

Abstract
The neuropathologic basis of in vivo cortical atrophy in clinical dementia syndromes remains poorly understood. This includes primary progressive aphasia (PPA), a language‐based dementia syndrome characterized by asymmetric cortical atrophy. The neurofibrillary tangles (NFTs) and amyloid‐ß plaques (APs) of Alzheimer's disease (AD) can cause PPA, but a quantitative investigation of the relationships between NFTs, APs and in vivo cortical atrophy in PPA‐AD is lacking. The present study measured cortical atrophy from corresponding bilateral regions in five PPA‐AD participants with in vivo magnetic resonance imaging scans 7–30 months before death and acquired stereologic estimates of NFTs and dense‐core APs visualized with the Thioflavin‐S stain. Linear mixed models accounting for repeated measures and stratified by hemisphere and region (language vs. non‐language) were used to determine the relationships between cortical atrophy and AD neuropathology and their regional selectivity. Consistent with the aphasic profile of PPA, left language regions displayed more cortical atrophy (P = 0.01) and NFT densities (P = 0.02) compared to right language homologues. Left language regions also showed more cortical atrophy (P < 0.01) and NFT densities (P = 0.02) than left non‐language regions. A subset of data was analyzed to determine the predilection of AD neuropathology for neocortical regions compared to entorhinal cortex in the left hemisphere, which showed that the three most atrophied language regions had greater NFT (P = 0.04) and AP densities (P < 0.01) than the entorhinal cortex. These results provide quantitative evidence that NFT accumulation in PPA selectively targets the language network and may not follow the Braak staging of neurofibrillary degeneration characteristic of amnestic AD. Only NFT densities, not AP densities, were positively associated with cortical atrophy within left language regions (P < 0.01) and right language homologues (P < 0.01). Given previous findings from amnestic AD, the current study of PPA‐AD provides converging evidence that NFTs are the principal determinants of atrophy and clinical phenotypes associated with AD.
Keywords: Alzheimer's disease, amyloid‐ß plaques, cortical atrophy, neurofibrillary tangles, primary progressive aphasia
Introduction
Magnetic resonance imaging (MRI) is a reliable and quantitative method for detecting structural changes in the brain during life. The basis of these anatomical changes is not well understood at the neurobiological level, especially in neurodegenerative diseases that cause cortical atrophy and dementia. The neuropathologic contributors to MRI‐based cortical atrophy have been extensively examined in individuals clinically diagnosed during life with the amnestic dementia of the Alzheimer‐type (DAT) and pathologically diagnosed post‐mortem with Alzheimer's disease (DAT‐AD). Amyloid‐ß plaques (APs) and neurofibrillary tau tangles (NFTs) are the primary histopathologic hallmarks of AD neuropathology. While both appear to contribute to neurodegenerative processes in DAT‐AD, NFTs may be the stronger pathologic correlate of antemortem cortical atrophy 16. In addition to cortical atrophy, neocortical and hippocampal NFTs have also been linked to the severity and progression of cognitive decline in DAT‐AD 3, 4. Taken together, these findings strongly indicate that in comparison to APs, the location and extent of NFTs more consistently correspond to the anatomy of cortical atrophy and cognitive decline characteristic of DAT‐AD. However, the neuropathologic basis of cortical atrophy remains unclear outside of those with DAT‐AD. AD neuropathology is known to cause distinct types of non‐amnestic dementia 1, 21, 44, 53, 54, 66, 80 with each clinical syndrome displaying common and variant‐specific atrophy patterns 58. The purpose of the current study was to determine if NFTs could also be the primary driver of cortical atrophy in a non‐amnestic variant of AD known as primary progressive aphasia (PPA).
PPA with AD neuropathology (PPA‐AD) is the aphasic variant of AD, a syndrome characterized by a gradual dysfunction of language processing with initial preservation of other cognitive domains such as memory 48, 51. AD is the pathologic diagnosis in approximately 40% of individuals with PPA 42, 53, 54, 68. MRI has revealed focal and asymmetric cortical atrophy that is concentrated in language regions of the language dominant hemisphere in PPA 63, 64, 66. Similar to the underpinnings of memory impairment and medial temporal lobe atrophy typical of DAT‐AD, recent studies suggest that the language deficits and atrophy of language cortices characteristic of PPA‐AD are associated with NFT densities 21, 38. These early findings in PPA‐AD, in combination with studies of DAT‐AD, provide converging evidence that NFTs are possibly a common and major contributor to cortical atrophy across the distinct clinical variants of AD.
Previous studies examining DAT‐AD and PPA‐AD participants presented with several shortcomings that may have clouded the reported relationships between AD neuropathology and in vivo atrophy. For example, many of these investigations included wide interval ranges (intervals = ~1–8 years) between the final MRI scan and death 11, 12, 14, 16, 18, 34, 37, 38, 40, 41, 74, 79, 81, a potential problem since rates of atrophy and neuropathologic accumulations have yet to be clarified and atrophy likely worsened over the longer intervals. Measurements of atrophy were often confined to the hippocampus 11, 12, 14, 18, 34, 37, 38, 41, 74, 79, whole‐brain volume 14, 18, 40, 41, 74, or ventricular volumes 18, 40, 41, 74, making it difficult to generalize the neuropathologic–anatomic relationships across brain regions. Select studies conducted larger analyses within 3–4 regions 12, 38, but their neuropathologic data were only collected unilaterally, a limitation also shared by studies of fewer regions 11, 34, 40, 79, 81. Bilateral examinations of multiple regions remain uncommon yet are critical to elucidating the relationship between neuropathology and atrophy, especially in clinical syndromes such as PPA that present with such stark hemispheric asymmetries.
NFTs and APs vary greatly across anatomical regions, gyral depth, and cortical layers in AD brains 2, 7, 61. However, previous DAT‐AD and PPA studies rarely obtained representative quantitative estimates of histopathologic markers. Qualitative evaluations of post‐mortem neuropathology and MRI that were collected with vague anatomical correspondence have likely led to inaccurate interpretations of neuropathologic–anatomic relationships. For example, atrophy has been assessed visually 11, 81, and neuropathologic accumulation has been determined by ranking 14, 34, 37, 38, Braak stage 14, 18, 37, 40, 74, 79, 81, or other semi‐quantitative methods 14, 38, 40 using only a few, if not single, post‐mortem sections.
