

Abstract
Objective
Mitral regurgitation (MR) developing concomitant with ischemic cardiomyopathy is a frequently diagnosed lesion, for which an optimal therapeutic strategy is lacking. The contribution of MR to the ongoing cardiac remodeling from myocardial ischemia (MI) remains controversial. We developed a novel experimental model in which myocardial ischemia and severe MR can be independently introduced, to study the role of MR in chronic remodeling of the ischemic heart.
Methods
98 rats were induced with MI+MR (group 1), or MI(group 2), or MR(group 3), or sham surgery(group 4). MR was introduced by inserting a needle into the anterior mitral leaflet via the ventricular apex in a beating heart. MI was introduced by ligating the left coronary artery. Biweekly ultrasounds were performed after surgery, and invasive hemodynamics were performed in some rats at 2, 10 and 20 weeks.
Results
At 2 weeks post-surgery, end diastolic volume in the ischemic hearts with MR was 432±103μl, compared to 390±76.3μl in ischemic hearts without MR (10.76% difference). By 20 weeks, these volumes were significantly higher in the former group at 767±246μl, compared to 580±85μl in the later (32.24% difference). End systolic volume in the ischemic hearts with MR was 147±46.8μl, comparable to 147±45.7μl without MR, at 2 weeks. By 20 weeks, the volumes increased to 357±136.4μl in the former group, compared to 271±82.3μl in the latter group (31.73% difference).
Conclusion
MR in ischemic hearts significantly increased end diastolic and end systolic volumes of the left ventricle, indicating adverse cardiac remodeling and worse systolic function.
Graphical Abstract
The adverse effects of severe mitral regurgitation on the remodeling of the ischemic hearts after a myocardial infarction were investigated in adult rats. Mitral regurgitation exacerbated adverse cardiac remodeling in the post-infarction rat hearts, compared to similar hearts that did not have any mitral regurgitation.
INTRODUCTION
Mitral regurgitation (MR) is diagnosed in >30% of the patients with ischemic cardiomyopathy, and is associated with accelerated heart failure and increased risk of mortality(1–4). However, the physiological basis for this increased clinical morbidity and mortality is not well described. MR reduces systolic afterload by providing a low resistance path for ejected blood volume to flow out of the left ventricle (LV), whereas it elevates the preload and end diastolic pressure (EDP). Thus, one would expect that MR would have minimal adverse effect and maybe a positive effect on systolic function for a prolonged period (as it is in primary MR from myxomatous mitral valve disease) but have a more pronounced adverse effect on the diastolic chamber function and geometry. Such a hypothesis stands true in isolated/primary MR(5), however, in MR associated with ischemic cardiomyopathy, the effect of MR on the LV chamber is confounded by the ischemic injury to the myocardium, and associated adaptation to injury. The interplay between MR and the adversely remodeling LV are unclear, and lack of structured knowledge in this area is evident from lack of equipoise in the clinical management and timing of MR repair in such patients.
Longitudinal studies in patients with MR and ischemic cardiomyopathy demonstrate a clear negative role of MR in worsening cardiac function and increasing mortality(4,6). However, surgical trials that studied the impact of correction MR in ischemic cardiomyopathy, have not demonstrated significant improvement in cardiac function or survival(7). Sub-set analysis of patients from these trials demonstrated that an LV end diastolic diameter (LVEDD) less than 65mm is predictive of functional recovery after MR surgery, indicating a possible cut-off LV size/volume where the elevated wall stress from the dilated chamber overpowers the stress from MR (8). The transcatheter mitral valve repair trials, COAPT and MITRA-FR, report similar findings with patients with smaller LVs responding better to MR correction(9,10). These clinical studies are however limited to an instantaneous time point in the course of the disease and cannot capture the impact of the natural history of the disease until the treatment.
Experimental models in animals can be useful in understanding the role of MR in ventricular remodeling, by allowing separation and control of the severity of MR and ventricular ischemia. Ovine and swine models of ventricular ischemia with a postero-lateral infarction leading to MR have been reported that mimic the human condition(11,12), but the challenge of separating the two lesions continues in these models. Beeri and Beaudoin reproduced an extracardiac atrio-ventricular conduit model initially reported by Braunwald (13–15), but the flow dynamics of such a conduit fail to mimic that of a mitral regurgitant orifice. Specifically, such a conduit abolishes isovolumetric relaxation phase of the cardiac cycle, impacting the cardiac hemodynamics. Furthermore, these studies have been performed in larger animal models, in which physiological and molecular biology studies are somewhat limited in scope.
In this study, we overcame these challenges by developing a rodent model of MR and MI, wherein MR severity and MI size are controllable(16). Using this rodent model, we investigated the role of MR in the adverse remodeling of an ischemic LV, and compared against age and weight matched animals that only had an MI without any MR. Two additional groups with pure MR and sham surgery were used as controls, with age and weight matching as well. The impact of each stressor on cardiac remodeling was assessed with serial and longitudinal cardiac function assessments over 20 weeks of the rodent’s life, that is equivalent to 10 human years. Changes in LV geometry, function, and systolic and diastolic indices were measured and presented.
METHODS
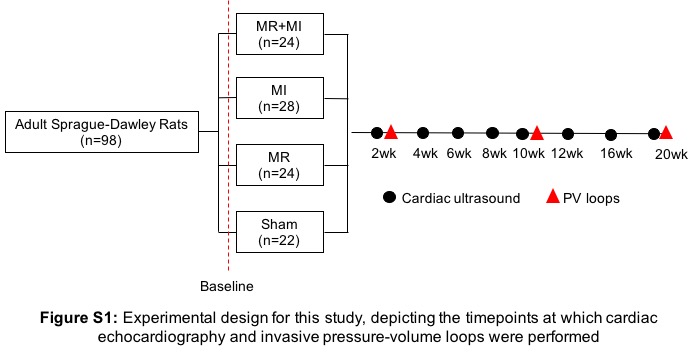
Experimental design (Figure S1)
Male Sprague-Dawley rats (N=102 rats, 4 deaths leading to N=98 completing the study) weighing 300–350g (Envigo Inc, Indiana, USA) were used. Procedures were approved by the Institutional Animal Care and Use committee (IACUC), and aseptic techniques were used. Rats were assigned to four groups: (Group 1) MI+MR; (Group 2) MI only; (Group 3) MR only; and (Group 4) surgical sham. Rats were survived after surgery and followed to a total of 20 weeks, with echocardiography at 2-week intervals until termination, and invasive hemodynamic measurements at 2, 10 and 20 weeks. Sample sizes were estimated to test the hypothesis that the rats with MR+MI will have a 25% higher end diastolic volume (EDV) compared to those with an MI alone at 20 weeks, with 90% power and an alpha of 0.05.
