

Abstract
Tufted angioma (TA) is a rare vascular tumor characterized by histologic tufts of proliferating capillaries that occurs in infancy or early childhood, with a poorly understood pathogenesis. Though benign, TA can be associated with the Kasabach-Merritt syndrome (KMS), a life-threatening consumptive coagulopathy and thrombocytopenia. Here, we explored the genetic mechanism underlying a case of TA associated with KMS via targeted sequencing of laser capture micro-dissected lesion and blood DNA, and identified a somatic, activating GNA14 mutation specific to the tumor. Our findings support aberrant GNA14 activation underlies the pathogenesis of TA associated with KMS.
BRIEF REPORT
Tufted angioma (TA) is a rare, benign vascular tumor of infancy that lies within the spectrum of hemangioendotheliomas alongside kaposiform hemangioendothelioma (KHE).1 TAs clinically present as violaceous, indurated or nodular plaques, and histologically show confluent lobules or “cannonballs” of spindled endothelial cells with slit-like lumina embedded in a fibrotic background.1 Disease progression varies considerably, with some demonstrating spontaneous regression or focal hyperhidrosis and hypertrichosis.1 Up to 10% of TA are associated with the Kasabach-Merritt syndrome (KMS), a consumptive coagulopathy characterized by profound thrombocytopenia, hypofibrinogenemia, and microangiopathic anemia with up to 24 percent mortality.1; 2 The underlying pathobiology of TA and its varying co-occurrence with KMS are poorly understood.
Here, we investigated the genetic mechanism of TA complicated by KMS by assessing for previously implicated mutations. The Yale Human Investigation Committee approved this study, which was conducted according to the Declaration of Helsinki Principles.
A 5-day-old boy presented with a congenital 14×4cm non-blanching patch on the right lateral torso and flank, which progressively increased in size, turned darker red, and became firm to palpation at 4 months (Figure 1A). The biopsy revealed superficial lobules of aggregated endothelial cells and an increased number of dilated and angulated thin-walled vessels in the dermis. Deeper in the dermis, dense and diffuse vessels infiltrated collagen bundles with small vascular spaces, consistent with TA (Figure 1B). Labs revealed thrombocytopenia with a platelet count of 84 × 1,000/μL (ref 219 – 465 × 1,000/ μL), decreased fibrinogen with a nadir of <70 mg/dL (ref 150–434 mg/dL), and elevated D-Dimers to 15.81 mg/L (ref 0.17 – 0.54 mg/L), consistent with KMS. 2mg/kg/day prednisone was started for 3 months with slow taper, and the lesion faded and softened. The lesion was stable at 18 months of age when his family moved to China and was lost to follow-up.
Figure 1. Tufted angioma associated with KMS with an underlying somatic GNA14 mutation.
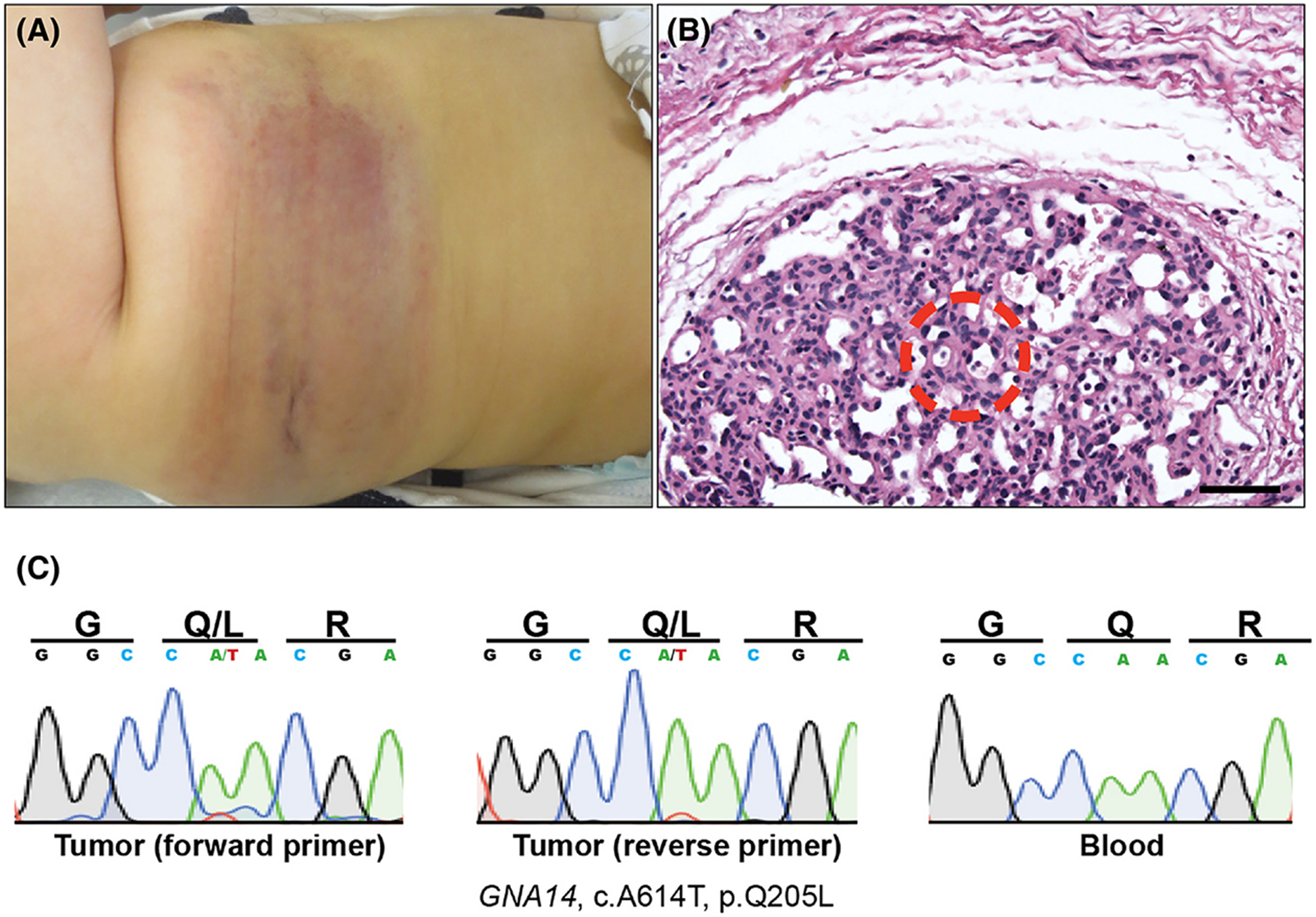
(A) A violaceous 14×4cm non-blanching patch is observed on the right lateral torso and flank, which was present since birth. (B) Lobular vascular proliferation composed of tightly packed capillaries lined by cytologically-bland endothelial cells (20X magnification, scale bar = 150μm). Area from where LCM was employed to isolated tumor-specific DNA is shown (red circle). (C) Sanger sequencing of tumor (trace data from both primers) and blood control shows GNA14 p.Q205L mutation in tumor only.