The current study sought to address these methodological limitations by using stereologic quantitation of AD neuropathology and surface‐based measurements of in vivo cortical atrophy in a well‐characterized cohort of PPA‐AD participants with MRI scans relatively close to death (7–30 months). Due to asymmetric and region‐specific atrophy, PPA represents an ideal dementia model for bilateral, multiregional investigations that are not commonly conducted in other neurodegenerative diseases. The focal degenerative patterns observed in PPA brains provided a unique opportunity to tease apart the relative contributions that pathologic lesions make to compromised vs. relatively spared regions within the same brains (eg, left vs. right hemisphere; language vs. non‐language regions). FreeSurfer neuroimaging software was utilized for the dual purpose of measuring cortical atrophy and providing a reliable methodology for delineating 14 bilateral regions involved in the cognitive domains of language, memory, and vision. Post‐mortem coronal sections were matched to in vivo coronal MRI visualized in each FreeSurfer‐derived region, ensuring anatomical correspondence between atrophy measurements and stereologic quantification of NFTs and APs.
The primary aim of this investigation was to determine if densities of AD neuropathology predicted in vivo cortical atrophy in PPA‐AD. A secondary aim was to assess the selective vulnerability of the language network in PPA‐AD by confirming that AD neuropathology and cortical atrophy were asymmetric and had a predilection for language regions in comparison to non‐language regions. Considering previous findings in PPA‐AD and DAT‐AD, we expected larger densities of NFTs, not APs, to be associated with greater cortical atrophy in language regions of the language‐dominant hemisphere in PPA‐AD.
Materials and Methods
Participants
The current study included five right‐handed PPA participants with AD as the primary neuropathologic diagnosis, each of whom had obtained at least one structural MRI scan acquired close to death. Each PPA patient was enrolled through the PPA Research Program at the Mesulam Cognitive Neurology and Alzheimer's Disease Center at the Northwestern University Feinberg School of Medicine. The diagnosis of PPA and subtypes was based on previously described guidelines which includes at least a 2‐year history of progressive, isolated impairments of speech or language functions 27, 48, 50. Two PPA participants were clinically diagnosed with the logopenic variant, one with the agrammatic variant and two were unclassifiable by extant criteria. All five PPA participants received a postmortem diagnosis of “high” AD neuropathologic change based on the published consensus criteria 33, 55. The Northwestern University Internal Review Board approved this study and all participants gave informed consent to their involvement and brain donations. Characteristics of PPA cases are presented in Table 1.
Table 1.
Characteristics of PPA participants. Abbreviations: APOE = apolipoprotein E; P = paraformaldehyde; F = formalin; G = agrammatic; L = logopenic; U = unclassifiable.
| PPA‐AD Case # | PPA subtype | Education (years) | Age at onset (years) | Age at scan (years) | Age at death (years) | Symptom duration (years) | Scan/death interval (years) | Post‐mortem interval (hours) | Fixative | APOE status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L | 16 | 56 | 61 | 62 | 4.7 | 0.61 | 6 | F | 3,3 |
| 2 | U | 12 | 55 | 62 | 64 | 7.9 | 2.14 | 14 | F | 3,3 |
| 3 | L | 15 | 67 | 74 | 76 | 8.6 | 2.00 | 8 | P | 3,4 |
| 4 | U | 14 | 51 | 59 | 61 | 9.4 | 2.54 | 19 | P | 3,4 |
| 5 | G | 18 | 72 | 76 | 78 | 6.2 | 2.54 | 28 | P | 3,4 |
MRI acquisition
Structural MRI scans were acquired from all PPA and healthy control participants on a 3T Siemens TIM Trio scanner using a 12‐channel birdcage head coil at the Northwestern University Center for Translational Imaging. A T1‐weighted 3D MPRAGE sequence included the following: repetition time = 2300 ms, echo time = 2.91 ms, inversion time = 900 ms, field of view = 256 mm, flip angle = 9° and 1 mm3 voxel resolution collected over 176 sagittal slices. The mean interval of time between the last scan and death (SDI) was 1.96 ± 0.8 years; range 0.61–2.54 years (Table 1).
MRI analysis
Structural MRI scans were preprocessed using FreeSurfer (v5.1.0, http://surfer.nmr.mgh.harvard.edu), a well‐validated software suite capable of detecting morphometric properties of neuroanatomy at submillimeter resolution 19. Topological surface errors were removed with manual iterative corrections based on established guidelines 73. Cortical thickness was calculated by measuring the distance between surface representations of the white–gray boundary and pial–CSF boundary 15. The asymmetry and regional severity of cortical atrophy was determined using whole‐brain and regional analyses in the five PPA participants in comparison to 35 previously described healthy controls 64. Prior to generating cortical atrophy data, we confirmed that the two groups were not different in age (PPA group: median 62 and range 59–76; control group: median 62 and range 50–74; Wilcoxon rank‐sum P = 0.23) and education (PPA group: median 15 and range 12–18; control group: median 16 and range 11–20; Wilcoxon rank‐sum P = 0.38).
The whole‐brain vertex‐wise assessment of cortical atrophy involved the generation of a cortical surface heat map at the PPA group level to visualize areas of peak cortical thinning in comparison to the control group (Figure 1A). The region of interest (ROI) analysis included measuring the cortical size of each ROI using mean cortical thickness and converting the thickness values to z‐scores derived from regional thickness measurements collected from the healthy control group. Mean cortical thicknesses from each ROI in each participant were converted to standardized z‐scores (representing the magnitude of cortical atrophy in each ROI) using the mean (μ) and standard deviation (σ) from age‐matched healthy controls:
Figure 1.