Surgical Procedure
Detailed materials and methods to reproduce this model are provided in the online supplement. Rats were weighed and sedated with 5% isoflurane in 100% oxygen. A 16G angiocath was used for endotracheal intubation and mechanical ventilation (Inspira AV – Harvard apparatus, 66 breaths/min) (Figure 1A). Electrocardiogram and body temperature were continuously monitored. Gentamycin (6mg/kg, SQ) and Rimadyl (2.5mg/kg, SQ) were administered prior to surgery, and incision site was cleaned and sterile draped. An 8Fr intracardiac echo probe (AcuNav, Biosense Webster) was used for transesophageal cardiac imaging, while a vascular probe (Vivid I, GE medical systems) was used for transthoracic cardiac imaging. A left thoracotomy was performed at the 4th or 5th intercostal space, and a pericardiotomy was performed. In group 1, a ligature was placed on the left coronary artery at ~5mm from the ostia with a 6–0 prolene, to induce an MI that included the anterior and lateral walls of the LV (Figure 1B). MI was confirmed from paling of the myocardium and electrocardiogram abnormalities (ST segment elevation and/or T-wave inversion) (Figure 1C). After 25–30 minutes of stable cardiac hemodynamics, a purse string suture was placed on the LV apex and a 23G needle was inserted into the LV and advanced into the anterior MV leaflet (Figure 1D) to create severe MR (Figure 1E–G). In group 2, only a MI was induced with the coronary ligature at a similar location, and a purse string suture was placed on the apex. In group 3-control, only MR was introduced, whereas in group 4, a sham surgery was performed with a purse string suture on the apex. The thoracotomy was closed in layers and a temporary chest tube was inserted to evacuate the thoracic cavity of air and effusions. Rats were gradually weaned from anesthesia, and Lasix were administered to treat any pulmonary congestion. Carprofen (SQ, 5 mg/kg, post-op days 1–5) and Buprenex (SQ, 0.05 mg/kg, post-op days 1–5) were administered in the post-operative phase.
Figure 1:

(A) Photograph of a rat placed in right lateral recumbent position, with a left thoracotomy performed between intercostal spaces to access the left ventricular apex. An 8 Fr transesophageal probe is placed for real time image guidance, using which a 23G needle is inserted into the beating heart to puncture the mitral valve leaflet. (B) Example photograph of a rat through the same incision that received a myocardial infarct, by ligation of the left coronary artery. (C) Changes in electrocardiogram before and after coronary ligation, indicating S-T segment elevation. (D) Echocardiographic image of the needle inserted into the left ventricle and through the mitral valve leaflet; (E) Photograph of the puncture hole that was made in the anterior leaflet by the needle; (F) Color doppler image of severe mitral regurgitation through the needle hole in the mitral leaflet; (G) Continuous wave doppler depicting systolic flow reversal through the mitral valve (yellow arrow).
Echocardiographic Imaging
Lesion severity was confirmed at 2 weeks post-surgery for inclusion in the study. MR severity was measured as jet area to left atrial area on B-mode, color Doppler, transesophageal echocardiographic images. MI size was measured from wall motion scoring on echo at the 2 week timepoint, and confirmed by photographing the hearts at termination and measuring their infarct size with planimetry. At biweekly intervals, both transthoracic and transesophageal echocardiography were performed to measure LV chamber dimensions and function. End diastolic volume (EDV), end systolic volume (ESV), ejection fraction (EF) were measured on 3-chamber cardiac views, and fractional shortening (FS) on multiple short axis slices and then averaged.
Invasive hemodynamics
At termination, a 1.9 Fr admittance probe was inserted into the LV (FTH-1912B-8018, Transonic Scisense, London, CA) and fifty consecutive pressure-volume loops were obtained and averaged for parameter estimation using Labscribe 3.0 (iWorx systems, Dover, NH). Measurements were repeated with preload reduction by inferior vena cava compression and a 12 sec respiratory apnea.
Euthanasia
Animals were heparinized and injected with 1cc of Euthasol to arrest the heart in diastole. Heart and lungs were excised, washed in saline, weighed and photographed.
Biological assays
All assays were performed on tissue samples from remote region of the heart. Part of the tissue stored in 10% non-buffered formalin and dehydrated in graded concentrations of ethanol were used for Hematoxylin and Eosin (H&E) and Trichrome staining. Tissue portions that were snap frozen were used to extract RNA using the RiboPure RNA purification kit (AM1924, Thermo Fisher) and the concentration and purity determine with a spectrophotometer. 0.5mg of total RNA from each sample was reverse transcribed with a reverse transcription mix, and the real time quantitative PCR reaction was performed in a 96 well plate array consisting of rodent specific cardiac remodeling genes. Gene array data is represented as fold regulation for each gene calculated on a logarithmic scale using the Qiagen online software, and only the significantly altered genes are reported.
Statistical methods
All data analysis was performed with GraphPad Prism 6.0. Normality of data was tested with D’Agostino-Pearson normality test. Normal data is presented as mean ± standard error, whereas non-normal data is presented as median with interquartile ranges. Two-way ANOVA was used to compare groups, with a p<0.05 considered to be significantly different. Non-parametric data was analyzed using a Mann Whitney test with Dunn’s correction. Temporal changes in EDV and ESV were compared with a mixed effects model with time as a factor, with multiple comparisons performed with Tukey’s multiple comparisons test. The p-values for the gene array data were calculated based on a Student’s t-test of the 2^(-Delta CT) values for each gene, with a p<0.05 considered to be significantly different compared to the sham group at the same time-point.
RESULTS
Baseline characteristics, lesion reproducibility and variation
Baseline EDV and ESV were comparable between the groups (Figure 2A–B), confirming equivalent cardiac dimensions and function in these age, weight and sex matched groups. Severe MR was confirmed and was comparable in the MR+MI group (44.5±10.6%) and MR group (43.7±12.6%) (Figure 2C). Planimetry derived myocardial infarct size after termination was comparable between the MR+MI and MI groups (Figure 2D). One animal in the MR+MI group, two in the MI group and one in the sham group died in the follow-up period.
Figure 2:

(A) Comparable end diastolic volume at baseline in the four experimental groups; (B) Comparable end systolic volume at baseline in the four experimental groups; (C) Comparable severe mitral regurgitation in the MR+MI and MR groups; (D) Comparable infarct size between animals in MR+MI and MI groups.
Structural Cardiac Chamber Remodeling
Representative sections from hearts explanted at 2,10 and 20 weeks from each group are shown in Figure 3A, with corresponding M-mode echocardiographic images shown in Figure 3B. At 2 weeks, LV volumes were elevated compared to sham in MI and MR+MI groups only, whereas wall thinning was not evident in any part of the myocardium. Heart weights were significantly higher in the MR (1480±131mg, p<0.05) and MR+MI (1570±130mg, p<0.005) groups but not the MI group (1380±130mg, p=0.62), compared to the sham (1310±60 mg), at this timepoint. At 10 weeks, chamber cavity size was quite larger in the MI and MR+MI groups, and only slightly elevated in the MR group, compared to the sham. Heart weights in the MR+MI (1840±270mg, p<0.05) and MR group (1730±230mg, p=0.06) were significantly elevated, but not in the MI group (1460±70mg, p=0.99), compared to sham (1430±140mg). At 20 weeks, the LV walls were thinner in the MR+MI and MI groups, compared to sham and MR groups. Hearts weights were not significantly different in any of the groups, compared to sham.
Figure 3:

(A) Representative photographs of intact and sectioned hearts explanted at 2 weeks, 10 weeks and 20 weeks after surgery in MR+MI, MI, MR and sham groups; (B) Representative M-mode echocardiographic images in each group depicting rapid LV dilatation in the MR+MI and MR groups compared to the other groups.
