

Abstract
Allostery is a ubiquitous biological mechanism in which a distant binding site is coupled to and drastically alters the function of a catalytic site in a protein. Allostery provides a high level of spatial and temporal control of the integrity and activity of biomolecular assembles composed of proteins, nucleic acids, or small molecules. Understanding the physical forces that drive allosteric coupling is critical to harnessing this process for use in bioengineering, de novo protein design, and drug discovery. Current microscopic models of allostery highlight the importance of energetics, structural rearrangements, and conformational fluctuations, and in this review, we discuss the synergistic use of solution NMR spectroscopy and computational methods to probe these phenomena in allosteric systems, particularly protein-nucleic acid complexes. This combination of experimental and theoretical techniques facilitates an unparalleled detection of subtle changes to structural and dynamic equilibria in biomolecules with atomic resolution, and we provide a detailed discussion of specialized NMR experiments as well as the complementary methods that provide valuable insight into allosteric pathways in silico. Lastly, we highlight two case studies to demonstrate the adaptability of this approach to enzymes of varying size and mechanistic complexity.
Keywords: Allostery, NMR, Molecular dynamics, Protein dynamics, Community network analysis
Introduction
Allostery is a fundamental biomolecular regulatory mechanism characterized by communication between spatially distinct sites within a protein. The binding of an allosteric effector (i.e., peptide, small molecule) modulates substrate binding affinity (Kd, K-type) and/or enzymatic activity (Vmax, V-type) by altering the structure and/or dynamics of the protein matrix (Fenton 2008). The idea that subtle conformational motions affect the energetic landscape of a protein to transmit chemical information has evolved with experimental technology, as novel allosteric systems do not always conform to classical paradigms from phenomenological models (Koshland Jr. et al. 1966; Monod et al. 1965). Coupled to this observation is the notion that amino acid “networks” intrinsic to the protein are activated by endogenous or exogenous stimuli (Fig. 1). These allosteric pathways present an opportunity for fine-tuning or controlling biological responses; thus, ensemble models of allostery, where proteins sample microstates along a free energy continuum (Motlagh et al. 2014), have replaced a purely structural view of discrete conformational changes. However, a unifying model for all allosteric systems remains elusive. Ensemble models describe differing proteins with the same thermodynamic parameters, but such models generally exclude communicative pathways between active and regulatory sites, even though such a connection is necessary from an experimental point-of-view. Coupled communication organizes the active and allosteric sites of enzymes for substrate binding and mediates proper functionality. Despite advancements in biochemical and biophysical probes, the complexity of these mechanisms is such that allosteric pathways remain largely uncharacterized, especially in high molecular weight proteins.
Fig. 1.

Allosteric pathways are composed of amino acid nodes that rely on the binding of a substrate or activator molecule to engage the network, often by stimulating local or global flexibility of the protein structure. Alteration of the allosteric pathway, shown here as a point mutation or the introduction of a non-competitive inhibitor, can abolish connections made by critical nodes, resulting in attenuated structural flexibility and catalytic activity. Hijacking these routes of chemical information transfer for distal control of protein function is a promising therapeutic approach
Identifying critical nodes along these pathways is desirable in drug discovery and tailored therapeutic design, and it is critical to engage a multitude of techniques, both complementary and orthogonal, to fully investigate allosteric mechanisms. Here, we highlight synergistic solution nuclear magnetic resonance (NMR) and computational studies used to elucidate structural and dynamic changes resulting from allosteric signaling. NMR is highly sensitive to subtle changes in protein structure and is extremely powerful for quantifying dynamic equilibria on a wide range of timescales (ps–sec). NMR is also the preferred method to validate computational predictions in ligand screening/docking and molecular dynamics (MD) simulations. Advanced computational techniques such as community network analysis and eigenvector centrality (EC) have become essential for the prediction and validation of allosteric pathways (Negre et al. 2018b; Rivalta et al. 2012), particularly since sophistication of modern computational tools expands the range of dynamic timescales that can be reliably probed, allowing access to slower dynamics utilized by large enzyme complexes for long-range communication.
Although other structural techniques such as free-electron laser crystallography can probe dynamic processes on timescales similar to those of NMR (Mizohata et al. 2018; Nango et al. 2016)), its connection to MD simulations is not as well-established and crystallography still requires multiple static snapshots to infer solution-like behavior. Cryo-electron microscopy (EM), by contrast, is adept at probing dynamics in very large complexes (Kujirai et al. 2018), but lacks the atomistic resolution of NMR, the ability to quantitate motional timescales, and is not well-suited to studies of biomolecules < 40 kDa. NMR is able to accurately quantitate both the ensemble structure and dynamics across many timescales, and its coupling to MD simulations to drastically improve the identification and characterization of allostery in protein complexes > 50 kDa is well-established. These studies, aided by modern experimental practices such as perdeuteration (Venters et al. 1996), transverse relaxation-optimized spectroscopy (TROSY) (Pervushin et al. 1997), sparse isotopic labeling (Tugarinov et al. 2006; Tugarinov and Kay 2003), 15N-detection (Takeuchi et al. 2016), and non-uniform sampling (NUS) (Barna et al. 1987; Delaglio et al. 2017), have facilitated NMR studies of much larger systems by preserving signal-to-noise and deconvoluting crowded spectra (Grishaev et al. 2005).
Scope of the review
In this review, we discuss advances in solution NMR and computation that facilitate characterization of allosteric pathways and lead to functional insights. A large number of elegant reports utilizing these methods have proven the efficiency of a combined NMR/computational approach (Fizil et al. 2018; Jensen et al. 2014; Turupcu et al. 2019). These studies have paved the way for the investigation of systems of increasing size and complexity, such as protein-nucleic acid complexes where allosteric effects are at the basis of mechanistic function. Here, we focus on two important protein-nucleic acid complexes—DNA polymerase β and the Clustered Regularly Interspaced Palindromic Repeats (CRISPR)-Cas9 —as case studies to detail how multi-domain structures transmit allosteric signals. In these systems, the complexity of the allosteric response is being tackled through a highly integrated approach, harnessing solution NMR and advanced MD methodologies, in combination with network models derived from graph theory (Fig. 2).
Fig. 2.

Integration of experimental and computational methods for studies of protein allostery. NMR spin relaxation experiments are conducted in parallel with MD simulations to identify clusters of amino acids exhibiting flexibility and inter-residue correlations (via network analysis and centrality). The data are pooled and compared to identify overlapping sites of flexibility that may be relevant to an allosteric pathway. Site-directed mutagenesis is conducted in vitro and in silico, and the effect on the structure, dynamic signal and correlations, and functional response are quantified. The process is iterated (i.e., carried out on mutants) to pinpoint changes in the pathway as well as critical allosteric nodes, after which allosteric modulators, such as drug-like small molecules, can be introduced into the system and tested biochemically
Solution NMR studies of protein dynamics and allostery
Conformational rearrangements occur frequently during enzymatic mechanisms, often as a result of ligand binding where the associated conformational changes can be rate-limiting to catalysis (Benkovic and Hammes-Schiffer 2003; Lisi and Loria 2017; Watt et al. 2007; Whittier et al. 2013). Thus, characterization of relevant dynamics associated with these transitions is essential for understanding the biochemical mechanism. The equilibria between energetically similar apo (ligand-free) and liganded enzymatic states can be characterized by numerous biophysical techniques; however, in many cases, it is unclear whether the enzyme in question is intrinsically capable of sampling the relevant conformations required for allosteric activation. To address this question in molecular detail, experimental and computational approaches that can accurately quantitate multi-timescale protein motions are required (Yuan et al. 2015). It is now well-established that allosteric communication contains enthalpic and entropic thermodynamic components (Tsai et al. 2009), the latter of which is strongly influenced by conformational fluctuations (Henzler-Wildman and Kern 2007; Tsai et al. 2008; Volkman et al. 2001; Wand 2001). Well-vetted NMR methods for the study of protein dynamics have been reported (Dyson and Wright 2004; Loria et al. 2008; Palmer 3rd 2004; Palmer III 2015), and we will briefly describe how these methods can be used in concert with molecular simulations to probe biological processes that access weakly populated conformational states (Baldwin and Kay 2009; Loria et al. 2008; Mittermaier and Kay 2009; Palmer 3rd 2004; Palmer III 2015) and equilibrium dynamics that contribute to the configurational entropy of the system (Igumenova et al. 2006; Mittermaier and Kay 2006; Trbovic et al. 2009).
Solution NMR spectroscopy is performed under physiologically relevant conditions and is capable of determining atomic resolution protein structures, capturing site-specific conformational changes, and quantifying protein motions spanning the ps–sec timescales. The contributions of many laboratories to the application of novel NMR experiments and elucidation of mechanisms by which proteins propagate chemical signals have demonstrated the diversity of allosteric systems, though only a few examples can be discussed in detail. Novel examples of dynamically-driven allostery continue to surface where NMR experiments and/or molecular dynamics (MD) simulations have been used to establish the mechanism (Dhulesia et al. 2008; Hwang et al. 2004; Jacoby et al. 1996; Jarymowycz and Stone 2008; Kalodimos 2011; Liu et al. 2008; Wiesner et al. 2006; Zhang et al. 2012; Fuentes et al. 2004; Masterson et al. 2010; Petit et al. 2009; Coyne and Giedroc 2013; Lee 2015; Chakravorty et al. 2013; Shi and Kay 2014).
NMR probes local and global biomolecular structure and dynamics (Shi and Kay 2014) (Velyvis et al. 2007, 2009; Lipchock and Loria 2010) to illuminate pathways of allosteric cross-talk (Figs. 1 and 3). Dynamic processes specifically related to allostery occur primarily in the μs–ms time regime, though contributions of faster (ps–ns) timescale motions have been noted, particularly as it relates to the optimization of the protein scaffold for effector binding and quantitation of configurational entropy in protein systems (Capdevila et al. 2017; Caro et al. 2017; Tzeng and Kalodimos 2012). The suite of NMR spin relaxation experiments designed to probe these time regimes have been used to interrogate internal protein motions (Akke and Palmer 1996; Loria et al. 1999a; Mulder et al. 2002) (Grey et al. 2003; Massi et al. 2005), protein folding (Hill et al. 2000; Korzhnev et al. 2004c; Tang et al. 2006), ligand binding events (Mittag et al. 2003; Tolkatchev et al. 2003), and enzymatic mechanisms (Cole and Loria 2002; Beach et al. 2005; Kovrigin and Loria 2006a, b; Kempf and Loria 2002; Berlow et al. 2007). The theoretical details of these solution NMR experiments are discussed in the next section, as are useful NMR observables that have powerful synergy with computation.
Fig. 3.