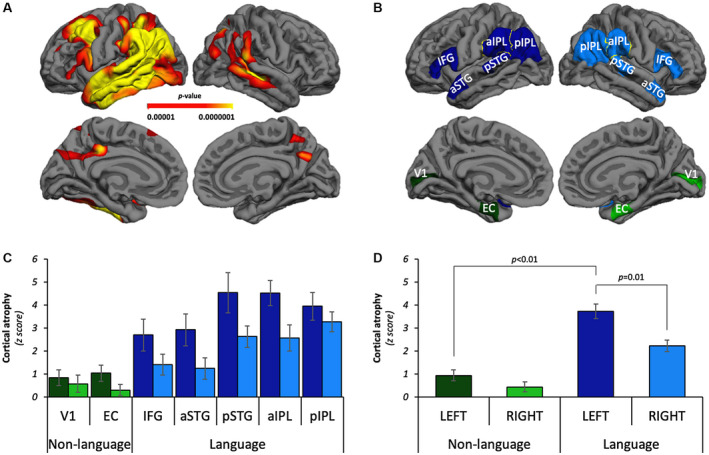
In vivo cortical atrophy was asymmetric and selectively greater in language ROIs in PPA‐AD. A. Heat map identifying peak sites of cortical atrophy in the PPA‐AD group relative to the healthy control group on lateral and medial reconstructions of a template cortical surface in FreeSurfer. Cortical thinning was more prominent in the left hemisphere language network, especially the temporal and parietal lobes. FDR correction set to 0.0001. B. Left language ROIs (dark blue), right language homologues (blue), left non‐language ROIs (dark green), right non‐language ROIs (green) displayed bilaterally on lateral and medial reconstructions of a template cortical surface in FreeSurfer. Inferior frontal gyrus (IFG), anterior superior temporal gyrus (aSTG), posterior superior temporal gyrus (pSTG), anterior inferior parietal lobule (aIPL), posterior inferior parietal lobule (pIPL), entorhinal cortex (EC), primary visual cortex (V1). C. The regional distribution of cortical atrophy across language and non‐language domains bilaterally. Note that tests were performed with regions combined (eg, left vs. right, language vs. non‐language; see D). Left language ROIs = dark blue bars; right language homologues = blue bars; left non‐language ROIs = dark green bars; right non‐language ROIs = green bars. D. The PPA‐AD group had significantly more cortical atrophy in the left language regions compared to right language homologues (P = 0.01), but asymmetry was not observed within the non‐language regions. Cortical atrophy was significantly greater in the language regions compared to the non‐language regions in the left hemisphere (P < 0.01).
Regions of interest
A total of 14 a priori ROIs were examined in each PPA case, 7 per hemisphere (Figure 1B). ROIs involved multiple cognitive domains, including language ROIs implicated in the PPA clinical profile, and non‐language ROIs representing relatively spared cognitive functions (primary sensory and memory areas) over most of the disease duration. Within the left language‐dominant hemisphere, five ROIs were language related and two were non‐language related. The contralateral homologue of each ROI was used to investigate hemispheric asymmetry. The bilateral “language ROIs” consisted of the inferior frontal gyrus (IFG), anterior superior temporal gyrus (aSTG), posterior superior temporal gyrus (pSTG), anterior inferior parietal lobule (aIPL) and posterior inferior parietal lobule (pIPL). The bilateral “non‐language ROIs” were the memory‐related entorhinal cortex (EC) and the primary visual cortex (V1). While the EC is known to be highly vulnerable to AD neuropathology and atrophy early in DAT‐AD 7, 26, 72, this region has not been thoroughly investigated in PPA, a dementia in which memory is not the salient deficit early in the disease course. The extent to which neuropathology and atrophy impact memory regions like the EC in PPA‐AD will provide novel information regarding the integrity of this region in the context of the heavily compromised language regions. In contrast, primary sensory regions such as V1 are typically less vulnerable to AD neuropathology and atrophy in most clinical presentations of AD (ie, common exceptions include posterior cortical atrophy and early onset DAT‐AD) 10, 32, 45, 58, 61, 78. V1 has also received little experimental attention in PPA, but it was anticipated to display little to no neuropathology and atrophy in PPA‐AD.
Each a priori ROI was initially delineated in the native space of each subject using the Desikan–Killiany atlas 17 available in FreeSurfer, which reliably subdivides the human cortex into gyral‐based ROIs 20. FreeSurfer tools were utilized to make small modifications to three atlas‐generated regions to construct the IFG, aSTG and pSTG. The IFG region required the merging of the “pars opercularis” and “pars triangularis” regions, while a precise division of the “superior temporal gyrus” along its longest axis created three equal parts, including an anterior segment (aSTG) and a posterior segment (pSTG), (Figure 1B).
Tissue processing
Brains were cut into ~1–2‐cm coronal blocks and fixed in either 10% formalin for 2 weeks or 4% paraformaldehyde for 30–36 hours at 4ºC, and then submerged into an increasing concentration gradient of sucrose (10%–40%) for cryoprotection (Table 1). Coronal blocks containing ROIs were cut into 40‐μm thick coronal sections and every 24th section was collected for staining procedures and stereologic quantitation. The number of sections available for each ROI was limited to the thickness of the coronal block and the anatomic range defined by FreeSurfer‐generated ROIs. While the EC was a consistently smaller anatomical ROI that resulted in the collection of 5–7 sections, neocortical areas were on average larger and resulted in the collection of 6–16 sections depending on the ROI.
In a 1:24 series of tissue, APs and NFTs were visualized with the Thioflavin‐S stain (1%), which recognizes ß‐pleated sheet protein conformations associated with mature AD neuropathology. Thioflavin‐S‐positive NFTs (fluorescent cytoplasmic fibrils that resembled the size and shape of somas) and dense‐core APs (extracellular coronas of dystrophic neurites that surrounded a central mass of brighter fluorescence) were selectively quantified (Figure 4). These particular NFTs and APs (as opposed to pre‐NFTs and diffuse APs) represent the pathologic hallmarks of AD and bear close relationships to neurodegenerative processes that likely underlie cortical atrophy 6, 24, 25, 47, 77. Another 1:24 series of adjacent tissue sections was processed immunohistochemically for neuronal nuclear protein (NeuN, mouse monoclonal; EMD Millipore; 1/2000), which selectively stains neuronal somas and thus the full extent of gray matter, permitting reliable anatomic identification and correspondence to Thioflavin‐S‐positive sections.
Figure 4.

Representative photomicrographs of the differential distributions of APs and NFTs across hemispheres and regions. NFTs accumulated more in language ROIs of the language‐dominant (left) hemisphere. APs had similar accumulations across hemispheres regardless of ROI. Top row is the aIPL, a language ROI; bottom row is the EC, a non‐language ROI. Images acquired at a magnification of 20x. Scale bars set to 50 μm. The closed arrow denotes an AP; the open arrow denotes an NFT.
Anatomic correspondence between in vivo and postmortem ROIs
Given that the major aim of the study was to relate in vivo cortical atrophy to post‐mortem AD neuropathology, it was critical that both data sets were reliably acquired from the same anatomical regions. A precise correspondence was achieved between brain regions during life and after death by initially visualizing each FreeSurfer‐generated ROI on high‐resolution coronal slices in Freeview, FreeSurfer's visualization tool. Next, coronal slices in Freeview were matched to every available coronal section of post‐mortem tissue immunohistochemically stained with NeuN to guide the boundaries drawn on the post‐mortem tissue. The NeuN‐positive tissue then directed the consistent placement of regional boundaries in the parallel series of Thioflavin‐S‐positive tissue, restricting stereological quantitation of AD markers to the same anatomical regions where cortical atrophy was measured during life (Figure 2).
