

 This work is licensed under a
This work is licensed under a Abstract
Medical treatments for corticotrophinomas are limited, and we therefore investigated the effects of epigenetic modulators, a new class of anti-tumour drugs, on the murine adrenocorticotropic hormone (ACTH)-secreting corticotrophinoma cell line AtT20. We found that AtT20 cells express members of the bromo and extra-terminal (BET) protein family, which bind acetylated histones, and therefore, studied the anti-proliferative and pro-apoptotic effects of two BET inhibitors, referred to as (+)-JQ1 (JQ1) and PFI-1, using CellTiter Blue and Caspase Glo assays, respectively. JQ1 and PFI-1 significantly decreased proliferation by 95% (P < 0.0005) and 43% (P < 0.0005), respectively, but only JQ1 significantly increased apoptosis by >50-fold (P < 0.0005), when compared to untreated control cells. The anti-proliferative effects of JQ1 and PFI-1 remained for 96 h after removal of the respective compound. JQ1, but not PFI-1, affected the cell cycle, as assessed by propidium iodide staining and flow cytometry, and resulted in a higher number of AtT20 cells in the sub G1 phase. RNA-sequence analysis, which was confirmed by qRT-PCR and Western blot analyses, revealed that JQ1 treatment significantly altered expression of genes involved in apoptosis, such as NFκB, and the somatostatin receptor 2 (SSTR2) anti-proliferative signalling pathway, including SSTR2. JQ1 treatment also significantly reduced transcription and protein expression of the ACTH precursor pro-opiomelanocortin (POMC) and ACTH secretion by AtT20 cells. Thus, JQ1 treatment has anti-proliferative and pro-apoptotic effects on AtT20 cells and reduces ACTH secretion, thereby indicating that BET inhibition may provide a novel approach for treatment of corticotrophinomas.
Keywords: bromo and extra terminal domain, histone code reader, pituitary, Cushing’s disease, neuroendocrine
Introduction
Corticotrophinomas represent >10% of all surgically removed pituitary adenomas, which are the most commonly encountered intracranial neoplasms that are identified in >25% of unselected autopsies and approximately 20% of the population undergoing intracranial imaging (Daly et al. 2009). Corticotrophinomas are associated with hypersecretion of adrenocorticotropic hormone (ACTH), which leads to excessive production of glucocorticoids by the adrenal cortex, and the resulting hypercortisolemia causes Cushing’s disease, whose clinical features include obesity, redistribution of adipose tissue, muscle atrophy with preclinical myopathy, diabetes mellitus, hypertension, osteoporosis, subfertility, skin thinning, depression, psychosis and increased susceptibility to infection (Daly et al. 2009, Ntali et al. 2013, Yates et al. 2015). Corticotrophinomas are therefore a cause of ACTH-dependent Cushing’s disease, which is also referred to as pituitary-dependent Cushing’s syndrome (Ntali et al. 2013). Corticotrophinomas, which are neuroendocrine tumours (NETs) of pituitary, are usually microadenomas (i.e. <10 mm in diameter) and often are too small to be detected by radiological imaging (e.g. MRI or computerised tomography (CT) scans) or identifiable at surgery (Cuevas-Ramos et al. 2016).
The treatment of choice for corticotrophinomas is transsphenoidal resection, which results in remission rates of 70–90% for microadenomas (Cuevas-Ramos et al. 2016). However, mortality rates of 1–2% are reported to be associated with transsphenoidal resection, and long-term (10 years) recurrence rates of ~20% following transsphenoidal resection indicate that a long-term cure is achieved in only ~60–80% of adults with corticotrophinomas (Daly et al. 2009, Cuevas-Ramos et al. 2016). Pharmacological treatments are available for patients for whom transsphenoidal surgery has not been successful in removing the corticotrophinomas and these include inhibitors of steroidogenesis (e.g. metyraprone, ketoconazole, mitotane, etomidate and osilodrostat); glucocorticoid antagonists (e.g. mifepristone); dopamine agonists such as cabergoline; and somatostain analogues such as pasireotide (Cuevas-Ramos et al. 2016). However, these current medical treatments for corticotrophinomas have limited efficacy, and thus, there is a clinically unmet need for improved pharmacological treatments for corticotrophinomas, especially for those occurring in patients who have contraindications for surgery or have had unsuccessful surgery.
