

Abstract
We recently identified a pathway underlying immune activation in hypertension. Proteins oxidatively modified by reactive isolevuglandin (isoLG) accumulate in dendritic cells (DCs). Prostaglandin E2 (PGE2) has been implicated in the inflammation associated with hypertension. We hypothesized that PGE2 via its EP3 receptor contributes to DC activation in hypertension. EP3−/− mice and wild type littermates were exposed to sequential hypertensive stimuli involving an initial 2-week exposure to the nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME) in drinking water, followed by a 2-week washout period, and a subsequent 4% high salt diet for 3 weeks. In wild type mice, this protocol increased systolic pressure from 123 ± 2 to 148 ± 8 mmHg (p<0.05). This was associated with marked renal inflammation and a striking accumulation of isoLG adducts in splenic DCs. However, the increases in blood pressure, renal T cell infiltration and DC isoLG formation were completely prevented in EP3−/− mice. Similar protective effects were also observed in wild type mice that received intracerebroventricular injection of a lentiviral vector encoding shRNA targeting the EP3 receptor. Further in vitro experiments indicated that PGE2 also acts directly on DCs via its EP1 receptors to stimulate intracellular isoLG formation. Together, these findings provide new insight into how EP receptors in both the central nervous system and peripherally on dendritic cells promote inflammation in salt-induced hypertension.
Keywords: Animal Models of Human Disease, Autonomic Nervous System, Basic Science Research, Inflammation, Hypertension, Reactive Oxygen Species
Graphical Abstract
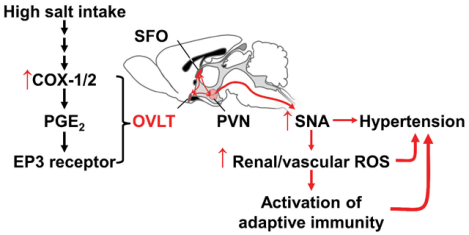
INTRODUCTION
In the last decade, increasing evidence has implicated a role of inflammation and immune cells in the genesis of hypertension. Myeloid cells, T cells, and B cells have been observed in the kidneys and vasculature of both hypertensive humans and in animals with experimental hypertension. Mice lacking T cells are protected against angiotensin II and deoxycorticosterone acetate (DOCA)-salt hypertension, and deletion of monocytes, which give rise to macrophages and dendritic cells (DCs), completely prevents angiotensin II-induced hypertension.1, 2 Deletion of T cells in the Dahl salt-sensitive rat blunts the hypertension and markedly reduces the renal damage associated with salt feeding.3
Research from our group has defined a potential mechanism by which antigen presenting cells contribute to hypertension. We have found that hypertension caused by angiotensin II, DOCA-salt hypertension and Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME)/high salt induce an increase in production of reactive oxygen species in CD11c positive antigen presenting cells, leading to oxidation of polyunsaturated fatty acids and formation of isolevuglandins.4 These highly reactive products covalently modify self-proteins, which in turn seem to be immunogenic.4, 5 We showed that isolevuglandin (isoLG)-modified peptides are presented in the context of MHC1.4 Loading DCs with isoLG-modified proteins promotes their ability to activate T cells, and scavenging IsoLGs prevents hypertension, T cell activation and end-organ damage.4 Interestingly, we have found that angiotensin II does not directly stimulate isoLG-adduct formation, but that other factors in the hypertensive milieu likely contribute. These include catecholamines released from sympathetic nerves and increased extracellular sodium.5, 6
A potent stimulus for DC activation is prostaglandin E2 (PGE2), the predominant prostaglandin metabolite of arachidonic acid. PGE2 enhances DC maturation and is highly effective in priming naïve T cells.7, 8 PGE2 also stimulates DC expression of C-C chemokine receptor type 7, which enhances homing of these cells to lymph nodes where they interact with T cells.9 Of note, PGE2 stimulates expression of co-stimulatory molecules including CD80, CD86 and CD40.8 Relevant to this, CD80 and CD86 are critical for development of hypertension.10 There are four known E prostanoid (EP) receptors of PGE2, termed EP1 through EP4, and the role of these on DC maturation and function seems to vary. EP1 and EP3 receptors stimulate Flt3 expression on hematopoietic progenitor cells and via signaling through STAT3, stimulates survivin expression and in turn enhances survival of DC-committed progenitor cells.11 PGE2 stimulates chemotaxis of human monocyte derived DCs early in their development.
In addition to stimulation of DCs, PGE2 signaling has major effects on vascular function and blood pressure control. Clinically, treatment with nonsteroidal anti-inflammatory agents, which inhibit not only PGE2 synthesis but also production of vasodilator prostaglandins, is associated with hypertension. The effects of PGE2 on blood pressure seem highly dependent on the EP receptors activated. Guan et al. showed that blockade of EP1 lowered blood pressure in spontaneously hypertensive rats, and that genetic deletion of EP1 markedly blunted angiotensin II-induced hypertension.12 Likewise, Chen et al. showed that knockout of EP3 in mice reduces baseline blood pressure and markedly decreases angiotensin II-induced hypertension.13 Recently we showed that while PGE2 alone evokes minimal contraction of mouse femoral arteries, in the presence of angiotensin II, it causes striking increases in tone.14 This potentiating effect is absent in mice lacking EP3 receptors, but not in mice lacking EP1 receptors.14 Pretreatment with the EP3 receptor antagonist DG-041 prevented angiotensin II priming of constriction to angiotensin II.14
Given the role of PGE2 in blood pressure control and DC differentiation, it is compelling to hypothesize that it has a role in modulating DC immunogenicity in hypertension. In this regard, Zhang et al. showed that salt feeding stimulates cyclooxygenase (COX) 2 and PGE synthase mRNA expression in renal macrophage/ monocytes and DCs, and that the major prostanoid made by these cells is PGE2.15 Thus, PGE2 produced by myeloid cells could have autacoid actions on adjacent cells in the kidney and vasculature where monocyte-derived cells accumulate in hypertension.
In the present study, we examined the specific role of the EP3 receptor in DC activation in a model of salt-induced hypertension. We tested the hypothesis that PGE2 can stimulate isoLG-adduct formation and maturation parameters in DCs. We further examined the role of EP1 and EP3 receptors in modulation of the response of DCs to PGE2.
METHODS
An extended methods section is available in the Online Data Supplement. The authors declare that all supporting data are available within the article and its online supplementary files.
Animals:
Wild type C57BL/6 mice were obtained from Jackson Laboratories and were studied at 3 months of age. Generation of EP3 receptor deficient (EP3−/−) mice on a C57BL/6 background has been previously described.16 Hypertension was induced by adding the nitric oxide synthase inhibitor L-NAME to the drinking water (0.5 mg/mL) for 2 weeks, followed by a 2-week washout and subsequently feeding a high-salt diet (4% NaCl) for 3 weeks. Conscious blood pressure measurement using telemetry was performed as previously described.17, 18 For DC adoptive transfer, 106 splenic DCs were obtained from wild type mice by magnetic isolation, and injected into recipient EP3−/− mice via tail vein as previously described.6 Power spectral analyses of blood pressure and heart rate were performed using the HemoLab software suite version 20.7 as previously described.19 To knockdown EP3 receptor expression in the brain, mice underwent a surgical procedure during which either a lentiviral vector expressing shRNA targeting EP3 gene or scrambled control sequences were administered via intracerebroventricular (ICV) injection. At study termination, mice were euthanized by exposure to CO2. The Institutional Animal Care and Use Committee approved all experimental protocols.