Figure 2.
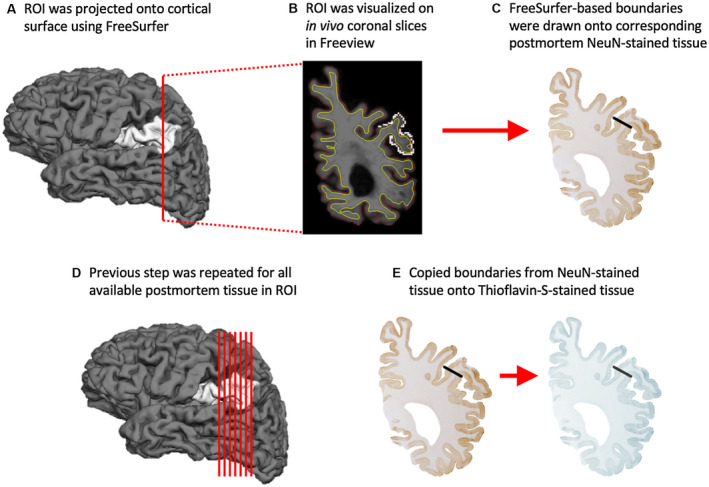
Anatomic correspondence between in vivo and post‐mortem ROIs. Standardized procedure for matching ROIs used in neuroimaging and post‐mortem analyses. In brief, each FreeSurfer‐generated ROI (A) was initially visualized on high‐resolution MRI scans in the coronal plane (B) before they were matched to every available coronal section of post‐mortem tissue immunohistochemically stained with NeuN (C). The ROI boundaries drawn onto all available post‐mortem NeuN‐positive tissue (D) were then copied onto the parallel series of Thioflavin‐S‐positive tissue (E), restricting stereological quantitation of AD markers to the same anatomical regions where cortical atrophy was measured during life. ROI is the left aIPL.
Stereologic design
Unbiased stereologic quantitation was carried out on every 24th section containing each ROI. NFTs and dense‐core APs were quantified at a final magnification of 60x across all cortical layers of each ROI using a stereological analysis performed on a workstation equipped with a Nikon Eclipse E800 microscope, motorized stage, and stereology software (StereoInvestigator v11.07, MBF Bioscience). The optical fractionator probe was used to estimate the populations of NFTs and APs from all available sections per ROI, with sampling grid dimensions that varied by anatomical region to produce a coefficient of error ≤0.15 71. The size of the counting frame was kept at 125 µm2, and a disector height of 16 µm with guard zones of 2 µm was set in order to ensure that quantification only occurred in the area of the tissue in which there was optimal staining and to prevent biases related to sectioning artifacts. Section thickness was measured at each counting site to calculate an average section thickness used in estimating populations of NFTs and APs. Densities of NFTs and APs were used for statistical analyses. Densities of NFTs and APs per mm3 were calculated by taking the estimated population using number weighted section thickness and dividing by the planimetry volume in each ROI.
Statistical analyses
FreeSurfer software was used to generate the cortical thinning heat maps of each hemisphere by contrasting the cortical thickness of the PPA brains against the healthy control group. A general linear model on every vertex along the cortical surface calculated the differences in cortical thickness between groups. Areas of peak cortical thinning (ie, atrophy) were detected and visualized after applying a stringent false discovery rate (FDR) threshold of 0.0001 to adjust for multiple comparisons 22.
All analyses were conducted using linear mixed models accounting for repeated measures. To assess if cortical atrophy displayed hemispheric asymmetry in each regional domain (language and non‐language), the relationship between atrophy and hemisphere was evaluated after stratifying by the type of region. To evaluate if atrophy was different between language and non‐language regions, the association between atrophy and type of region was determined in only the left hemisphere. To assess the hemispheric asymmetry of NFT and AP densities in each regional domain, the following relationships were evaluated after stratifying by the type of region: NFT density and hemisphere, and AP density and hemisphere. To examine the language region selectivity of AD neuropathology, the associations that NFT and AP densities each have with type of region were carried out in only the left hemisphere. These two models were then repeated but excluded V1, IFG, and aSTG in order to determine the predilection of NFT and AP densities for the most atrophied neocortical regions (pSTG, aIPL, pIPL) relative to the entorhinal cortex. In addition, models were used to evaluate the relationship between NFT and AP densities in left language regions and in contralateral language homologues. Lastly, models were used to evaluate the relationships that cortical atrophy had with NFT and AP densities in left language regions and in contralateral language homologues. Adjusters varied by model: when cortical atrophy was compared to NFT and AP densities, models were adjusted for age at death and SDI; when cortical atrophy was related to hemisphere or type of region, models were adjusted for age at scan and SDI; when NFT or AP densities were the outcomes, models were adjusted for age at death and post‐mortem interval. Significance was set to P < 0.05 for all comparisons using SAS software v9.4; SAS Institute.
Results
Hemispheric and language region selectivity of cortical atrophy
Cortical atrophy measured at the whole‐brain level or by ROI was consistent with the patterns of atrophy previously reported from PPA‐AD or PPA with suspected AD neuropathology 21, 53, 66. An FDR of 0.0001 was used to identify peak cortical thinning using vertex‐wise thickness analyses across the whole brain of the PPA‐AD group (Figure 1A). As expected, cortical atrophy was left lateralized including the entire lateral temporal lobe, posterior aspects of the frontal lobe excluding the motor cortex, and most of the parietal lobe with sparing of the superior parts of the superior parietal lobule and precuneus. Bilateral cortical thinning was restricted to the inferior precuneus and the temporoparietal junction extending into the superior temporal sulcus. There was no significant cortical thinning observed in most medial regions or most of the occipital lobe, including the bilateral non‐language regions EC and V1 at the whole‐brain group level.
Regional quantification of cortical atrophy mirrored the patterns of atrophy observed at the whole‐brain level (Figure 1C). ROI analyses of cortical atrophy demonstrated differences between hemispheres and between language and non‐language domains. Left language regions displayed greater cortical atrophy compared to right language homologues (left language regions estimated mean: 3.74, right homologues estimated mean: 2.23; P = 0.01), but no asymmetry was observed in the non‐language regions (Figure 1D; Table S1.1). In the left hemisphere, cortical atrophy was significantly greater in the language regions compared to the non‐language regions (language regions estimated mean: 3.74, non‐language regions estimated mean: 0.95; P < 0.01), (Figure 1D; Table S1.1). The greatest cortical atrophy was observed in the regions comprising the left temporoparietal junction (pSTG, aIPL, and pIPL).
