). Statistical analyses were carried out using GraphPad Prism 8 (San Diego, CA, USA) software. Differences were considered to be statistically significant for P values ≤ 0.05. All results are expressed as mean ± SEM t test was performed to evaluate the statistical difference between two groups. A one-way ANOVA with uncorrected Fisher’s Least Significant Difference (LSD) test was used to evaluate statistical differences between multiple groups. Statistical details of experiments can be found in the figure legends.
DATA CODE AND AVAILABILITY
This study did not generate/analyze datasets/code.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Normal human kidney | Urologic Oncology Branch, CCR, NCI | N/A |
| Mouse heart tissue (from an athymic female mouse) | Animal vivarium, CCR, NCI | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LDH inhibitor NCI-006 | NCI, this manuscript | N/A |
| Metformin hydrochloride (in vitro use) | Sigma-Aldrich | Cat. # PHR1084 |
| IACS-010759 | NCATS | N/A |
| Metformin (in vivo use) | NIH vet pharmacy | N/A |
| Critical Commercial Assays | ||
| CellTiter-Glo | Promega | Cat. # G7571 |
| NAD/NADH Quantitation kit | Sigma-Aldrich | Cat. # MAK037–1KT |
| Lactate Colorimetric/Fluorometric Assay Kit | Abcam | Cat. # ab65330 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat. # 103015–100 |
| Experimental Models: Cell Lines | ||
| MIA PaCa-2 | ATCC | Cat. # ATCC CRL-1420 |
| HT29 | ATCC | Cat. # ATCC HTB-38 |
| HEK293-T | ATCC | Cat. # ATCC ACS-4500 |
| Experimental Models: Organisms/Strains | ||
| Female athymic nude mice | Frederick Cancer Research Center Animal Production Facility |
N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| ImageJ 1.49v | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
Highlights.
Specific LDH inhibition in vivo reduces growth rate of glycolytic tumors
Depth and duration of tumor LDH inhibition can be monitored in real time by HP-MRSI
LDH inhibition in vivo redirects pyruvate to support oxidative phosphorylation
Inhibiting mitochondrial complex 1 and LDH enhances durability of anti-tumor response
ACKNOWLEDGMENTS
This research was supported with funding from both the National Cancer Institute Experimental Therapeutics (NExT) Program and from the Intramural Research Program of the National Cancer Institute.
Footnotes
DECLARATION OF INTERESTS
D.U., G.R., B.T.M., X.H., A.J., W.M., A.S., D.J.M., C.V.D., A.G.W., V.M.D.-U., and L.M.N. are inventors on a patent related to this work.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.039.
REFERENCES
- Albers MJ, Bok R, Chen AP, Cunningham CH, Zierhut ML, Zhang VY, Kohler SJ, Tropp J, Hurd RE, Yen YF, et al. (2008). Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 68, 8607–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, and Golman K (2003). Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 100, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colón M, Dodson CS, Gilbert SA, Greshock J, Jing J, et al. (2013). Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CI (1956). The calculation of microbial assays. Bacteriol. Rev 20, 243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, Kwong M, Hong R, Gao M, Del Nagro C, et al. (2016). Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol 12, 779–786. [DOI] [PubMed] [Google Scholar]
- Brisson L, Bański P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, Van Hée VF, Vazeille T, Tardy M, Falces J, et al. (2016). Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 30, 418–431. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Dölle C, Felici R, and Ziegler M (2012). The NAD metabolome—a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752. [DOI] [PubMed] [Google Scholar]
- Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al. (2015). Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 112, E4410–E4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci. Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, et al. (2011). Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med 51, 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P, Le A, Vander Jagt DL, Tsukamoto T, Martinez GV, Dang CV, and Gillies RJ (2013). Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Res. 73, 4190–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, and Leder P (2006). Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434. [DOI] [PubMed] [Google Scholar]
- Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, and Viollet B (2010). Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granchi C, Paterni I, Rani R, and Minutolo F (2013). Small-molecule inhibitors of human LDH5. Future Med. Chem 5, 1967–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DK, Orton MR, Mariotti E, Boult JK, Panek R, Jafar M, Parkes HG, Jamin Y, Miniotis MF, Al-Saffar NM, et al. (2013). Model free approach to kinetic analysis of real-time hyperpolarized 13C magnetic resonance spectroscopy data. PLoS ONE 8, e71996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK (2012). Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev 64 (Suppl), 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, Kaipparettu BA, and Levine H (2019). Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 116, 3909–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JP, Datta T, Pretsch W, Charles DJ, and Dörmer P (1987). Mechanisms of compensation of hemolytic anemia in a lactate dehydrogenase mouse mutant. Exp. Hematol 15, 664–670. [PubMed] [Google Scholar]
