

Abstract
Prior studies reported that haploinsufficiency of the transcription factor ETS-1 is renoprotective in Dahl salt-sensitive rats, but the mechanism is unclear. Here, we tested whether ETS-1 is involved in hypertension-induced renal microvascular pathology and autoregulatory impairment. Hypertension was induced in salt-sensitive rats and salt-sensitive rats that are heterozygous with one wild-type or reference allele of Ets1 (SSEts1+/−) by feeding a diet containing 4% sodium chloride for one week. Increases in blood pressure did not differ. However, phosphorylated ETS-1 increased in afferent arterioles of hypertensive salt-sensitive rats, but not in hypertensive SSEts1+/− rats. Afferent arterioles of hypertensive salt-sensitive rats showed increased monocyte chemotactic protein-1 expression and infiltration of CD68 positive monocytes/macrophages. Isolated kidney microvessels showed increased mRNA expression of vascular cell adhesion molecule, intercellular adhesion molecule, P-selectin, fibronectin, transforming growth factor-β, and collagen I in hypertensive salt-sensitive rats compared with hypertensive SSEts1+/− rats. Using the in vitro blood-perfused juxtamedullary nephron preparation, pressure-mediated afferent arteriolar responses were significantly blunted in hypertensive salt-sensitive rats compared to hypertensive SSEts1+/− rats. Over a 65-170 mm Hg pressure range tested baseline arteriolar diameters averaged 15.1 μm and remained between 107% and 89% of baseline diameter in hypertensive salt-sensitive rats vs. 114% and 73% in hypertensive SSEts1+/− rats (significantly different). Thus, ETS-1 participates in renal arteriolar pathology and autoregulation and thereby is involved in hypertension-mediated kidney injury in salt-sensitive rats.
Keywords: chronic kidney disease, salt, inflammation, fibrosis
Graphical Abstract
Introduction
Hypertensive nephropathy is one of the most common causes of chronic kidney disease (CKD) and is the second leading cause of end-stage kidney disease (ESKD), producing over a quarter of all cases of ESKD in the U.S.1, 2 Despite improvements in the treatment of hypertension, the percentage of hypertensive patients who ultimately develop ESKD has remained constant (0.5 to 1%)2, suggesting that new approaches are needed to ameliorate the morbid kidney consequences of hypertension. A hallmark of human hypertensive nephropathy is a thickening of the vascular walls from the arcuate arteries down to the afferent arterioles; pathologic changes in the afferent arterioles is particularly conspicuous.3–5 These vascular changes promote glomerulosclerosis from ischemia, accumulation of extracellular matrix and podocyte loss with proteinuria. Despite extensive investigation, it remains unclear why some hypertensive patients develop end-organ kidney damage, while others do not. Because of the multiple variables – use and type of antihypertensive medications, degree of hypertension over time, duration of hypertension, etc. – that may in turn affect kidney function, the molecular pathogenesis of the arteriolar pathology in hypertensive nephropathy has not been explained. Insight into the molecular mechanisms underlying renovascular pathobiology in hypertension may therefore be gained through the study of animal models.
The Dahl salt-sensitive (SS) rat is a well-characterized model of hypertension and kidney injury. Because the onset, level of hypertension, and subsequent reduction in renal function are rapid, reproducible and constant, this animal model is useful for studying the pathogenesis of hypertension-induced kidney disease.6, 7 Within one week of feeding a high-salt diet, SS rats uniformly develop hypertension that is accompanied by progressive loss of renal function and glomerular injury.6–9 This response to hypertension differs substantially from the spontaneously hypertensive rat, which develops only mild proteinuria and kidney damage over an extended period of hypertension.10
Advances in genetic manipulation have permitted deletion of specific genes of interest in the rat11 and thereby provide a more detailed characterization of the pathogenesis of underlying causes of CKD such as hypertensive nephropathy. Avian Erythroblastosis Virus E26 Oncogene Homolog-1 (abbreviated ETS-1) is a transcription factor that is encoded within a quantitative trait locus (QTL) that is located on rat chromosome 8 and has linkage to albumin excretion and kidney damage.12, 13 ETS-1 is a founding member of a group of transcription factors that contain a DNA-binding site characterized by a winged helix-turn-helix motif.14 Because our initial studies showed that a dominant-negative peptide inhibitor of ETS-1 reduced glomerular injury and interstitial fibrosis in hypertensive SS rats,15 SS rats with an inactivating mutation of one Ets-1 allele (SSEts1+/− rats, previously also abbreviated ES13) were generated for additional study. Kidneys from SSEts1+/− rats were protected from subsequent hypertensive injury.13 Accordingly, we tested the hypothesis that ETS-1 plays an important role in hypertension-induced renal microvascular pathology and dysregulation of afferent arteriolar autoregulatory behavior.
Results
Despite radio-telemetry-monitored blood pressures that did not differ, kidney injury was detected in hypertensive SS rats, but was abrogated in hypertensive SSEts1+/− rats.
SS rats with an inactivating mutation of a single Ets1 allele, termed SSEts1+/− rats, were confirmed as described.13 SS and SSEts1+/− rats were maintained on 0.3% NaCl diet and then switched to a 4% NaCl diet for one week. At baseline, systolic blood pressure (SBP) and diastolic blood pressure (DBP) did not differ between SS and SSEts1+/− rats. One week on the 4% NaCl diet increased both daytime and nighttime SBP and DBP in SS and SSEts1+/− rats to levels that did not differ between the groups (n=6/each, p-value >0.05) (Figure 1A). Heart rates did not differ between SS and SSEts1+/− rats on either diet (data not shown).
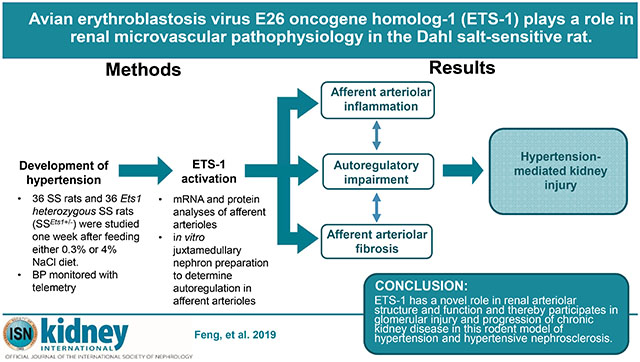
Figure 1. Renal dysfunction was present in the hypertensive SS rats, but was abrogated in the SSEts1+/− rats despite similar increases in telemetry blood pressures.
A, SSEts1+/− and the control SS rats were maintained on a 0.3% NaCI diet and then fed a 4% NaCI diet for one week. BP was monitored by radio-telemetry. At baseline, mean systolic blood pressure (SBP) and diastolic blood pressure (DBP) did not differ between SS (Solid line) and SSEts1+/− (Dashed line) rats. One week of high salt intake significantly increased SBP and DBP in both SS and SSEts1+/− rats (*p-value < 0.05, n=6); the increase in SBP and DBP did not differ (p-value >0.05, HT SS vs. HT SSEts1+/−, n=6) between SSEts1+/− and SS rats. B, With the development of hypertension, mean serum creatinine concentrations of the HT SS and HT SSEts1+/− groups were higher than both Pre-HT groups; however, mean serum creatinine levels were less in the HT SSEts1+/− group, compared with HT SS (#p-value <0.05, n=8). C, After one week of high salt intake, HT SS rats also showed a significant increase in the urinary excretion of albumin as compared with the other three groups of rats (#p-value < 0.05, n=8).