Hemispheric and language region selectivity of AD neuropathology
Densities of NFTs and APs displayed different distributions across hemispheres and ROIs (Figure 3A and B). NFT densities were greater in the left language regions compared to the right language homologues (left language regions estimated mean: 5395, right homologues estimated mean: 3318; P = 0.02), but not in the bilateral non‐language regions (Figures 3C and 4; Table S1.2). AP densities did not show any significant asymmetry when models were stratified to the language and non‐language regions (Figures 3D and 4; Table S1.2). NFT densities were significantly greater in language regions compared to non‐language regions within the left hemisphere (language regions estimated mean: 5395, non‐language regions estimated mean: 2032; P = 0.02), (Figures 3C and 4; Table S1.2). Left language regions showed a non‐significant trend for having greater AP densities than left non‐language regions (language regions estimated mean: 845, non‐language regions estimated mean: 490; P = 0.05), (Figure 3D; Table S1.2).
Figure 3.
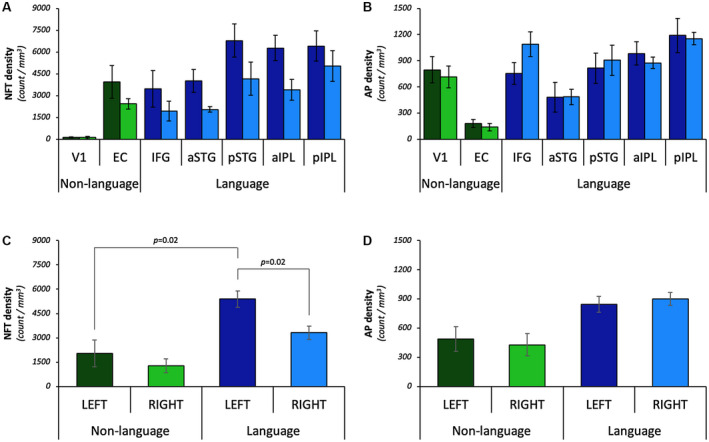
Only NFT densities displayed hemispheric asymmetry and language region selectivity in PPA‐AD. The regional distribution of NFTs (A) and APs (B) across language and non‐language domains bilaterally. Note that tests were performed with regions combined (eg, left vs. right, language vs. non‐language; see C and D). Left language ROIs = dark blue bars; right language homologues = blue bars; left non‐language ROIs = dark green bars; right non‐language ROIs = green bars. C. NFT densities were greater in the left language regions compared to their right hemisphere homologues (P = 0.02); non‐language regions did not harbor asymmetric densities of NFTs. NFT densities were significantly greater in language regions compared to non‐language regions within the left hemisphere (P = 0.02). D. AP densities displayed a non‐significant trend for language selectivity (P = 0.05) and did not show significant asymmetry.
To test whether NFT and AP accumulation had a predilection for the most atrophied language regions compared to the EC (an early site of structural and AD neuropathologic change in DAT‐AD), a subset of data was analyzed in the left hemisphere that excluded V1, IFG, and aSTG. These analyses showed that the three most atrophied language regions (ie, pSTG, aIPL, pIPL) had greater mean densities of NFTs (language regions estimated mean: 6492, EC estimated mean: 3940; P = 0.04) and APs (language regions estimated mean: 996, EC estimated mean: 182; P < 0.01) compared to the EC (Table S1.3).
Relationships between NFTs, APs, and cortical atrophy
NFT densities were not associated with AP densities in the language regions or in right language homologues (Table S1.4). Cortical atrophy had a positive relationship with NFT densities in the left language regions (parameter estimate 0.000453; P < 0.001), (Figure 5A) and their contralateral homologues (parameter estimate 0.000375; P < 0.001), (Table S1.5). Cortical atrophy was not associated with AP densities in the language regions or in right language homologues (Figure 5B; Table S1.5).
Figure 5.
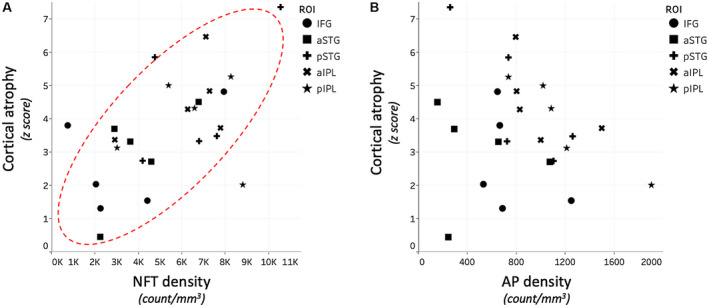
In vivo cortical atrophy was related to NFTs, but not APs, in PPA‐AD. A. Cortical atrophy showed a positive relationship with NFT densities within the left language regions (P < 0.01). B. Cortical atrophy was not related to AP densities in the regions examined.
Discussion
Cortical atrophy might be an intermediate feature of disease progression, emerging after a substantial accumulation of neuropathology, but preceding or concurrent to prominent clinical deficits 35, 36. NFTs appear to be the stronger correlate of cortical atrophy in DAT‐AD 16, but it is not clear if this relationship is present in non‐amnestic presentations of AD. The current study sought to determine the relationships between in vivo cortical atrophy, AP densities, and NFT densities in PPA‐AD. Based on a quantitative analysis of MRI scans relatively close to death and stereologic estimations of AD neuropathology lacking in previous studies of PPA‐AD or DAT‐AD, we found that NFTs, not APs, showed a distribution reflective of the asymmetric aphasic profile that was also significantly associated with cortical atrophy. We show for the first time that NFTs were commensurate with cortical atrophy inside and outside the language network of PPA‐AD (Figure 5A), which is consistent with the NFT and atrophy relationship measured by tau‐PET (positron emission tomography) in individuals with PPA 39, 56, 59, 82. Moreover, the asymmetry of only NFTs agrees with previous clinicopathologic relationships reported by our group and others which have shown that NFTs have an anatomical distribution that is mostly concordant with the atrophy and clinical symptoms characteristic of the PPA phenotype 21, 38.