Left Ventricular Diastolic Function
Figure 4A depicts the longitudinal changes in end diastolic volume in the sham (black), MR (green), MI (blue) and MR+MI (red) groups over the 20 weeks after surgery. Longitudinal changes in EDV over time were significant, per two-way repeated measures ANOVA with time as a factor (p<0.0001). In the sham group, EDV was significantly elevated at 6 weeks post-surgery compared to the baseline (p=0.008), and at every timepoint thereafter until termination (p<0.05 compared to the baseline). In the MR group, EDV was significantly elevated at 2 weeks post-surgery compared to the baseline (p=0.0005) and was significantly higher at every subsequent timepoint compared to the baseline (p<0.0001). In the MI group, EDV was significantly higher at 4 weeks post-surgery compared to the baseline (p=0.004) and at all subsequent timepoints. In the MR+MI group, EDV was significantly elevated by 2 weeks post-surgery, and at all timepoints thereafter until termination. A summary of all comparisons in each group, at every timepoint are reported in Table S1. When comparing EDV in each experimental group to the sham data at the same timepoint, EDV in the MR group was significantly higher in the 12th week (p=0.0076) and thereafter, by the 20th week in the MI group (p=0.0392), and 6th week onwards in the MR+MI group (p<0.05). Figure 4B depicts the changes in the LV end diastolic filling pressure (EDP) at 2, 10 and 20 weeks in each group. At 2 weeks, EDP was significantly higher in the three experimental groups, compared to sham. By 20 weeks, EDP reduced significantly in all the groups, compared to the 2-week timepoint, but remained significantly higher than the sham at the 20-week timepoint. Left Ventricular stiffness, measured as the slope of the end-diastolic pressure volume relationship (EDPVR) (Figure 4C), was significantly lower in the MR group at 2 weeks, and in MR and MR+MI groups at 20 weeks. Tau-Glantz, a measure of active diastolic relaxation (Figure 4D) was significantly elevated in the MR, MI and MR+MI groups at all the timepoints, compared to sham. At 2 weeks, Tau-Glantz was higher in MR+MI compared to sham and MR groups (p<0.05), whereas such increase was not observed in MR and MI groups. At 10 and 20 weeks, Tau-Glantz was significantly higher in the three groups compared to sham at each timepoint.
Figure 4:

(A) Temporal changes in end diastolic volume in the four groups of animals over 20 weeks after surgery; (B) Left ventricular end diastolic pressure measured using an invasive catheter at 2, 10 and 20 weeks after surgery in each group; (C) Slope of end diastolic pressure volume relationship (EDPVR) at 2, 10 and 20 weeks after surgery in each group; (D) Tau-Glantz measured at 2, 10 and 20 weeks in each group. (In all figures * indicates statistical difference compared to other experimental groups at the same timepoint, with color of the * indicating the group compared with. In all figures # indicates statistical difference compared to baseline in the same experimental group).
Left Ventricular Systolic Function
Figure 5A depicts the temporal changes in LV end systolic volume all the experimental groups, over the 20-week time period. Longitudinal changes in ESV over time were significant, per two-way repeated measures ANOVA with time as a factor (p<0.0001). In the sham group, ESV did not change between timepoints. In the MR group, ESV was significantly elevated at 4 weeks post-surgery compared to the baseline (p=0.0254) and remained significantly higher at every subsequent timepoint compared to the baseline (p<0.0001). In the MI group, ESV was significantly higher at 2 weeks post-surgery compared to the baseline (p=0.0029) and at all subsequent timepoints (p<0.0001). In the MR+MI group, ESV was significantly elevated by 2 weeks post-surgery (p=0.0185), and at all timepoints thereafter until termination (p<0.0001). A summary of all comparisons in each group, at every timepoint are reported in Table Sl. When comparing groups to the sham at the same timepoint, ESV in the MR group was different than sham by the 12th week (p=0.03), in the MI group was higher by the 8th week (p=0.0072) and thereafter, and in the MR+MI group by the 6th week (p=0.034) and thereafter. Figure 5B depicts the temporal changes in ejection fraction in each group. When compared to the respective pre-surgical baseline data, EF did not change in the sham group, reduced significantly at 16 weeks post-surgery in the MR group (p=0.03) and thereafter, by the 2nd week in the MI group (p=0.0314) and thereafter, and the 4th week and thereafter in the MR+MI group (p=0.0145). Similar trends were observed in fractional shortening as well, as depicted in Figure 5C. Impaired contractility in the MI and MI+MR groups, early after surgery is further validated by reduced end systolic pressure depicted in Figure 5D.
Figure 5:

(A) Temporal changes in end systolic volume in the four groups of animals over 20 weeks after surgery; (B) Ejection fraction changes over 20 weeks in each of the four groups; (C) Temporal changes in fractional shortening over 20 weeks in each experimental group; (D) End systolic pressure at 2, 10 and 20 weeks after surgery in each experimental group ( In all figures * indicates statistical difference compared to other experimental groups at the same timepoint, with color of the * indicating the group compared against. # indicates statistical difference compared to baseline in the same experimental group).