Epigenetic-targeting compounds are a new class of anti-tumour drugs, and one such family of small molecule bromo and extra-terminal domain (BET) inhibitors, which target the bromodomains (BRDs) of the protein family members BRD2, BRD3, BRD4 and BRDT that bind acetylated residues on histones that regulate gene expression, and particularly those of tissue-specific genes (Filippakopoulos et al. 2010), have been shown in preclinical in vitro and in vivo studies to have efficacy in a number of tumour types including pancreatic neuroendocrine tumours, glioma, nuclear protein in testis (NUT)-midline carcinoma, leukaemias and renal cell carcinoma (Beesley et al. 2014, Coude et al. 2015, Ishida et al. 2017a , b , Leal et al. 2017, Lines et al. 2017, Wu et al. 2017). Moreover, in order to determine if BET inhibitors may also represent an effective novel therapy for corticotrophinomas in reducing proliferation and increasing apoptosis of these pituitary cells, we first investigated the mouse corticotroph tumour cell line AtT20 for expression of the BET protein family members and then the effects of the BET inhibitors JQ1 and PFI-1 on proliferation, apoptosis and ACTH secretion by these pituitary cells.
Materials and methods
Cell lines, in vitro assays and compounds
AtT20 murine pituitary corticotroph tumour cells were purchased from the American Type Culture Collection (ATCC) (CCL-89); murine cells that were used as a human corticotroph tumour cell line is not currently available. AtT20 cells are small, round, adherent cells that have a doubling time of approximately 1–2 days and were originally isolated from a LAF1 mouse pituitary tumour (Buonassisi et al. 1962). Cells were cultured in DMEM media, supplemented with 10% foetal calf serum (FCS) (Sigma-Aldrich), maintained at 37°C, 5% (vol/vol) CO2 and tested for mycoplasma using the MycoAlert kit (Lonza). PFI-1, (+)-JQ1 (henceforth JQ1) and its inactive control compound ((-)-JQ1, henceforth JQ1-) were suspended and diluted in dimethyl sulfoxide (DMSO, Sigma-Aldrich), as previously described (Lines et al. 2017). Both compounds were obtained from the Structural Genomics Consortium (SGC, University of Oxford), and further details on the structure and specificity for each compound are available at https://www.thesgc.org/chemical-probes. Octreotide (Sigma-Aldrich) was suspended and diluted in distilled water. Untreated and vehicle (DMSO-only)-treated AtT20 cells were used as controls. For all studies, cells underwent trypsin treatment, before the cell number was determined by trypan blue staining and counting using a haemocytometer. Proliferation, apoptosis and senescence assays were performed in 96-well plates with 5000 cells seeded per well, 24 h before drug treatment. For cell cycle analysis, 50,000 cells were seeded per well in 24-well plates, 24 h before drug treatment. Cell viability, as an indication of cell proliferation, was assessed using the CellTiter Blue Cell Viability assay (Promega), whereby 20 µL of CellTiter Blue reagent was added per well, incubated for 2 h at 37°C, 5% (vol/vol) CO2 and the fluorescent outputs read on a CytoFluor microplate reader (PerSeptive Biosystems, MA, USA) at 530 nm excitation and 580 nm emission (Eachkoti et al. 2014). Cell death by apoptosis was evaluated using the CaspaseGlo 3/7 assay (Promega), whereby 75 µL of CaspaseGlo reagent was added per well, incubated for 1 h at room temperature and the luminescent outputs read on a Turner Biosystems luminometer. Cellular senescence was determined using the Cellular Senescence Assay (SA-β-gal staining) kit (Cell Biolabs, CA, USA) (Lee et al. 2015), 96 h after drug treatment. Cells were visualised using an Eclipse E400 microscope (Nikon), and images were captured using a DXM1200C digital camera and NIS-Elements BR 2.30 software (both Nikon). The percentage of positively stained cells were counted, per total cells, from n = 20 10× magnification fields. Cell cycle progression was determined using proidium iodide (PI) (Sigma-Aldrich) staining, 48 h after drug treatment. Cells were fixed in 100% ethanol, washed with PBS and DNA stained by incubation with PI solution (10 µg/mL PI, 200 µg/mL RNase, 0.1% Triton-X, 150 mM sodium chloride; diluted in PBS) for 30 min at room temperature. Intensity of PI staining was detected on a FACS calibur flow cytometer (Becton Dickinson, NJ, USA) using Cellquest pro software. Peaks of fluorescence, representing DNA content, were used to evaluate the percentage of cells at each cell cycle phase, using FloJo software, as previously described (Lines et al. 2017).