Solution NMR is an ideal tool to probe biomolecular structure and dynamics. a Interaction of a protein with a binding partner (i.e., substrate or inhibitor) can cause a conformational transition that alters the local chemical environments of nearby amino acids or domains, perturbing NMR chemical shifts. b Conformational exchange motions can be readily measured with NMR by sampling peak intensities as a non-equilibrium state undergoes relaxation. Changes in peak intensities as a function of a variable relaxation delay provide signatures of flexible amino acids within a protein matrix. c Protein motions can be measured by NMR across the entire timescale of biology. Critically for this review, NMR experiments developed to probe slow timescale (μs-ms) dynamics are discussed in detail below
Chemical shift perturbations
The resonant frequency (ω0), or Larmor precession, of an NMR-active nucleus is dependent on the local magnetic field (B), with contributions from the external field (B0) and local electronic environment (Bi), where B = B0 + Bi. In practice, resonant frequencies are referenced to a standard frequency in order to quantitate the chemical shift (δ, in parts-per-million or Hertz) and minimize the contribution of B0. Thus, the chemical shift is extremely sensitive to changes in the local electronic environment and can be measured with high precision. Small changes in protein structure caused by ligand binding or a transition between inactive and active states (T → R in allosteric nomenclature) are easily detectable with NMR chemical shifts. In a simple two-site exchange model describing the interaction of an enzyme with an allosteric effector, NMR-detectable nuclei sample two unique structural (and magnetic) environments as the allosteric effector binds/unbinds. The populations of each state, pT (tense or inactive) and pR (relaxed or active), have chemical shifts δT and δR, respectively. The chemical shift difference between these two states is given by Δω = |δR- δT| and the rate of chemical exchange between states T and R is given by kex = kT → R + kR → T. In the case of fast-to-intermediate exchange between conformers, where kex ≥ Δω, the observed chemical shift (δobs = PRδR + PTδT) is a population weighted average of the T and R states, where pR/T are the equilibrium populations of each conformational state and δR/T are their respective NMR chemical shifts (Fig. 4). Under slow exchange conditions (kex˂ Δω), two resonances are observed and the equilibrium populations are proportional to the peak volumes. Thus, changes in NMR chemical shifts for any allosteric system broadly represent a shift in the T-to-R equilibrium.
Fig. 4.
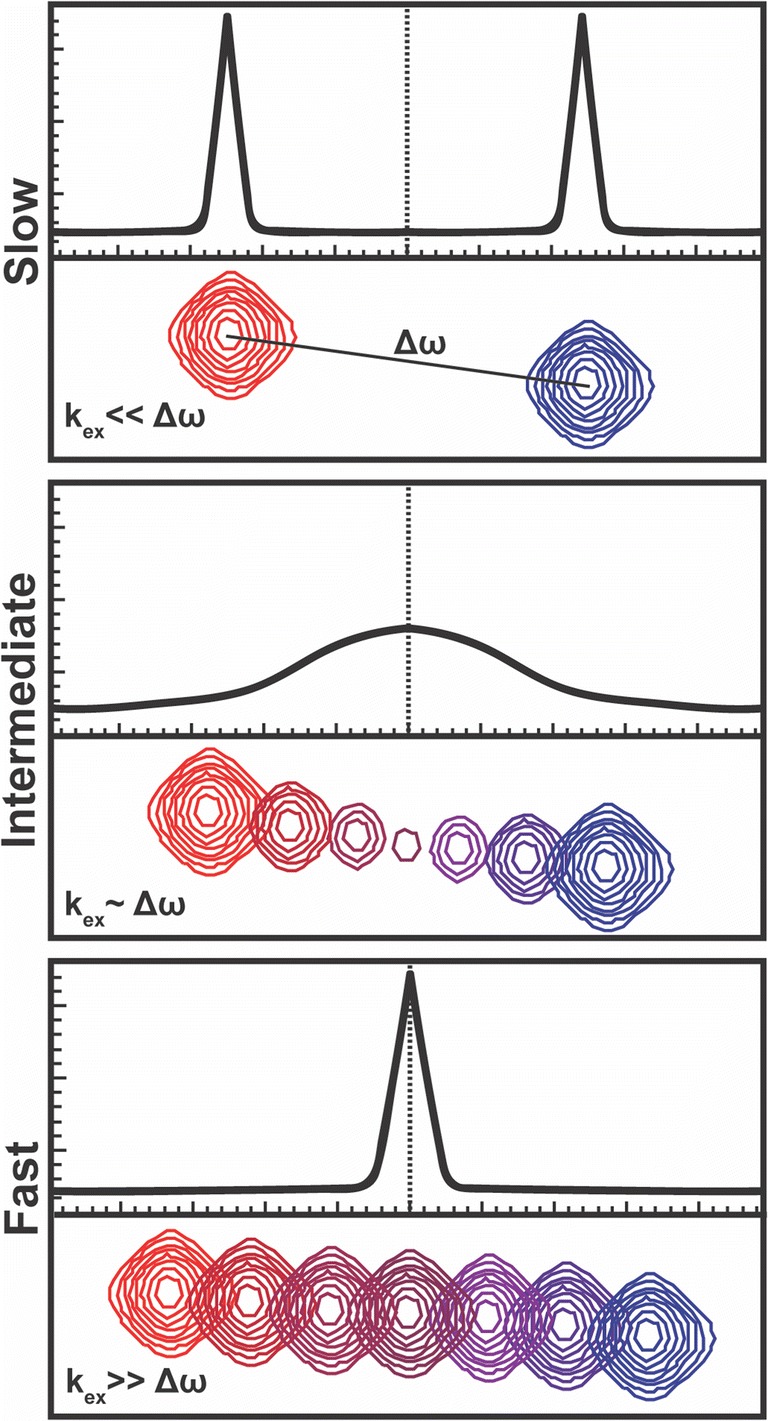
Exchange regimes dictate NMR chemical shift behavior based on the relationship between kex and Δω. For three distinguishable time regimes: slow (top), intermediate (middle), and fast (bottom), the 1D projection of a 1:1 mixture is shown for a 2D titration series. Analysis of the NMR lineshapes provide information about kex and Δω
Using this theoretical model of NMR chemical shifts, several methods have been developed to identify allosteric residues in proteins (Cui et al. 2017). One example, Chemical Shift Covariance Analysis (CHESCA) first reported by Melacini and coworkers (Selvaratnam et al. 2011), analyzes the chemical shifts of multiple allosteric states of a protein to identify pairwise correlations in covariant residues that compose an allosteric network. Alternatively, protocols to fit NMR lineshapes in a series of 2D spectra collected as allosteric effectors are titrated into protein samples have been described to extract both thermodynamic (K and ΔG) and kinetic information (kex, kT → R, and kR → T) (Kovrigin 2012; Shinya et al. 2017; Waudby et al. 2016). The sensitivity of these methods to communication between active and allosteric sites is limited by the assumption that extrinsic factors such as buffer conditions, temperature, and pH have little effect on the allosteric ensemble, as well as by the fact that the chemical shift itself contains contributions from ligand binding, structural reshuffling at protein interfaces, and small pKa shifts as hydrogen bond networks are altered. Coupling NMR chemical shift analysis to molecular simulations (vide infra) provides a solution to the inherent complexities of a purely chemical shift-based treatment of allosteric equilibria by utilizing MD trajectories to visualize structural states (i.e., conformational changes) along the dynamic ensemble.
Fast timescale conformational motions (ps–ns)
Several thorough reviews of NMR spin relaxation have been given (Cavanagh et al. 2007; Lisi and Loria 2016a, b; Palmer 3rd 2004; Palmer 3rd and Massi 2006), so here we present a focused overview of useful experiments for biological investigations of allostery. Motions on the ps–ns timescale, which are faster than the rotational diffusion of a protein, are examined to quantitate equilibrium fluctuations in bond vectors of individual amino acids and changes in chemical shift anisotropy or dipolar interactions between nuclei (Clore et al. 1990; Peng and Wagner 1992; Ishima and Nagayama 1995; Mandel et al. 1995; Farrow et al. 1995; d’Auvergne and Gooley 2003; Cole and Loria 2003). Fast timescale dynamics associated with entropically driven allostery have been described for several systems (Lisi et al. 2016; Tzeng and Kalodimos 2012; Wand 2017) and are a critical component of the free energy landscape that regulates the population of allosterically activated (i.e., R) and inactive (T) states. Pico-nanosecond motions are readily measured by NMR and classical MD, providing an avenue to synergistically probe the changing energy landscapes and three-dimensional structures associated with protein flexibility. For a heteronuclear spin-1/2 pair, i.e., an amide proton-nitrogen (1H-15N) of the protein backbone, longitudinal (Sz,R1) and transverse (Sx/y,R2) relaxation of non-equilibrium magnetization to Boltzmann equilibrium are described by (Abragam 1961)
| 1 |
| 2 |
and the steady-state heteronuclear nuclear overhauser effect (NOE) is given by
| 3 |
where ωI and ωS are the Larmor frequencies of the I (1H) and S (15N) nuclei, c = (2/15)Δσ2ωS2, γI and γS are the gyromagnetic ratios of the I (1H) and S (15N) nuclei, Δσ is the chemical shift anisotropy of the S (15N) nucleus and σNOE is the cross-correlation relaxation term from I → S (1H → 15N). The contribution of μs–ms conformational motion to transverse relaxation is contained in Rex, which is negligible in many cases. The dipolar coupling constant (d) in the R1 and R2 cases is described by Eq. 4,
| 4 |
where μo is the permeability of free space, ħ is Planck’s constant divided by 2π, γI and γS are again the gyromagnetic ratios of the I (1H) and S (15N) nuclei, and 〈rIS〉 is the average bond length between I and S. The spectral density function describing overall and internal bond vector fluctuations is written as
| 5 |
where τc is the rotational correlation time of the biomolecule, and τe is the effective correlation time for internal motions. S2 is a generalized order parameter ranging from zero-to-one that is commonly related to the configurational entropy of protein bond vectors, where lower values indicate heightened flexibility (Akke et al. 1993; Li et al. 1996; Yang and Kay 1996). Qualitative assessments of conformational entropy through the average order parameter, 〈S2〉, have been described by Bracken et al. (Kneller et al. 2002) using the product of experimentally measured R1 and R2 relaxation rates
| 6 |
where 〈R1R2〉 is the mean value and R1R2max is the calculated maximum value. Thus, R1R2 values below the mean of the data correspond to sites with lower order parameters (i.e., heightened flexibility).
Characterization of the amino acid side chain dynamics (i.e., in methyl groups of hydrophobic residues such as Ile-δCH3, Leu-δCH3, and Val-γCH3) is an increasingly popular probe in large allosteric systems with dense backbone 1H-15N NMR spectra. Fast motions of methyl-containing side chains can be characterized by 1H-1H dipolar cross-correlated relaxation rate constant, η, expressed in Eq. 7 and described in studies of malate synthase G (Tugarinov et al. 2003), ribonuclease (Gill et al. 2019), and fatty acid binding protein (Liu et al. 2003).
| 7 |
Here, rHH is the distance between methyl protons, γH is the proton gyromagnetic ratio, and τc is once again the rotational correlation time. is the second Legendre polynomial in which θaxis,HH is the angle between the 3-fold symmetry axis of the methyl group and the vector between two methyl protons (i.e., 90°). The order parameter S2 describes the amplitude of the equilibrium fluctuations at the methyl symmetry axis. and are the relaxation rates for the single quantum 1H coherences for fast and slow relaxation, respectively.