Assessment of serum creatinine and albuminuria revealed that renal dysfunction was present in the HT SS rats, but was abrogated in the HT SSEts1+/− rats despite similar increases in blood pressures. While mean serum creatinine concentration and mean albumin excretion rate did not differ between SS and SSEts1+/− rats maintained on the 0.3% NaCl diet, after one week of high salt intake, mean serum creatinine concentration and mean urinary albumin/creatinine ratio increased in HT SS rats, and was higher than observed in HT SSEts1+/− rats (p-value <0.05, n=8) (Figures 1B and 1C).
ETS-1 was activated in kidney microvasculature of HT SS, but not HT SSEts1+/− rats.
Total RNA was extracted from dissected kidney microvessels and relative expression of ETS-1 was measured using Sybr Green RTPCR method with primers that recognized Ets1 mRNA in SS rats and mRNA of the mutant gene present in the SSEts1+/− rats. The development of hypertension did not alter Ets1 mRNA expression between HT SS and HT SSEts1+/− (1.10 ± 0.11 vs 0.88 ± 0.13; p value > 0.05). Immunofluorescence staining, however, demonstrated an increase in phospho-ETS-1(T38) in nuclei specifically of smooth muscle in kidney microvessels of HT SS rats, compared with nuclear phospho-ETS-1(T38) levels in microvessels of HT SSEts1+/− rats. Threonine phosphorylation of this site has been shown increased activity of ETS-1.14, 16, 17 Basal levels of phospho-ETS-1(T38)-positive cells were low in Pre-HT SS, Pre-HT SSEts1+/−, and in HT SSEts1+/− rats (Figures 2A and 2B).
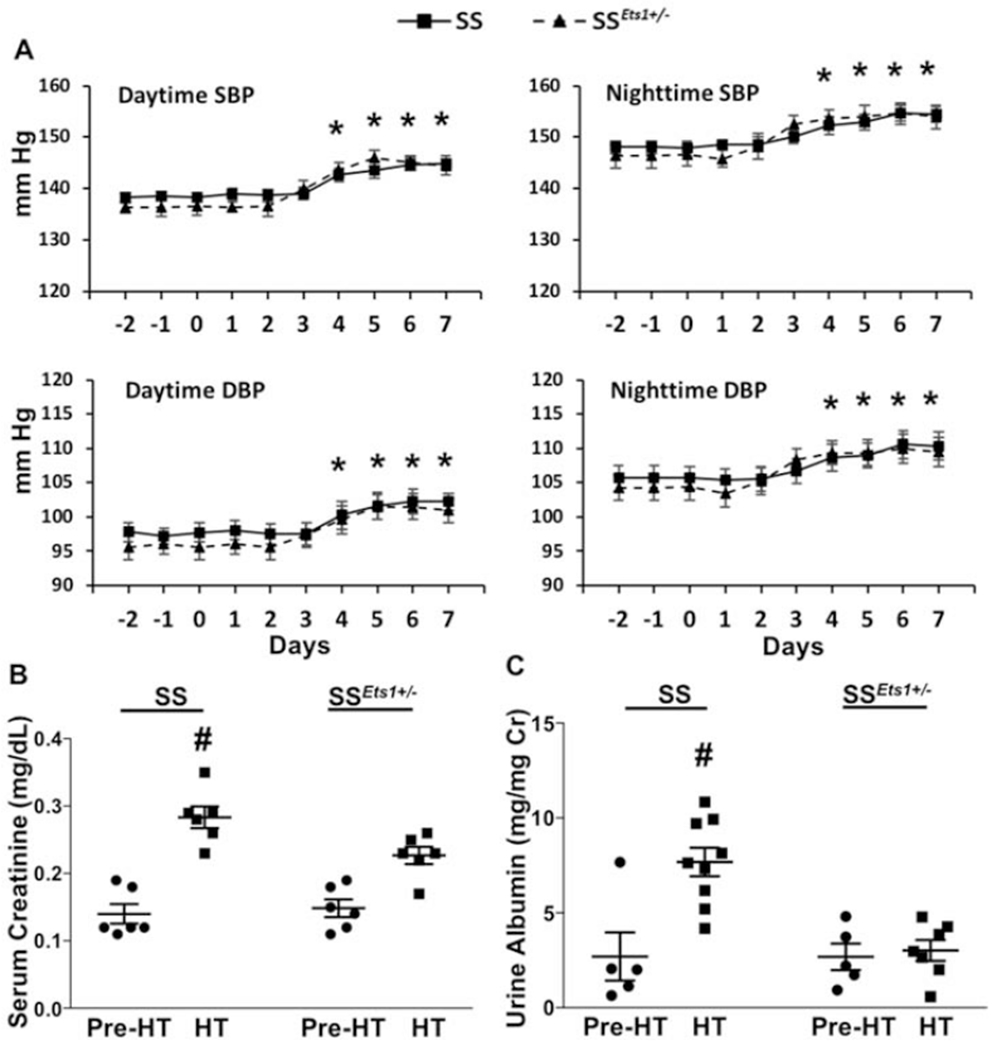
Figure 2. ETS-1 activity increased in kidney microvasculature of HT SS rats, compared with HT SSEts1+/− rats.
A, Immunofluorescence microscopy using alpha-smooth muscle actin (SMA) antibody (Green), which outlined the afferent arteriole, and phospho-ETS-1(T38) antibody (Red), which detected activated ETS-1, was performed in the four groups of rats (Pre-HT SS, HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/−). Basal levels of phospho-ETS-1(T38)-positive cells were low in both Pre-HT SS and SSEts1+/− rats (Upper panel, left). An increase in phospho-ETS-1(T38) staining in the nuclei of smooth muscle cells of the afferent arteriole and microvessels in the kidney of the HT SS rats was observed in both transverse (upper panel, middle) and longitudinal (upper panel, right) views of the afferent arteriole, while the observed numbers of phospho-ETS-1(T38)-positive cells were decreased in the HT SSEts1+/− group. B, Quantification based on determining the ratio of phospho-ETS-1(T38)-positive and -negative cells, which were detected by Alexa Flour 594 staining, in the afferent arteriole showed a dramatic increase in ETS-1 activation in the HT SS group. *p-value < 0.05 versus HT SSEts1+/−, Pre-HT SS and Pre-HT SSEts1+/− groups; n=6 animals in each group. G, glomerulus.
Expression of CCL2 in kidney microvasculature increased in HT SS, but not SSEts1+/−, rats and was accompanied by peri-microvasculature infiltration of macrophages.
We have previously confirmed chemokine CCL2 is transcriptionally activated by ETS-1.13 Here we measured relative mRNA expression of CCL2 in kidney microvessels and quantified macrophage infiltration, identified by ED1-positive staining, surrounding the kidney microvessels in the four groups of rats. Basal mRNA level of kidney microvascular CCL2 was lower in the Pre-HT SSEts1+/− rats compared to that in the Pre-HT SS rats (p-value <0.05, n=6), and increased dramatically in microvessels of HT SS rats (p-value <0.05, n=6). Kidney microvascular CCL2 mRNA levels did not increase in HT SSEts1+/− rats (Figure 3). This increase was accompanied by increased infiltration of ED1-positive macrophages around the kidney microvessels of HT SS rats, compared with in HT SSEts1+/− rats (p-value < 0.05, n=6).