Flow cytometry:
To examine renal inflammatory cell infiltration, single cell suspensions were prepared and analyzed by flow cytometry as previously described.18 IsoLG adducts in DCs were determined by intracellular staining using a single chain antibody D11 as described previously.4 This antibody accurately detects isoLG attached to lysines in peptides and proteins independent of adjacent amino acid sequence.
Primary culture of DCs:
DCs were positively selected from the spleen using an autoMACS separator and CD11c magnetic beads (Miltenyi Biotech). The purity of these was confirmed to be >95% by flow cytometry. Splenic CD11c+ cells were placed in 24-well plates at a density of 5×105 per well and cultured in 500 μL RPMI1640 medium supplemented with 10% fetal bovine serum for 24 hours. PGE2, EP1 receptor antagonist SC-51322, EP3 receptor antagonist DG-041, EP1 or EP3 receptor agonists 17-phenyl-trinor-PGE2 (Cayman Chemical, Ann Arbor, MI), sulprostone (Cayman Chemical, Ann Arbor, MI), MB-28767 (a gift from Dr. M.P.L. Caton Rhone-Poulenc), and ONO-AE-248 were applied as indicated below. DG-041 and ONO-AE-248 were provided by the Vanderbilt Institute of Chemical Biology Synthesis Core.
Real-time PCR:
After euthanasia, brains were quickly removed from mice after L-NAME/high salt and immediately frozen on dry ice. Tissue samples from subfornical organ (SFO), organum vasculosum laminae terminalis (OVLT), paraventricular nucleus (PVN), and cortex were punched in a cryostat according to coordinates taken from a mouse atlas.20 With total RNA and then cDNA obtained from samples, the expression of COX-1, COX-2, EP1, EP2, EP3 and EP4 receptors, as well as β-actin as a control gene, were measured using TaqMan real-time PCR.
Confocal Microscopy:
For superoxide detection, kidneys were rapidly removed after euthanasia, immersed in optimal cutting temperature media, and frozen in dry ice. Thirty-μm sections were obtained and used for detecting superoxide by DHE as described previously.21
Immunohistochemistry:
Five-micron sections were obtained from formalin fixed, paraffin embedded kidneys. Collagen was visualized and quantified by Masson Trichrome blue staining as previously described.4, 6
Statistics:
Data are expressed as mean ± SEM. To compare the effect of EP3 receptor deficiency in hypertension, two-way analysis of variance ANOVA was used. For telemetry blood pressure measurements over time, two-way ANOVA with repeated-measures was employed, followed with a Bonferroni post hoc test when significance was indicated. P values are reported in the figures and a value less than 0.05 was considered statistically significant.
RESULTS
Role of EP3 Receptor in L-NAME/High Salt-induced Hypertension:
Consistent with previous studies, the L-NAME/high salt protocol caused hypertension in wild type mice.18 However, EP3 receptor deficiency significantly attenuated this response (Figure 1A). Heart rate was similarly elevated in both wild type and EP3−/− mice during L-NAME and high salt diet feeding. Spectral power analysis of the blood pressure and heart rate variability however showed that the ratio of low-frequency to high-frequency heart rate oscillations was significantly increased after L-NAME/high salt in wild type mice, but not in EP3−/− mice (Figure 1B and S1B). Likewise, the L-NAME/high salt protocol increased the low-frequency/high-frequency ratio in the blood pressure variability of wild type, but not EP3−/− mice (Figure 1C and S1C). In keeping with this, urinary norepinephrine was elevated in wild type mice but not in mice lacking EP3 receptors during hypertension (Figure 1D), further supporting that EP3 deficiency blunts sympathetic outflow.
Figure 1:
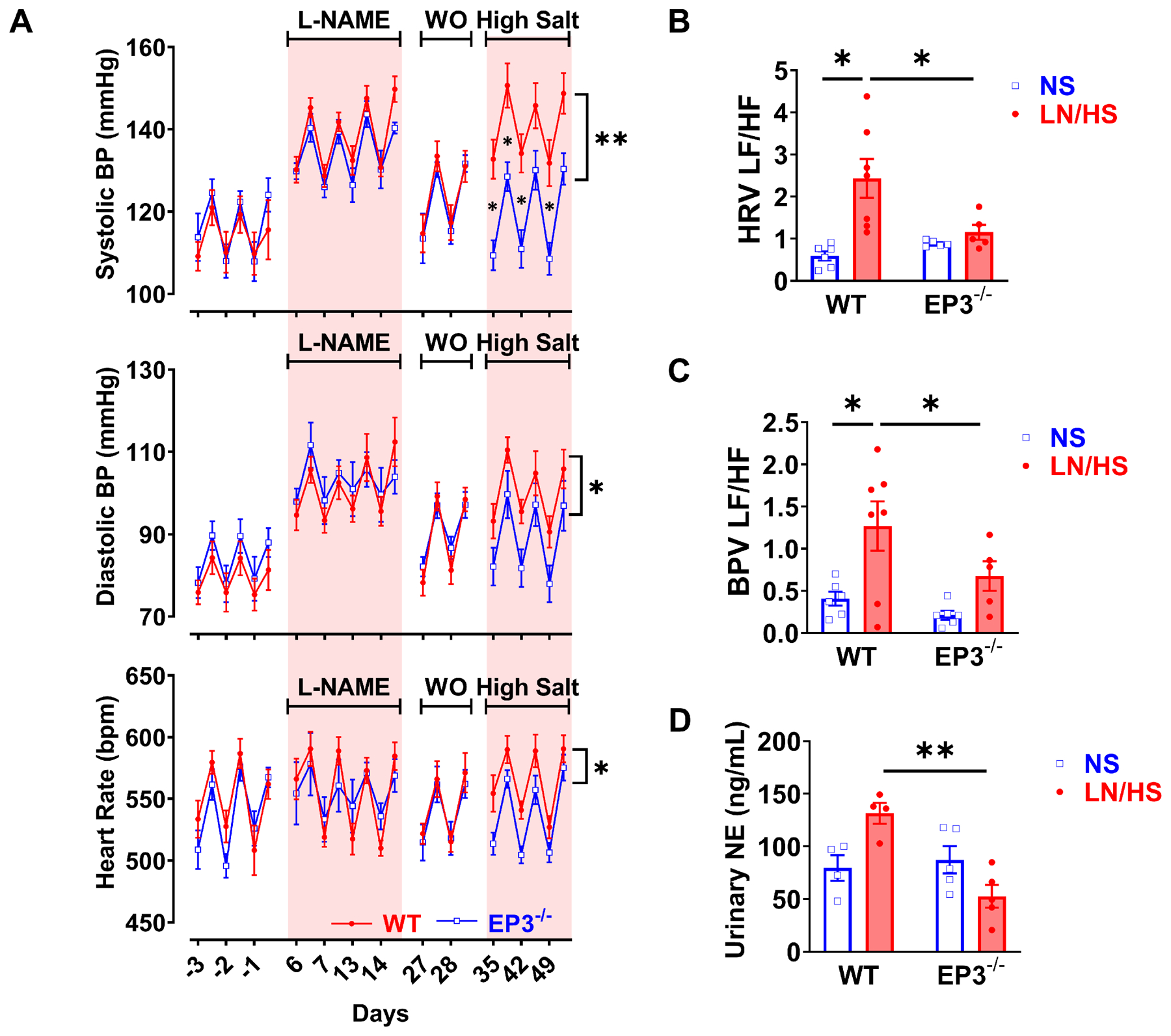
Effects of EP3 deficiency on L-NAME/high salt induced hypertension and autonomic function. Wild type and EP3−/− mice received 0.5 mg/mL L-NAME (LN) in the drinking water, followed by a 2-week washout (WO) and subsequently a 4% high-salt diet. Systolic blood pressure (BP), diastolic BP and heart rate (panels A) were measured by telemetry. Power spectral analysis of blood pressure and heart rate variabilities at night time are shown in B and C. Spot urine was collected at the end of high salt treatment, and urinary norepinephrine (NE) was determined by HPLC as shown in D. Blood pressure and heart rate data were analyzed by area under the curve followed with t test, data from the power spectral analysis and urinary norepinephrine were analyzed with 2-way ANOVA and Bonferroni post-hoc multiple comparisons. n=5 to 9 in each group. *P<0.05, **P<0.01.