Quantitative real-time PCR (qRT-PCR)
Total RNA from AtT20 cells was extracted using the miRVana kit (Ambion, Thermo Fisher Scientific) and 1 µg used to generate cDNA using the Quantitect RT kit (Qiagen), as described (Lines et al. 2017). Gene-specific Quantitect primers (Qiagen) were used for qRT-PCR reactions, which utilised the Quantitect SYBR green kit (Qiagen), on a RotorGene 5, as described (Lines et al. 2017). Each test sample was normalized to the geometric mean of reference genes glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and calnexin (Canx). The relative expression of target cDNA in all qRT-PCR studies was determined using published methods (Pfaffl 2001).
RNA-sequence (RNA-Seq) analysis
Confluent AtT20 cells were treated with 1 µM JQ1- or JQ1 for 48 h before RNA extraction using the RNeasy kit (Qiagen). RNA sequencing was performed on three independent experimental replicates for each cell line and treatment. Following polyA-enrichment and library preparation, 50 bp paired-end sequencing was performed (Illumina 2500 machine, rapid mode, 2 lanes). Reads were aligned to the mouse reference genome (GRCm37) using TopHat2 (Kim et al. 2013) and duplicate reads were removed (Picard Tools MarkDuplicates). Approximately 22–30 million high-quality reads per sample were mapped uniquely to Ensembl-annotated genes; gene counts were summarised using HTSeq (Anders et al. 2015) and filtered to exclude genes with fewer than ten reads on average per sample. The filtered set of genes were further analysed in the R statistical software (https://www.r-project.org/) to characterise data quality and sample clustering patterns. Data normalisation and differential expression analysis, comparing JQ1 and JQ1- treatment conditions, were performed using the edgeR package (Robinson et al. 2010). Genes with adjusted P value <0.05 and showing a fold change >2 in either direction were considered significant. Identification of altered cellular pathways was undertaken using QIAGEN Ingenuity® Pathway Analysis (IPA®, QIAGEN, www.qiagen.com/ingenuity). Gene-set enrichment analysis (GSEA) against MSigDB (software.broadinstitute.org/gsea/msigdb) gene-sets (v.6.2) was performed using the fgsea package in R/Bioconductor (http://bioconductor.org/packages/fgsea/) after mapping mouse EntrezIDs to human using the biomaRt package (Durinck et al. 2009, Sergushichev 2016).
Western blot analysis
AtT20 cells were lysed in NP40 lysis buffer: 250 mM NaCl, Tris 50 mM (pH 8.0), 5 mM EDTA, 0.5% NP-40 (v/v) and 2× Protease inhibitor tablets (Roche), as described (Lines et al. 2017). Lysates were prepared in 4× Laemmli loading dye (BioRad) boiled at 95°C for 5 min, resolved using 6% or 10% SDS-PAGE gel electrophoresis, transferred to polyvinylidene difluoride membrane and probed with primary antibodies (Brd2 – D89B4, Cell Signalling; Brd3 – 61489, Active motif, La Hulpe, Belgium; Brd4 – PA5-51550, Thermo Fisher Scientific; NFKβ1 (ab32360), cIAP2 (ab23423), Src (ab47405), POMC (ab94446) and SSTR2 (ab134152) all - Abcam; Calnexin – ab2301, Millipore; and Gapdh – ab181603, AbCam) and HRP-conjugated secondary antibody (sc-2357, Santa Cruz Biotechnology) prior to visualisation using Pierce ECL Western Blotting substrate (Thermo Fisher Scientific), as described (Lines et al. 2017). Calnexin or Gapdh protein expression was used as a loading control, and lysates from human bronchopulmonary neuroendocrine cells (H727) were used as a positive control (Lines et al. 2017). Densitometry analysis was performed by calculating the number of pixels per band using ImageJ software. Data were represented as the number of pixels of the protein band, relative to the number of pixels of the corresponding Gapdh or Calnexin band.