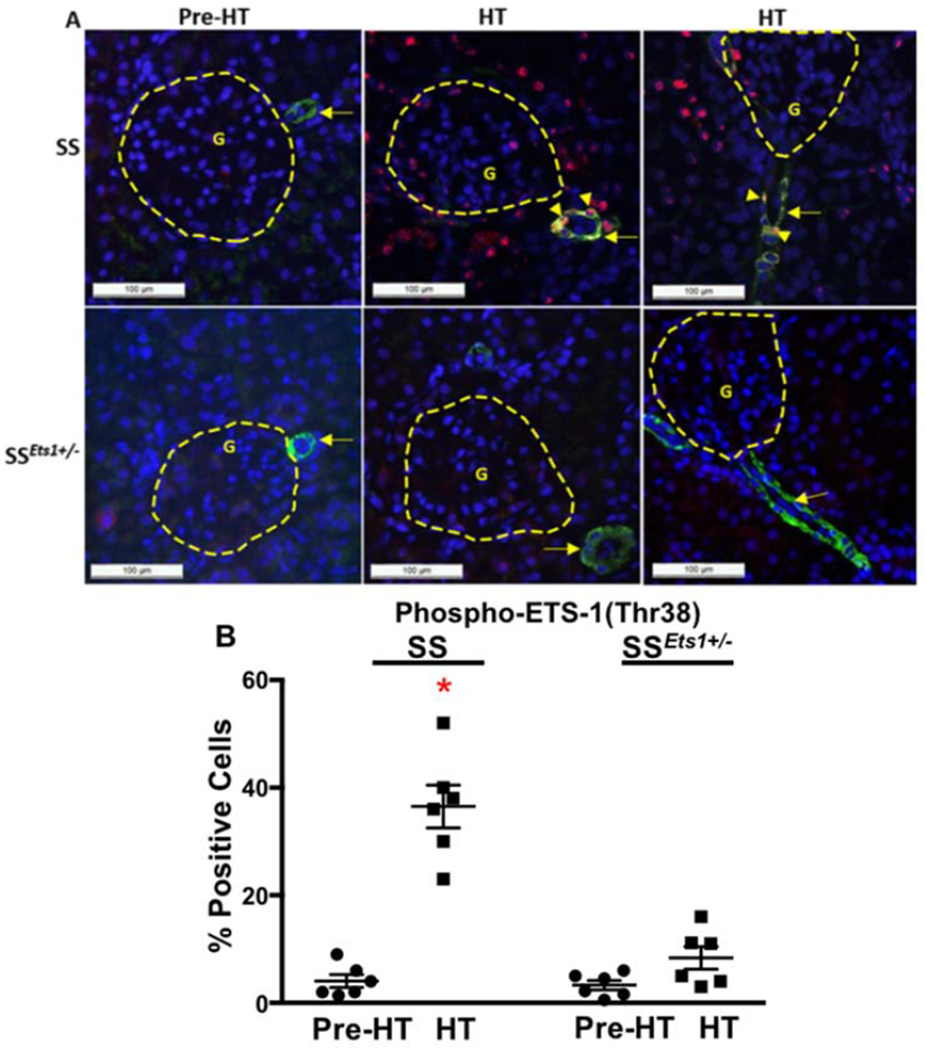
Figure 3. Expression of CCL2 in kidney microvasculature increased in HT SS rats and was accompanied by peri-microvascular accumulation of macrophages.
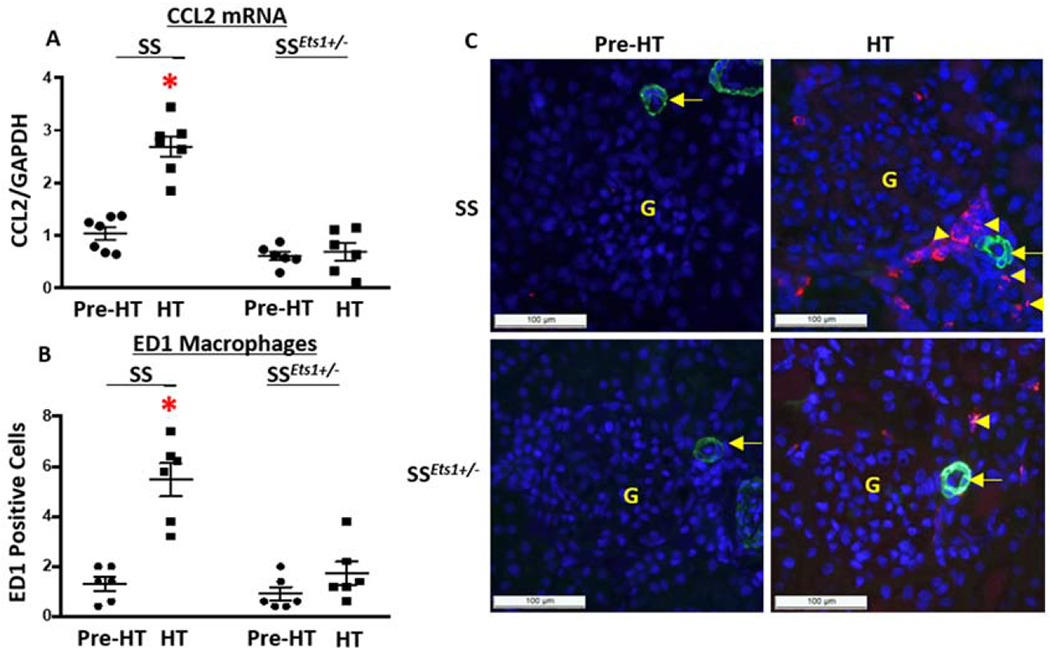
A, Basal relative level of mRNA of CCL2 in dissected kidney microvessels of Pre-HT SS rats was slightly increased when compared with relative mRNA levels from microvessels of Pre-HT SSEts1+/− rats. Kidney microvascular CCL2 further increased in HT SS and was significantly higher than that in the HT SSEts1+/− rats. *p-value < 0.05 versus Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− groups; n=6 animals in each group B and C, Immunofluorescence microscopy using alpha-smooth muscle actin (SMA) antibody (Green) to outline the afferent arteriole and ED1 antibody (Red) to stain macrophages showed low basal levels of ED1-positive macrophages (Red) in both Pre-HT SS and SSEts1+/− rats, but increased around the kidney microvasculature in HT SS rats, compared with the HT SSEts1+/− rats. *p-value < 0.05 versus Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− groups; n=6 animals in each group. G, glomerulus.
Fibronectin increased in kidney microvasculature of HT SS rats, but was abrogated in SSEts1+/− rats.
We and others previously observed significant remodeling of kidney microvessels in Pre-HT and HT SS rats, compared with their Sprague-Dawley counterpart.6, 18–20 Here we quantified relative mRNA expression of the fibrotic molecule, fibronectin, and co-localized protein expression with immunofluorescence microscopy. Mean basal level of fibronectin mRNA was less in Pre-HT SSEts1+/− rats compared to that observed in Pre-HT SS rats (Figure 4A). Fibronectin mRNA increased within one week in the HT SS rats and was significantly higher than that in the HT SSEts1+/− rats. Immunofluorescence studies showed fibronectin deposited around the kidney microvessels and in the interstitium (Figure 4B).
Figure 4. Fibronectin increased in kidney microvasculature of hypertensive SS, but not SSEts1+/−, rats.