Effects of EP3 Receptor Deficiency on Renal Injury:
Superoxide production in both renal tubular cells and the vasculature contributes to hypertension. As measured by dihydroethidium staining, superoxide production was increased in the kidneys of mice with L-NAME/high salt hypertension, particularly in vascular endothelial cells. This was absent in mice lacking EP3 receptors (Figure 2A and 2B). Renal collagen deposition, as evidenced by Masson’s Trichrome staining, was increased in wild type, but not EP3−/− mice (Figure 2C and 2D).
Figure 2:
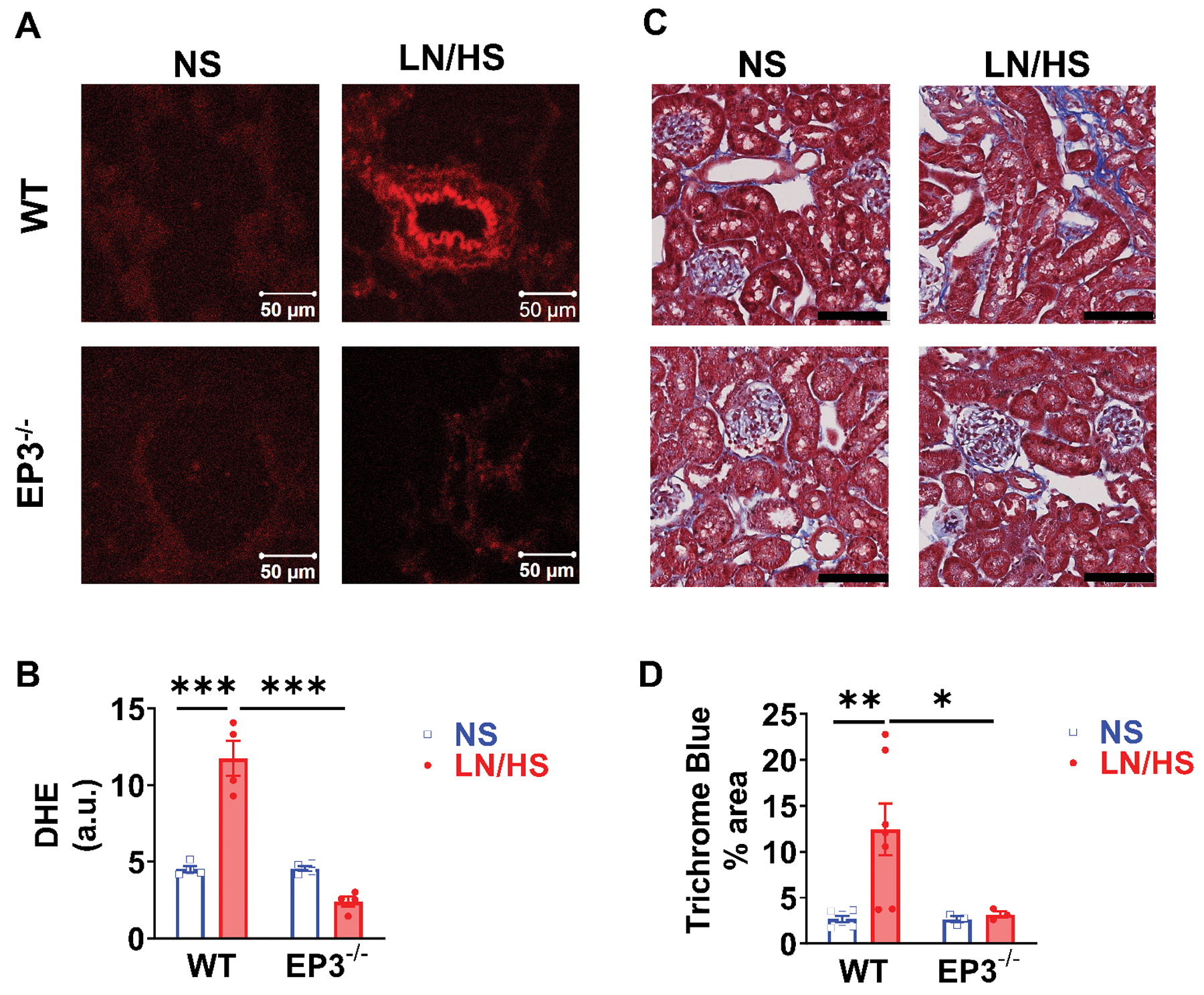
Effects of EP3 deficiency on renal oxidative stress and renal fibrosis in response to L-NAME/high salt (LN/HS) induced hypertension. DHE fluorescence was visualized at 530 nm to 560 nm and relative fluorescence intensity was quantified in (A) and (B). Collagen was identified using Masson’s trichrome blue stain and quantified in (C) and (D). Data were analyzed using 2-way ANOVA and post-hoc multiple comparisons. *P<0.05, **P<0.01, n= 4 to 8 in each group. Black lines denote 100 μm.
Effect of EP3 Receptor Deficiency on Renal Inflammation and T-Cell Activation in L-NAME/High Salt-induced Hypertension:
Flow cytometry of single cell suspensions of the kidney showed that L-NAME/high salt induced a 2 to 3-fold increase in total renal leukocytes, total T cells and both CD4+ and CD8+ T cells, as well as IFN-γ and IL-17a produced from T cells, and that this was prevented in EP3−/− mice (Figure 3). Further, L-NAME/high salt increased both CD44high/CD62Lhigh central and CD44high/CD62Llow effector memory T cell formation in the kidney and this was also blunted in EP3−/− mice (Figure S2).