ACTH enzyme-linked immunosorbant assay (ELISA)
AtT20 cells were cultured in 24-well plates in 1 mL media. Cells were counted, harvested and media collected at 0, 24, 48 and 72 h after JQ1- or JQ1 treatment. Harvested cells were lysed and used for Western blot analysis. An ELISA was performed on the cell media to assess for ACTH secretion. Briefly, media was diluted 1:3 in distilled water and 200 μL added per well of a mouse/rat ACTH ELISA kit (Origene, MD, USA), incubated for 4 h with biotinylated and enzyme-labelled antibodies, before visualising with 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate and reading at 405 nM on a Pherastar FS (BMG LabTech, Ortenberg, Germany). Data were normalised to 0 h ACTH levels and corrected for cell number per well to avoid bias through alterations in proliferation and apoptosis.
Statistical analysis
Data were analysed using one-way or two-way ANOVA correcting for multiple comparisons or Dunnett’s test for a single control group, as previously described (Walls et al. 2012, Gorvin et al. 2013). For in vitro data, comparisons were relative to vehicle, and for the ACTH ELISA and RNA-Seq data, comparisons were relative to JQ1- treatment. RNA-Seq data were analysed using statistical models implemented in the edgeR package (Robinson et al. 2010). RNA-Seq results are reported with adjusted P values, after correcting raw P values for multiple testing using and Benjamini–Hochberg’s method (Benjamini & Hochberg 1995) to control the false discovery rate (FDR) at 5%.
Results
AtT20 cells express members of the BET family, with Brd2 being the most abundant
Evaluation of the expression of the four BET family members Brd2, Brd3, Brd4 and Brdt in AtT20 cells by quantitative real-time PCR (qRT-PCR) revealed that Brd2 is the most abundant member with 7.7-fold higher expression than Brd3 (P < 0.0005) and 2.1-fold higher expression than Brd4 (P < 0.0005) (Fig. 1A). Brd4 was the second most abundant BET family member, with 3.7-fold (P < 0.0005) higher expression than Brd3 (Fig. 1A). Expression of Brdt, which is testis specific, was not detected after 40 cycles of the qRT-PCR. Western blot analysis confirmed expression of BRD2, BRD3 and BRD4 proteins in AtT20 cells (Fig. 1B).
Figure 1.