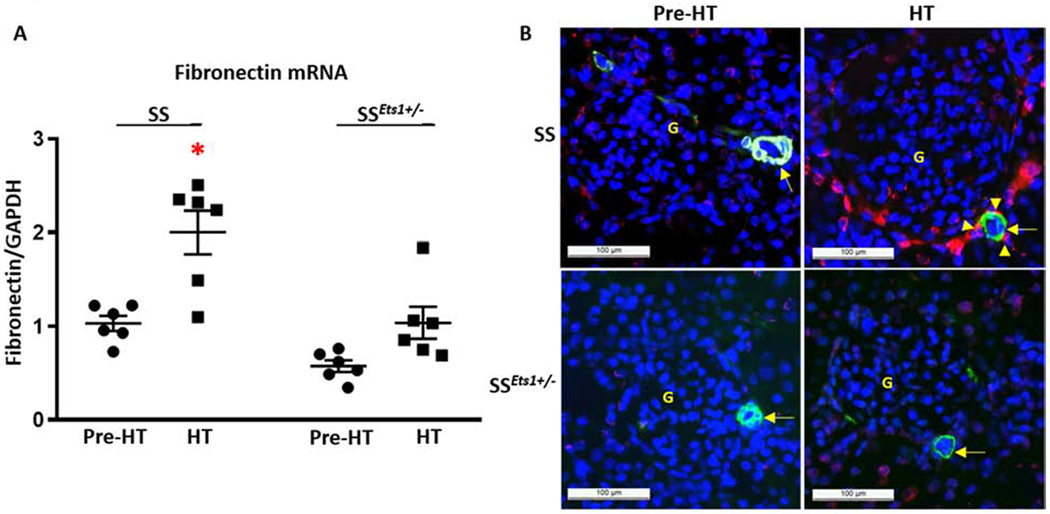
A, Basal relative mRNA level of fibronectin was higher in Pre-HT SS rats compared with Pre-HT SSEts1+/− rats. Fibronectin mRNA level further increased in the HT SS rats, and was significantly higher than that observed in the microvessels of HT SSEts1+/− rats (*p-value <0.05 vs Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− groups; n=6 animals in each group). B, Immunofluorescence microscopy using alpha-smooth muscle actin (SMA) antibody (Green) to outline the afferent arteriole and antibody to fibronectin (Red) showed deposition of fibronectin around the kidney microvessels of HT SS rats. G, glomerulus.
Pro-inflammatory and fibrotic molecules increased in kidney microvasculature of HT SS, but not SSEts1+/−, rats.
In addition to CCL2 (Figure 3) and fibronectin (Figure 4), we determined relative mRNA expression of several additional molecules known to be regulated by ETS-1.13,21–25 Relative microvascular mRNA expression of VCAM, ICAM and P-selectin increased 1.6-, 1.9- and 3.7-fold, respectively, in HT SS rats, but did not change in HT SSEts1+/− rats (Figure 5A). Similarly, relative microvascular mRNA expression of TGF-β, collagen I, and osteopontin increased 1.5-, 1.6- and 2.3-fold, respectively, in HT SS rats, but did not increase in HT SSEts1+/− rats (p-value <0.05 vs HT SSEts1+/−, N=6) (Figure 5B).
Figure 5. Mean steady-state relative mRNA levels of pro-inflammatory and fibrotic molecules were increased in kidney microvasculature of HT SS, but not SSEts1+/−, rats.
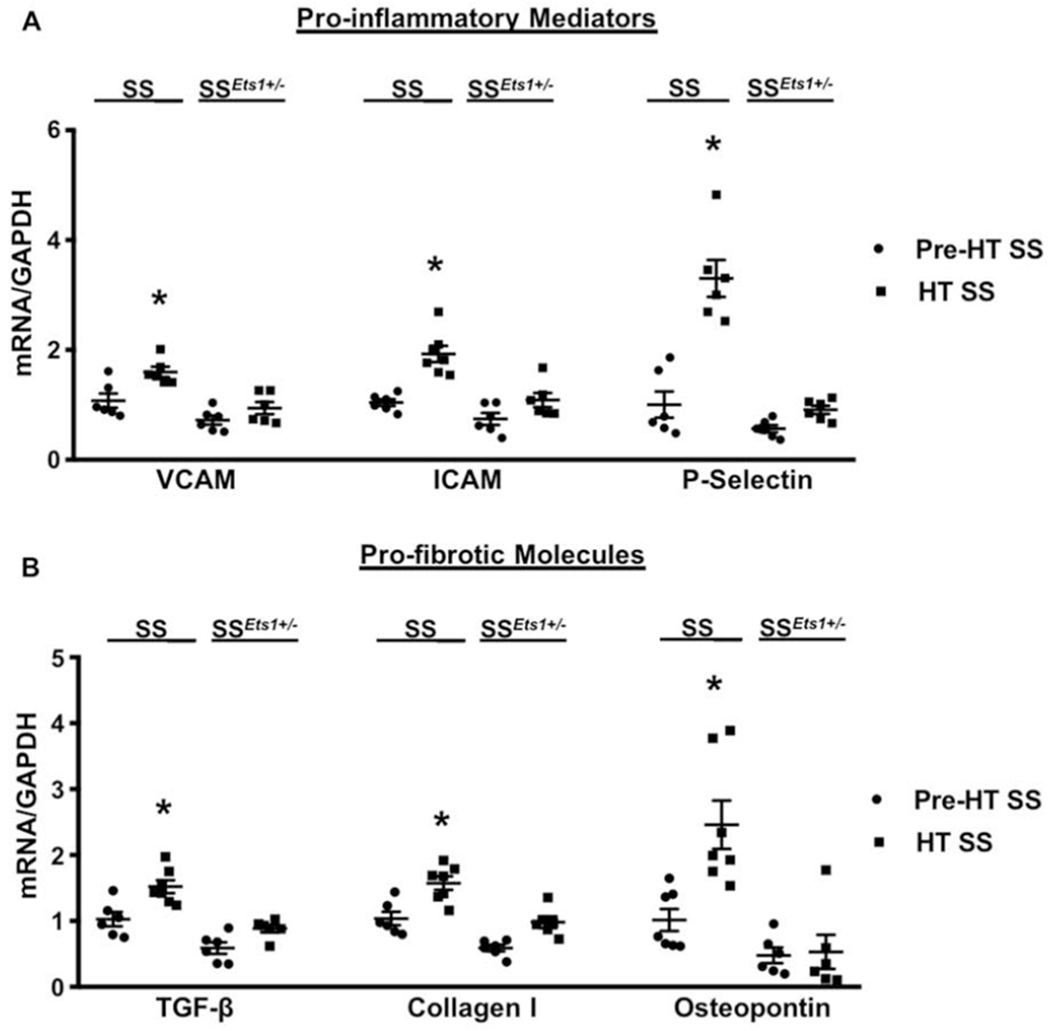
A, Kidney microvascular mRNA expression of inflammatory mediators, VCAM, ICAM and P-selectin increased 1.6-, 1.9- and 3.7-fold, respectively, in HT SS rats and were significantly higher than levels observed in the microvessels of Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− rats. *p-value < 0.05 versus Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− groups; n=6 animals in each group. B, Kidney microvascular mRNA expression of fibrotic molecules, TGF-β, collagen I and osteopontin increased 1.5-, 1.6- and 2.3-fold, respectively, in HT SS rats and were greater than corresponding levels in microvessels of Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− rats. *p-value < 0.05 versus Pre-HT SS, Pre-HT SSEts1+/−, and HT SSEts1+/− groups; n=6 animals in each group.
Haploinsufficiency of ETS-1 improved renal autoregulation in Dahl HT SS Rats.
Baseline diameters of afferent arterioles were similar across the four groups of rats (p-value >0.05) (Figure 6A). Afferent arterioles from Pre-HT SS and Pre-HT SSEts1+/− rats exhibited normal autoregulatory behavior. Decreasing renal perfusion pressure from 100 to 65 mm Hg increased afferent arteriolar diameter to 118 ± 2% and 119 ± 4% of baseline diameter (p-value <0.05, Figure 6B), respectively. Stepwise increases in perfusion pressure to 170 mm Hg decreased arteriolar diameter to 75.4 ± 3% and 72.9 ± 4% of baseline diameter (p-value <0.05), respectively. In contrast, pressure-mediated afferent arteriolar responses were markedly impaired in HT SS rats. Baseline diameter averaged 15.1 ± 1.2 μm and remained between 89 ± 4% and 107 ± 3% (p-value < 0.05 vs. Pre-HT SS) of baseline over the 65-170 mm Hg pressure range tested (Figure 6B). Importantly, renal autoregulation remained intact in HT SSEts1+/− rats. Baseline diameter averaged 15.6 ± 0.9 μm, and decreasing renal perfusion pressure from 100 to 65 mm Hg resulted in a diameter increase to 114 ± 2%, while increasing perfusion pressure to 170 mm Hg resulted in a pressure-dependent vasoconstriction to 73 ± 3% (p-value <0.05 vs HT SS).