Slower timescale motions (μs–ms)
Molecular motions occurring on the μs–ms timescale are associated with conformational changes that participate in allosteric transitions and can be commensurate with rates of enzymatic catalysis (Whittier et al. 2013). Transitions between inactive and active enzymatic states (i.e., T ⇄ R where kex = kT → R + kR → T) cross significant energy barriers, modulate the isotropic chemical shift of the atoms involved, broaden the associated NMR resonances, and increase the transverse relaxation rate constant (R2) by an amount Rex where R2 = R20 + Rex and R20 is the “motion-free” transverse relaxation rate that describes motion at or near the Larmor frequency. In the fast exchange limit, Rex = pTpRΔω2/kex, where Δω is the chemical shift difference between states T and R, pT/R are the equilibrium populations of the (assumed) two conformations, and kex is the exchange rate constant of the dynamic process. Relaxation experiments probing μs–ms dynamics have been widely employed to investigate allosteric mechanisms in kinases (Masterson et al. 2010; Srivastava et al. 2014), molecular machines, and DNA binding proteins (Capdevila et al. 2017; Vise et al. 2005).
Micro-millisecond conformational exchange is characterized by relaxation-compensated Carr-Purcell-Meiboom-Gill (rcCPMG) (Loria et al. 1999a, b) and/or off-resonance rotating frame relaxation (R1ρ) experiments (Akke and Palmer 1996; Deverell et al. 1970), both of which quantify the rate of conformational exchange (kex), the equilibrium populations of each conformer (pa,pb), and the chemical shift difference between conformers (∆ω) (Lisi and Loria 2016a). The measured value of Rex in the rcCPMG experiment depends on τcp, the repetition rate of a 180° pulse (i.e., [τ-180-τ]n) (Carver and Richards 1972; Jen 1978; Davis et al. 1994) or the effective field strength ωe in the R1ρ experiment (Deverell et al. 1970). The variation of the measured transverse relaxation rate (R2(1/τcp)) with 180° pulse spacing (τcp) in the single quantum rcCPMG experiment applied to backbone amide groups in biomolecules (Loria et al. 1999a) is expressed in Eq. 8 (Carver and Richards 1972; Jen 1978; Davis et al. 1994)
| 8 |
where and are the intrinsic (i.e., motion-free) transverse relaxation rates of the two sites and D± and η± are
| 9 |
and ; .
Equation 8 is required when conformational motion is in the slow-to-intermediate exchange regime (kex≤ ∆ω); however, under fast exchange conditions (kex> ∆ω), the expression simplifies to (Luz and Meiboom 1963)
| 10 |
where ϕex= pApBΔω2 (Ishima and Torchia 1999; Millet et al. 2000; Kovrigin et al. 2006; Lisi and Loria 2016a). Adaptation of the rcCPMG experiment for multiple quantum (MQ) relaxation dispersion studies of Ile-δCH3, Leu-δCH3, and Val-γCH3 methyl groups (Korzhnev et al. 2004a, b; Isaacson et al. 2007) has facilitated NMR investigations of large multimeric proteins (Fig. 5) and machines.
Fig. 5.

13CH3-labeling of Met-Ile-Leu-Val-Ala-Thr residues can deconvolute NMR spectra of large proteins and effectively resolve amino acid types by chemical shift. NMR studies of small subdomains within larger proteins and complexes remain popular, and the ability to use 13C-methyl labeling to build up NMR spectra of large complexes from smaller fragments can aid in the assignment and interpretation of NMR data
Molecular motions occurring faster than can be quantified by rcCPMG experiments (~ 105 s−1) are probed by off-resonance R1ρ (Akke and Palmer 1996; Trott and Palmer 3rd 2002)
| 11 |
where effective magnetic fields for conformers A and B are given by and , and the offsets from the radio-frequency (RF) carrier for resonances A and B, and population-averaged resonances are represented as ωA, ωB, and ωiso, respectively. The RF field strength ω1 has a tilt angle θ = arctan (ω1/ωiso), and in the fast exchange limit, the R1ρ experiment is expressed in Eq. 12
| 12 |
where
| 13 |
The slower molecular motions probed by rcCPMG and R1ρ can be accessed, to some extent, by long MD simulations that require high computing power. Pushing computation further into the micro-millisecond regime is a new frontier and would allow for a more direct comparison with NMR data on the most critical timescale for allostery.
Computational approaches for elucidation of allosteric pathways
Molecular dynamics simulations
To date, MD simulations have been powerful for investigations of conformational dynamics and subtle fluctuations associated with allostery (Dokholyan 2016; Guo and Zhou 2016; Wagner et al. 2016; Wodak et al. 2019). Notably, the group of Luthey-Schulten combined MD simulations with graph theory (Sethi et al. 2009), first describing intricate allosteric responses as a communication network. In large biomolecular complexes, i.e., a multi-domain protein with nucleic acid elements, the conformational landscape is characterized by slow dynamical motions, thereby affecting the transmission of the allosteric response over longer timescales (Kern and Zuiderweg 2003). The synergistic use of MD and NMR to quantify contributions of fast timescale dynamics to protein function and allosteric regulation, most notably via order parameters (S2), is well-documented (Salvi et al. 2016; Wand 2001, 2017). However, a remarkable constraint to MD simulations in capturing allosteric responses of large biomolecular systems has been the difficulty of quantifying slow timescale (μs–ms) dynamics with computation. The recently introduced the application of Gaussian-accelerated MD (GaMD) (Miao et al. 2015) has now enabled unconstrained enhanced sampling that captures μs–ms timescale events, facilitating a stronger degree of complementarity between simulation and NMR in studies of dynamic allostery.
GaMD adds a harmonic boost potential that smoothes the potential energy surface of the simulation system, accelerating the transitions between low-energy states. For a system of N atoms at positions , when the system potential is lower than a threshold energy E =Vmax, the energy surface is modified by adding a boost potential as
| 14 |
where k is the harmonic force constant. The two adjustable parameters, E and k, are determined from classical MD to collect the maximum (Vmax), minimum (Vmin), average (Vavg), and standard deviation (σV) of the system potential energies. When E is set to the lower bound E = Vmax, k0 can be calculated as
| 15 |
where σ0 is a user-specified upper limit (i.e., 10 kBT) for accurate energetic reweighting. Alternatively, when the threshold energy E is set to its upper bound,
| 16 |
with calculated between 0 and 1. The boost potential follows a near-Gaussian distribution, allowing for robust sampling, while ensemble canonical average is reached by reweighting each point in the configuration space on the modified potential by the strength of the Boltzmann factor of the bias energy, exp[βΔV(rt(i))] at that particular point. GaMD has been successfully employed to describe slow timescale biomolecular processes in remarkably large biological systems, such as the CRISPR-Cas9 complex (Palermo 2019; Palermo et al. 2017a; Ricci et al. 2019) and G protein-coupled receptors (Miao and McCammon 2016), as well as the intracellular signaling pathway of a medically relevant G protein mimetic nanobody (Miao and McCammon 2018). GaMD is highly effective at preserving the information arising from slow timescale motion that is necessary for tracing allosteric signals in large biomolecular complexes, which has been typically carried out solely through network models.
Network models
Network models are efficient analyses to characterize allosteric effects (Dokholyan 2016; Guo and Zhou 2016; Sethi et al. 2009; Wagner et al. 2016; Wodak et al. 2019) that rely on correlation matrices that inform whether spatially distant sites in a protein are coupled in motion over dynamic trajectories. Generalized correlation (GCij) methods (Lange and Grubmuller 2006) are often employed to capture non-collinear correlations between residue pairs (i, j) based on Shannon’s entropy and describe both linear and non-linear coupled motions. Based on GCij, a dynamical network model is built describing the system as a graph of nodes (i.e., amino acid Cα), connected by edges, with a distance weighted according to wij= − log GCij. The resulting weighted graph is used to structure the system’s correlations through community network analysis (CNA) (Sethi et al. 2009), which defines groups of highly correlated residues and the strength of their interconnectivity to quantify chemical information flow. Residues clustered together by CNA have shown similar conformational exchange parameters in NMR experiments (Lisi et al. 2016; Rivalta et al. 2012). Further, the reshuffling of dynamic community networks upon allosteric ligand binding in MD simulations has informed NMR studies of allosteric pathways (Lisi et al. 2017).
To extract the optimal pathways for information transfer across protein networks, several approaches have been proposed. In a seminal paper (Sethi et al. 2009), Luthey-Schulten and coworkers analyzed the weighted graph by computing the “shortest pathways” connecting pairs of functionally important residues. In these weighted dynamic networks, the “shortest pathways” were calculated using the Floyd-Warshall algorithm (Floyd 1962), which sums the lengths (wij) of all edges of various pathways connecting two distant residues and identifies the route displaying the shortest total length. Another strategy utilizes the Dijkstra algorithm (Dijkstra 1959), which is widely employed in graph theory, particularly in cartography, to determine shortest routes of travel. This algorithm uses the GCij coefficients as a metric to identify routes composed by inter-node connections (wij), that minimize the total distance (and maximize correlation) between amino acids. Thus, the Dijkstra algorithm optimizes motional momentum transport that facilitates efficient signaling between selected residues. Clusters of flexible residues identified in NMR experiments that fall along the computationally derived route of information transfer are likely to be directly involved in the allosteric mechanism.
Eigenvector centrality
One of the cornerstones of network theory is the concept of centrality (Doshi et al. 2016), i.e., the relative influence of a node or cluster of nodes to the network (Alvarez-Socorro et al. 2015; Borgatti 2005; Hanke and Foraita 2017; Newman 2010). A recently proposed method harnesses the so-called eigenvector centrality (EC) (Negre et al. 2018b), where the EC of a node, ci, is defined as the sum of the centralities of all nodes that are connected to it by an edge, Aij where the edges Aij are elements of the adjacency matrix A (based on GCij), and λ is the eigenvalue associated with the eigenvector composed by ci elements. This approach relies on assigning functional dynamics to the major collective mode of the system (i.e., the first eigenvector of A), and hence, λ is the highest eigenvalue of A. The EC estimation quantifies the degree of connectivity of each amino acid or nucleobase in the system, measuring how well the nodes of the protein network are connected to other well-connected nodes. This enriches the information derived from betweenness centrality (BC) metrics, which measures instead how information flows between nodes (or edges) in a network (Sethi et al. 2009). Distinct from other centrality descriptors, EC also serves as a measure of the connectivity against a fixed scale when normalized and can reliably compare the connectivity of a system following allosteric effector binding. This concept has been especially critical to identifying targets for site-directed mutagenesis and inhibitor docking within allosteric networks. Residues with high EC may also be dynamic transducers in proteins, where mutations at these sites stimulate or attenuate protein motions (Lisi et al. 2017; Negre et al. 2018b). EC is also valuable to obtain the main mode of collective correlation responsible for the allosteric signal, beyond the capabilities of standard principal component methods.