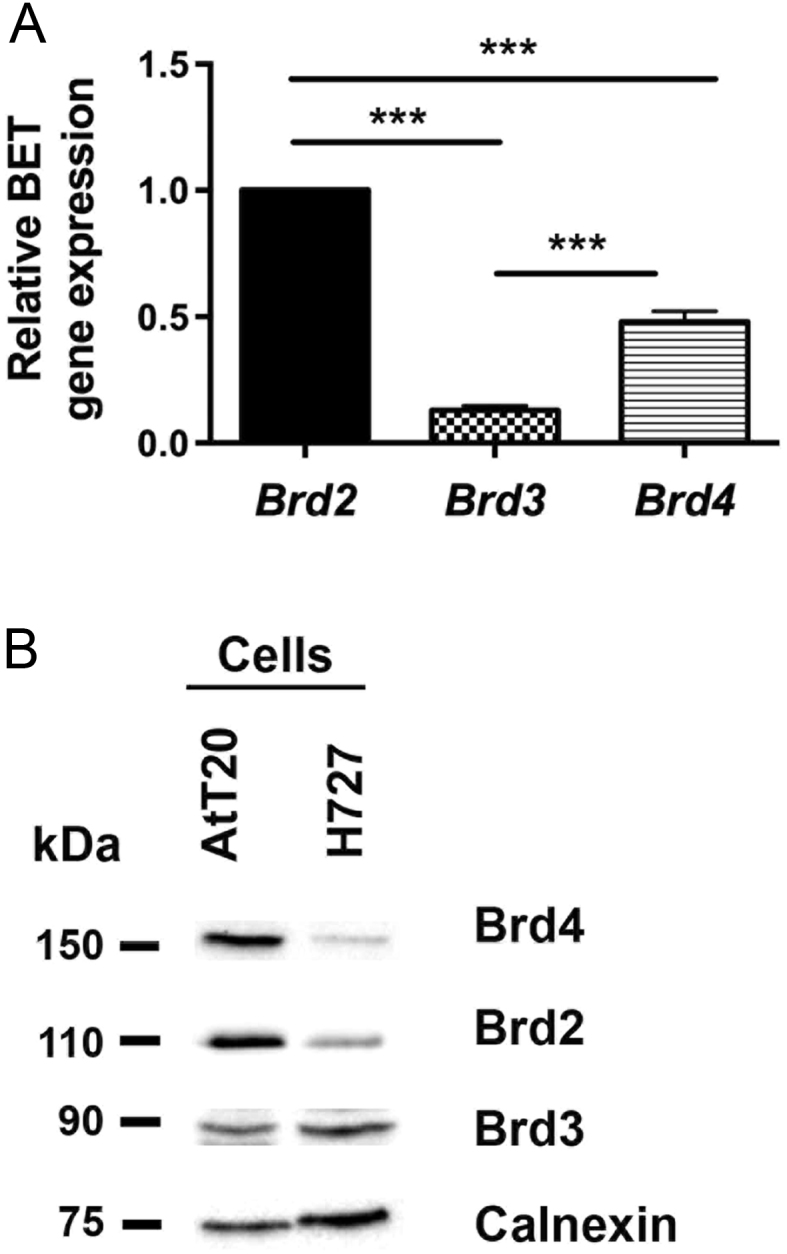
Expression of BET family members in AtT20 cells. The mRNA expression of the BET family members Brd2, Brd3 and Brd4 was examined in AtT20 cells by qRT-PCR (A). Data from four cell passages are represented relative to Brd2 expression; ***P < 0.0005. Western blot analysis was used to confirm expression of BRD2, BRD3 and BRD4 proteins (B), with representative data from four cell passages shown; H727 cell lysate was used as a positive control, and calnexin (a housekeeper gene) was used as a loading control.
JQ1 and PFI-1 reduce proliferation of AtT20 cells
The effect of JQ1 and PFI-1 on proliferation of AtT20 cells was evaluated after 96 h treatment with 1 µM of each compound and compared to proliferation after treatment with the negative control JQ1-, DMSO (vehicle only) and untreated cells. One µM was selected for both compounds as this dose had previously been demonstrated to have efficacy in cell models, without eliciting non-specific effects (summary of drug properties, toxicity and efficacy available from https://www.thesgc.org/chemical-probes). JQ1 and PFI-1 significantly reduced proliferation by 95% (P < 0.0005) and 43% (P < 0.0005), respectively (Fig. 2A). Further investigation of proliferation using dose escalation (concentrations of 20 nM, 50 nM, 100 nM, 500 nM and 1 µM) studies revealed JQ1 to significantly reduce proliferation of AtT20 cells with a half maximal effective concentration (EC50) value of 13.7 nM, whereas the EC50 value for PFI-1 was much higher at 940 nM (Fig. 2B). For all subsequent experiments 1 µM of JQ1 and PFI-1 was used, as the higher dose would ensure maximal cellular responses for both compounds, and also at this dose the negative control compound (JQ1-) did not significantly alter cell proliferation. In addition, JQ1 significantly reduced AtT20 cell proliferation as early as 24 h after treatment (by 39%, P < 0.005), whereas PFI-1 only significantly reduced proliferation (by 36%, P < 0.05) 72 h after treatment, when compared to control treated cells (Fig. 2C). The effects of JQ1 were maintained following removal of the compound from the culture media, such that 96 h after removal of JQ1 from the culture media, the AtT20 cells had not resumed proliferation, whereas the proliferation of cells treated with PFI-1 had increased by 7.7-fold and 20.2-fold for untreated cells (Fig. 2D).