Figure 6. Haploinsufficiency of ETS-1 improved renal microvasculature autoregulation in SS rats.
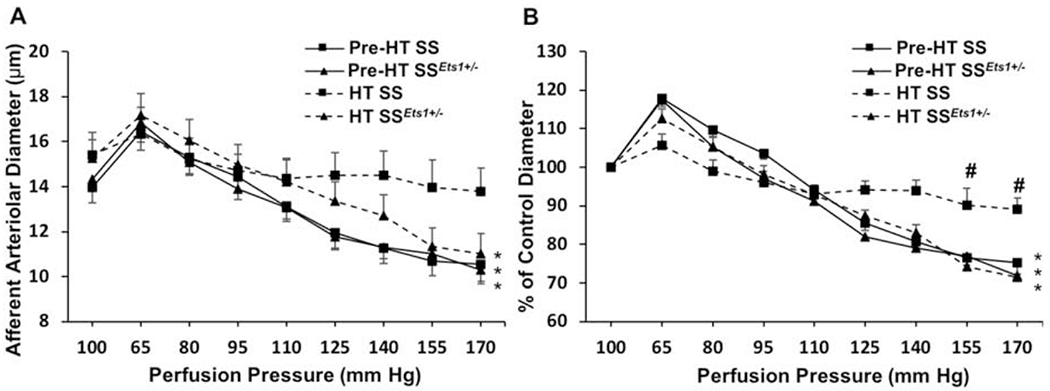
Afferent arteriolar autoregulation was assessed using the in vitro blood-perfused juxtamedullary nephron preparation. A: The afferent arteriolar response to alterations in renal perfusion pressure was measured in SS rats treated with a 0.3% salt diet (Pre-HT SS, squares with a solid line), SS rats that are heterozygous with one wild-type or reference allele of Ets1 (SSEts1+/− rats) fed with a 0.3% salt diet (Pre-HT SSEts1+/−, triangles with solid line), SS rats treated with 4% high salt diet (HT SS, squares with a dashed line), or SSEts1+/− rats fed with a 4% salt diet (HT SSEts1+/−, triangles with a dashed line) over 7 days. B: Data were normalized as percent of the control diameter at 100 mm Hg. Values were expressed as mean ± SEM. *p-value < 0.05 versus control diameter in the same group; #p-value < 0.05 versus Pre-HT SS kidneys at the same perfusion pressure; n=6.
Discussion
ETS-1 is the prototypical member of the ETS family of transcription factors that regulate a diverse collection of genes involved in cell growth and differentiation, inflammation and fibrosis.16, 17, 26 Although originally thought to be involved solely in the development and function of lymphoid-derived cells,27, 28 excellent work by Zhan, et al.,22 demonstrated that expression of ETS-1 in vascular smooth muscle and endothelial cells was critically important in angiotensin II-mediated thickening of the arterial wall and perivascular fibrosis. The present study adds to understanding the function of ETS-1 in the vascular biology of the kidney microvasculature. New findings of the study demonstrated a role for ETS-1 in an arteriolar pathological process that was accelerated by the development of hypertension in the Dahl SS rat. The pathological changes coincided with the activation of ETS-1, which promoted inflammatory and fibrotic events in preglomerular microvessels. Prior sophisticated studies demonstrated a direct role specifically for increased renal perfusion pressure as the proximate cause of kidney infiltration of immune cells that included macrophages.29, 30 By focusing on kidney microvasculature, our studies not only fit well with these reports but also provide new results. Importantly, temporally associated with the early arteriolar inflammatory and fibrotic changes was development of afferent arteriolar autoregulatory impairment and glomerular injury with increased serum creatinine concentration and albuminuria. The effects were abrogated in SSEts1+/− rats, which demonstrated haploinsufficiency of Ets1.13 Certainly, the renoprotection afforded by reduction of activity of ETS-1 in the microvasculature does not necessarily mean that an abnormally increased ETS-1 activity is responsible for the phenotype. Regardless, the studies uncovered a novel role of ETS-1 in renal arteriolar structure and function. Along with the demonstration that Ets1 is located within a QTL that associated with albuminuria and kidney damage,12, 13 the combined findings support the hypothesis that ETS-1 participates in the microvascular autoregulatory impairment and associated glomerular injury and progression of chronic kidney disease in this rodent model of hypertension and hypertensive nephrosclerosis.
Prior studies have shown that renal microvascular remodeling developed slowly in the prehypertensive phase in SS rats but was greatly accelerated with the development of hypertension.6, 18, 19 Morphometric analysis demonstrated an increase in wall thickness of the interlobular arteries and arterioles with the development of hypertension. These changes in wall thickness appeared to be related to increased numbers of cells and excess matrix deposition.6 The present study found that vascular pathology developed rapidly when SS rats became hypertensive, with the renal microvasculature of SS rats showing increased expression of inflammatory markers that included CCL2, VCAM-1, ICAM-1, and P-Selectin (Figures 3 and 5). The experiments also demonstrated increased expression of fibrosis markers that included fibronectin, TGF-β, collagen I, and osteopontin in renal microvasculature (Figures 4 and 5). Consistent with prior studies showing a role for Ets1 in vascular pathobiology,22 SSEts1+/− rats did not show significant changes in the renal microvasculature despite increases in blood pressure that did not differ from SS rats (Figure 1). These data were also consistent with prior studies that concluded that the hypertensive nephropathy of SS rats was a disorder of the vascular remodeling process involving vascular smooth muscle.20 This prior study showed enhanced signaling through the epidermal growth factor receptor in vascular smooth muscle cells of SS rats,31 although other signaling molecules, such as TGF-β, have been suggested to play an important function in the development of hypertensive nephropathy of SS rats.32, 33
The kidney microvasculature of HT SS rats showed increased activity of ETS-1, as demonstrated by an increase in threonine phosphorylation of the N-terminus of the molecule by the Ras-Extracellular Signal-Regulated Kinase (ERK) pathway.14, 16, 17 Therefore, a growth factor-mediated or stress-activated signaling pathway involving ERK regulated the function of ETS-1 by post-translational modification. The present study demonstrated that with development of hypertension the afferent arterioles of SS rats showed an increase in the numbers of cells that expressed both phospho-ETS-1(T38) and alpha-smooth muscle actin (Figure 2), indicating activation of ETS-1 in vascular smooth muscle cells.