LV Pressure-Volume Loops
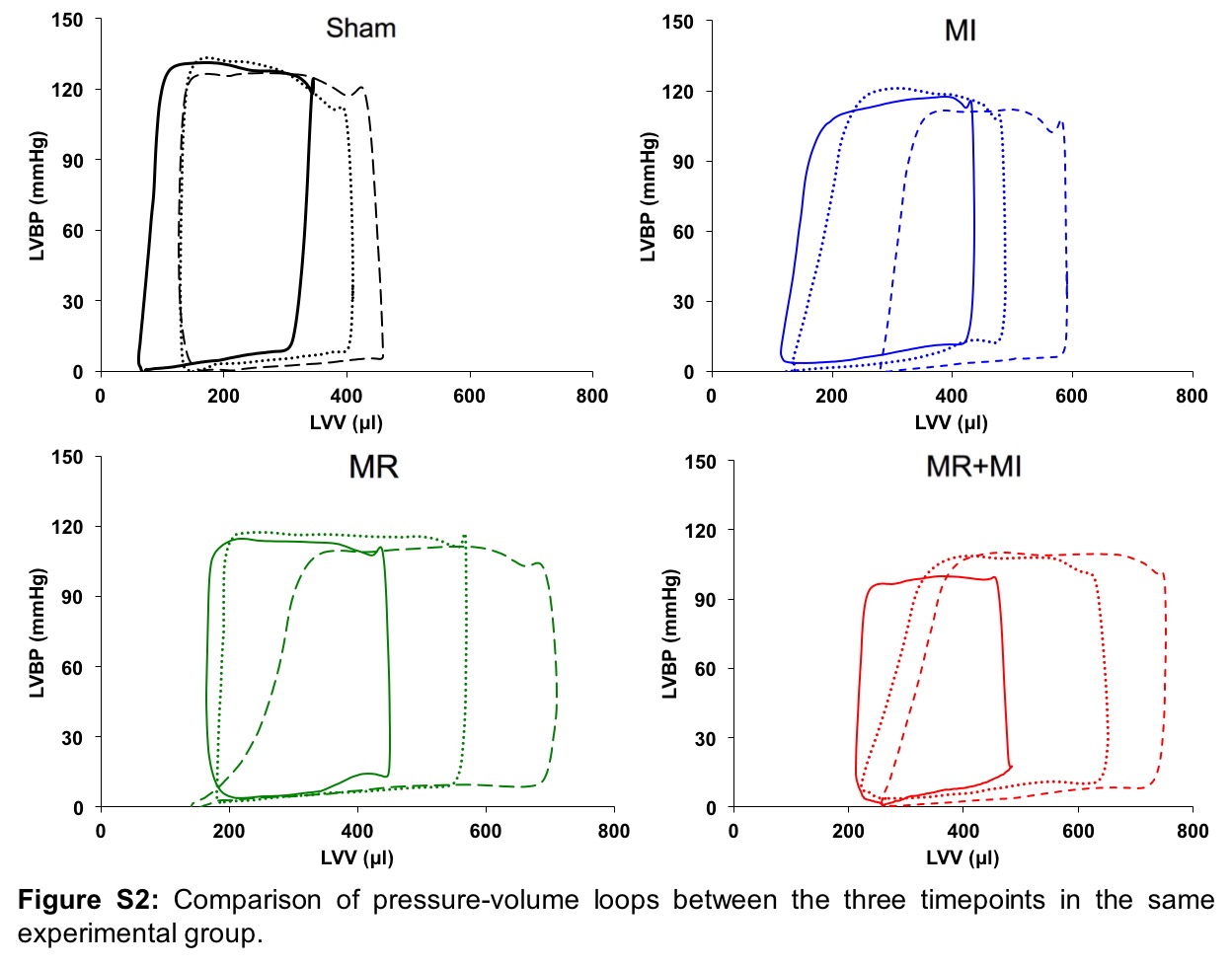
Changes in LV pressure-volume loops between groups at the same timepoint, and between timepoints in the same group are shown in Figure 6A. A rightward shift is observed in the three disease groups compared to the sham, with MR+MI demonstrating the most rightward shift as early as 2 weeks and persisted thereafter until 20 weeks. By 20 weeks, the pressure-volume loop in the MR and MR+MI was widest, indicating higher stroke volume, but also higher stroke work to eject the volume. Such widening of the loops was exclusively observed in MR and MR+MI, but not the MI or sham groups, indicating that such widening is a result of volume overload from MR. Figure 6B depicts the differences in end systolic pressure volume relationship between the groups, with the worst systolic contractility in the MR+MI group, followed by the MI and then MR at all the three timepoints. Figure 6C depicts the end diastolic pressure volume relationship compared between the groups. At 2 weeks, the groups with MR demonstrated a significant rightward shift compared to sham and MI, indicating increased LV compliance to accommodate the volume overload. By 10 weeks, the differences were normalized, whereas at 20 weeks, a leftward shift was observed in the MR group, indicating possible stiffening of the LV chamber. Such a leftward shift was not observed in the MR+MI group at 20 weeks. Derived indices from the pressure volume loops are reported in Table 2. Figure 7A depicts the rate of pressure development in early systole, indicating worst indices in the MR+MI group as early as 2 weeks. By 20 weeks, all experimental groups had poor rate of pressure development, indicating contractile dysfunction. Figure 7B depicts the rate of pressure decay during late systole to early diastole, with poor indices in the three groups as early as 2 weeks and persisting thereafter until the 20 weeks compared to the sham. Rate of pressure decay was worst in the MR+MI group compared to MR and MI at 2 weeks, and MR at 10 weeks. Rate of pressure build up after normalization to diastolic ventricular volume demonstrated poor contractility in all the experimental groups at 2, 10 and 20 weeks, as shown in Figure 7C. Stroke work was largest in the MR group compared to the sham at all the timepoints, as shown in Figure 7D. In the MR+MI group, stroke work increased significantly by 10 weeks and remained high at 20 week. Diastolic wall stress was highest in the MR+MI group at 2 weeks and was significantly higher compared to the three other groups, as shown in Figure 7E. It reduced with time in the MR+MI, and by 20 weeks even through the overall diastolic wall stress was lower, it was significantly higher than sham in all the three experimental groups. These changes in diastolic wall stress, parallel changes in end diastolic pressure (EDP). Figure 7F depicts the systolic wall stress, which was highest in the MI group at 20 weeks, compared to sham. Reduced systolic wall stress in the MR+MI group at 20 weeks may be attributed to reduced end systolic pressure from poor contractility.
Figure 6:

(A) Averaged pressure-volume loops of data acquired in each group of animals at 2, 10 and 20 weeks after surgery; (B) End systolic pressure volume relationship at 2, 10 and 20 weeks in each experimental group, depicting a rightward shift in all the groups compared to the sham; (C) Changes in end diastolic pressure volume relationship at 2, 10 and 20 weeks in each experimental group. Compared to sham (black line), a rightward shift indicates increased LV capacitance.
Table 2:
Invasive hemodynamic measurements and calculations in each group at 2, 10 and 20 weeks after the surgery
| 2 week | 10 week | 20 week | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sham | MR | MI | MR+MI | Sham | MR | MI | MR+MI | Sham | MR | MI | MR+MI | |
| HR (bpm) | 320.4±20.03 | 313.1±24.08 | 315.6±14.24 | 315.7±30.75 | 307.8±13.96 | 306.5±8.99 | 292.8±20.89 | 312.6±31.32 | 339.6±25.12 | 309.3±29.84 | 208.9±30.62 | 320.3±27.68 |
| ESP (mm Hg) | 122.6±5.94 | 113.2±11.69 | 105.7±6.72* | 98.48±8.61*# | 133.5±4.04 | 117.6±7.79* | 115.7±10.51* | 104.9±5.03* | 122.5±7.19 | 115.7±12.58 | 121.7±13.51 | 111.8±7.42 |
| EDP (mm Hg) | 9.99±3.15 | 15.68±2.83* | 15.12±0.64* | 20.24±2.50*#$ | 8.96±1.57 | 9.40±2.59 | 15.67±3.26*# | 13.72±1.54 | 4.11±1.23 | 10.23±3.59* | 9.37±4.18* | 9.74±3.73* |
| dP/dt max(mmHg/s) | 8915±1036 | 8228±608 | 7867±693 | 6619±1267* | 10477±984.1 | 9324±882.6 | 7427±943.1*# | 6567±896.9*# | 10988±2240 | 8760±1409* | 8709±1771* | 8349±1274* |
| dP/dt min(mmHg/s) | (11575)±1647 | (8995)±1388* | (8847)±910* | (5602)±1069*#$ | (12982)±1455 | (9579)±2111* | (7572)±1492* | (6511)±966.3* | (10953)±1437 | (8508)±1991* | (8380)±906.6* | (7621)±1591* |
| CO (ul/min) | 58884±13959 | 72691±11853 | 43455±7190# | 61779±25214 | 60661±7116 | 104814±39005* | 39802±5961# | 90628±16083$ | 112776±12180 | 109366±21186 | 71778±20271*# | 85515±44200 |
| SW (mJoules) | 3.19±0.99 | 3.35±0.51 | 2.17±0.22 | 2.39±0.75 | 3.84±0.73 | 5.90±1.79 | 2.95±0.34# | 4.72±1.00 | 5.12±0.66 | 5.44±1.11 | 3.51±0.89*# | 4.03±1.86# |
| maxPwr (mWatt) | 18.88±22.73 | 88.02±85.58 | 40.63±18.34 | 149.5±31.22*# | 24.96±20.03 | 25.69±26.99 | 65.43±51.73 | 57.8±22.98 | 15.15±10.19 | 51.34±42.79 | 104.2±80.05 | 89.42±75.96 |
| EA(mmHg/ul) | 0.6995±0.2073 | 0.5037±0.0933 | 0.7997±0.1829 | 0.5919±0.2566 | 0.6878±0.1055 | 0.3977±0.1841* | 0.8797±0.1595# | 0.374±0.0771*# | 0.3756±0.0385 | 0.3722±0.0914 | 0.5819±0.1508# | 0.583±0.3968 |