While the clinical syndromes of DAT and PPA might share the same underlying AD neuropathology, the mechanisms involved and the sequence of events leading to atrophy and clinical impairment might differ. Given that regions with more neuropathology may represent earlier sites of disease manifestation and neurodegeneration, our findings in PPA‐AD suggest that the distribution of NFTs violates the Braak staging of neurofibrillary accumulation typically observed in DAT‐AD. More specifically, we quantitatively showed in PPA‐AD that mean NFT densities were significantly greater in three neocortical language regions of the left temporoparietal junction compared to the entorhinal cortex. Evidence for limbic‐to‐neocortical Braak staging of NFTs comes from cross‐sectional post‐mortem examinations made in different clinical stages of DAT‐AD. The earliest known sites of NFT formation in DAT‐AD are in allocortical areas such as the entorhinal cortex 7, basal forebrain 23, 52, 70 or brainstem nuclei 28, 29, 60, 69. In contrast, APs may follow a neocortical‐to‐limbic propagation in DAT‐AD with an origin in the isocortex of frontal, parietal or temporal lobes 3, 76.
There is emerging evidence that neocortical areas may be the earliest sites of neuropathology and neurodegeneration in non‐amnestic variants of AD such as PPA‐AD 62. The preponderance of AD neuropathology to left language regions in our PPA‐AD cohort supports the possibility of a neocortical pathogenesis, and is consistent with longitudinal studies showing that the language network exhibits the earliest and most severe atrophy in PPA 63, 64. Therefore, it could be the case that neuropathology began in the most vulnerable language regions that define the PPA phenotype, compromising their integrity before propagating to neighboring regions based on the patterns of connectivity and susceptibility 8, 9, 13, 46, 49. This theoretical trans‐synaptic spread across neocortical‐to‐limbic regions could explain the unique distribution of AD neuropathology and cortical atrophy we observed in our PPA‐AD cohort, as well as the progression of impairments from language to non‐language cognitive domains across successive stages of disease course. Longitudinal PET studies and post‐mortem examinations of individuals with PPA at earlier preclinical stages can help elucidate the neuropathologic origin and stereotypical propagation hypothesized in PPA.
Cortical atrophy differentiated language from non‐language regions in the left hemisphere which was most consistent with the distribution of NFT densities. However, both NFT and AP densities were significantly greater in the left temporoparietal junction compared to the left EC. The factors that contribute to the concentration of both AD neuropathologic markers to these select language regions are not yet clear, but a synergistic interaction may have driven their prominence. For example, it has been hypothesized in DAT‐AD 30 that an Aß‐Tau interactive model might better predict neuropathologic change and cognitive deficits compared to the serial amyloid cascade model 31 or dual‐pathway models 75 put forth previously for typical AD. Our observations in PPA‐AD suggest that the language network is distinctly vulnerable to structural and AD neuropathologic changes and may have been the site of a possible feedback loop promoting the greatest neurodegeneration. It is important to note that in the present study in PPA‐AD, NFT and AP densities were not related to each other in the language regions and NFT densities greatly outnumbered AP densities in all neocortical and entorhinal regions except the visual cortex. In fact, the NFT‐to‐AP ratio was almost twofold higher in the left language regions compared to their contralateral homologues. Thus, the dissimilar densities and distributions of NFTs and APs across the cortex may also reflect independent pathogenic mechanisms by which each pathology makes different contributions to neurodegenerative processes in PPA‐AD.
While language regions displayed elevated densities of APs and more so NFTs, the relatively less vulnerable non‐language regions in PPA‐AD displayed unique patterns that were not observed in language regions. For example, V1 was the least atrophic and contained the smallest densities of NFTs, but its AP densities were comparable to AP densities in almost all language regions (Figures 1C, and 3A,B). In contrast, entorhinal cortex contained the smallest densities of APs, but its NFT densities were comparable to NFT densities in the anterior language regions (ie, IFG, aSTG), (Figure 3A,B). These observations indicate that V1 had a greater AP‐to‐NFT ratio while the entorhinal cortex had a greater NFT‐to‐AP ratio. The clinical significance of these opposing patterns in non‐language regions remains unclear in our PPA‐AD cohort. All PPA‐AD participants had intact vision that permitted the completion of the neuropsychological battery and none reported visual impairments beyond the need for corrective lenses. In relation to entorhinal cortex integrity, some memory impairments eventually surfaced in a few PPA cases, but spatial navigation was not formally assessed after diagnosis. Nevertheless, all PPA‐AD participants had primary deficits disproportionately greater in the language domain, which was concordant with NFT and severity of atrophy.
Concomitant factors may have influenced the relationship between AD neuropathology and cortical atrophy. We have recently shown that the same PPA‐AD cohort included in this study had significant microglial activation in the white matter that was associated with gray matter atrophy 57. In addition to glial‐mediated inflammation, upstream intermediates of NFTs and APs may have caused, at least in part, the neurodegenerative patterns. There is evidence that soluble precursors of insoluble NFTs (tau oligomers) and APs (amyloid‐ß oligomers) track disease progression 43 and have neurotoxic, and potentially synergistic capacities that operate independent of their downstream counterparts (ie, NFTs and APs) 5. Consistent with most observations made in DAT‐AD 16, the distributions of dense‐core APs did not mirror the neurodegenerative processes in PPA‐AD. Since it has been shown that APs do not parallel disease progression in DAT‐AD 3, one potential reason for this discordance could be that the accumulation or clearing of APs occur at rates dissimilar to the rate of atrophy in the time elapsed between the final MRI scan and death. Neuropil threads, ghost tangles, pretangles, and diffuse plaques are other pathologic features that may also affect gray matter composition and should be investigated in future studies.
The participants that met inclusion criteria for this study resulted in a small cohort that were only male and limited our statistical models. It will be important to replicate these findings in an investigation of a larger population with balanced sexes that could permit multivariate models adjusted for additional demographics. However, the small number of PPA‐AD participants permitted an extensive and quantitative analysis of post‐mortem tissue that could be systematically compared to MRI scans close to death. Genetic, developmental, or other acquired risk factors might confer region‐specific vulnerabilities to disease in PPA 67. For example, learning disability has been shown to be closely associated with PPA patients and their first‐degree relatives compared to other dementia and control groups 65. While the current PPA‐AD cohort had no known genetic risk factors, three participants had a personal (cases 3 and 5) and/or family (cases 2, 3, and 5) history of learning disability. Additionally, two participants had a family history of either AD (case 5) or Parkinson's disease (case 1).
In summary, the current investigation demonstrated that AD neuropathology had a predilection for left language regions that incurred the most atrophy in PPA‐AD. Only NFTs accumulated in accordance with both the hemispheric and language selectivity characteristics of PPA‐AD and were significantly related to cortical atrophy in most cortical regions examined. Since NFTs appear to be more closely associated with reductions in gray matter across the clinical spectrum of AD, it is likely that NFTs play a prominent role in the neuronal loss and/or shrinkage underlying cortical atrophy. Future studies are necessary to identify the cellular changes associated with cortical atrophy and AD neuropathology in PPA.