Renovascular autoregulatory responses to changes in blood pressure permit a consistent renal blood flow and glomerular filtration rate.34, 35 Roman’s laboratory demonstrated that hypertensive Dahl S rats had an impaired ability to autoregulate renal blood flow in the normal range of renal perfusion pressure.36 Besides a myogenic response, tubuloglomerular feedback (TGF) has been shown to contribute to renal autoregulation,37, 38 but prior studies demonstrated an intact TGF response in SS rats.39 Takenaka, et al.,40 used the isolated perfused hydronephrotic preparation to show that Dahl salt-resistant and prehypertensive SS rats had comparable myogenic responses in kidney microvessels. In contrast, after three weeks on a high-salt diet, when the systemic blood pressures were very high (SBP 202 ± 4 mmHg), the myogenic response of kidney microvessels was markedly impaired in SS rats.40 In the present study, SS rats developed a rapid and reproducible hypertension-induced defect in microvascular autoregulatory responses, with the afferent arterioles failing to exhibit pressure-induced vasoconstriction (Figure 6). Autoregulatory impairment of the afferent arteriole therefore accompanied the changes in renal microvascular pathology. This finding is relevant, since this renoprotective process has been suggested to prevent the transmission of increased arterial pressure into the glomerular capillaries and when impaired, permits glomerular hypertension and subsequent glomerular injury.41–45 By demonstrating a reduction in renal function with an increase in albuminuria (Figure 1) in SS rats but not SSEts1+/− rats, which had preserved afferent arteriole autoregulatory responses (Figure 6), our study provided direct corroboration of impaired afferent arteriolar autoregulation and kidney injury, which had been originally derived from other models of kidney disease.41–43
In summary, the current studies uncovered a novel function of ETS-1 in renal microvascular pathology and autoregulation and the development of hypertension-associated kidney damage in the SS rat. Because the arteriolar pathology and autoregulatory impairment developed in the same time frame, an unanswered question is whether the hypertension-associated arteriolar pathology produced the autoregulatory impairment, or vice versa. Nevertheless, by providing a focus on the molecular pathogenesis of the early renal microvascular disease and impaired autoregulatory response to hypertension, the findings have potentially brought us closer to understanding the molecular basis of hypertensive nephropathy. The observations may provide new opportunities for exploration of early diagnosis of impaired renal autoregulation, and for development of novel therapeutic strategies that protect against this common mediator of CKD and ESKD.
Methods
Animals
This study was performed in accordance with the recommendations in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at the University of Alabama at Birmingham approved the project. Studies were conducted using Dahl salt-sensitive rats (SS/JrHsd/Mcw, abbreviated SS),9, 46–48 and Dahl SS/JrHsd/Mcw rats that have a deletion mutation in one Ets1 (SSEts1+/− rats, previously also abbreviated ES13). The original SS and SSEts1+/− strains of rats were generous gifts from Dr. Aron Geurts, Medical College of Wisconsin. SSEts1+/− rats were genetically identical to the littermate SS rats, except for a heterozygotic gene mutation of Ets1 introduced using genome engineering with Zinc-Finger Nucleases (ZFN),11 which targeted exon 3 of rat Ets1 gene and resulted in an 8 base-pair (ATTGGGTG) deletion mutation.13 Litters never contained homozygous SSEts1−/− rats, likely indicating embryonic lethality. Littermate SS rats served as the controls in these experiments. Rat breeders and weanlings were fed purified AIN-76A rodent diet (Dyets Inc., Bethlehem, PA) containing 0.3% NaCl and tap water ad libitum until study.
Genotyping and Confirmation of Ets-1 mutation
For genotyping, Taqman probes that recognized the 8-bp deletion sequence (mutant probe) or wild-type sequence (wild-type probe) were designed. The wild-type Taqman probe was 5’-CATCACCCAATCCC-3’ and the Taqman probe to detect the mutation in Ets-1 was 5’- CAGCCCACATCCCG-3’. The genotypes were determined as wild type (homozygous SS), heterozygous (SSEts1+/−) or homozygous for the mutation, depending upon whether the samples were positive with only wild-type probes, positive with both wild-type and mutant probes, or positive with only mutant probes, respectively, as previously described.13 DNA samples of rats in the study were amplified with real time PCR and used to determine genotype (Transnetyx, Cordova, TN). The forward primer sequence was 5’-GCAGTGGACAGAAACCCATGT-3’ and the reverse sequence was 5’-TGCTGCTCCATTCATACAGAACTTC-3’.
Experiments were performed using 9- to 12-week old, age-matched male SSEts1+/− rats and littermates (SS). Because a subtle variation in blood pressure might impact outcomes in our study and because of the need to monitor blood pressure in awake, unrestrained animals, continuous monitoring of blood pressures was performed using radio-telemetry.49, 50 Rats were anesthetized using 2% isoflurane and an implantable radio-telemetry transmitter (HD-S10; DSI, St. Paul, MN) was inserted into the left femoral artery to continuously record awake, unrestrained BP.51 Rats were allowed 7 days to recover and were then divided into 4 groups (N=6-8 rats per group) and fed either a control diet containing 0.3% NaCl (AIN-76A Rodent Diet, Dyets, Inc.) (groups termed Pre-HT SS and Pre-HT SSEts1+/−) or a 4% NaCl diet for one week (AIN-76A Rodent Diet, Dyets, Inc.) to induce hypertension (groups termed HT SS and HT SSEts1+/−). BP was continuously monitored in awake, unrestrained rats throughout the 7-day study.
On day 6 of high-salt feeding, some rats of the four experimental groups were placed in metabolic cages for 24 hours to collect urine to determine concentrations of albumin and creatinine. Urinary albumin concentrations were determined by ELISA (Cat. E110-125, Bethyl, Montgomery, TX) and normalized against urinary creatinine concentration (Cat. DICT-500, BioAssay System, Hayward, CA). Rats were anesthetized and blood was collected for determination of serum creatinine using liquid chromatography tandem mass spectrometry (Waters 2795 LC-MS/MS; Conquer Scientific, San Diego, CA)52 and the kidneys were harvested for physiology, histology and molecular analyses.
The in vitro blood-perfused juxtamedullary nephron (JMN) preparation
Autoregulatory behavior of afferent arterioles was assessed in the four experimental groups (Pre-HT SS, Pre-HT SSEts1+/−, HT SS and HT SSEts1+/−, N=12 rats per group), using the in vitro blood-perfused JMN technique as described previously.53, 54 Briefly, two identical rats were anesthetized with pentobarbital sodium intraperitoneally (50 mg/kg body weight) for each JMN experiment. The right kidney was cannulated and continuously perfused with Tyrode’s buffer (Sigma-Aldrich, St. Louis, MO) containing 5.2% bovine serum albumin (BSA, Calbiochem, Billerica, MA). Perfusate blood was collected via a carotid artery cannula from the kidney donor and an identical blood donor and centrifuged to obtain the plasma and erythrocytes for kidney perfusion (hematocrit of ~33%). The inner cortical surface of the right kidney was exposed and the ends of the intrarenal arteries and arterial branches were tied with 100 nylon suture to restore renal perfusion pressure. After completion of the dissection, the kidney was switched to the reconstituted blood and the image of the kidney was displayed on a video monitor via a high-resolution NC-70 Newvicon video camera (DAGE-MTI) and recorded on DVD for later analysis. The inner arteriole diameter was measured every 12 seconds at a single site using a calibrated image-shearing monitor (Model 908, Vista Electronics, Valencia, CA) and was calculated from the average of all diameter measurements collected during the final 2 min of each treatment period.
Kidney microvessel isolation for mRNA expression analysis
Briefly, rats were anesthetized with sodium pentobarbital for retrograde perfusion through the abdominal aorta. The kidneys were perfused with 5.2% BSA and removed in ice-cold physiological salt solution. Medulla and intrarenal and arcuate arteries were removed and cortical tissue was gently pressed through a nylon membrane sieve (100-μm pore size; BioDesign, Inc., Carmel, NY). The kidney tissue was transferred into a RNAlater™ stabilization solution (Invitrogen, Thermo Fisher Scientific, Waltham). Segments of interlobular arteries with attached afferent arterioles were identified and collected by microdissection under a stereoscope for mRNA extraction.
Real-Time, quantitative RT-PCR analysis of inflammatory and fibrotic gene expression in kidney microvessels
Total RNA was extracted from isolated kidney microvessels with TRIzol (Invitrogen) and treated with DNAase I to remove genomic DNA and then purified with use of an RNA purification kit (Invitrogen). The protein- and DNA-free RNA was reverse transcribed to cDNA with use of the Superscript IV (Invitrogen). cDNA was amplified by PCR in the Roche LightCycler480 (Roche) for 40 cycles using SYBR GREEN method (Applied Biosystems) and specific primers (Table 1), relative RNA levels were calculated with the PCR threshold cycles software and a standard equation (Applied Biosystems). mRNA expression was normalized against GAPDH for each sample, then standardized to the group of Pre-HT SS as 1.