Reliability of simulated ensembles with respect to NMR experiments
One critical aspect of a combined NMR-computational approach lies in assessing the reliability of the simulated ensemble with respect to the NMR experiment. A useful way to gain insight into how well the structural and dynamical features captured by MD represent NMR data is to compute the NMR chemical shifts based on the conformational ensemble obtained via MD simulations. A straightforward way to predict the 1H, 13C, and 15N chemical shifts from an ensemble of structures derived via MD simulations relies on the use of the SHIFTX2 code (Han et al. 2011), which employs ensemble machine learning combined with a mixed sequence, structure-based method. The SHIFTX2 algorithm has been trained and tested with high-resolution X-ray structures (< 2.1 Å) and verified chemical shifts assignments. SHIFTX2 combines two ensemble machine learning alghoritms: bagging and boosting. Bagging algorithm trains “base learners” (i.e., the individual learning algorithms of the ensemble) from a random sample of the original dataset, averaged over predictions of all the individual base learners. In contrast, boosting algorithm trains subsequent base learners on mistakes of the previous base learner. SHIFTX2 further implements an alghorithm to select the optimal set of features (i.e., χ2 and χ3 angles, solvent accessibility, H-bond geometry, pH, temperature). By applying SHIFTX2 over MD simulation runs, one can generate average chemical shifts that are comparable to experimental values, thereby providing an assessment of how a structural ensemble derived from MD relates to that of NMR at the molecular level. It should be noted, however, that the comparison of computed and experimental chemical shifts does not probe allosteric effects directly. Indeed, chemical shifts are a local property, susceptible to changes in the electronic environment, as well as local events such as substrate binding and shifts in amino acid pKa. Hence, allosteric signaling itself is described through the analysis of the slow- and fast-timescale dynamics captured through solution NMR and simulations. Nonetheless, SHIFTX2 remains an important tool to predict which amino acids may be most sensitive to changes in a dynamic ensemble.
Case study 1: DNA polymerase β (pol β)
DNA polymerases are a well-established family of enzymes that have been extensively studied for their role in replicating and maintaining genomic DNA. The fidelity of DNA polymerases, or ability to select and insert correct nucleotides from an intracellular pool (i.e., A, T, G, C) during catalysis, is essential for multicellular organisms. Polymerases rely on various mechanisms for fidelity, and larger replicative polymerases contain accessory proofreading domains that confirm the inserted nucleotide bases are not mismatched. However, the maintenance of fidelity for smaller polymerases lacking these accessory domains is poorly understood. In this case study, we highlight the 39 kDa DNA polymerase (pol) β that lacks a proofreading domain and functions in the mammalian base excision repair (BER) pathway (Krokan and Bjoras 2013; Wallace et al. 2012). Pol β is composed of two domains with distinct enzymatic functions, a deoxyribose phosphate (dRP) lyase domain (8 kDa) and a nucleotide transferase domain (31 kDa) composed of a DNA binding subdomain (thumb), catalytic subdomain (palm), and nucleotide selection subdomain (fingers). Within short patch BER, pol β identifies and repairs gapped DNA by recruiting and inserting a deoxynucleoside triphosphate (dNTP). The ability of pol β to accurately discern matched (correct) versus mismatched (incorrect) base pairs is integral to its function, highlighted by the fact that over 30% of human cancers have mutations in the gene encoding pol β (Marsden et al. 2017; Starcevic et al. 2004). Many of these cancer-linked mutations lead to a loss of function; however, several mutations maintain near wild-type (WT) catalytic efficiency with vastly altered fidelity and have been termed “mutator variants.” Expression of pol β mutants within healthy mammalian cells has been shown to cause cellular transformation and metastasis (Marsden et al. 2017; Murphy et al. 2012; Wallace et al. 2012); thus, there is great interest in elucidating the mechanistic underpinnings of the pol β selection process, particularly the allosteric component by which binding of the appropriate nucleotide is regulated. Several reviews (Barakat et al. 2012; Beard and Wilson 2006, 2014; Hakem 2008; Yamtich and Sweasy 2010) have rigorously described the known molecular mechanism and structure of pol β and related polymerases; therefore, we will only highlight important NMR and computational studies that have improved the understanding of allosteric regulation underlying its nucleotide selection.
Foundational understanding of four precatalytic states of pol β; apo enzyme, gapped DNA bound (binary), as well as two ternary states (gapped DNA + dNTP), in “open” and “closed” forms, has been established through X-ray crystallography and Förster Resonance Enenergy Transfer (FRET) (Towle-Weicksel et al. 2014). The ternary states are distinguished by 7–10 Å closure of the C-terminal nucleotide selection domain around the active site complex (Fig. 6a). However, time-averaged snapshots of these complexes in open and closed conformations do not inform the allosteric mechanism of nucleotide selection (Freudenthal et al. 2012; Sawaya et al. 1997). FRET studies explored global motions of the precatalytic transitions of pol β following dNTP binding to the binary (i.e., DNA-bound) enzyme (Towle-Weicksel et al. 2014) and suggested an induced fit mechanism for allosteric control of pol β, where the allosteric effector, a matched dNTP, causes a conformational change and subsequent non-covalent mechanistic step that regulates catalysis (Towle-Weicksel et al. 2014). This obligate non-covalent step has been suggested to be the binding of a magnesium ion or an additional conformational change. Recent NMR and computational investigations have provided further insight into intra- and intermolecular structural and dynamical processes underlying nucleotide selection and pol β activation.
Fig. 6.

a X-ray crystal structures of open (black) and closed (gray) pol β. DNA molecules are shown in orange shades. b NMR chemical shift signatures of the open (black, WT without nucleotide), closed (gray, WT with matched nucleotide), and “off-pathway” (purple, I260Q mutant with mismatched nucleotide) structural states of pol β. c Schematic representation of chemical shift vector analysis reported by Liptak et al., accounting for various degrees of enzymatic closure around matched and mismatched nucleotides in WT and I260Q mutant pol β. WT pol β adopts its native, closed structure upon matched dNTP binding (black, gray), while the cancer-associated I260Q variant populates altered structures with various dNTPs (red, blue, green) that deviate from the WT pathway. The normalized vector magnitude was calculated as ∆δn = ∆δexptl/∆δref, where ∆δref is the WT trajectory. The degree of I260Q pol β closure and its overall structure relative to WT pol β is summarized on the correlation plot (right). I260Q data points deviating from the 1,1-cross-section display either altered structures (horizontal) or altered levels of enzymatic closure (vertical). Dots are colored according to the I260Q:DNA complexes indicated in the vector diagram at left
Due to the size of pol β (~ 40 kDa), early NMR studies utilized sparse 1H,13C-methyl labeling of methionine residues to probe DNA binding to the apo enzyme and subsequent binding of a non-hydrolyzable dNTP to the binary complex. This work highlighted specific residues within pol β that participate in the formation of binary and ternary complexes as well as spectral signatures of the “open” and “closed” allosteric states sampled in the presence of mismatched and matched dNTPs, respectively (Bose-Basu et al. 2004). More recent 1H-15N experiments probing backbone amides highlighted activation of the pol β allosteric network upon matched dNTP binding due to changes in the flexibility of the protein backbone (Berlow et al. 2012; Loria et al. 2008), where NMR relaxation dispersion revealed specific amino acids in apo and binary pol β exhibiting millisecond timescale motions. Interestingly, several of these dynamic sites are coincident with mutations found in certain cancers (Starcevic et al. 2004), suggesting that flexibility of pol β precatalytic states, allosteric regulation of pol β, and its role in disease may be linked (Berlow et al. 2012; Moscato et al. 2016).
Additional support for an allosteric network in pol β comes from MD simulations of cancer-associated mutants with disrupted hydrogen bonding between N279 and a bound dCTP (C = cytosine) that showed strongly attenuated catalytic efficiencies (kcat/KM) despite the fact that N279 does not make direct contact with the primer or α-phosphate of dCTP (Martinek et al. 2007). Corresponding kinetic studies note the overall change in free energy (ΔG) more closely approximates ΔG of dNTP binding than that of the catalytic reaction (Xiang et al. 2006), supporting a mechanism in which pol β is preorganized for dNTP binding and subsequent allosteric changes propagate to improve or impede pol β catalytic efficiency. In addition to the importance of N279 as an allosteric modulator of pol β, computational studies have established the importance of magnesium ions in both the active site and the allosteric network in general (Palermo et al. 2015). Computational modeling of pre- and post-chemistry steps by Schlick and coworkers showed that in the absence of magnesium, dCTP triphosphate oxygens interact with different amino acid side chains than in the presence of magnesium and suggested that closure of the nucleotide selection domain around the active site requires two magnesium ions (Yang et al. 2004). Opening of the enzyme post-chemistry is concomitant with the release of one magnesium ion from the active site in MD simulations.
Recent NMR and FRET experiments by Loria, Sweasy, and coworkers on an I260Q pol β mutant that retains catalytic activity but with greatly diminished dNTP discrimination, assessed the allosteric role of the hydrophobic hinge connecting the nucleotide selection and catalytic subdomains (Liptak et al. 2018; Starcevic et al. 2005). In order to understand how dNTP binding altered the structure (i.e., the degree of enzymatic closure) of I260Q relative to the WT enzyme, Liptak et al. used a vector analysis method to measure the “on” and “off” pathway trajectories of colinear NMR chemical shifts (Fig. 6b, c). In this approximation, trajectories of NMR chemical shifts that deviated from those of WT pol β, quantified by the angle between WT and I260Q vectors (θ, Fig. 6c, left), were suggestive of an altered closed conformation of the enzyme, distinct from that WT pol β. Further, diminished magnitudes of linear I260Q chemical shifts suggested this mutation and altered structure hindered the ability of pol β to sample a fully closed structure (Fig. 6c, right). Diminished closure in the nucleotide selection subdomain in the presence of dNTP mismatches is known to be important for fidelity of the WT enzyme, as addition of a mismatched nucleotide does not induce large chemical shift perturbations in its 1H-13CH3 NMR spectrum. However, addition of a mismatched nucleotide to the I260Q mutant induces large chemical shift perturbations in the nucleotide selection domain, many of which fall along the pathway to closure seen for WT pol β in the presence of a matched nucleotide. The chemical shift vector methodology in Fig. 6c allowed for identification of specific residues that contribute to the pol β fidelity based on their ability (or lack thereof) to sample closed conformations in various dNTP-bound states.
The use of NMR and computation in the study of DNA pol β fidelity was crucial to refining the current view of allostery that governs the transferase activity of the enzyme. Initial X-ray crystallographic work highlighted the induced fit of a dNTP into the active site via closure of the nucleotide selection domain, while NMR and computation clarified the molecular details of the process, namely the regions of pol β that composed an allosteric network and displayed the flexibility necessary to alter the open/closed ensemble. In the current model of pol β nucleotide selection, allosteric activation is achieved by formation of a Watson-Crick pair between the incoming nucleotide and templating base, which is controlled by the affinity of pol β for matched/mismatched nucleotides. A chemical signal is then propagated through the hydrophobic hinge to the active site, where the incorporation of magnesium ions modulates catalytic function. Since enzymatic catalysis (kcat, kpol) is affected by preceding steps in this information transfer, pol β appears to display mixed K-type and V-type allostery.