- Kurhanewicz J, Bok R, Nelson SJ, and Vigneron DB (2008). Current and potential applications of clinical 13C MR spectroscopy. J. Nucl. Med 49, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, and Dang CV (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 107, 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Adler AS, Shang Y, Hunsaker T, Truong T, Peterson D, Torres E, Li L, Haley B, Stephan JP, et al. (2012). An integrated genomic screen identifies LDHB as an essential gene for triple-negative breast cancer. Cancer Res. 72, 5812–5823. [DOI] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, and Saavedra E (2007). Energy metabolism in tumor cells. FEBS J. 274, 1393–1418. [DOI] [PubMed] [Google Scholar]
- Mott BT, Eastman RT, Guha R, Sherlach KS, Siriwardana A, Shinn P, McKnight C, Michael S, Lacerda-Queiroz N, Patel PR, et al. (2015). High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci. Rep. 5, 13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, et al. (2013). Metabolic imaging of patients with prostate cancer using hyperpolarized [1–13C]pyruvate. Sci. Transl. Med 5, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M (2014). Overcoming drug development bottlenecks with repurposing: repurposing biguanides to target energy metabolism for cancer treatment. Nat. Med 20, 591–593. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, and Galluzzi L (2018). Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretsch W, Merkle S, Favor J, and Werner T (1993). A mutation affecting the lactate dehydrogenase locus Ldh-1 in the mouse. II. mechanism of the LDH-A deficiency associated with hemolytic anemia. Genetics 135, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai G, Brimacombe KR, Mott BT, Urban DJ, Hu X, Yang SM, Lee TD, Cheff DM, Kouznetsova J, Benavides GA, et al. (2017). Discovery and optimization of potent, cell-active pyrazole-based inhibitors of lactate dehydrogenase (LDH). J. Med. Chem 60, 9184–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Matsumoto S, Takakusagi Y, Matsuo M, Morris HD, Lizak MJ, Munasinghe JP, Devasahayam N, Subramanian S, Mitchell JB, and Krishna MC (2015). 13C-MR spectroscopic imaging with hyperpolarized [1-13C]pyruvate detects early response to radiotherapy in SCC tumors and HT-29 tumors. Clin. Cancer Res. 21, 5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggins BT, Matsuo M, White AO, Saito K, Munasinghe JP, Sourbier C, Yamamoto K, Diaz V, Takakusagi Y, Ichikawa K, et al. (2018). Hyperpolarized [1-13C]-pyruvate magnetic resonance spectroscopic imaging of prostate cancer in vivo predicts efficacy of targeting the Warburg effect. Clin. Cancer Res. 24, 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Schafer XL, Ambeskovic A, Spencer CM, Land H, and Munger J (2016). Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 17, 821–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolková K, Plecitá-Hlavatá L, Bellance N, Benard G, Rossignol R, and Ježek P (2011). Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol 43, 950–968. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Warburg O, and Negelein KPE (1924). Ueber den stoffwechsel der tumoren. Biochem. Z 152, 319–344. [Google Scholar]
- Ward RA, Brassington C, Breeze AL, Caputo A, Critchlow S, Davies G, Goodwin L, Hassall G, Greenwood R, Holdgate GA, et al. (2012). Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J. Med. Chem 55, 3285–3306. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, and Chandel NS (2015). Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 107, 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. (2014). Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 19, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung C, Gibson AE, Issaq SH, Oshima N, Baumgart JT, Edessa LD, Rai G, Urban DJ, Johnson MS, Benavides GA, et al. (2019). Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth in preclinical models of Ewing sarcoma. Cancer Res. 79, 5060–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ždralević M, Brand A, Di Ianni L, Dettmer K, Reinders J, Singer K, Peter K, Schnell A, Bruss C, Decking SM, et al. (2018). Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem 293, 15947–15961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut ML, Yen YF, Chen AP, Bok R, Albers MJ, Zhang V, Tropp J, Park I, Vigneron DB, Kurhanewicz J, et al. (2010). Kinetic modeling of hyperpolarized 13C1-pyruvate metabolism in normal rats and TRAMP mice. J. Magn. Reson 202, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, and Guppy M (2004). Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res. Commun 313, 459–465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.