Table 1.
Primers for Real-time PCR analysis.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Ets1* | AGGCATTGTGGGTAATAACA | AAGTCCACTGTCGCTGTCTC |
| Ccl2 | CAGGTGTCCCAAAGAAG | CAAAGGTGCTGAAGTCC |
| Fibronectin | GCCTTCAACTTCTCCTGTGA | GTTGCAAACCTTCAATGGTC |
| VCAM | GGGGATTCCGTTGTTCT | CAGGGCTCAGCGTCAGT |
| ICAM | CAAACGGGAGATGAATGG | TGGCGGTAATAGGTGTAAAT |
| P-selectin | AATGAAATCGCTCACCTC | TTATTGGGCTCGTTGTCT |
| Tgfb1 | CTACTGCTTCAGCTCCACAGAGA | ACCTTGGGCTTGCGACC |
| Collagen I | CCAGCCGCAAAGAGTCTACA | AGGCCATTGTGTATGCAGCT |
| Osteopontin | AGGAGTTTCCCTGTTTCTG | GTCTTCCCGTTGCTGTC |
| GAPDH | ATTCTTCCACCTTTGATGC | TGGTCCAGGGTTTCTTACT |
Of note, the primers used to amplify Ets1 mRNA also amplify the mRNA of the mutant gene.
Immunofluorescence staining
Five-micron-thick kidney sections were prepared from paraffin-embedded tissues. After deparaffinization and antigen retrieval, the sections were rinsed in phosphate-buffered saline. The sections were then incubated with rabbit or mouse primary antibodies to phosphorylated ETS-1 (44-1104G, Invitrogen), monocyte/macrophage marker CD68 (ED1, Bio-Rad) or fibronectin (ab2413, Abcam) at 4°C overnight. Another primary antibody to smooth muscle actin (Mouse, 1A4, Invitrogen or Rabbit, ab5694, Abcam) was also used to outline the vessel in the kidney sections. The sections were washed and incubated with the respective secondary antibodies conjugated with either Alexa Fluor 488 (green, Invitrogen) or Alexa Fluor 594 (red, Invitrogen). Counterstaining of the nucleus was achieved by mounting sections with hardset mounting media containing DAPI (blue, Vector Laboratories). Negative controls by omission of primary antibody were included in each experiment. Images were acquired using a Leica DM6000 epifluorescence microscope (Leica Microsystems, Bannockburn, IL) with a Hamamatsu ORCA ER cooled CCD camera and Simple PCI software (Compix, Inc, Cranberry Township, PA).
Statistical Analyses
Data are expressed as the mean ± SEM. Blood pressure data were analyzed using a two-way ANOVA for repeated measures. All other data were analyzed by two-way ANOVA. When the overall F test result of ANOVA was significant, a Holm-Sidak post-hoc test was also performed. A p-value <0.05 assigned statistical significance.
Translational Statement.
Hallmarks of hypertensive nephropathy include microvascular pathology and glomerulosclerosis. This study demonstrated a function for ETS-1 in the early microvascular inflammatory and fibrotic pathological changes and autoregulatory impairment in hypertensive Dahl salt-sensitive rats. The observations may provide new opportunities for exploration of early diagnosis of impaired renal autoregulation in hypertension, and for development of novel therapeutic strategies that protect against this common mediator of CKD and ESKD.
Acknowledgments
We thank Dr. Aron Geurts of Medical College of Wisconsin for the SSEts1+/− rat. We thank UAB-UCSD O’Brien Center for creatinine measurements and the Heflin Center for Genomic Science at UAB for the use of Roche LightCycler480 384-well Real Time PCR.
This work was supported in part by an American Heart Association Scientist Development Grant (15SDG25760063), a Department of Veterans Affairs Program Project Award (1 1IP1 BX001595), a Merit Award (1 I01 CX001326) from the United States (U.S.) Department of Veterans Affairs Clinical Sciences R&D (CSRD) Service, a National Institutes of Health George M. O’Brien Kidney and Urological Research Centers Program (P30 DK079337), NIH R56 HL128285 (to D.X.), NIH R01 DK106500 (to Z.G.), NIH R01 DK044628 (to E.W.I.), a UAB Anderson Innovation Award, and a UAB School of Medicine AMC21 Multi-PI Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest/Disclosure Statement
None
References:
- 1.National Center for Health Statistics. Health US. 2011: Table 51. End-stage renal disease patients, by selected characteristics: United States, selected years 1980–2010. Centers for Disease Control and Prevention. [Google Scholar]
- 2.U.S. Renal Data System. Researcher’s Guide to the USRDS Database. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease, Bethesda, MD, 2017. [Google Scholar]
- 3.Castleman B and Smithwick RH. The relation of vascular disease to the hypertensive state; based on a study of renal biopsy from one hundred hypertensive patients. JAMA. 1943;121:1256–1261. [Google Scholar]
- 4.Castleman B and Smithwick RH. The relation of vascular disease to the hypertensive state; the adequacy of the renal biopsy as determined from a study of 500 patients. N Engl J Med. 1948;239:729–32. [DOI] [PubMed] [Google Scholar]
- 5.Sommers SC, Relman AS and Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol. 1958;34:685–715. [PMC free article] [PubMed] [Google Scholar]
- 6.Chen PY, St John PL, Kirk KA, et al. Hypertensive nephrosclerosis in the Dahl/Rapp rat. Initial sites of injury and effect of dietary L-arginine supplementation. Lab Invest. 1993;68:174–84. [PubMed] [Google Scholar]
- 7.Chen PY and Sanders PW. L-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J Clin Invest. 1991;88:1559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahl LK, Heine M and Tassinari L. Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature. 1962;194:480–2. [DOI] [PubMed] [Google Scholar]
- 9.Zicha J, Dobesova Z, Vokurkova M, et al. Age-dependent salt hypertension in Dahl rats: fifty years of research. Physiol Res. 2012;61 Suppl 1:S35–87. [DOI] [PubMed] [Google Scholar]
- 10.Garrett MR, Dene H and Rapp JP. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J Am Soc Nephrol. 2003;14:1175–87. [DOI] [PubMed] [Google Scholar]
- 11.Geurts AM, Cost GJ, Freyvert Y, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrett MR, Joe B and Yerga-Woolwine S. Genetic linkage of urinary albumin excretion in Dahl salt-sensitive rats: influence of dietary salt and confirmation using congenic strains. Physiol Genomics. 2006;25:39–49. [DOI] [PubMed] [Google Scholar]
- 13.Feng W, Chen B, Xing D, et al. Haploinsufficiency of the Transcription Factor Ets-1 Is Renoprotective in Dahl Salt-Sensitive Rats. J Am Soc Nephrol. 2017;28:3239–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupsky CM, Gentile LN, Donaldson LW, et al. Structure of the Ets-1 pointed domain and mitogen-activated protein kinase phosphorylation site. Proc Natl Acad Sci U S A. 1998;95:12129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng W, Chumley P, Prieto MC, et al. Transcription factor avian erythroblastosis virus E26 oncogen homolog-1 is a novel mediator of renal injury in salt-sensitive hypertension. Hypertension. 2015;65:813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang BS, Hauser CA, Henkel G, et al. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol. 1996;16:538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wasylyk B, Hagman J and Gutierrez-Hartmann A. Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem Sci. 1998;23:213–6. [DOI] [PubMed] [Google Scholar]
- 18.Khan NJ, Hampton JA, Lacher DA, et al. Morphometric evaluation of the renal arterial system of Dahl salt-sensitive and salt-resistant rats on a high salt diet. I. Interlobar and arcuate arteries. Lab Invest. 1987;57:714–23. [PubMed] [Google Scholar]