Case study 2: CRISPR-Cas9
The CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats–associated protein 9) system is crucial to prokaryotic organisms as an adaptive immune response against invading bacteriophages (Doudna and Charpentier 2014; Jinek et al. 2012) and is widely utilized as a genome editing tool in biotechnology and medicine (Adli 2018). Cas9 is a 160 kDa RNA-guided endonuclease that creates double-strand breaks in DNA upon site-specific recognition and binding of a short 2–5 nucleotide Protospacer Adjacent Motif (PAM) that precedes the cleavage site (Fig. 7). The multi-domain Cas9 enzyme is composed of a large recognition lobe (REC) that accommodates the formation of a RNA:DNA hybrid through three subdomains (REC1-3), and a nuclease lobe including two domains HNH and RuvC that cleave the DNA strand complementary to the guide RNA and the non-complementary strand, respectively.
Fig. 7.

Structure of the Sp CRISPR-Cas9 system, composed of the endonuclease Cas9 in complex with a guide RNA and a target DNA. The Cas9 enzyme is shown in molecular surface, highlighting the HNH (green), RuvC (blue), and REC (gray) domains with different colors. The RNA (orange) as well as the target DNA strand (TS, cyan) and the non-target DNA strand (NTS, violet) are shown as ribbons. A close-up view of the PAM-binding region is shown on the right
The spatially separated, yet functionally connected recognition and nuclease sites of Cas9 implied an allosteric relay, which was initially probed by the insertion of exogenous PDZ domains into the native Cas9 structure by Doudna and coworkers, highlighting regions of the enzyme as “hotspots” (Oakes et al. 2016). Biochemical studies by Doudna and coworkers (Sternberg et al. 2014, 2015) have indicated that allostery in Cas9 synchronizes DNA binding, recognition, and concerted double-stranded cleavage. Early computational investigations revealed that allosteric cross-talk between the HNH and RuvC catalytic domains is essential for activation of concerted DNA cleavage (Palermo et al. 2017b) and more recently, Chen and colleagues revealed by MD that motions of the REC lobe govern the conformational changes of the HNH domain toward cleavage (Chen et al. 2017). While X-ray structures have shown “open” (apo) or “closed” (RNA/DNA-bound) snapshots of Cas9 (Huai et al. 2017; Jiang and Doudna 2015; Jinek et al. 2014; Nishimasu et al. 2014), it is widely hypothesized that Cas9, like other allosteric proteins (Koshland Jr. et al. 1966; Lukin et al. 2003; Monod et al. 1965; Yuan et al. 2015) populates numerous microstates between these structures (Dagdas et al. 2017).
In this respect, the nature of allosteric communication within this protein-nucleic acid complex is poorly understood, though recent investigations have provided intricate detail about how nucleic acid binding information is transmitted across the multi-domain Cas9 structure to control the activation of the catalytic HNH nuclease. HNH displays a remarkable conformational mobility, observed by cryo-EM (CryoEM; EMD-3277) to be at lower resolution (8–10 Å) than the overall structure (6 Å) (Jiang et al. 2016). The first all-atom MD simulation of Cas9 (Palermo et al. 2016) also highlighted the “striking” flexibility of HNH with respect to the remaining protein subunits that enable HNH to rapidly sample its activated state primed for DNA cleavage. HNH activation is dependent on the conformational dynamics of the REC lobe of Cas9, where allosteric function relies on the opening of the REC3 region (amino acids 480–718) to integrate the incoming RNA:DNA hybrid (Chen et al. 2017). This interaction is thought to propagate a biological signal to REC2 (amino acids 180–308), which directly contacts HNH, enabling its approach to the DNA cleavage site on the complementary strand. In spite of extensive structural characterization of Cas9, high-resolution snapshots of its fully activated state have yet to be observed, perhaps due to the inherent flexibility of HNH when engaged in enzymatic function. However, early computational studies employing targeted approaches and accelerated MD have provided the first structural information on the activated complex, including the structure of an activated HNH domain (Palermo et al. 2017a; Zuo and Liu 2017). Extensive MD simulations have also revealed that the REC1-3 regions collectively move with HNH during its activation (Palermo et al. 2018), supporting the “allosteric hypothesis” of Chen and colleagues (Chen et al. 2017). However, the nature of the allosteric control that REC exerts on HNH and the mechanism of its signal transduction remains unclear. Each of these functionalities relies on molecular motion and is thus amenable to characterization by synergistic NMR and MD approaches. Thus, the most recent biophysical probes of Cas9 are integrating solution NMR (Lisi and Loria 2016a) and MD or novel GaMD (Miao et al. 2015) with network models (Sethi et al. 2009) and newly developed centrality analysis methods (Negre et al. 2018a) to track the molecular motions at the core of the allosteric communication between REC, HNH, and RuvC.
Since coupling of NMR data to all-atom MD simulations has significantly increased our understanding of dynamic allostery in smaller protein-nucleic acid complexes (Adhireksan et al. 2017; Lisi and Loria 2016a; Wodak et al. 2019), these methodologies are being extended to the Cas9 machinery (Belato et al. 2019), building on outcomes of prior MD simulations (Palermo 2019; Palermo et al. 2016, 2017a, b, 2018; Ricci et al. 2019; Zuo and Liu 2017). Several domains critical to Cas9 allosteric control have been studied by 1H-15N and 1H-13CH3 NMR (Belato 2019) (Fig. 8) in order to generate fingerprints of the key players in Cas9 allostery and connect with previous computational work highlighting the dynamic interplay of HNH, REC2, and REC3. NMR spin relaxation experiments reveal multi-timescale dynamics within this axis of Cas9, which may be involved in information transfer for concerted cleavage of the two DNA strands. Regions of fast (ps–ns) dynamics are consistent with those proposed by early MD studies of Cas9 allostery, but further investigation of these dynamic properties is required to decipher the mechanism of communication between these domains.
Fig. 8.

NMR spectra of critical domains of Cas9 implicated in its allosteric mechanism. 1H-15N correlation spectra of backbone amide residues are shown for the REC2 and REC3 regions, while a 1H-13CH3 methyl spectrum reporting on Ile, Leu, Val, Ala, Met, and Thr side chains is shown for HNH. NMR studies of the structure and dynamics of these critical domains are required to confirm the allosteric relay proposed by MD simulations
The role of the PAM sequence is also intriguing, as its binding initiates DNA association and cleavage, triggering inter-dependent molecular motions of the Cas9 domains (Palermo et al. 2017b; Sternberg et al. 2014). Specifically, PAM has been identified as an “allosteric activator” of Cas9 function from MD simulations performed of Cas9 binding to a ‘5–TGG–3’ PAM sequence (i.e., Cas9–PAM) (Anders et al. 2014), and on its analogue, crystallized without PAM (Nishimasu et al. 2014). A total of ~ 13 μs of aggregate MD runs probing the impact of PAM binding on the dynamics of Cas9 revealed that PAM induces an “open–to–close” conformational transition, as indicated by principal component analysis. This conformational change agrees with the transition hypothesized for nucleic acid binding (Jiang et al. 2016; Palermo et al. 2017a) and reveals that PAM is essential in the process. Analysis of coupled motions through a generalized correlation (GC) method revealed that PAM binding significantly strengthens the motional correlations of Cas9, particularly at HNH, inducing a shift in its conformational dynamics consistent with the function of other allosteric effectors (Guo and Zhou 2016). Notably, by introducing a per-domain correlation score (Csi) matrix that accumulates (and normalizes) the GCij coefficients over each domain, high inter-domain correlations are observed for the motions of HNH and RuvC. This dynamic cross-talk is not observed in the absence of PAM, indicating that PAM binding induces the coupled dynamics of HNH and RuvC (Sternberg et al. 2015). In the presence of PAM, HNH also correlates strongly with the α-helical REC lobe, supporting a direct signal transfer (Chen et al. 2017) and suggesting that PAM acts as a long-range allosteric effector (Sternberg et al. 2014).
To further understand the role of PAM in the allosteric network, community network analysis (Rivalta et al. 2012; Sethi et al. 2009) was applied to structure Cas9 correlations (Fig. 9a, described above in computational approaches). This analysis shows that in the absence of PAM (Fig. 9a, top panel), a high fragmentation of the community structure is observed, which is then reduced upon PAM binding (Fig. 9a, bottom panel). Thus, in the absence of PAM, communication between regions of Cas9 is weak, hampering facile signal transduction. Indeed, upon PAM binding, the communities reorganize (evidenced by changes in the edge betweenness, i.e., thicker bonds) to strongly couple REC2 (community 4), HNH (8), and RuvC (1), a sign of increased correlation or signal transfer and supporting the role of PAM as an effector of inter-domain cross-talk (Sternberg et al. 2014). These community networks were analyzed to determine the route(s) of information transfer between HNH and RuvC. The allosteric “pathways” between residues computed from the dynamic network as a sum of their edge lengths, identifying the most likely signaling routes (i.e., the pathways exhibiting the shortest edge lengths) (Rivalta et al. 2012; Sethi et al. 2009). This analysis revealed that the PAM-mediated allosteric signaling is likely to be transduced through the L1/L2 flexible loops, connecting HNH and RuvC (Fig. 9b). This is consistent with the origin of a dynamic pathway throughout HNH determined by MD (Fig. 9c), and also agrees well with previous experimental evidence, suggesting an allosteric signal transport across REC–HNH–RuvC (Chen et al. 2017). This conclusion is supported by previous structural studies suggesting that conformational changes in L1 and L2 could transmit information about HNH activation to RuvC (Jiang et al. 2016). The importance of single amino acids within these loops was quantified through an analysis of the node betweenness (i.e., the number of shortest pathways that cross the node). This study indicated that residues displaying the highest node betweenness, Q771 and E584 (within L1 and L2, respectively) engage in interactions with K775 and R905, forming essential edges of communication between HNH and RuvC. K775A or R905A mutations were shown to alter Cas9 selectivity (Chen et al. 2017; Slaymaker et al. 2016), opening an avenue for controlling nuclease activity through its allosteric signaling. Overall, these collective studies indicate that PAM binding induces a tight cross-talk between HNH and RuvC (Palermo et al. 2017b; Sternberg et al. 2014, 2015). However, data collected to this point do not yet clarify how the effect of PAM binding propagates to the nuclease domains. This open question is currently driving additional integrative NMR and computational research, with the goal of deciphering the mechanism of information transfer between the PAM-binding site and the HNH and RuvC domains.
Fig. 9.