| PVA (mJoules) | 6.049±1.919 | 5.554±2.608 | 3.946±0.603 | 4.37±2.234 | 5.513±1.049 | 8.444±2.035 | 5.396±2.147 | 7.091±1.877 | 6.973±0.765 | 7.996±2.267 | 6.435±2.196 | 6.77±2.128 |
| PE (mJoules) | 2.859±1.588 | 2.207±2.602 | 1.774±0.492 | 1.981±1.997 | 1.67±0.65 | 2.54±1.54 | 2.44±2.02 | 2.37±1.34 | 1.85±0.587 | 2.55±1.91 | 2.92±1.90 | 2.73±2.18 |
| Efficiency (%) | 54.53±11.39 | 76.28±32.95 | 60.49±10.79 | 69.07±39.14 | 76.79±11.26 | 74.37±16.79 | 68.78±35.24 | 78.07±13.55 | 73.96±8.25 | 74.27±18.58 | 65.06±21.53 | 65.62±22.76 |
| ESPVR | 0.79±0.34 | 0.64±0.24 | 0.44±0.19 | 0.28±0.05* | 0.97±0.19 | 0.46±0.10* | 0.32±0.02* | 0.29±0.08* | 0.81±0.25 | 0.52±0.15* | 0.39±0.14* | 0.38±0.08* |
| EDPVR | 0.0118±0.0022 | 0.0073±0.0012$ | 0.0156±0.0100 | 0.0069±0.0030$ | 0.0095±0.0018 | 0.0063±0.0030 | 0.0107±0.0037 | 0.0074±0.0019 | 0.0078±0.0017 | 0.0058±0.0031 | 0.0077±0.0055 | 0.0092±0.0058 |
| TauW | 7.985±0.85 | 9.817±1.13 | 9.736±0.99 | 12.51±1.71*#$ | 7.617±0.38 | 8.636±0.63 | 10.75±1.63*# | 10.85±0.26*# | 7.466±0.76 | 9.584±1.56* | 9.712±1.54* | 9.561±1.70* |
| TauG | 10.51±0.90 | 12.57±0.73 | 13.24±1.63 | 16.98±3.52*#$ | 9.84±0.74 | 16.59±8.62 | 15.84±3.19 | 17.18±3.36 | 9.57±0.81 | 13.39±3.47* | 13.17±2.63 | 14.28±4.46* |
| PRSW | 87.82±14.52 | 73.5±9.36 | 67.4±11.26* | 55.23±12.81* | 103.4±4.48 | 83.1±16.12 | 62.69±11.59*# | 78.45±1.86* | 113±27.51 | 85.07±14.62* | 72.67±14.51* | 64.98±13.56* |
| dPmax/EDV | 25.53±12.03 | 21.54±11.19 | 23.86±8.07 | 20.65±10.55 | 21.16±4.71 | 13.9±4.45 | 13.01±2.32 | 8.98±1.08 | 22.4±5.38 | 15.09±5.50* | 14.85±4.40* | 14.24±5.34* |
Comparison between groups at the same timepoint
compared to sham
compared to MR
compared to MI
Figure 7:

Temporal changes in cardiac functional indices at 2, 10 and 20 weeks after surgery in the four experimental groups: (A) dP/dt max in the left ventricle; (B) dP/dt min in the left ventricle; (C) dP/dt max normalized to mean end diastolic volume in that group at that timepoint; (D) Stroke work; (E) Wall stress at end diastole in the equatorial region of the left ventricle; (F) Wall stress at end systole in the equatorial region of the left ventricle.
Histological assessment of the myocardium
Figure 8 depicts representative H&E and trichrome staining of the explanted hearts at 2, 10 and 20-week timepoints. Hematoxylin and eosin staining did not demonstrate any significant differences in gross morphology between the samples at any of the timepoints. However, in trichrome stains, MI hearts had higher fibrosis (blue color) compared to any of the other groups. Such extent of fibrosis was not observed in the MI+MR group, though some sparsely distributed fibrotic tissue was evident.
Figure 8:

(Top) Hematoxylin and eosin staining of the myocardial explants at 20 weeks; (Bottom) Trichrome staining of the myocardial explants at 20 weeks
Gene expression
Table 3 summarizes the significantly upregulated and downregulated genes in each group compared to the sham group. In the MR group, some cardiac extracellular matrix genes were significantly downregulated at both 2 and 20 weeks. Ecm1 is the only gene significantly upregulated at 2 weeks. In the MI only group, several fibrosis genes were highly upregulated by 2 and 20 weeks, confirming myocardial fibrosis on the histological sections. Most of the upregulated genes at these time points included collagen isoforms and matrix metalloproteinases, metallopeptidases, and their inhibitors (TIMPs). In the MR+MI group, significantly altered genes were distinct from both MR only and MI only groups. Integrin coding genes were significantly downregulated by 20 weeks in this group, potentially indicating a mechanosensory pathway trigger.
Table 3:
Differential gene expression in the myocardium at 2 weeks and 20 weeks after inducing the lesion, compared to sham
| MR 2 week | MR 20 week | ||||||
| Gene | Description | Fold regulation | p-value | Gene | Description | Fold regulation | p-value |
| Cdh1 | Cadherin 1 | −5.71 | 0.014 | Col4a3 | Collagen, type IV, alpha 3 | −1.52 | 0.008 |
| Cntn1 | Contactin 1 | −4.17 | 0.047 | Ctnna2 | Catenin (cadherin associated protein), alpha 2 | −4.35 | 0.012 |
| Col2a1 | Collagen, type II, alpha 1 | −7.95 | 0.0038 | Lamb2 | Laminin, beta 2 | −1.88 | 0.02 |
| Ecm1 | Extracellular matrix protein 1 | 2.1 | 0.022 | Mmp11 | Matrix metallopeptidase 11 | −2.41 | 0.0074 |
| Pecam1 | Platelet/endothelial cell adhesion molecule 1 | −2.19 | 0.049 | ||||
| Selp | Selectin P | −8.76 | 0.029 | ||||
| MI 2 week | MI 20 week | ||||||
| Gene | Description | Fold regulation | p-value | Gene | Description | Fold regulation | p-value |
| Adamts2 | ADAM metallopeptidase with thrombospondin type 1 motif, 2 | 3.38 | 0.047 | Adamts8 | ADAM metallopeptidase with thrombospondin type 1 motif, 8 | 3.17 | 0.0011 |
| Col2a1 | Collagen, type II, alpha 1 | −5.07 | 0.024 | Col1a1 | Collagen, type I, alpha 1 | 2.57 | 0.045 |
| Col5a1 | Collagen, type V, alpha 1 | 2.42 | 0.04 | Col3a1 | Collagen, type III, alpha 1 | 2.02 | 0.017 |
| Itga3 | Integrin, alpha 3 | −3.59 | 0.048 | Col4a3 | Collagen, type IV, alpha 3 | −2.14 | 0.039 |
| Itgb3 | Integrin, beta 3 | −2.09 | 0.046 | Col8a1 | Collagen, type VIII, alpha 1 | 5.34 | 0.0053 |
| Pecam1 | Platelet/endothelial cell adhesion molecule 1 | −5.04 | 0.032 | Ctgf | Connective tissue growth factor | 3.65 | 0.0104 |
| Fbln1 | Fibulin 1 | 2.03 | 0.0176 | ||||
| Fn1 | Fibronectin 1 | 3.27 | 0.0013 | ||||