Figure 3:
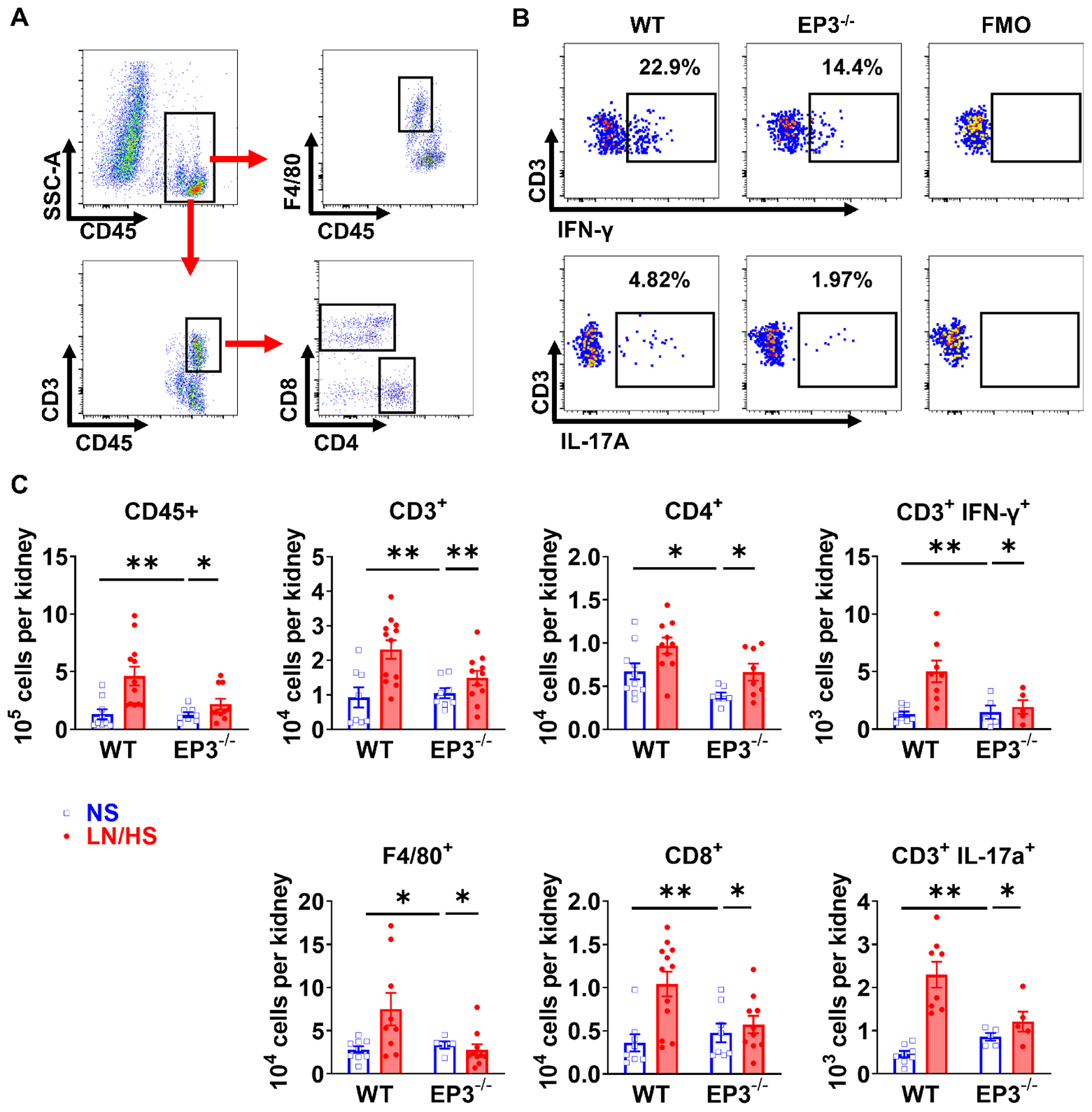
Effects of EP3 deficiency on renal leukocyte and T-cell infiltration. Mice underwent L-NAME/high salt (LN/HS) protocol as in figure 1. Flow cytometry gating strategy of kidneys are shown in (A). Live singlet cells were gated for total leukocytes (CD45+), monocytes/macrophages (F4/80+), total T cells (CD3+), CD4+ and CD8+ T cells. Representative intracellular staining for IFN-γ and IL-17A in CD3+ T cells from wild type and EP3−/− mice, as well as respective fluorescence-minus-one (FMO) controls are shown in (B). Mean data are shown in (C). Data were analyzed using 2-way ANOVA followed by Bonferroni post-hoc test, n=5 to 12 in each group. *P<0.05, **P<0.01.
Effect of EP3 Receptors on Dendritic Cell Activation in L-NAME/High Salt-induced Hypertension:
Because DCs play a critical role in activating T cells and contribute to hypertension, we performed additional studies to determine if EP3 receptors modulate DC phenotype. The L-NAME/high salt protocol increased amount of isoLG adducts in DCs by approximately 50% from the spleen of wild type animals, but not in DCs of EP3−/− mice (Figure 4B). During DC maturation, surface expression of the co-stimulatory molecules CD80 and CD86 is increased and we have shown these B7 ligands are essential for hypertension.5,10 Consistent with these previous findings, L-NAME/high salt increased surface expression of CD86 in DCs of WT but not EP3−/− mice (Figure 4C).
Figure 4:
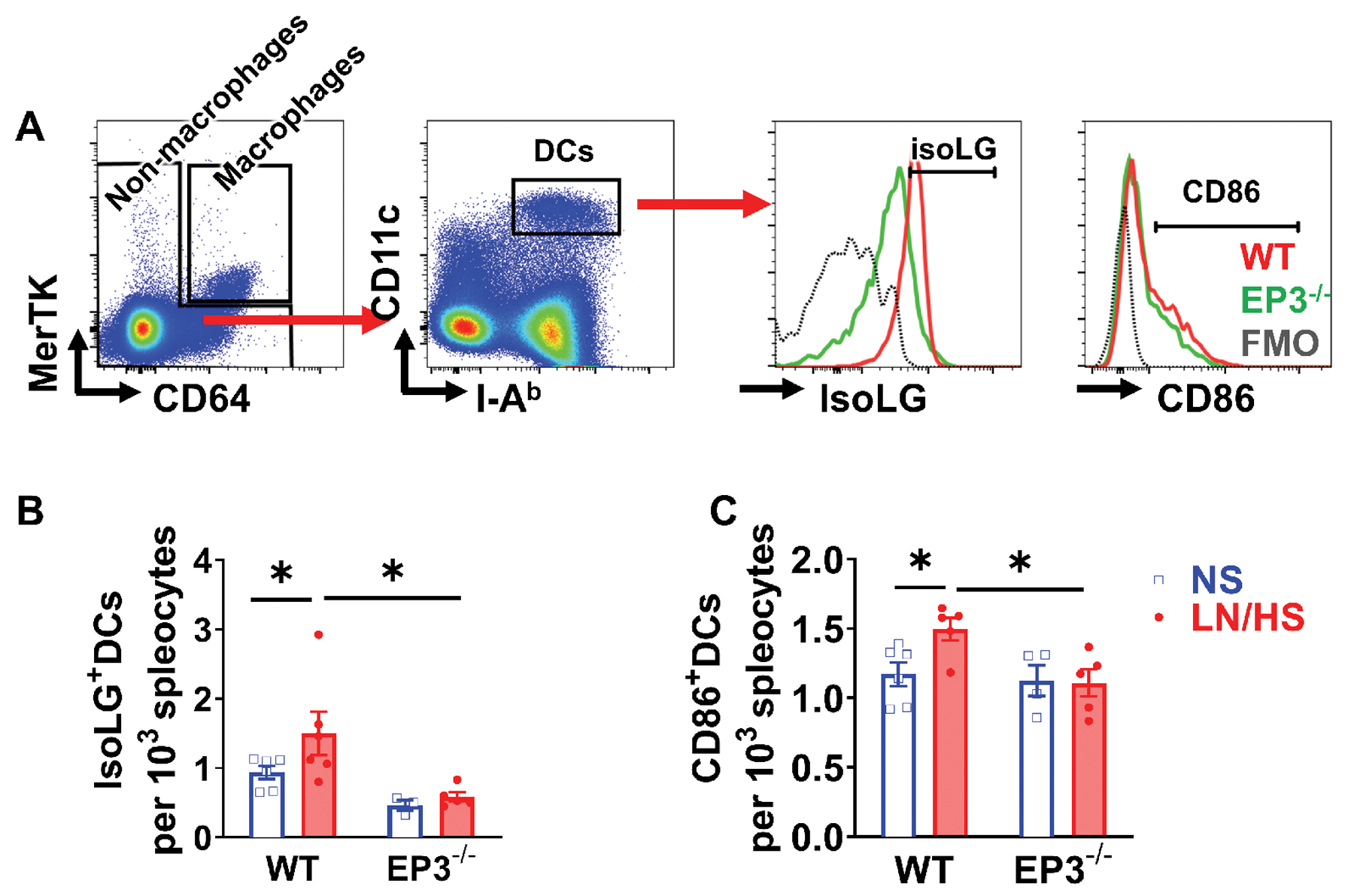
Effects of EP3 deficiency on dendritic cell activation and isoLG adduct formation in L-NAME/high salt (LN/HS) induced hypertension. DCs were gated after exclusion of macrophages, and expression of costimulatory molecules CD86, and intracellular isoLG-protein adducts were measured. Representative results are shown in (A), and mean data are shown in (B) and (C). Fluorescence minus one controls (FMO) were shown in black dashed lines. Data were analyzed using 2-way ANOVA. *P<0.05, n= 4 to 6 in each group.