Figure 2.

Effects of BET inhibitors JQ1 and PFI-1 on proliferation of AtT20 cells. JQ1 and PFI-1 significantly reduced proliferation of AtT20 cells, when compared to untreated (UT) cells, DMSO-only treated cells and JQ1-treated cells. Proliferation was assessed using CellTiter Blue assays after 96 h treatment with 1 µM of each compound. Control treatments are represented by black bars and test compound treatment by white bars (A). Dose escalation studies using 20 nM, 50 nM, 100 nM, 500 nM and 1 µM JQ1 or PFI-1 and their effects on proliferation of AtT20 cells after 96 h treatment. The EC50 value is indicated by a solid line. Responses to JQ1 (filled squares and solid line), JQ1- (filled circles and broken line) and PFI-1 (filled triangles and broken line) are shown (B). The efficacy of the BET inhibitors JQ1 and PFI-1 was further examined by time course analysis of cell proliferation after a single treatment with 1 µM JQ1 or PFI-1. Cells were treated with 1 µM JQ1-, JQ1 or PFI-1 and proliferation measured every 24 h and up to 96 h following treatment. Control treatments are represented by dashed lines and drug treatments by solid lines (C). After 96 h treatment with 1 µM JQ1-, JQ1 or PFI-1, compounds were removed (indicated by the arrow) and AtT20 cells culture in standard culture media. Proliferation was measured every 24 h for the following 96 h (D). For all experiments, significance is relative to DMSO vehicle only treatment with (A) ***P < 0.0005 and in (B, C and D) PFI-1 statistics represented by the symbol § and JQ1 statistics by *; §/*P < 0.05, §§/**P < 0.005 and §§§/***P < 0.0005. For all experiments untreated (UT), vehicle only (DMSO) and JQ1- were used as negative controls, and experiments were performed in n = 4 biological replicates, with four technical replicates per experiment.
JQ1, but not PFI-1, increases apoptosis of AtT20 cells
To assess the ability of JQ1 and PFI-1 to induce apoptosis, the cells were treated with compounds and analysed by Caspase Glo and cell cycle analysis. JQ1 treatment, compared to vehicle control treatment, significantly increased apoptosis, assessed using the Caspase Glo assay, of AtT20 cell lines by 2.4-fold (P < 0.0005) 24 h after treatment and up to 53.2-fold (P < 0.0005) 96 h after treatment (Fig. 3A). In contrast, PFI-1 had no significant effect on the rate of apoptosis at the concentration used (Fig. 3A). Furthermore, cell cycle analysis using flow cytometry and propidium iodide staining revealed significantly more (by 2-fold, P < 0.05) cells in the sub G1 phase of the cell cycle, indicative of apoptosis, when treated with JQ1, when compared to control treated cells (Fig. 3B and C), whereas PFI-1 treatment had no effect on the cell cycle phase of AtT20 cells (Fig. 3B and C). Neither JQ1 nor PFI-1 treatment significantly altered cellular senescence of AtT20 cells, when compared to control treated cells assessed by β-galactosidase staining (Supplementary Fig. 1, see section on supplementary materials given at the end of this article). These results indicate that JQ1 reduces AtT20 cell proliferation through a major pro-apoptotic effect, rather than arresting cells in the G1-G2/M phases of the cell cycle.
Figure 3.