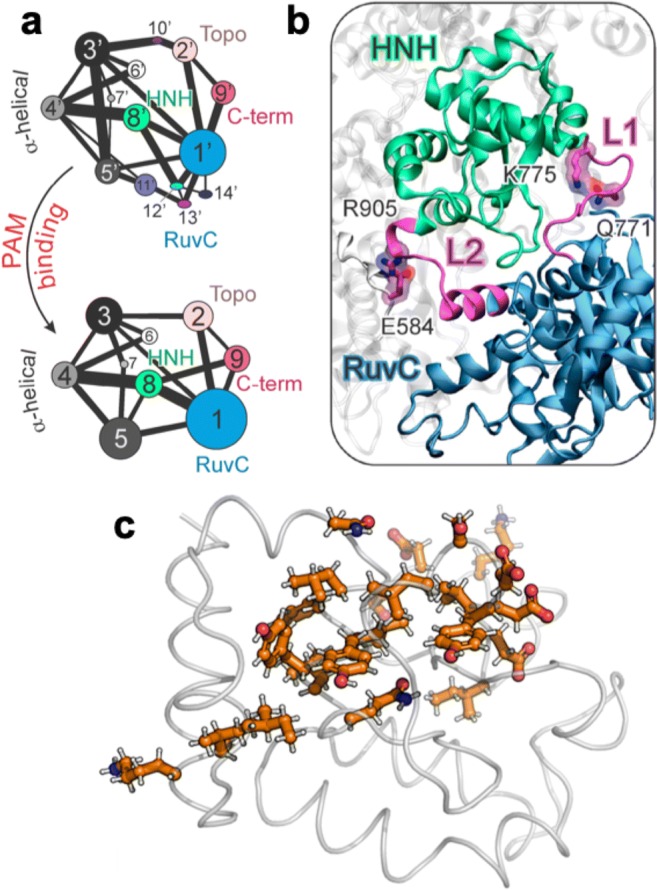
a Community network analysis of the Cas9 structure in the absence (top) and presence (bottom) of PAM (DNA). Bond thickness between communities represents the strength of the intercommunication. b Allosteric pathway captured from the dynamic network, showing biological information flow between the HNH (green) and RuvC (blue) nucleases utilizes the L1 and L2 loops. Residues displaying the highest node betweenness, Q771 and E584, engage in interactions with K775 and R905, forming essential edges of communication between HNH and RuvC. c Proposed network of flexibile residues computed from dynamical network models of Cas9 after optimizing motional correlation between residues adjacent to RuvC and REC2. Portions of this figure are reproduced with permission from Palermo et al. J. Am. Chem. Soc. 2017, 139, 16028. Copyright American Chemical Society
Summary
Allostery in biomolecular assemblies provides a high level of functional control over vital biological processes. The combination of experimental solution NMR with in silico techniques can elucidate the mechanisms by which allosteric control is maintained in nucleoprotein assemblies. The highlighted case studies illustrate a broad range of molecular size and structural complexity that can be explored with these methodologies.
Acknowledgments
G.P. acknowledges funding from the National Science Foundation (NSF), as this material is based upon work supported by the National Science Foundation under Grant No. CHE-1905374. GPL acknowledges funding from the COBRE Center for Computational Biology of Human Disease (NIGMS P20GM109035).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abragam A. Principles of nuclear magnetism. Oxford: Clarendon Press; 1961. [Google Scholar]
- Adhireksan Z, Palermo G, Riedel T, Ma Z, Muhammad R, Rothlisberger U, Dyson PJ, Davey CA. Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat Commun. 2017;8:14860. doi: 10.1038/ncomms14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1911. doi: 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akke M, Palmer AG. Monitoring macromolecular motions on microsecond-millisecond time scales by R1r-R1 constant-relaxation-time NMR spectroscopy. J Am Chem Soc. 1996;118:911. [Google Scholar]
- Akke M, Brüschweiler R, Palmer AG. NMR order parameters and free energy: an analytic approach and application to cooperative Ca2+ binding by calbindin D9k. J Am Chem Soc. 1993;115:9832–9833. [Google Scholar]
- Alvarez-Socorro AJ, Herrera-Almarza GC, Gonzalez-Diaz LA. Eigencentrality based on dissimilarity measures reveals central nodes in complex networks. Sci Rep. 2015;5:17095. doi: 10.1038/srep17095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Barakat KH, Gajewski MM, Tuszynski JA. DNA polymerase beta (pol beta) inhibitors: a comprehensive overview. Drug Discov Today. 2012;17:913–920. doi: 10.1016/J.Drudis.2012.04.008. [DOI] [PubMed] [Google Scholar]
- Barna JCJ, Laue ED, Mayger MR, Skilling J, Worrall SJP. Exponential sampling, an alternative method for sampling in two-dimensional Nmr experiments. J Magn Reson. 1987;73:69–77. doi: 10.1016/0022-2364(87)90225-3. [DOI] [Google Scholar]
- Beach H, Cole R, Gill M, Loria JP. Conservation of μs-ms enzyme motions in the apo- and substrate-mimicked state. J Am Chem Soc. 2005;127:9167–9176. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Structure and mechanism of DNA polymerase beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Structure and mechanism of DNA polymerase beta. Biochemistry. 2014;53:2768–2780. doi: 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belato HB, East KW, Lisi GP. (1)H, (13)C, (15)N backbone and side chain resonance assignment of the HNH nuclease from streptococcus pyogenes CRISPR-Cas9. Biomol Nmr Assign. 2019;13:367–370. doi: 10.1007/s12104-019-09907-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Berlow RB, Igumenova TI, Loria JP. Value of a hydrogen bond in triosephosphate isomerase loop motion. Biochemistry. 2007;46:6001–6010. doi: 10.1021/bi700344v. [DOI] [PubMed] [Google Scholar]
- Berlow RB, Swain M, Dalal S, Sweasy JB, Loria JP. Substrate-dependent millisecond domain motions in DNA polymerase beta. J Mol Biol. 2012;419:171–182. doi: 10.1016/j.jmb.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgatti SP. Centrality and network flow. Soc Networks. 2005;27:55–71. doi: 10.1016/j.socnet.2004.11.008. [DOI] [Google Scholar]
- Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase beta. Biochemistry. 2004;43:8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- Capdevila DA, Braymer JJ, Edmonds KA, Wu H, Giedroc DP. Entropy redistribution controls allostery in a metalloregulatory protein. Proc Natl Acad Sci U S A. 2017;114:4424–4429. doi: 10.1073/pnas.1620665114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro JA, Harpole KW, Kasinath V, Lim J, Granja J, Valentine KG, Sharp KA, Wand AJ. Entropy in molecular recognition by proteins. Proc Natl Acad Sci U S A. 2017;114:6563–6568. doi: 10.1073/pnas.1621154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver JP, Richards RE. General two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. 2007. [Google Scholar]
- Chakravorty DK, Parker TM, Guerra AJ, Sherrill CD, Giedroc DP, Merz KMJ. Energetics of zinc-mediated interactions in the allosteric pathways of metal sensor proteins. J Am Chem Soc. 2013;135:30–33. doi: 10.1021/ja309170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1101/160036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- Cole R, Loria JP. Evidence for flexibility in the function of ribonuclease A. Biochemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- Cole R, Loria JP. FAST-modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- Coyne HJ, Giedroc DP. Backbone resonance assignments of the homotetrameric (48 kD) copper sensor CsoR from Geobacillus thermodenitrificans in the apo- and Cu(I)-bound states: insights into copper-mediated allostery. Biomol Nmr Assign. 2013;7:279–283. doi: 10.1007/s12104-012-9428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui DS, Beaumont V, Ginther PS, Lipchock JM, Loria JP. Leveraging reciprocity to identify and characterize unknown allosteric sites in protein tyrosine phosphatases. J Mol Biol. 2017;429:2360–2372. doi: 10.1016/j.jmb.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Auvergne EJ, Gooley PR. The use of model selection in the model-free analysis of protein dynamics. J Biomol NMR. 2003;25:25–39. doi: 10.1023/A:1021902006114. [DOI] [PubMed] [Google Scholar]
- Dagdas YS, Chen JS, Sternberg SH, Doudna JA. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv. 2017;3:eaao002. doi: 10.1126/sciadv.aao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis D.G., Perlman M.E., London R.E. Direct Measurements of the Dissociation-Rate Constant for Inhibitor-Enzyme Complexes via the T1ρ and T2 (CPMG) Methods. Journal of Magnetic Resonance, Series B. 1994;104(3):266–275. doi: 10.1006/jmrb.1994.1084. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Walker GS, Farley KA, Sharma R, Hoch JC, Arbogast LW, Brinson RG, Marino JP (2017) Non-uniform sampling for all: more NMR spectral quality, Less Measurement Time. Am Pharm Rev 20 [PMC free article] [PubMed]
- Deverell C, Morgan RE, Strange JH. Chemical exchange by nuclear magnetic relaxation in the rotating frame. Mol Phys. 1970;18:553–559. [Google Scholar]
- Dhulesia A, Gsponer J, Vendruscolo M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J Am Chem Soc. 2008;130:8931–8939. doi: 10.1021/ja0752080. [DOI] [PubMed] [Google Scholar]
- Dijkstra EW. Numer Math. 1959;1:269–271. [Google Scholar]
- Dokholyan NV. Controlling allosteric networks in proteins. Chem Rev. 2016;116:6463–6487. doi: 10.1021/acs.chemrev.5b00544. [DOI] [PubMed] [Google Scholar]
- Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci U S A. 2016;113:4735–4740. doi: 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Unfolded proteins and protein folding studied by NMR. Chem Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Zhang O, Szabo A, Torchia DA, Kay LE. Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fizil A, Sonderegger C, Czajlik A, Fekete A, Komaromi I, Hajdu D, Marx F, Batta G. Calcium binding of the antifungal protein PAF: structure, dynamics and function aspects by NMR and MD simulations. PLoS One. 2018;13:e0204825. doi: 10.1371/journal.pone.0204825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RW. Algorithm-97 - shortest path. Commun Acm. 1962;5:345. doi: 10.1145/367766.368168. [DOI] [Google Scholar]
- Freudenthal BD, Beard WA, Wilson SH. Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure. 2012;20:1829–1837. doi: 10.1016/j.str.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and Intramolecular signaling in a PDZ domain. J Mol Biol. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Gill ML, Hsu A, Palmer AG. Detection of chemical exchange in methyl groups of macromolecules. J Biomol NMR. 2019;73:443–450. doi: 10.1007/s10858-019-00240-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey MJ, Wang C, Palmer AG., 3rd Disulfide bond isomerization in basic pancreatic trypsin inhibitor: multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J Am Chem Soc. 2003;125:14324–14335. doi: 10.1021/ja0367389. [DOI] [PubMed] [Google Scholar]
- Grishaev A, Wu J, Trewhella J, Bax A. Refinement of multidomain protein structures by combination of solution small-angle X-ray scattering and NMR data. J Am Chem Soc. 2005;127:16621–16628. doi: 10.1021/ja054342m. [DOI] [PubMed] [Google Scholar]
- Guo J, Zhou HX. Protein allostery and conformational dynamics. Chem Rev. 2016;116:6503–6515. doi: 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27:589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke M, Foraita R. Clone temporal centrality measures for incomplete sequences of graph snapshots. BMC Bioinformatics. 2017;18:261. doi: 10.1186/s12859-017-1677-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Hill RB, Bracken C, DeGrado WF, Palmer AG., 3rd Molecular motions and protein folding: characterization of the backbone dynamics and folding equilibrium of alpha D-2 using C-13 NMR spin relaxation. J Am Chem Soc. 2000;122:11610–11619. doi: 10.1021/ja001129b. [DOI] [Google Scholar]
- Huai G, Li G, Yao R, Zhang Y, Cao M, Kong L, Jia C, Yuan H, Chen H, Lu D, Huang Q. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat Commun. 2017;8:1–9. doi: 10.1038/s41467-017-01496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc Natl Acad Sci U S A. 2004;101:9618–9623. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igumenova TI, Frederick KK, Wand AJ. Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem Rev. 2006;106:1672–1699. doi: 10.1021/cr040422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson RL, Simpson PJ, Liu M, Cota E, Zhang X, Freemont P, Matthews S. A new labeling method for methyl transverse relaxation-optimized spectroscopy NMR spectra of alanine residues. J Am Chem Soc. 2007;129:15428–15429. doi: 10.1021/ja0761784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishima R, Nagayama K. Protein backbone dynamics revealed by quasi spectral density function analysis of amide N-15 nuclei. Biochemistry. 1995;34:3162–3171. doi: 10.1021/bi00010a005. [DOI] [PubMed] [Google Scholar]