Blood and archived tumor samples were evaluated. Laser capture microdissection (LCM) was employed to isolate gDNA specific to lesional vessels (Figure 1B), and sequencing was performed for all exons of the Ras subfamily, BRAF, GNAQ, GNA11, and GNA14 per previously reported protocols.3 In so doing, we identified a somatic GNA14 c.614A>T (p.Q205L) mutation specific to the tumor (Figure 1C).
A case of TA and a case of sporadic pyogenic granuloma without KMS, as well as one case of KHE with KMS, were previously reported with p.Q205L mutations in GNA14, suggesting GNA14 activation underlies tumorigenesis.3 However, why only some tumors present with KMS despite arising via the same genetic mutation remains elusive.3 GNA14 activation via the p.Q205L mutation was shown to specifically upregulate the pERK-MAPK pathway, providing potential targets for therapy.3 Counterintuitively, treatment with sirolimus, an mTOR inhibitor with feedback activation of MAPK, has reported tumor shrinkage and amelioration of KMP in a KHE.4; 5 KHE or TA without GNA14 mutation may instead have activation of pAkt-mTOR pathway, with differential response to mTOR versus MAPK inhibition. Our findings further implicates the MAPK pathway in TA and TA associated with KMS, and highlights MAPK inhibition as an important strategy against hemangioendotheliomas refractory to currently available treatment options.1
ACKNOWLEDGEMENTS
This work was supported by Doris Duke Charitable Foundation Clinical Scientist Development Award and a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)/NIH (5R01AR062111) to KAC. YHL is supported by the Medical Scientist Training Program at Yale University (NIH/NIGMS T32 GM007205), and is a recipient of the Clinical Mentorship Award from the Doris Duke Charitable Foundation.
Footnotes
Research was conducted at Yale School of Medicine in New Haven, Connecticut, USA
All authors consent to manuscript publication. There are no conflict of interests to disclose.
The Yale Human Investigation Committee approved this study, which was conducted according to Declaration of Helsinki Principles. The consented subject provided archived tumor and blood samples.
REFERENCES
- 1.Osio A, Fraitag S, Hadj-Rabia S. Clinical spectrum of tufted angiomas in childhood: A report of 13 cases and areview of the literature. JAMA Dermatol 2010; 146(7): 758–763. [DOI] [PubMed] [Google Scholar]
- 2.Croteau SE, Liang MG, Kozakewich HP et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr 2013; 162: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim YH, Bacchiocchi A, Qiu J et al. GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am J Hum Genet 2016; 99: 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid I, Klenk AK, Sparber-Sauer M, Koscielniak E, Maxwell R, Haberle B. Kaposiform hemangioendothelioma in children: a benign vascular tumor with multiple treatment options. World J Pediatr 2018; 14: 322–329. [DOI] [PubMed] [Google Scholar]
- 5.Carracedo A, Ma L, Teruya-Feldstein J et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118: 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]