| Itgb4 | Integrin, beta 4 | 3.35 | 0.007 | ||||
| Lama2 | Laminin, alpha 2 | −1.91 | 0.047 | ||||
| Lamc1 | Laminin, gamma 1 | 1.23 | 0.039 | ||||
| Mmp11 | Matrix metallopeptidase 11 | −2.07 | 0.0035 | ||||
| Mmp15 | Matrix metallopeptidase 15 | −1.95 | 0.045 | ||||
| Ncam1 | Neural cell adhesion molecule 1 | 3.46 | 0.0049 | ||||
| Postn | Periostin, osteoblast specific factor | 5.36 | 0.0028 | ||||
| Spp1 | Secreted phosphoprotein 1 | 4.28 | 0.004 | ||||
| Timp1 | TIMP metallopeptidase inhibitor 1 | 2.28 | 0.0049 | ||||
| Timp2 | TIMP metallopeptidase inhibitor 2 | 1.34 | 0.04 | ||||
| Vcan | Versican | 1.95 | 0.0067 | ||||
| Vtn | Vitronectin | −4.47 | 0.0036 | ||||
| MR + MI 2wk | MR + MI 20wk | ||||||
| Gene | Description | Fold regulation | p-value | Gene | Description | Fold regulation | p-value |
| Cdh1 | Cadherin 1 | −5.71 | 0.027 | Col4a1 | Collagen, type IV, alpha 1 | −1.31 | 0.017 |
| Col2a1 | Collagen, type II, alpha 1 | −7.21 | 0.016 | Col4a3 | Collagen, type IV, alpha 3 | −2.77 | 0.04 |
| Col3a1 | Collagen, type III, alpha 1 | 4.69 | 0.038 | Itga2 | Integrin, alpha 2 | −2.74 | 0.03 |
| Col5a1 | Collagen, type V, alpha 1 | 2.19 | 0.05 | Itgav | Integrin, alpha V | −1.84 | 0.039 |
| Ecm1 | Extracellular matrix protein 1 | 3.67 | 0.003 | Lamb2 | Laminin, beta 2 | −2.11 | 0.026 |
| Emilin1 | Elastin microfibril interfacer 1 | 3.09 | 0.005 | Mmp11 | Matrix metallopeptidase 11 | −5.03 | 0.0069 |
| Thbs2 | Thrombospondin 2 | 2.06 | 0.046 | Sparc | Secreted protein, acidic, cysteine-rich (osteonectin) | −1.3 | 0.026 |
| Vcan | Versican | 2.81 | 0.038 | ||||
DISCUSSION
Experimental model
MR creation with a needle stick to the anterior mitral leaflet was reproducible. MR severity is precise by using the same needle size, providing good control of the lesion in this model. Performing this procedure with a single needle pass to induce MR requires training in both imaging and surgical technique, which we established in multiple rats prior to this study. Despite controlling for the size of the leaflet hole, some degree of variability in MR is expected as the transmitral gradient in systole determines the regurgitant flow through the hole, which cannot be controlled. MI was reproducible as well, by ligating the coronary artery at the same level in all the animals. Placing the ligature 5mm from the ostium created an antero-lateral infarct, indicating that the left circumflex artery or the septal artery were not distal to the ligature. Kainuma et al. elegantly demonstrated in their rat angiographic data that ligating the left main artery 2mm from its ostium carries a high risk of including the circumflex and septal artery, which validates our approach. This experimental model does not mimic the clinical mitral valve etiologies of primary MR from mitral valve prolapse, or mitral valve deformation leading to secondary MR in LV failure. However, it faithfully represents the volume overload effect that the LV experiences in the setting of either forms of MR, which is the focus of this study. Thus, the advantages of separating the MR and the MI in this model and the novel experimental design it allows (Graphical abstract), provide unique benefits that would not be available in models that mimic the clinical mitral valve lesions.
Impact of MR on LV Mass, Hypertrophy and Dilatation
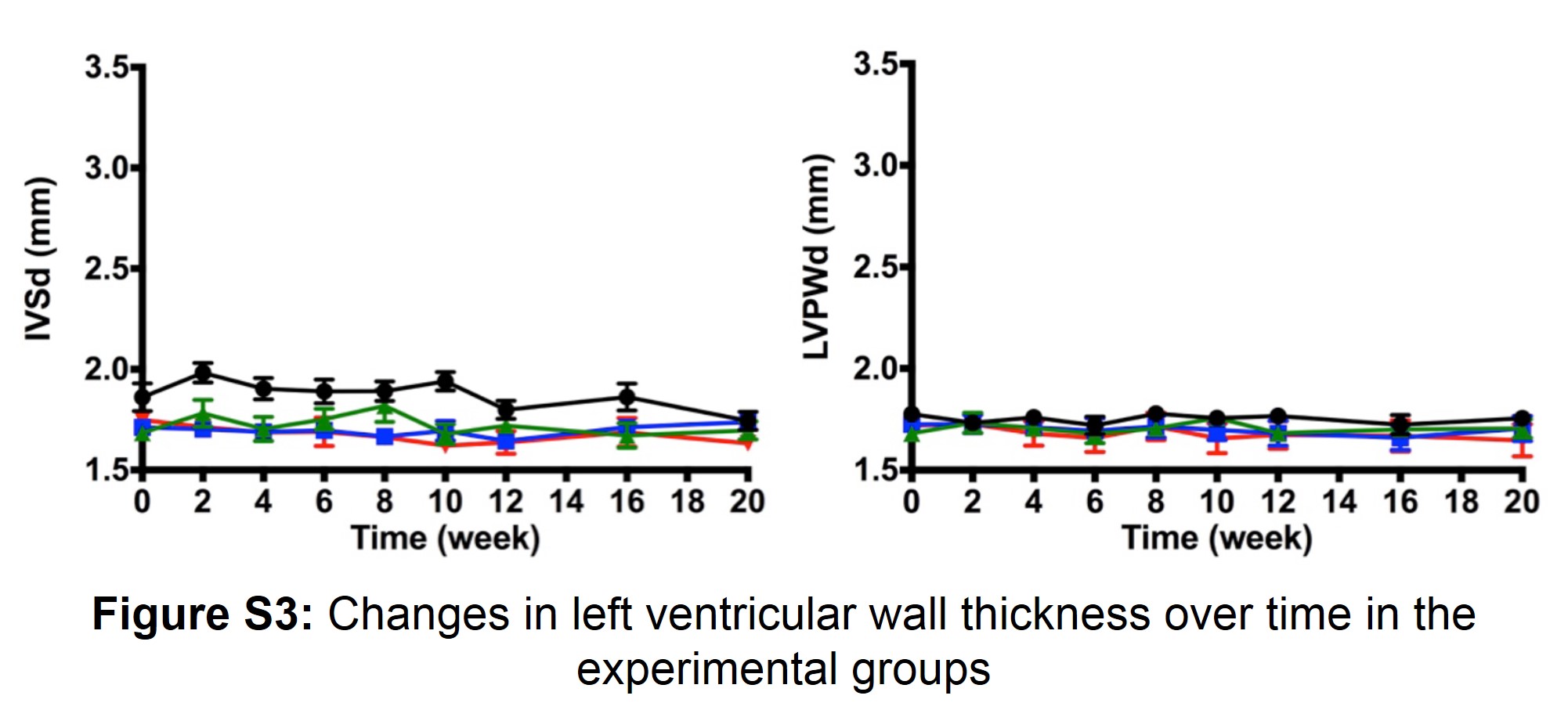
Heart weight was significantly higher in the MR+MI and MR groups compared to the MI only group, and sham groups at 2 weeks. These differences are seemingly preserved over the 20-week period, without a significant change in the rate of growth. The volume overload from MR seemed to trigger an early hypertrophic response, which may cause an increase in cardiomyocyte mass and thus heart weight. Changes in the LV wall thickness in these rats was minimal (Figure S3), but large increase in LV end diastolic volume were observed in animals in the MR+MI and MR groups. This increase in volume despite preserved wall thickness, may be attributed to the elongation of cardiomyocytes without changes in cross-sectional area, and serial addition of sarcomeres in the setting of MR. Gerdes et al. demonstrated such cardiomyocyte elongation in volume overload after an aorto-caval fistula(17). Interestingly such changes seem to be preserved or dominant when MR is associated with an ischemic LV, where the ischemia also contributes to cardiac hypertrophy and failure. This study only demonstrates these findings at the chamber level, and further studies are required to validate the findings at the cellular level. Another significant finding of this study is that the end diastolic volume is higher in MR+MI and MR groups, early and persistently through the entire follow-up duration, than MI or sham groups. Interpreting the volume data alongside the end-diastolic pressure volume relationship (EDPVR), a rightward shift in this relationship was observed at 2 weeks’ measurements but lost thereafter. It is likely that dilatation of the LV in the setting of MR may involve an early rise in myocardial compliance, which may involve some early structural changes in the myocardium(16). Despite such changes, end diastolic pressure was not normalized in the hearts with MR, providing a constant trigger for further remodeling and ultimately very large LV chamber volumes at 20 weeks. In MR+MI and MR groups, a significant reduction in end diastolic pressure was measured from 2 to 20 weeks, but not in the other groups.