We performed additional experiments to determine the role of EP3 receptors on DCs versus those on other cells in L-NAME/high salt-induced hypertension in EP3−/− animals. We isolated 106 splenic DCs isolated from wild type donors and transferred them to EP3 receptor deficient recipient mice. When subjected to L-NAME/high salt, these chimeric mice were still protected from hypertension during high salt diet, similar to EP3−/− mice, indicating EP3 receptors on DCs do not play a role in hypertension, while those on other somatic cells are likely critical (Figure S3).
Effect of EP3 Receptors in the Central Nervous System in L-NAME/High Salt-induced Hypertension:
Because the power spectral analyses indicated a role of EP3 receptor in modulating autonomic control of blood pressure and heart rate, we collected brain samples from mice under normal salt diet, after 2 weeks of L-NAME, as well as after completion of L-NAME/high salt protocol. The expression of cyclooxygenases and EP receptors, determined by real-time PCR, is shown in the heat map in Figure 5A, as well as Figure S4 and S5. COX-2 expression was significantly upregulated in the OVLT of mice that received L-NAME/high salt, but not in the SFO or PVN. Interestingly, among EP receptors, EP3 expression was greatest in the OVLT, and was downregulated following L-NAME/high salt. We therefore hypothesized that EP3 receptors in the OVLT, and possibly other circumventricular organs, mediate salt sensitive hypertension in L-NAME/high salt model. Expression of the EP3 receptor in circumventricular organs of wild type mice was knocked down by EP3 shRNA via ICV injection. As assessed by radiotelemetry, a ~15mmHg reduction of systolic blood pressure was observed in mice that had ICV injection of shRNA against EP3 receptor, compared to mice that received control vector injection during the high salt phase of the L-NAME/high salt protocol (Figure 5B). Power spectral analysis also revealed that these mice also had reduced sympathetic outflow during the high salt phase, as indicated by lower frequency to high frequency ratios in both blood pressure and heart rate oscillations. Silenced expression of EP3 receptor was confirmed in the OVLT and SFO (Figure 5D). Flow cytometry of splenic DCs indicated a marked attenuation of isoLG-adducted proteins (Figure 6A), and this was associated with alleviated tissue infiltration of total leukocyte, total T cells, F4/80+ monocytes/macrophages, CD4+ and CD8+ T cell subsets (Figure 6D and Figure S6).
Figure 5:
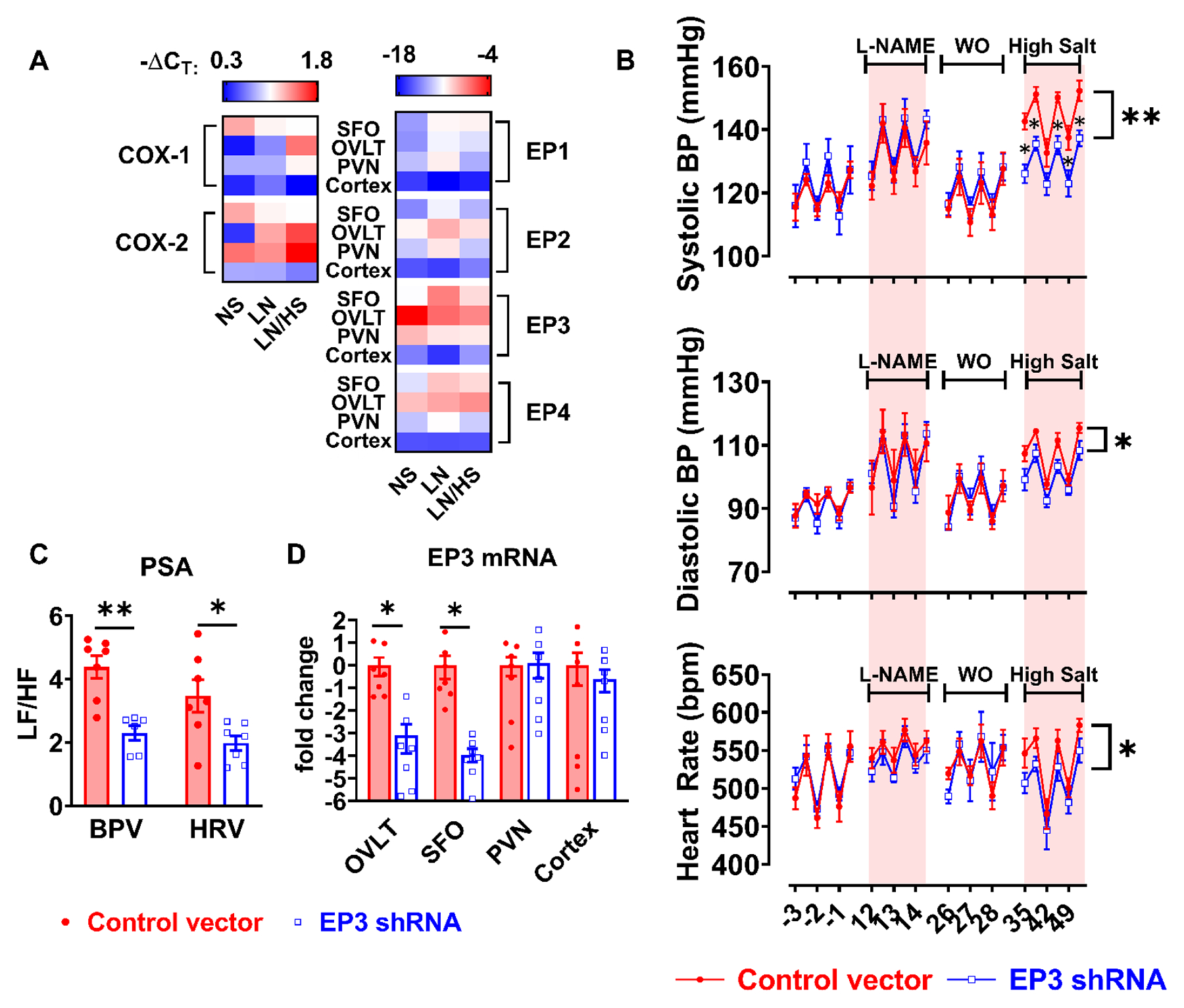
Role of central EP3 receptors in L-NAME/high salt induced hypertension. Punch biopsies of the subfornical organ (SFO), organum vasculosum laminae terminalis (OVLT), paraventricular nucleus (PVN) and cortex were collected from mice after normal salt (NS), L-NAME only (LN), and L-NAME/high salt (LN/HS). Gene expression of COX-1, COX-2, and PGE2 receptors (EP1-EP4) was determined by real time PCR shown in (A). Beta-actin was used as control gene to calculate ΔCT, n=3 to 9 in each group. Knockdown EP3 receptor was accomplished by ICV injection of 107 lentivirus vectors encoding shRNAs targeting EP3 gene or scrambled control sequences. After recovery from surgery, mice were subjected to L-NAME/high salt protocol, and systolic blood pressure (BP), diastolic BP and heart rate were recorded using telemetry shown in (B). Data were analyzed by area under the curve followed with t test. Power spectra analyses (PSA) of blood pressure and heart rate variability (BPV and HRV) are shown in panel (C). After animals were sacrificed, EP3 receptor mRNA levels in OVLT, SFO, PVN, and cortex were determined by real time PCR shown in (D). Data were analyzed by multiple t tests, *P<0.05, **P<0.01, n=7 in both groups.