- Ishima R, Torchia DA. Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J Biomol NMR. 1999;14:369–372. doi: 10.1023/a:1008324025406. [DOI] [PubMed] [Google Scholar]
- Jacoby E, Hua QX, Stern AS, Frank BH, Weiss MA. Structure and dynamics of a protein assembly. 1H-NMR studies of the 36 kDa R6 insulin hexamer. J Mol Biol. 1996;258:136–157. doi: 10.1006/jmbi.1996.0239. [DOI] [PubMed] [Google Scholar]
- Jarymowycz VA, Stone MJ. Remote changes in the dynamics of the phosphotyrosine-binding domain of insulin receptor substrate-1 induced by phosphopeptide binding. Biochemistry. 2008;47:13371–13382. doi: 10.1021/bi801096b. [DOI] [PubMed] [Google Scholar]
- Jen J. Chemical exchange and NMR T2 relaxation – the multisite case. J Magn Reson. 1978;30:111. [Google Scholar]
- Jensen MR, Zweckstetter M, Huang JR, Blackledge M. Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy. Chem Rev. 2014;114:6632–6660. doi: 10.1021/cr400688u. [DOI] [PubMed] [Google Scholar]
- Jiang F, Doudna JA. The structural biology of CRISPR-Cas systems. Curr Opin Struct Biol. 2015;30:100–111. doi: 10.1016/j.sbi.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang FG, Taylor DW, Chen JS, Kornfeld JE, Zhou KH, Thompson AJ, Nogales E, Doudna JA. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016;351:867–871. doi: 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:12479971–12479911. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalodimos CG. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011;20:773–782. doi: 10.1002/pro.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf JG, Loria JP. Theory and applications of protein dynamics from solution NMR. Cell Biochem Biophys. 2002;37:187–211. doi: 10.1385/cbb:37:3:187. [DOI] [PubMed] [Google Scholar]
- Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Kneller JM, Lu M, Bracken C. An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc. 2002;124:1852–1853. doi: 10.1021/ja017461k. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. Probing slow dynamics in high molecular weight proteins by methyl-trosy NMR spectroscopy: application to a 723-residue enzyme. J Am Chem Soc. 2004;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kay LE. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J Am Chem Soc. 2004;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE. Low-populated folding intermediate of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430:586–590. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Kovrigin EL. NMR line shapes and multi-state binding equilibria. J Biomol NMR. 2012;53:257–270. doi: 10.1007/s10858-012-9636-3. [DOI] [PubMed] [Google Scholar]
- Kovrigin EL, Loria JP. Characterization of the transition state of functional enzyme dynamics. J Am Chem Soc. 2006;128:7724–7725. doi: 10.1021/ja061435a. [DOI] [PubMed] [Google Scholar]
- Kovrigin EL, Loria JP. Enzyme dynamics along the reaction coordinate: critical role of a conserved residue. Biochemistry. 2006;45:2636–2647. doi: 10.1021/bi0525066. [DOI] [PubMed] [Google Scholar]
- Kovrigin EL, Kempf JG, Grey MJ, Loria JP. Faithful estimation of dynamics parameters from CPMG relaxation dispersion measurements. J Magn Reson. 2006;180:93–104. doi: 10.1016/j.jmr.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujirai T, Ehara H, Fujino Y, Shirouzu M, Sekine SI, Kurumizaka H. Structural basis of the nucleosome transition during RNA polymerase II passage. Science. 2018;362:595–598. doi: 10.1126/science.aau9904. [DOI] [PubMed] [Google Scholar]
- Lange OF, Grubmuller H. Generalized correlation for biomolecular dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- Lee AL. Contrasting roles of dynamics in protein allostery: NMR and structural studies of CheY and the third PDZ domain from PSD-95. Biophys Rev. 2015;7:217–226. doi: 10.1007/s12551-015-0169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Raychaudhuri S, Wand AJ. Insights into the local residual entropy of proteins provided by NMR relaxation. Protein Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipchock JM, Loria JP. Nanometer propagation of millisecond motions in V-type allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak C, Mahmoud MM, Eckenroth BE, Moreno MV, East K, Alnajjar KS, Huang J, Towle-Weicksel JB, Doublie S, Loria JP, Sweasy JB (2018) I260Q DNA polymerase beta highlights precatalytic conformational rearrangements critical for fidelity. Nucleic Acids Res. 10.1093/nar/gky825 [DOI] [PMC free article] [PubMed]
- Lisi GP, Loria JP. Solution NMR spectroscopy for the study of enzyme allostery. Chem Rev. 2016;116:6323–6369. doi: 10.1021/acs.chemrev.5b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, Loria JP. Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function. Prog Nucl Magn Reson Spectrosc. 2016;92-93:1–17. doi: 10.1016/j.pnmrs.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, Loria JP. Allostery in enzyme catalysis. Curr Opin Struct Biol. 2017;47:123–130. doi: 10.1016/j.sbi.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Lisi GP, Manley GA, Hendrickson H, Rivalta I, Batista VS, Loria JP. Dissecting dynamic allosteric pathways using chemically related small-molecule activators. Structure. 2016;24:1155–1166. doi: 10.1016/j.str.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, East KW, Batista VS, Loria JP. Altering the allosteric pathway in IGPS suppresses millisecond motions and catalytic activity. Proc Natl Acad Sci U S A. 2017;114:E3414–E3423. doi: 10.1073/pnas.1700448114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Zheng Y, Cistola DP, Yang D. Measurement of methyl 13C-1H cross-correlation in uniformly 13C-, 15N-, labeled proteins. J Biomol NMR. 2003;27:351–364. doi: 10.1023/a:1025884922203. [DOI] [PubMed] [Google Scholar]
- Liu JX, Zhang JH, Yang YS, Huang HD, Shen WQ, Hu Q, Wang XS, Wu JH, Shi YY. Conformational change upon ligand binding and dynamics of the PDZ domain from leukemia-associated rho guanine nucleotide exchange factor. Protein Sci. 2008;17:1003–1014. doi: 10.1110/ps.073416508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria JP, Rance M, Palmer AG., 3rd A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. doi: 10.1021/ja983961a. [DOI] [Google Scholar]
- Loria JP, Rance M, Palmer AG., 3rd A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999;15:151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Lukin JA, Kontaxis G, Simplaceanu V, Yuan Y, Bax A, Ho C. Quaternary structure of hemoglobin in solution. Proc Natl Acad Sci U S A. 2003;100:517–520. doi: 10.1073/pnas.232715799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz Z, Meiboom S. Nuclear Magnetic Resonance (N.M.R.) Study of the protolysis of trimethylammonium ion in aqueous solution. Order of the reaction with respect to solvent. J Chem Phys. 1963;39:366. [Google Scholar]
- Mandel A, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- Marsden CG, Dragon JA, Wallace SS, Sweasy JB. Base excision repair variants in cancer. DNA Repair Enzymes: Cell, Molecular, and Chemical Biology. 2017;591:119–157. doi: 10.1016/bs.mie.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinek V, Bren U, Goodman MF, Warshel A, Florian J. DNA polymerase beta catalytic efficiency mirrors the Asn279-dCTP H-bonding strength. Febs Lett. 2007;581:775–780. doi: 10.1016/j.febslet.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi F, Grey MJ, Palmer AG., 3rd Microsecond timescale backbone conformational dynamics in ubiquitin studied by NMR R1rho relaxation experiments. Protein Sci. 2005;14:735–742. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Cheng C, Yu T, Tonelli M, Kornev A, Taylor SS, Veglia G. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6:821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao YL, McCammon JA. Graded activation and free energy landscapes of a muscarinic G-protein-coupled receptor. P Natl Acad Sci USA. 2016;113:12162–12167. doi: 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao YL, McCammon JA. Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. P Natl Acad Sci USA. 2018;115:3036–3041. doi: 10.1073/pnas.1800756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Feher VA, McCammon JA. Gaussian accelerated molecular dynamics: unconstrained enhanced sampling and free energy calculation. J Chem Theory Comput. 2015;11:3584–3595. doi: 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG., 3rd The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc. 2000;122:2867–2877. doi: 10.1021/ja993511y. [DOI] [Google Scholar]
- Mittag T, Schaffhausen B, Gunther UL. Direct observation of protein-ligand interaction kinetics. Biochemistry. 2003;42:11128–11136. doi: 10.1021/bi0347499. [DOI] [PubMed] [Google Scholar]
- Mittermaier AK, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- Mittermaier AK, Kay LE. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Mizohata E, Nakane T, Fukuda Y, Nango E, Iwata S. Serial femtosecond crystallography at the SACLA: breakthrough to dynamic structural biology. Biophys Rev. 2018;10:209–218. doi: 10.1007/s12551-017-0344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Moscato B, Swain M, Loria JP. Induced fit in the selection of correct versus incorrect nucleotides by DNA polymerase beta. Biochemistry. 2016;55:382–395. doi: 10.1021/acs.biochem.5b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder FA, Hon B, Mittermaier AK, Dahlquist FW, Kay LE. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2002;124:1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Donigan KA, Jaeger J, Sweasy JB. The E288K colon tumor variant of DNA polymerase beta is a sequence specific mutator. Biochemistry. 2012;51:5269–5275. doi: 10.1021/bi3003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nango E, Royant A, Kubo M, Nakane T, Wickstrand C, Kimura T, Tanaka T, Tono K, Song C, Tanaka R, Arima T, Yamashita A, Kobayashi J, Hosaka T, Mizohata E, Nogly P, Sugahara M, Nam D, Nomura T, Shimamura T, Im D, Fujiwara T, Yamanaka Y, Jeon B, Nishizawa T, Oda K, Fukuda M, Andersson R, Bath P, Dods R, Davidsson J, Matsuoka S, Kawatake S, Murata M, Nureki O, Owada S, Kameshima T, Hatsui T, Joti Y, Schertler G, Yabashi M, Bondar AN, Standfuss J, Neutze R, Iwata S. A three-dimensional movie of structural changes in bacteriorhodopsin. Science. 2016;354:1552–1557. doi: 10.1126/science.aah3497. [DOI] [PubMed] [Google Scholar]
- Negre CFA, Morzan UN, Hendrickson HP, Pal R, Lisi GP, Loria JP, Rivalta I, Batista VS. Eigenvector centrality distribution for characterization of protein allosteric pathways. Proc Natl Acad Sci U S A. 2018;115:12201–12208. doi: 10.1073/pnas.1810452115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre CFA, Morzan UN, Hendrickson HP, Pal R, Lisi GP, Loria JP, Rivalta I, Ho J, Batista VS. Eigenvector centrality for characterization of protein allosteric pathways. Proc Natl Acad Sci U S A. 2018;115:E12201–E12208. doi: 10.1073/pnas.1810452115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M (2010) Networks: an introduction. Oxford University Press, Inc.