Conflict of Interest
The authors have no conflicts of interest to declare.
Supporting information
Acknowledgments
We would like to thank the participants and their families for making this study possible. We are also grateful for the technical assistance provided by Adam Martersteck, Aneesha Nilakantan, PhD, Allison Rainford, Farzan Rahmani and Derin Cobia, PhD, as well as the assistance with data collection provided by Benjamin Rader, MS, and Mallory Ward, MS. This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS) (NS095652), NINDS (NS085770), NINDS (NS075075), National Institute on Aging (NIA) (T32 AG20506), NIA (AG056258), Northwestern Alzheimer's Disease Center (NIA, AG13854), National Institute on Deafness and Other Communication Disorders (NIDCD) (DC008552) and the Florane and Jerome Rosenstone Fellowship.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K et al (2007) Focal cortical presentations of Alzheimer's disease. Brain 130:2636–2645. [DOI] [PubMed] [Google Scholar]
- 2. Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex 1:103–116. [DOI] [PubMed] [Google Scholar]
- 3. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42:631. [DOI] [PubMed] [Google Scholar]
- 4. Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL et al (1995) Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol 52:81–88. [DOI] [PubMed] [Google Scholar]
- 5. Bloom GS (2014) Amyloid‐β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71:505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bobinski M, Wegiel J, Wisniewski HM, Tarnawski M, Reisberg B, de Leon MJ et al (1996) Neurofibrillary pathology–correlation with hippocampal formation atrophy in Alzheimer disease. Neurobiol Aging 17:909–919. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl 165:3–12. [DOI] [PubMed] [Google Scholar]
- 9. Brettschneider J, Tredici KD, Lee VMY, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Broe M, Hodges JR, Schofield E, Shepherd CE, Kril JJ, Halliday GM (2003) Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60:1005–1011. [DOI] [PubMed] [Google Scholar]
- 11. Burton EJ, Barber R, Mukaetova‐Ladinska EB, Robson J, Perry RH, Jaros E et al (2009) Medial temporal lobe atrophy on MRI differentiates Alzheimer's disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain 132:195–203. [DOI] [PubMed] [Google Scholar]
- 12. Burton EJ, Ladinska EBM, Perry RH, Jaros E, Barber R, O'Brien JT (2012) Quantitative neurodegenerative pathology does not explain the degree of hippocampal atrophy on MRI in degenerative dementia. Int J Geriatr Psychiatry 27:1267–1274. [DOI] [PubMed] [Google Scholar]
- 13. de Calignon A, Polydoro M, Suárez‐Calvet M, William C, Adamowicz DH, Kopeikina KJ et al (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Csernansky JG, Hamstra J, Wang L, McKeel D, Price JL, Gado M et al (2004) Correlations between antemortem hippocampal volume and postmortem neuropathology in AD subjects. Alzheimer Dis Assoc Disord 18(4):190–195. [PubMed] [Google Scholar]
- 15. Dale AM, Fischl B, Sereno MI (1999) Cortical surface‐based analysis. NeuroImage 9:179–194. [DOI] [PubMed] [Google Scholar]
- 16. Dallaire‐Théroux C, Callahan BL, Potvin O, Saikali S, Duchesne S (2017) Radiological‐pathological correlation in Alzheimer's disease: systematic review of antemortem magnetic resonance imaging findings. J Alzheimers Dis 57:575–601. [DOI] [PubMed] [Google Scholar]
- 17. Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D et al (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 31:968–980. [DOI] [PubMed] [Google Scholar]
- 18. Erten‐Lyons D, Dodge HH, Woltjer R, Silbert LC, Howieson DB, Kramer P et al (2013) Neuropathologic basis of age‐associated brain atrophy. JAMA Neurol 70:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fischl B, Dale AM (2000) Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH et al (2004) Automatically parcellating the human cerebral cortex. Cerebral Cortex 14:11–22. [DOI] [PubMed] [Google Scholar]
- 21. Gefen T, Gasho K, Rademaker A, Lalehzari M, Weintraub S, Rogalski E et al (2012) Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Genovese CR, Lazar NA, Nichols T (2002) Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage 15:870–878. [DOI] [PubMed] [Google Scholar]
- 23. Geula C, Nagykery N, Nicholas A, Wu C‐K (2008) Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J Neuropathol Exp Neurol 67:309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP et al (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60:1495–1500. [DOI] [PubMed] [Google Scholar]
- 25. Gomez‐Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC et al (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41:17–24. [DOI] [PubMed] [Google Scholar]
- 26. Gomez‐Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16:4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gorno‐Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al, editors (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grinberg LT, Rub U, Ferretti REL, Nitrini R, Farfel JM, Polichiso L et al (2009) The dorsal raphe nucleus shows phospho‐tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathol Appl Neurobiol 35:406–416. [DOI] [PubMed] [Google Scholar]
- 29. Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging 28:327–335. [DOI] [PubMed] [Google Scholar]
- 30. Han P, Shi J (2016) A theoretical analysis of the synergy of amyloid and tau in Alzheimer's disease. J Alzheimers Dis 52:1461–1470. [DOI] [PubMed] [Google Scholar]
- 31. Hardy JA, Higgins GA (1992) Alzheimers‐disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 32. Hof PR, Archin N, Osmand AP, Dougherty JH, Wells C, Bouras C et al (1993) Posterior cortical atrophy in Alzheimer's disease: analysis of a new case and re‐evaluation of a historical report. Acta Neuropathol 86:215–223. [DOI] [PubMed] [Google Scholar]
- 33. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jack CR, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC et al (2002) Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 58:750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS et al (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW et al (2008) Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 63:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Josephs KA, Dickson DW, Murray ME, Senjem ML, Parisi JE, Petersen RC et al (2013) Quantitative neurofibrillary tangle density and brain volumetric MRI analyses in Alzheimer's disease presenting as logopenic progressive aphasia. Brain Lang 127:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Josephs KA, Martin PR, Botha H, Schwarz CG, Duffy JR, Clark HM et al (2018) [18F]AV‐1451 tau‐PET and primary progressive aphasia. Ann Neurol 83:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS et al (2008) Beta‐amyloid burden is not associated with rates of brain atrophy. Ann Neurol 63:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaur B, Himali JJ, Seshadri S, Beiser AS, Au R, McKee AC et al (2014) Association between neuropathology and brain volume in the Framingham Heart Study. Alzheimer Dis Assoc Disord 28:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knibb JA, Xuereb JH, Patterson K, Hodges JR (2006) Clinical and pathological characterization of progressive aphasia. Ann Neurol 59:156–165. [DOI] [PubMed] [Google Scholar]