Figure 6:
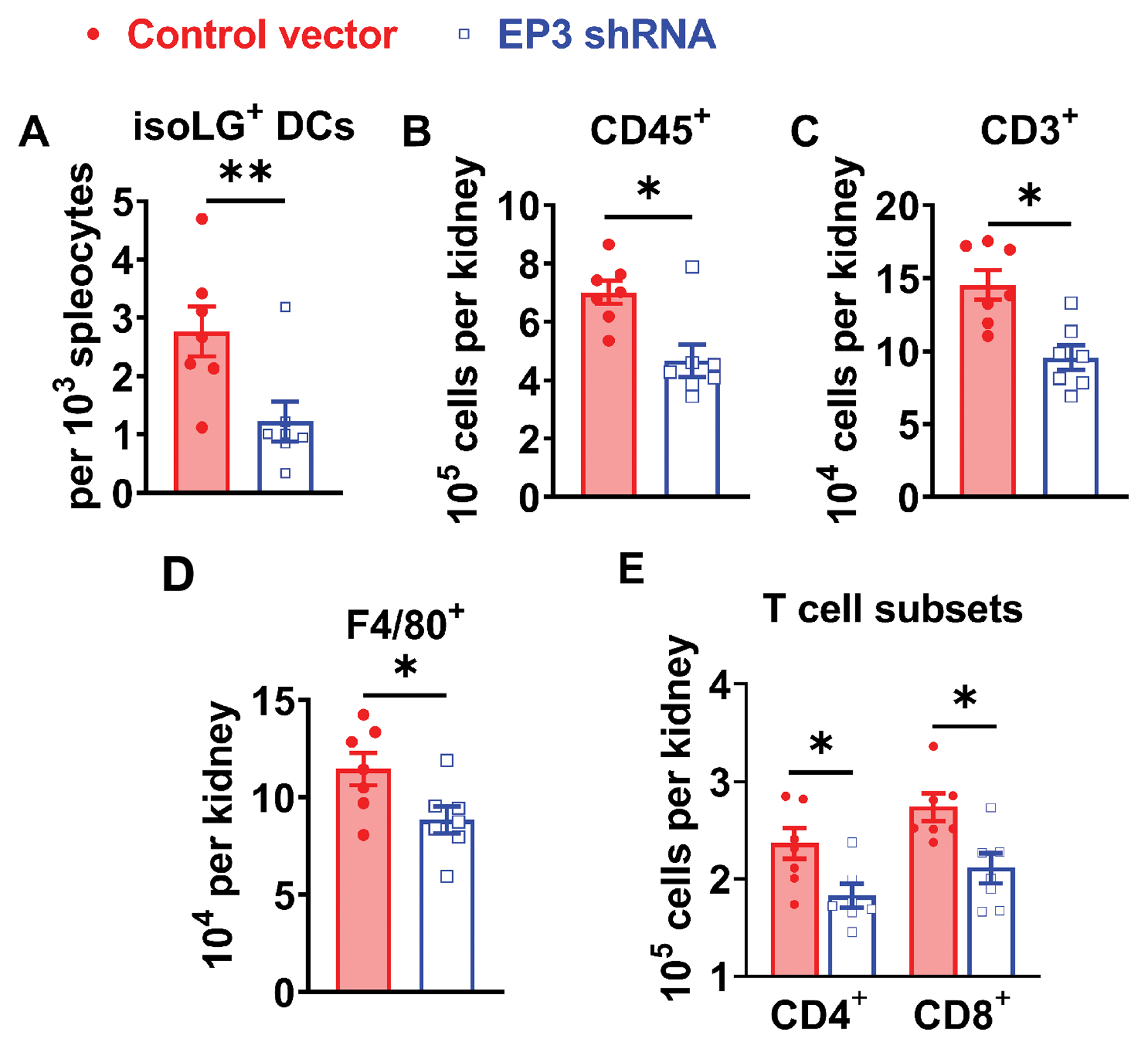
Role of central EP3 receptors in L-NAME/high salt induced renal inflammation. IsoLG adducts in splenic DCs were quantified by flow cytometry shown in (A). Total leukocytes (CD45+), total T lymphocytes (CD3+), monocytes/macrophages (F4/80+), and CD4+ and CD8+ T cell subsets in the kidney were quantified by flow cytometry shown in from panel B to E. Data were analyzed by student t tests. P<0.05, **P<0.01, n=7 in both groups.
Direct Effects of PGE2 on IsoLG formation in Dendritic Cells:
To further determine if PGE2 directly promotes isoLG adduct formation in DCs, we cultured mouse splenic DCs with increasing doses of PGE2 for 24 hours. As shown in Figure 6A, 50 nM PGE2 tripled isoLG adducts in DCs from baseline, and this is accompanied with upregulation of CD86 expression (Figure S7A and S7B). To further determine the EP receptor subtypes involved in these effects, splenic DCs were pretreated with EP1 or EP3 receptor antagonists before PGE2 was added. Interestingly, the EP1 receptor antagonist SC-51322 completely prevented the accumulation of isoLG adduct induced by PGE2, but EP3 receptor blocker DG-041 did not (Figure S7C). In a separate experiment, splenic DCs were stimulated with EP1 or EP3 receptor agonists. 17-phenyl-trinor-PGE2, which has similar affinity to mouse EP1 and EP3 receptors, increased isoLG adduct formation. However, sulprostone, MB-28767, and ONO-AE-248, which preferentially bind to EP3 receptors in mouse cells, did not affect isoLG adduct formation (Figure S7D).