- Nishimasu H, Ran FA, Hsu PD, Konemann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal Structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, Savage DF. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. 2016;34:646–651. doi: 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G. Structure and dynamics of the CRISPR-Cas9 catalytic complex. J Chem Inf Model. 2019;59:2394–2406. doi: 10.1021/acs.jcim.8b00988. [DOI] [PubMed] [Google Scholar]
- Palermo G, Cavalli A, Klein ML, Alfonso-Prieto M, Dal Peraro M, De Vivo M. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc Chem Res. 2015;48:220–228. doi: 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- Palermo G, Miao Y, Walker RC, Jinek M, McCammon JA. Striking plasticity of CRISPR-Cas9 and key role of non-target DNA, as revealed by molecular simulations. ACS Cent Sci. 2016;2:756–763. doi: 10.1021/acscentsci.6b00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G, Miao Y, Walker RC, Jinek M, McCammon JA. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc Natl Acad Sci U S A. 2017;114:7260–7265. doi: 10.1073/pnas.1707645114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G, Ricci CG, Fernando A, Basak R, Jinek M, Rivalta I, Batista VS, McCammon JA. Protospacer adjacent motif-induced allostery activates CRISPR-Cas9. J Am Chem Soc. 2017;139:16028–16031. doi: 10.1021/jacs.7b05313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G, Chen JS, Ricci CG, Rivalta I, Jinek M, Batista VS, Doudna JA, McCammon JA. Key role of the REC lobe during CRISPR-Cas9 activation by “sensing”, “regulating” and “locking” the catalytic HNH domain. Q Rev Biophys. 2018;51:e9. doi: 10.1017/S0033583518000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Palmer AG, 3rd, Massi F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106:1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- Palmer AG., III Enzyme dynamics from NMR spectroscopy. Acc Chem Res. 2015;48:457–465. doi: 10.1021/ar500340a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JW, Wagner G. Mapping of spectral density functions using heteronuclear NMR relaxation measurements. J Magn Reson. 1992;98:308–332. doi: 10.1016/0022-2364(92)90135-T. [DOI] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci CG, Chen JS, Miao Y, Jinek M, Doudna JA, McCammon JA, Palermo G. Deciphering off-target effects in CRISPR-Cas9 through accelerated molecular dynamics. ACS Cent Sci. 2019;5:651–662. doi: 10.1021/acscentsci.9b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalta I, Sultan MM, Lee NS, Manley G, Loria JP, Batista VS. Allosteric pathways in imidazole glycerol phosphate synthase. Proc Natl Acad Sci U S A. 2012;109:E1428–E1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi N, Abyzov A, Blackledge M. Multi-timescale dynamics in intrinsically disordered proteins from NMR relaxation and molecular simulation. J Phys Chem Lett. 2016;7:2483–2489. doi: 10.1021/acs.jpclett.6b00885. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci U S A. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical networks in tRNA: protein complexes. Proc Natl Acad Sci USA. 2009;106:6620–6625. doi: 10.1073/Pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Kay LE. Tracing an allosteric pathway regulating the activity of the HsIV protease. Proc Natl Acad Sci USA. 2014;111:2140–2145. doi: 10.1073/pnas.1318476111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinya S, Ghinet MG, Brzezinski R, Furuita K, Kojima C, Shah S, Kovrigin EL, Fukamizo T. NMR line shape analysis of a multi-state ligand binding mechanism in chitosanase. J Biomol NMR. 2017;67:309–319. doi: 10.1007/s10858-017-0109-6. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava AK, McDonald LR, Cembran A, Kim J, Masterson LR, McClendon CL, Taylor SS, Veglia G. Synchronous opening and closing motions are essential for cAMP-dependent protein kinase A signaling. Structure. 2014;22:1735. doi: 10.1016/j.str.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- Starcevic D, Dalal S, Sweasy J. Hinge residue Ile260 of DNA polymerase beta is important for enzyme activity and fidelity. Biochemistry. 2005;44:3775–3784. doi: 10.1021/bi047956x. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–6+. doi: 10.1038/Nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, Doudna JA. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527:110–113. doi: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Arthanari H, Imai M, Wagner G, Shimada I. Nitrogen-detected TROSY yields comparable sensitivity to proton-detected TROSY for non-deuterated, large proteins under physiological salt conditions. J Biomol NMR. 2016;64:143–151. doi: 10.1007/s10858-016-0015-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Grey MJ, McKnight J, Palmer AG, 3rd, Raleigh DP. Multistate folding of the villin headpiece domain. J Mol Biol. 2006;355:1066–1077. doi: 10.1016/j.jmb.2005.10.066. [DOI] [PubMed] [Google Scholar]
- Tolkatchev D, Xu P, Ni F. Probing the kinetic landscape of transient peptide-protein interactions by use of peptide 15(N) NMR relaxation dispersion spectroscopy: binding of an antithrombin peptide to human prothrombin. J Am Chem Soc. 2003;125:12432–12442. doi: 10.1021/ja021238l. [DOI] [PubMed] [Google Scholar]
- Towle-Weicksel JB, Dalal S, Sohl CD, Doublie S, Anderson KS, Sweasy JB. Fluorescence resonance energy transfer studies of DNA polymerase beta the critical role of fingers domain movements and a novel non-covalent step during nucleotide selection. J Biol Chem. 2014;289:16541–16550. doi: 10.1074/jbc.M114.561878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trbovic N, Cho J-H, Abel R, Friesner RA, Rance M, Palmer AG., 3rd Protein side-chain dynamics and residual conformational entropy. J Am Chem Soc. 2009;131:615–622. doi: 10.1021/ja806475k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Palmer AG., 3rd R1ρ Relaxation outside of the fast-exchange limit. J Magn Reson. 2002;154:157–160. doi: 10.1006/jmre.2001.2466. [DOI] [PubMed] [Google Scholar]
- Tsai Chung-Jung, del Sol Antonio, Nussinov Ruth. Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. Journal of Molecular Biology. 2008;378(1):1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Del Sol A, Nussinov R. Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol Biosyst. 2009;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Ile, Leu, and Val Methyl Assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H[bond]13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- Turupcu Aysegul, Bowen Alice M., Di Paolo Alexandre, Matagne André, Oostenbrink Chris, Redfield Christina, Smith Lorna J. An NMR and MD study of complexes of bacteriophage lambda lysozyme with tetra‐ and hexa‐N‐acetylchitohexaose. Proteins: Structure, Function, and Bioinformatics. 2019;88(1):82–93. doi: 10.1002/prot.25770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng SR, Kalodimos CG. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- Velyvis A, Yang YR, Schachman HK, Kay LE. A solution NMR study showing that active site ligands and nucleotides directly perturb the allosteric equilibrium in aspartate transcarbamoylase. Proc Natl Acad Sci USA. 2007;104:8815–8820. doi: 10.1073/pnas.0703347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velyvis A, Schachman HK, Kay LE. Application of methyl-TROSY NMR to test allosteric models describing effects of nucleotide binding to aspartate transcarbamoylase. J Mol Biol. 2009;387:540–547. doi: 10.1016/j.jmb.2009.01.066. [DOI] [PubMed] [Google Scholar]
- Venters RA, Farmer BTI, Fierke CA, Spicer LD. Characterizing the use of perdeuteration in NMR studies of large proteins: 13C, 15N and 1H assignments of human carbonic anhydrase II. J Mol Biol. 1996;264:1101–1106. doi: 10.1006/jmbi.1996.0699. [DOI] [PubMed] [Google Scholar]
- Vise PD, Baral B, Latos AJ, Daughdrill GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005;33:2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA, Amaro RE. Emerging computational methods for the rational discovery of allosteric drugs. Chem Rev. 2016;116:6370–6390. doi: 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand AJ. Dynamic activation of protein function: a view emerging from NMR spectroscopy. Nat Struct Biol. 2001;8:926–931. doi: 10.1038/nsb1101-926. [DOI] [PubMed] [Google Scholar]
- Wand AJ. Bringing disorder and dynamics in protein allostery into focus. Proc Natl Acad Sci U S A. 2017;114:4278–4280. doi: 10.1073/pnas.1703796114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc Natl Acad Sci U S A. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waudby CA, Ramos A, Cabrita LD, Christodoulou J. Two-dimensional NMR lineshape analysis. Sci Rep. 2016;6:24826. doi: 10.1038/srep24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittier SK, Hengge AC, Loria JP. Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science. 2013;341:899–903. doi: 10.1126/science.1241735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner S, Wybenga-Groot LE, Warner N, Lin H, Pawson T, Forman-Kay JD, Sicheri F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006;25:4686–4696. doi: 10.1038/sj.emboj.7601315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodak SJ, Paci E, Dokholyan NV, Berezovsky IN, Horovitz A, Li J, Hilser VJ, Bahar I, Karanicolas J, Stock G, Hamm P, Stote RH, Eberhardt J, Chebaro J, Dejaegere A, Cecchini M, Changeux JP, Bolhuis PJ, Vreede J, Faccioli P, Orioli S, Ravasio R, Yan L, Brito C, Wyart M, Gkeka P, Rivalta I, Palermo G, McCammon JA, Panecka-Hofman J, Wade RC, Di Pizio A, Niv MY, Nussinov R, Tsai CJ, Jang H, Padhorny D, Kozakov D, McLeish T. Allostery in its many disguises: from theory to applications. Structure. 2019;27:566–578. doi: 10.1016/j.str.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Oelschlaeger P, Florian J, Goodman MF, Warshel A. Simulating the effect of DNA polymerase mutations on transition-state energetics and fidelity: evaluating amino acid group contribution and allosteric coupling for ionized residues in human pol beta. Biochemistry. 2006;45:7036–7048. doi: 10.1021/bi060147o. [DOI] [PubMed] [Google Scholar]
- Yamtich J, Sweasy JB. DNA polymerase family X: function, structure, and cellular roles. Bba-Proteins Proteom. 2010;1804:1136–1150. doi: 10.1016/J.Bbapap.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Kay LE. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: application to protein folding. J Mol Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- Yang L, Arora K, Beard WA, Wilson SH, Schlick T. Critical role of magnesium ions in DNA polymerase beta’s closing and active site assembly. J Am Chem Soc. 2004;126:8441–8453. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Tam MF, Simplaceanu V, Ho C. New look at hemoglobin allostery. Chem Rev. 2015;115:1702–1724. doi: 10.1021/cr500495x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bouget-Bonnet S, Buck M. Combining NMR and molecular dynamics for insights into the allostery of small GTPase-protein interactions. Methods Mol Biol. 2012;796:235–259. doi: 10.1007/978-1-61779-334-9_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z, Liu J. Structure and dynamics of Cas9 HNH domain catalytic state. Sci Rep. 2017;7:17271. doi: 10.1038/s41598-017-17578-6. [DOI] [PMC free article] [PubMed] [Google Scholar]