- 43. Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, Platt B (2016) Soluble pre‐fibrillar tau and β‐amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol 132:875–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kramer JH, Miller BL (2000) Alzheimer's disease and its focal variants. Semin Neurol 20:447–454. [DOI] [PubMed] [Google Scholar]
- 45. Lewis DA, Campbell MJ, Terry RD, Morrison JH (1987) Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: a quantitative study of visual and auditory cortices. J Neurosci 7:1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C et al (2012) Trans‐synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Masliah E, Honer WG, Mallory M, Voigt M, Kushner P, Hansen L et al (1994) Topographical distribution of synaptic‐associated proteins in the neuritic plaques of Alzheimer's disease hippocampus. Acta Neuropathol 87:135–142. [DOI] [PubMed] [Google Scholar]
- 48. Mesulam MM (1982) Slowly progressive aphasia without generalized dementia. Ann Neurol 11:592–598. [DOI] [PubMed] [Google Scholar]
- 49. Mesulam MM (1999) Neuroplasticity failure review in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron 24:521–529. [DOI] [PubMed] [Google Scholar]
- 50. Mesulam MM (2001) Primary progressive aphasia. Ann Neurol 49:425–432. [PubMed] [Google Scholar]
- 51. Mesulam MM (2003) Primary Progressive Aphasia — A Language‐Based Dementia. N Engl J Med 349:1535–1542. [DOI] [PubMed] [Google Scholar]
- 52. Mesulam M (2004) The cholinergic lesion of Alzheimer's disease: pivotal factor or side show? Learn Memory 11:43–49. [DOI] [PubMed] [Google Scholar]
- 53. Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH (2014) Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain 137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mesulam M, Wicklund A, Johnson N, Rogalski E, Léger GC, Rademaker A et al (2008) Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 63:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2011) National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nasrallah IM, Chen YJ, Hsieh M‐K, Phillips JS, Ternes K, Stockbower GE et al (2018) 18F‐Flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med 59:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ohm DT, Kim G, Gefen T, Rademaker A, Weintraub S, Bigio EH et al (2018) Prominent microglial activation in cortical white matter is selectively associated with cortical atrophy in primary progressive aphasia. Neuropathol Appl Neurobiol 45:216–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ossenkoppele R, Cohn‐Sheehy BI, La Joie R, Vogel JW, Möller C, Lehmann M et al (2015) Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer's disease. Hum Brain Mapp 36:4421–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL et al (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139:1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parvizi J, Van Hoesen GW, Damasio A (2001) The selective vulnerability of brainstem nuclei to Alzheimer's disease. Ann Neurol 49:53–66. [DOI] [PubMed] [Google Scholar]
- 61. Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP (1985) Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA 82:4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Phillips JS, Da Re F, Dratch L, Xie SX, Irwin DJ, McMillan CT et al (2017) Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer's disease. Neurobiol Aging 63:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rogalski E, Cobia D, Harrison TM, Wieneke C, Weintraub S, Mesulam MM (2011) Progression of language decline and cortical atrophy in subtypes of primary progressive aphasia. Neurology 76:1804–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rogalski E, Cobia D, Martersteck A, Rademaker A, Wieneke C, Weintraub S et al (2014) Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology 83:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rogalski E, Johnson N, Weintraub S, Mesulam M (2008) Increased frequency of learning disability in patients with primary progressive aphasia and their first‐degree relatives. Arch Neurol 65:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rogalski E, Sridhar J, Rader B, Martersteck A, Chen K, Cobia D et al (2016) Aphasic variant of Alzheimer disease: clinical, anatomic, and genetic features. Neurology 87:1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rogalski E, Weintraub S, Mesulam MM (2013) Are there susceptibility factors for primary progressive aphasia? Brain Lang 127:135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rohrer JD, Rossor MN, Warren JD (2012) Alzheimer's pathology in primary progressive aphasia. Neurobiol Aging 33:744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rub U, Del Tredici K, Schultz C, Thal DR, Braak E, Braak H (2000) The evolution of Alzheimer's disease‐related cytoskeletal pathology in the human raphe nuclei. Neuropathol Appl Neurobiol 26:553–567. [DOI] [PubMed] [Google Scholar]
- 70. Sassin I, Schultz C, Thal DR, Rub U, Arai K, Braak E et al (2000) Evolution of Alzheimer's disease‐related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol 100:259–269. [DOI] [PubMed] [Google Scholar]
- 71. Schmitz C, Hof PR (2005) Design‐based stereology in neuroscience. Neuroscience 130:813–831. [DOI] [PubMed] [Google Scholar]
- 72. Schmitz TW, Nathan Spreng R, The Alzheimer's Disease Neuroimaging Initiative (2016) Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nat Commun 7:13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ségonne F, Pacheco J, Fischl B (2007) Geometrically accurate topology‐correction of cortical surfaces using nonseparating loops. IEEE Trans Med Imaging 26:518–529. [DOI] [PubMed] [Google Scholar]
- 74. Silbert LC, Quinn JF, Moore MM, Corbridge E, Ball MJ, Murdoch G et al (2003) Changes in premorbid brain volume predict Alzheimer's disease pathology. Neurology 61:487–492. [DOI] [PubMed] [Google Scholar]
- 75. Small SA, Duff K (2008) Linking Aβ and Tau in late‐onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 77. Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K et al (2002) Neurotoxic effects of thioflavin S‐positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc Natl Acad Sci USA 99:13990–13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Uylings HBM, de Brabander JM (2002) Neuronal changes in normal human aging and Alzheimer's disease. Brain Cogn 49:268–276. [DOI] [PubMed] [Google Scholar]
- 79. Vemuri P, Whitwell JL, Kantarci K, Josephs KA, Parisi JE, Shiung MS et al (2008) Antemortem MRI based STructural Abnormality iNDex (STAND)‐scores correlate with postmortem Braak neurofibrillary tangle stage. NeuroImage 42:559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Warren JD, Fletcher PD, Golden HL (2012) The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol 8:451–464. [DOI] [PubMed] [Google Scholar]
- 81. Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD et al (2008) MRI correlates of neurofibrillary tangle pathology at autopsy: A voxel‐based morphometry study. Neurology 71:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J et al (2017) Association of in vivo [18F]AV‐1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 74:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.