DISCUSSION
The current studies indicate that PGE2, by acting on the EP3 receptor, plays a critical role in initiating inflammation in a salt-sensitive model of hypertension as delineated in the scheme shown in Figure 6F. Our data indicate that mice lacking the EP3 receptor develop blunted hypertension during the salt feeding phase of the L-NAME/high salt challenge, and this is associated with reduced renal infiltration of T cells. Importantly, activation of DCs, as evidenced by surface expression of CD86 and accumulation of IsoLG-protein adducts is markedly reduced in mice lacking EP3 receptors. While EP3 receptors are widely distributed, our data also suggest a predominant effect of central EP3 receptors, as power analysis of heart rate and blood pressure variability and urinary norepinephrine suggested that sympathetic outflow is reduced in EP3−/− mice and central knockdown of this receptor using ICV injection of shRNA yielded a phenotype similar to its global deficiency. We also observed that the EP3−/− mice had reduced superoxide production in the kidney and were protected from developing renal fibrosis in hypertension. PGE2 also was found to have direct effects on DCs, causing reactive oxygen species (ROS) production and isoLG adducted protein formation. However, this is mediated by EP1 but not EP3 receptors in DCs.
PGE2 and the EP3 receptor have been previously shown to modulate blood pressure and hemodynamics in various animal models of hypertension. In keeping with our current findings, blockade of the EP3 receptor attenuates angiotensin II-induced vasoconstriction and its acute pressor response in mice.22 Activation of EP3 potentiates vasoconstriction of renal afferent arterioles,23 and EP3 receptor deficient mice have higher baseline renal blood flow than wild type mice.24 We have also shown that EP3 receptor promotes vasoconstriction through concurrent signaling with the angiotensin II type 1 receptor.14, 24
Data from our in vivo and in vitro experiments indicates that PGE2 likely modulates DC activation in hypertension through multiple mechanisms. PGE2 has prominent effects on ROS production and isoLG adduct accumulation in DCs in vitro. These direct effects of PGE2 seem to be mediated by EP1 activation, as they were prevented by the EP1 receptor antagonist SC-51322 and not by the EP3 receptor antagonist DG-041. The abrogated accumulation of isoLG adducts in DCs of EP3−/− mice is more likely to be caused by attenuated ROS production in the kidney. This is compatible with our previous finding that increased efferent renal sympathetic tone promotes ROS production in the kidney, leading to isoLG adduct accumulation in DCs and subsequent activation of adaptive immunity in hypertension.6
A major role of the EP3 receptor in the sympathetic nervous system is supported by the changes in power spectra of blood pressure and heart rate of EP3−/− mice, as well as the data from transduction of shRNA targeting EP3 receptor in the central nervous system. Recent studies from acute central administration of PGE2 and selective EP receptor subtype agonists and antagonists indicate that activation of EP3 in the PVN and rostral ventrolateral medulla stimulates sympathetic outflow and increase in blood pressure.25–27 Compared to other EP receptors, we found the EP3 receptor to be highly expressed in the OVLT. We also observed enhanced expression of COX-2 and a downregulation of EP3 receptor in the OVLT during the high salt phase in the L-NAME/high salt treated mice, suggesting that COX-2 dependent PGE2 production leads to activation of EP3 receptors and subsequent activation of sympathetic nervous system. Of note, the down regulation of EP3 receptor expression is possibly due to a negative feedback mechanism in response to long term overactivation of this G-protein-coupled receptor. A previous study by Wei et al. showed a similar upregulation of COX-2 in the SFO and PVN of rats that received SFO microinjection of tumor necrosis factor-α or interleukin-1β, suggesting that neuroinflammation may lead to feedforward systemic immune activation.28
It is particularly interesting that upregulation of COX-2 was only found in the OVLT as a result of the L-NAME/high salt protocol. As a part of the circumventricular organs in the brain, the OVLT has a highly permeable microvasculature and plays a pivotal role in sensing and regulating plasma osmolality.29 Kinsman et al. recently showed that OVLT neurons detect extracellular NaCl, and promote sympathoexcitation to raise in blood pressure.30 It is possible that COX-2 dependent PGE2 and EP3 signaling mediates OVLT pre-sympathetic neuronal activity in response to high salt feeding. It is of interest that angiotensin II-induced hypertension has been linked to ROS production the SFO, another site in the lamina terminalis, through COX-1 dependent EP1 receptor activation.31 Of note, recent studies have established a role of superoxide formation the OVLT in driving sympatho-excitation in angiotensin II-induced hypertension.32, 33
Our in vitro experiments suggest that EP1 receptor activation can also have direct effects on DCs. We observed a marked effect of EP1 stimulation of DCs in culture on isoLG adduct formation. Others have shown that blockade or deletion of the EP1 receptor reduces oxidative stress and hypertension development.12, 31 This direct EP1 effect on DCs and its role in hypertension requires future investigation, however our current data suggest that ROS production from tissue (e.g. renal vasculature) may play a more important role than DC intracellular ROS in immune activation in hypertension. We have previously shown that vascular ROS can promote formation of isoLGs in antigen presenting cells,34 and it is likely that a similar mechanism is operative in the case of salt-feeding, as indicated by our current findings. Monocytes and monocyte derived DCs are intimately related to the vasculature, and could either receive oxidant signals from adjacent vessels, or perhaps phagocytose oxidatively modified proteins made in adjacent vessels.
In conclusion, we found that PGE2 and the EP3 receptor are essential in the development of salt sensitive hypertension and activation of adaptive immune cells through activation sympathetic outflow. Further studies are needed to understand the mechanism by which high salt diet induce COX-2 expression in the central nervous system, which may lead to new therapeutic targets for treatment of salt-sensitive hypertension.
Perspectives
It is well accepted that PGE2 plays multiple roles in the cardiovascular system and inflammation, and many of its pro-hypertensive and pro-inflammatory effects are mediated by EP1 and EP3 receptors. We have demonstrated that salt-sensitive hypertension in L-NAME/high salt model requires PGE2 acting on EP3 receptors in the central nervous system, which leads to enhanced sympathetic outflow, peripheral ROS production, isoLG adducted protein accumulation in DCs and renal inflammation. PGE2 also directly stimulates ROS production and isoLG adduct formation in DCs through EP1 but not EP3 receptor. These observations provide evidence for a previously unrecognized role of EP3 receptor in hypertension and renal inflammation.
Supplementary Material
Novelty and Significance.
What is new:
We identify a previously unknown role of central EP3 receptors in modulating immune activation and hypertension.
What is relevant:
These findings provide new insight into understanding how the central nervous system regulates inflammation in hypertension.
Summary:
EP3 receptors in the brain, likely in the organum vasculosum of the lateral terminalis, are activated by PGE2 in hypertension and promote sympathetic outflow that in turn increases reactive oxygen species production in peripheral tissues, particularly the kidney. These events promote formation of isolevuglandin adducts, which seem to act as neoantigens in dendritic cells. Genetic deletion or shRNA mediated local knockdown of EP3 in the circumventricular organs prevents these effects and lowers blood pressure.