Contractile Function of the Heart
End systolic volume, which is inversely proportional to contractility, was higher in ischemic hearts with MR than those without MR. Compared to their respective pre-operative baseline, ESV was elevated in the MR+MI group within 4 weeks after surgery and remained significantly elevated. In the MI only group, ESV was not significantly elevated until 8 weeks after surgery. Ejection fraction and fractional shortening also followed similar trends, confirming that MR induces early contractile dysfunction as well, beyond diastolic dilatation. Exacerbated systolic dysfunction of the ischemic LV in the presence of MR is slightly counter intuitive based on existing knowledge, as MR provides a pop-off valve effect and thus should reduce the afterload and preserve contraction. Despite these favorable hemodynamic conditions, it is possible that the rapid increase in the chamber volume combined with minimal radial hypertrophy of the LV may lead to abnormal chamber wall stress, that is several magnitudes higher than the afterload reduction from MR. Urabe et al. also found that volume overload from MR leads to myofibrillar loss, which may be likely in the ischemic hearts as well. Cellular function and ultrastructural changes in this setting are thus warranted but were not within the scope of this work(18).
Relevance to prior preclinical studies
Data from this study should be considered in the context of earlier preclinical reports, some of which corroborate our findings and others that do not. Our findings agree with those of Beeri et al.(14) and Beaudoin et al(13), who created clinically significant volume overload on the ischemic heart using an extracardiac left ventricular-to-atrial conduit in sheep. Beeri and colleagues reported that persistent volume overload alongside a myocardial infarction elevated EDV and reduced ejection fraction, whereas early repair of such a volume overload reversed this deleterious remodeling. Beaudoin and colleagues in the same model, demonstrated that delayed correction of volume overload did not reverse the remodeling adequately, with a strong momentum towards cardiac remodeling to failure. Our findings do not agree with those reported by Matsuzaki et al.(19), in an ovine model of post-infarction IMR, wherein correcting IMR at 8 weeks post-infarction did not improve cardiac function or remodeling. This discrepancy may be attributed to multiple factors, but specifically to the clinically insignificant regurgitant volumes that develop in these models within 8 weeks. In our experience, despite a sufficiently large infarction of the LV via OM2 and OM3 occlusion, these animals often develop only mild to moderate MR fraction, and not the clinically significant MR volumes mimicked in this study. Data from Matsuzaki et al. confirm this finding by reporting that the end diastolic pressure (EDP) in animals with and without IMR correction are identical, despite similar LV sizes. Furthermore, an undersizing ring was used to correct IMR in this model, which we have demonstrated to impair LV systolic and diastolic mechanics and thus inhibit reverse remodeling(20).
Clinical relevance of the findings
Data from this study demonstrates that presence of MR has a negative effect on the ischemic LV, by dilating it rapidly and contributing to systolic dysfunction. Earlier correction of MR in these ischemic LVs may thus be warranted, but further translational studies in larger animal models and humans may be necessary to confirm this hypothesis.
Limitations
The experimental nature of this work limits extrapolation of data to clinical practice in some respects. In the clinical setting, MR can develop acutely after an infarction (i.e. from papillary muscle rupture) or at later stages when the infarcted/ischemic LV has remodeled and dilated to an extent that mitral valve closure becomes challenging. This study was limited to only early/acute onset MR, and further studies are required to investigate the effects of late onset MR after the infarct has matured and the LV has begun to remodel in shape and function. The extent of myocardial damage in patients with MR varies between patients, whereas it was held constant in this experimental study. Thus, this data represents a subset population with a relatively large myocardial infarction, but may not reflect those with diffused myocardial disease. Patients with functional mitral regurgitation after a myocardial infarction could present with other comorbid conditions such as tricuspid regurgitation or aortic stenosis, which were not mimicked in this study. This study focuses on changes in LV function, but understanding changes in LV mechanics using passive distension experiments and material testing could provide further insights into the pathophysiology.
CONCLUSION
Mitral regurgitation worsens cardiac remodeling and pump function of the ischemic left ventricle, by inducing rapid left ventricular dilatation and systolic contractile dysfunction.
Supplementary Material
Figure S1: Experimental design for this study, depicting the time points at which cardiac echocardiography and invasive pressure-volume loops were performed
{kind=link}
Figure S2: Changes in pressure-volume loops over time within the same experimental group
{kind=link}
Figure S3: Changes in left ventricular wall thickness over time in the experimental groups
{kind=link}
Figure S4: Representative image of a myocardial infarct from a randomly selected heart depicting involvement of the anterior and lateral walls, but not the septum.
{kind=link}
Video: Procedure video depicting induction of mitral regurgitation in the beating rat heart model
Online supplement: Detailed surgical techniques to induce mitral regurgitation in the beating rat heart
Table S1: Longitudinal comparisons of different timepoints within the same experimental group for end diastolic volume and end systolic volumes.
CENTRAL PICTURE. Ischemic hearts with mitral regurgitation (left) and without mitral regurgitation (right).

CENTRAL MESSAGE
Ischemic hearts with mitral regurgitation had persistently higher chamber volumes than those without mitral regurgitation. Chamber wall stress and stroke work were higher, potentially increasing myocardial metabolism.
Table 1:
Changes in body weight, heart weight, indexed heart weight and heart rate in each experimental group at 2, 10 and 20 weeks after surgery.
| MR+MI | MI | MR | Sham | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk | |
| Body Weight (g) | 418.3 ± 30.6 | 450.0 ± 33.1 | 476.7 ± 38.9 | 399.3 ± 37.9 | 441.9 ± 40.5 | 473.8 ± 28.9 | 416.7 ± 29.3 | 456.3 ± 46.9 | 488.3 ± 28.9 | 421.8 ± 24.6 | 455.7 ± 23.8 | 488.0 ± 27.0 |
| Heart Weight (g) | 1.57 ± 0.13** | 1.84 ± 0.27* | 1.81 ± 0.27 | 1.38 ± 0.13 | 1.46 ± 0.07 | 1.61 ± 0.10^ | 1.48 ± 0.13* | 1.73 ± 0.23 | 1.71 ± 0.29 | 1.31 ± 0.06 | 1.43 ± 0.14 | 1.62 ± 0.10^# |
| HW Index (g/kg) | 3.70 ± 0.28 | 3.99± 0.50 ** | 3.83 ± 0.43 | 3.59 ± 0.48 | 3.35 ± 0.20 | 3.45 ± 0.38 | 3.64 ± 0.48 | 3.55 ± 0.29 | 3.56 ± 0.65 | 3.22 ± 0.30 | 3.03 ± 0.16 | 3.33 ± 0.38 |
| HR (bpm) | 315.7 ± 30.7 | 312.6 ± 31.3 | 320.3 ± 27.7 | 315.6 ± 14.2 | 292.8 ± 20.9 | 308.9 ± 30.6 | 313.1 ± 24.1 | 306.5 ± 9.0 | 309.3 ± 29.8 | 320.4 ± 20.0 | 307.8 ± 14.0 | 339.6 ± 25.1 |
Values are mean ± SD.
indicates p<0.05 versus 2 week in the same experimental group
indicates p<0.05 versus 10 week in the same experimental group
p < 0.05 versus the sham group at the same time
p <0.005 versus the sham group at the same time
p<0.0001 versus the sham group at the same time
HW index = Heart Weight (g) / Body Weight (kg), HR = heart rate
PERSPECTIVE.