- 19.Hampton JA, Bernardo DA, Khan NA, et al. Morphometric evaluation of the renal arterial system of Dahl salt-sensitive and salt-resistant rats on a high salt diet. II. Interlobular arteries and intralobular arterioles. Lab Invest. 1989;60:839–46. [PubMed] [Google Scholar]
- 20.Wang PX and Sanders PW. Mechanism of hypertensive nephropathy in the Dahl/Rapp rat: a primary disorder of vascular smooth muscle. Am J Physiol Renal Physiol. 2005;288:F236–42. [DOI] [PubMed] [Google Scholar]
- 21.Feng W, Xing D, Hua P, et al. The transcription factor ETS-1 mediates proinflammatory responses and neointima formation in carotid artery endoluminal vascular injury. Hypertension. 2010;55:1381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan Y, Brown C, Maynard E, et al. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest. 2005;115:2508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abe K, Nakashima H, Ishida M, et al. Angiotensin II-induced osteopontin expression in vascular smooth muscle cells involves Gq/11, Ras, ERK, Src and Ets-1. Hypertens Res. 2008;31:987–98. [DOI] [PubMed] [Google Scholar]
- 24.Yockell-Lelievre J, Spriet C, Cantin P, et al. Functional cooperation between Stat-1 and ets-1 to optimize icam-1 gene transcription. Biochem Cell Biol. 2009;87:905–18. [DOI] [PubMed] [Google Scholar]
- 25.Hahne JC, Okuducu AF, Fuchs T, et al. Identification of ETS-1 target genes in human fibroblasts. Int J Oncol. 2011;38:1645–52. [DOI] [PubMed] [Google Scholar]
- 26.Lelievre E, Lionneton F, Soncin F, et al. The Ets family contains transcriptional activators and repressors involved in angiogenesis. Int J Biochem Cell Biol. 2001;33:391–407. [DOI] [PubMed] [Google Scholar]
- 27.Kola I, Brookes S, Green AR, et al. The Ets1 transcription factor is widely expressed during murine embryo development and is associated with mesodermal cells involved in morphogenetic processes such as organ formation. Proc Natl Acad Sci U S A. 1993;90:7588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartel FO, Higuchi T and Spyropoulos DD. Mouse models in the study of the Ets family of transcription factors. Oncogene. 2000;19:6443–54. [DOI] [PubMed] [Google Scholar]
- 29.Mori T, Polichnowski A, Glocka P, et al. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol. 2008;19:1472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans LC, Petrova G, Kurth T, et al. Increased Perfusion Pressure Drives Renal T-Cell Infiltration in the Dahl Salt-Sensitive Rat. Hypertension. 2017;70:543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying WZ and Sanders PW. Enhanced expression of EGF receptor in a model of salt-sensitive hypertension. Am J Physiol Renal Physiol. 2005;289:F314–21. [DOI] [PubMed] [Google Scholar]
- 32.Dahly AJ, Hoagland KM, Flasch AK, et al. Antihypertensive effects of chronic anti-TGF-beta antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R757–67. [DOI] [PubMed] [Google Scholar]
- 33.Murphy SR, Dahly-Vernon AJ, Dunn KM, et al. Renoprotective effects of anti-TGF-beta antibody and antihypertensive therapies in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2012;303:R57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aukland K and Oien AH. Renal autoregulation: models combining tubuloglomerular feedback and myogenic response. Am J Physiol. 1987;252:F768–83. [DOI] [PubMed] [Google Scholar]
- 35.Burke M, Pabbidi MR, Farley J, et al. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol. 2014;12:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roman RJ. Abnormal renal hemodynamics and pressure-natriuresis relationship in Dahl salt-sensitive rats. Am J Physiol. 1986;251:F57–65. [DOI] [PubMed] [Google Scholar]
- 37.Feldberg R, Colding-Jorgensen M and Holstein-Rathlou NH. Analysis of interaction between TGF and the myogenic response in renal blood flow autoregulation. Am J Physiol. 1995;269:F581–93. [DOI] [PubMed] [Google Scholar]
- 38.Schnermann J and Briggs JP. Tubuloglomerular feedback: mechanistic insights from gene-manipulated mice. Kidney Int. 2008;74:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlsen FM, Leyssac PP and Holstein-Rathlou NH. Tubuloglomerular feedback in Dahl rats. Am J Physiol. 1998;274:R1561–9. [DOI] [PubMed] [Google Scholar]
- 40.Takenaka T, Forster H, De Micheli A, et al. Impaired myogenic responsiveness of renal microvessels in Dahl salt-sensitive rats. Circ Res. 1992;71:471–80. [DOI] [PubMed] [Google Scholar]
- 41.Bidani AK, Griffin KA, Williamson G, et al. Protective importance of the myogenic response in the renal circulation. Hypertension. 2009;54:393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loutzenhiser R, Griffin KA and Bidani AK. Systolic blood pressure as the trigger for the renal myogenic response: protective or autoregulatory? Curr Opin Nephrol Hypertens. 2006;15:41–9. [DOI] [PubMed] [Google Scholar]
- 43.Loutzenhiser R, Griffin K, Williamson G, et al. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loutzenhiser R, Bidani AK and Wang X. Systolic pressure and the myogenic response of the renal afferent arteriole. Acta Physiol Scand. 2004;181:407–13. [DOI] [PubMed] [Google Scholar]
- 45.Loutzenhiser R, Bidani A and Chilton L. Renal myogenic response: kinetic attributes and physiological role. Circ Res. 2002;90:1316–24. [DOI] [PubMed] [Google Scholar]
- 46.Elijovich F, Weinberger MH, Anderson CA, et al. Salt Sensitivity of Blood Pressure: A Scientific Statement From the American Heart Association. Hypertension. 2016;68:e7–e46. [DOI] [PubMed] [Google Scholar]
- 47.Cowley AW Jr., Stoll M, Greene AS, et al. Genetically defined risk of salt sensitivity in an intercross of Brown Norway and Dahl S rats. Physiol Genomics. 2000;2:107–15. [DOI] [PubMed] [Google Scholar]
- 48.Dahl M, Kasanen A and Peltonen T. The effect of Segontin, Persantin and nitroglycerin on the coronary artery. Experimental studies with cinecoronarography. Ann Med Exp Biol Fenn. 1962;40:423–6. [PubMed] [Google Scholar]
- 49.Drueke TB and Devuyst O. Blood pressure measurement in mice: tail-cuff or telemetry? Kidney Int. 2019;96:36. [DOI] [PubMed] [Google Scholar]
- 50.Luft FC. Men, mice, and blood pressure: telemetry? Kidney Int. 2019;96:31–33. [DOI] [PubMed] [Google Scholar]
- 51.Feng W, Ying WZ, Aaron KJ, et al. Transforming growth factor-beta mediates endothelial dysfunction in rats during high salt intake. Am J Physiol Renal Physiol. 2015;309:F1018–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ying WZ, Allen CE, Curtis LM, et al. Mechanism and prevention of acute kidney injury from cast nephropathy in a rodent model. J Clin Invest. 2012;122:1777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inscho EW, Carmines PK and Navar LG. Juxtamedullary afferent arteriolar responses to P1 and P2 purinergic stimulation. Hypertension. 1991;17:1033–7. [DOI] [PubMed] [Google Scholar]
- 54.Guan Z, Singletary ST, Cook AK, et al. Sphingosine-1-phosphate evokes unique segment-specific vasoconstriction of the renal microvasculature. J Am Soc Nephrol. 2014;25:1774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]