Acknowledgements
We are grateful to the Translational Pathology Shared Resource and Vanderbilt Cell Imaging Shared Resource for the preparation and imaging the immunostaining slides, and to Vanderbilt Hormone Assay & Analytical Services Core for the measurements of urinary norepinephrine.
Sources of Funding
This work was supported by the National Institutes of Health Grants R35HL140016 and Program Project Grant P01HL129941 to D.G.H., R56HL127218, R01HL134895 to R.M.B, and American Heart Association Scientist Development Grant 17SDG33670829 to L.X.
ABBREVIATIONS
- ANOVA
Analysis of variance
- CD
cluster of differentiation
- COX
cyclooxygenase
- DC
dendritic cell
- DOCA
deoxycorticosterone acetate
- EP
E prostanoid
- ICV
intracerebroventricular
- isoLG
isolevuglandin
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- NOS
nitric oxide synthase
- OVLT
organum vasculosum of the laminae terminalis
- PGE2
prostaglandin E2
- PVN
paraventricular nucleus
- ROS
reactive oxygen species
- SFO
subfornical organ
Footnotes
Disclosures
None
REFERENCES
- 1.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme m-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381 [DOI] [PubMed] [Google Scholar]
- 3.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. J Clin Invest. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circ Res. 2015;117:547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalinski P Regulation of immune responses by prostaglandin e2. J Immunol. 2012;188:21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142 [DOI] [PubMed] [Google Scholar]
- 9.Luft T, Jefford M, Luetjens P, Toy T, Hochrein H, Masterman KA, Maliszewski C, Shortman K, Cebon J, Maraskovsky E. Functionally distinct dendritic cell (dc) populations induced by physiologic stimuli: Prostaglandin e(2) regulates the migratory capacity of specific dc subsets. Blood. 2002;100:1362–1372 [DOI] [PubMed] [Google Scholar]
- 10.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh P, Hoggatt J, Hu P, Speth JM, Fukuda S, Breyer RM, Pelus LM. Blockade of prostaglandin e2 signaling through ep1 and ep3 receptors attenuates flt3l-dependent dendritic cell development from hematopoietic progenitor cells. Blood. 2012;119:1671–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, Gao Y, Chen L, Zhang X, Davis LS, Wei M, Fan X, Carmosino M, Hao C, Imig JD, Breyer RM, Breyer MD. Antihypertensive effects of selective prostaglandin e2 receptor subtype 1 targeting. J Clin Invest. 2007;117:2496–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen D, Tang J, Wan Q, Zhang J, Wang K, Shen Y, Yu Y. E-prostanoid 3 receptor mediates sprouting angiogenesis through suppression of the protein kinase a/beta-catenin/notch pathway. Arterioscler Thromb Vasc Biol. 2017;37:856–866 [DOI] [PubMed] [Google Scholar]
- 14.Kraemer MP, Choi H, Reese J, Lamb FS, Breyer RM. Regulation of arterial reactivity by concurrent signaling through the e-prostanoid receptor 3 and angiotensin receptor 1. Vascul Pharmacol. 2016;84:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. 2015;125:4281–4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceddia RP, Lee D, Maulis MF, Carboneau BA, Threadgill DW, Poffenberger G, Milne G, Boyd KL, Powers AC, McGuinness OP, Gannon M, Breyer RM. The pge2 ep3 receptor regulates diet-induced adiposity in male mice. Endocrinology. 2016;157:220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of t-lymphocyte activation and vascular inflammation produced by angiotensin ii-induced hypertension. Circ Res. 2010;107:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the nadph oxidases in the subfornical organ in angiotensin ii-induced hypertension. Hypertension. 2013;61:382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franklin KBJ, Paxinos G. Paxinos and franklin’s the mouse brain in stereotaxic coordinates. Amsterdam: Academic Press, an imprint of Elsevier; 2013. [Google Scholar]
- 21.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125:1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, Miao Y, Zhang Y, Dou D, Liu L, Tian X, Yang G, Pu D, Zhang X, Kang J, Gao Y, Wang S, Breyer MD, Wang N, Zhu Y, Huang Y, Breyer RM, Guan Y. Inactivation of the e-prostanoid 3 receptor attenuates the angiotensin ii pressor response via decreasing arterial contractility. Arterioscler Thromb Vasc Biol. 2012;32:3024–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin e(2) on the renal afferent arteriole : Role of ep(3) and ep(4) receptors. Circ Res. 2000;86:663–670 [DOI] [PubMed] [Google Scholar]
- 24.Audoly LP, Ruan X, Wagner VA, Goulet JL, Tilley SL, Koller BH, Coffman TM, Arendshorst WJ. Role of ep(2) and ep(3) pge(2) receptors in control of murine renal hemodynamics. Am J Physiol Heart Circ Physiol. 2001;280:H327–333 [DOI] [PubMed] [Google Scholar]
- 25.Ariumi H, Takano Y, Masumi A, Takahashi S, Hirabara Y, Honda K, Saito R, Kamiya HO. Roles of the central prostaglandin ep3 receptors in cardiovascular regulation in rats. Neurosci Lett. 2002;324:61–64 [DOI] [PubMed] [Google Scholar]
- 26.Zhang ZH, Yu Y, Wei SG, Nakamura Y, Nakamura K, Felder RB. Ep(3) receptors mediate pge(2)-induced hypothalamic paraventricular nucleus excitation and sympathetic activation. Am J Physiol Heart Circ Physiol. 2011;301:H1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezq S, Abdel-Rahman AA. Rostral ventrolateral medulla ep3 receptor mediates the sympathoexcitatory and pressor effects of prostaglandin e2 in conscious rats. J Pharmacol Exp Ther. 2016;359:290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei SG, Yu Y, Zhang ZH, Felder RB. Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension. 2015;65:1126–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thrasher TN, Keil LC, Ramsay DJ. Lesions of the organum vasculosum of the lamina terminalis (ovlt) attenuate osmotically-induced drinking and vasopressin secretion in the dog. Endocrinology. 1982;110:1837–1839 [DOI] [PubMed] [Google Scholar]
- 30.Kinsman BJ, Simmonds SS, Browning KN, Stocker SD. Organum vasculosum of the lamina terminalis detects nacl to elevate sympathetic nerve activity and blood pressure. Hypertension. 2017;69:163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao X, Peterson JR, Wang G, Anrather J, Young CN, Guruju MR, Burmeister MA, Iadecola C, Davisson RL. Angiotensin ii-dependent hypertension requires cyclooxygenase 1-derived prostaglandin e2 and ep1 receptor signaling in the subfornical organ of the brain. Hypertension. 2012;59:869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collister JP, Taylor-Smith H, Drebes D, Nahey D, Tian J, Zimmerman MC. Angiotensin ii-induced hypertension is attenuated by overexpressing copper/zinc superoxide dismutase in the brain organum vasculosum of the lamina terminalis. Oxid Med Cell Longev. 2016;2016:3959087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishi T, Hirooka Y. Oxidative stress in the brain causes hypertension via sympathoexcitation. Front Physiol. 2012;3:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ 2nd, Madhur MS, Harrison DG. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:50–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.