Consensus is lacking regarding the contribution of MR to adverse remodeling of the ischemic left ventricle, thus an optimal strategy to manage MR is lacking. Data from this study demonstrates that MR has a rapid and persistent dilatory effect on the left ventricle, increasing the chamber volume significantly, and leading to elevated wall stress and stroke work, all of which can accelerate heart failure.
Acknowledgments
Funding
This work was partially funded by grants from American Heart Association (17POST33661278 and 14SDG20380081), the National Institutes of Health (1R01HL135145–01A1, 1R01HL133667–01A1, 1R01HL140325–01A1) and seed funding from the Carlyle Fraser Heart Center at Emory University Hospital Midtown, Atlanta, GA.
GLOSSARY
- MR
Mitral regurgitation
- MI
Myocardial infarction
- MV
Mitral valve
- MR
Mitral regurgitation
- EDV
End diastolic volume
- ESV
systolic volume
- EF
Ejection fraction
- ESPVR
End systolic pressure volume relationship
- EDPVR
End diastolic pressure volume relationship
- EDP
End diastolic pressure
- LV
Left ventricle
- LA
Left atrium
- TTE
Transthoracic echocardiography
- TEE
Transesophageal echocardiography
Footnotes
Conflicts of Interest
None relevant to this work
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Aronson D, Goldsher N, Zukermann R et al. Ischemic mitral regurgitation and risk of heart failure after myocardial infarction. Arch Intern Med 2006;166:2362–8. [DOI] [PubMed] [Google Scholar]
- 2.Bursi F, Enriquez-Sarano M, Jacobsen SJ, Roger VL. Mitral regurgitation after myocardial infarction: a review. The American journal of medicine 2006;119:103–12. [DOI] [PubMed] [Google Scholar]
- 3.Hillis GS, Moller JE, Pellikka PA, Bell MR, Casaclang-Verzosa GC, Oh JK. Prognostic significance of echocardiographically defined mitral regurgitation early after acute myocardial infarction. American heart journal 2005;150:1268–75. [DOI] [PubMed] [Google Scholar]
- 4.Grigioni F, Detaint D, Avierinos JF, Scott C, Tajik J, Enriquez-Sarano M. Contribution of ischemic mitral regurgitation to congestive heart failure after myocardial infarction. Journal of the American College of Cardiology 2005;45:260–7. [DOI] [PubMed] [Google Scholar]
- 5.Corporan D, Onohara D, Hernandez-Merlo R, Sielicka A, Padala M. Temporal changes in myocardial collagen, matrix metalloproteinases, and their tissue inhibitors in the left ventricular myocardium in experimental chronic mitral regurgitation in rodents. American journal of physiology Heart and circulatory physiology 2018;315:H1269–H1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigioni F, Enriquez-Sarano M, Zehr KJ, Bailey KR, Tajik AJ. Ischemic mitral regurgitation: long-term outcome and prognostic implications with quantitative Doppler assessment. Circulation 2001;103:1759–64. [DOI] [PubMed] [Google Scholar]
- 7.Smith PK, Puskas JD, Ascheim DD et al. Surgical treatment of moderate ischemic mitral regurgitation. The New England journal of medicine 2014;371:2178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone GW, Lindenfeld J, Abraham WT et al. Transcatheter Mitral-Valve Repair in Patients with Heart Failure. The New England journal of medicine 2018. [DOI] [PubMed] [Google Scholar]
- 9.Stone GW, Lindenfeld J, Abraham WT et al. Transcatheter Mitral-Valve Repair in Patients with Heart Failure. The New England journal of medicine 2018;379:2307–2318. [DOI] [PubMed] [Google Scholar]
- 10.Obadia JF, Messika-Zeitoun D, Leurent G et al. Percutaneous Repair or Medical Treatment for Secondary Mitral Regurgitation. The New England journal of medicine 2018;379:2297–2306. [DOI] [PubMed] [Google Scholar]
- 11.Shi W, Mclver BV, Kalra K et al. A Swine Model of Percutaneous Intracoronary Ethanol Induced Acute Myocardial Infarction and Ischemic Mitral Regurgitation. Journal of cardiovascular translational research 2017;10:391–400. [DOI] [PubMed] [Google Scholar]
- 12.Llaneras MR, Nance ML, Streicher JT et al. Large animal model of ischemic mitral regurgitation. The Annals of thoracic surgery 1994;57:432–9. [DOI] [PubMed] [Google Scholar]
- 13.Beaudoin J, Levine RA, Guerrero JL et al. Late repair of ischemic mitral regurgitation does not prevent left ventricular remodeling: importance of timing for beneficial repair. Circulation 2013;128:S248–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beeri R, Yosefy C, Guerrero JL et al. Early repair of moderate ischemic mitral regurgitation reverses left ventricular remodeling: a functional and molecular study. Circulation 2007;116:I288–93. [DOI] [PubMed] [Google Scholar]
- 15.Braunwald E, Welch GH Jr., Sarnoff SJ. Hemodynamic effects of quantitatively varied experimental mitral regurgitation. Circulation research 1957;5:539–45. [DOI] [PubMed] [Google Scholar]
- 16.Corporan D, Onohara D, Hernandez-Merlo R, Sielicka A, Padala M. Temporal Changes in Myocardial Collagen, Matrix Metalloproteinases and their Inhibitors in Experimental Chronic Mitral Regurgitation in Rodents. American journal of physiology Heart and circulatory physiology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerdes AM, Campbell SE, Hilbelink DR. Structural remodeling of cardiac myocytes in rats with arteriovenous fistulas. Laboratory investigation; a journal of technical methods and pathology 1988;59:857–61. [PubMed] [Google Scholar]
- 18.Urabe Y, Mann DL, Kent RL et al. Cellular and ventricular contractile dysfunction in experimental canine mitral regurgitation. Circulation research 1992;70:131–47. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzaki K, Morita M, Hamamoto H et al. Elimination of ischemic mitral regurgitation does not alter long-term left ventricular remodeling in the ovine model. Ann Thorac Surg 2010;90:788–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu D, McBride E, Wong K, Kalra K, Padala M. Undersized mitral annuloplasty impairs diastolic relaxation mechanics of the left ventricle in a swine model of chronic ischemic mitral regurgitation. 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Experimental design for this study, depicting the time points at which cardiac echocardiography and invasive pressure-volume loops were performed
Figure S2: Changes in pressure-volume loops over time within the same experimental group
Figure S3: Changes in left ventricular wall thickness over time in the experimental groups
Figure S4: Representative image of a myocardial infarct from a randomly selected heart depicting involvement of the anterior and lateral walls, but not the septum.
Video: Procedure video depicting induction of mitral regurgitation in the beating rat heart model
Online supplement: Detailed surgical techniques to induce mitral regurgitation in the beating rat heart
Table S1: Longitudinal comparisons of different timepoints within the same experimental group for end diastolic volume and end systolic volumes.