Effect of BET inhibition on apoptosis of AtT20 cells. Apoptosis of AtT20 cells was evaluated every 24 h for 96 h after a single treatment with 1 µM JQ1 or PFI-1. Apoptosis was measured by detection of caspase 3/7 activity (A). Cell cycle profiles were analysed after 48 h compound treatment using propidium iodide staining and flow cytometry (B). Gating for cell cycle phases is indicated on the histograms; FL2-A is the level of fluorescence resulting from the propidium iodide staining (C). UT is represented by black bars, DMSO represented by bars with diagonal lines, JQ1-represented by chequered bars, JQ1 by white bars and PFI-1 treatment by grey bars. For all experiments untreated (UT), vehicle only (DMSO) and JQ1- were used as negative controls, and experiments were performed in n = 4 biological replicates, with four technical replicates per experiment. Statistical significance is relative to DMSO vehicle only treatment with *P < 0.05 and ***P < 0.0005.
Expression of genes in proliferation- and apoptosis-associated pathways are significantly altered in AtT20 cells after JQ1 treatment
RNA-Seq analysis was performed to identify the genes that may be involved in the decreased proliferation (Fig. 2) and increased apoptosis (Fig. 3) of AtT20 cells after JQ1 treatment. A total of 2365 genes were found to be dysregulated (at 5% FDR and with greater than 2-fold change of expression level), and this comprised 751 genes that were significantly upregulated and 1614 genes that were significantly downregulated in JQ1 treated, compared to JQ1- control treated AtT20 cells. The most significantly highly up- and downregulated genes (Fig. 4A and B) included metastasis associated lung adenocarcinoma transcript 1 (Malat1), ChaC glutathione specific gamma-glutamylcyclotransferase 1 (Chac1), neuronal prntraxin 1 (Nptx1), fetuin B (Fetub), phosphodiesterase 1C (Pde1c) and fibroblast growth factor 21 (Fgf21). In addition, Myc, which is a widely reported anti-tumorigenic target of BRD4, was observed in the RNA-Seq data to be upregulated by 2.8-fold in JQ1-treated cells, when compared to JQ1-treated AtT20 cells (Fig. 4B), and this was confirmed by qRT-PCR (Supplementary Fig. 2). Furthermore 68% of genes were observed to be downregulated, while only 32% of genes were observed to be upregulated (Fig. 4B). However, the most highly dysregulated genes were not associated with specific anti-tumorigenic pathways, and GSEA was therefore performed. This indicated that there was a significant enrichment of genes associated with a reduction in hormone activity (FDR < 0.0002) and a significant enrichment of genes associated with increased hallmarks of apoptosis (FDR < 0.0298) (Fig. 4C). Therefore, we focussed on the downregulated genes associated with proliferation, apoptosis and hormone activity in all further analysis.
Figure 4.

Transcriptional dysregulation in JQ1-treated AtT20 cells. Heatmap of the top 50 significantly (P < 0.05) up- and downregulated genes following 48 h treatment with JQ1, including Fetub, Pde1c, Fgf21, Nptx1, Chac1 and Malat1. Data shown are fragments per kilobase of exon model per million reads mapped (FPKM) values from RNA-seq experiments, column normalized as indicated in the inset (A). Volcano plot of log2 fold changes of all genes indicates that there were a higher number of significantly downregulated than upregulated genes. The top ten up- or downregulated (lowest P values) are highlighted in red and blue respectively. Myc, typically downregulated following BET targeting with JQ1 is also highlighted in the plot (B). GSEA identified significant (P < 0.05) enrichment of the GO_HORMONE_ACTIVITY (top) and HALLMARK_APOPTOSIS (bottom) gene-sets from MSigDB. While the hormone activity signature was found to be down-regulated, the apoptosis signature was found to be upregulated. The plots show the running sum for the molecular signature database gene set within the AtT20/JQ1 data including the maximum enrichment score and the leading edge subset of enriched genes (C).
To further identify genes specifically associated with apoptosis pathways, IPA was performed on the 2365 dysregulated genes. In total, 12 apoptosis-associated genes were observed (eight upregulated and four downregulated) in JQ1-treated cells, compared to JQ1- control treated cells (Table 1 and Supplementary Fig. 3). The downregulation of these apoptosis-associated genes was validated using qRT-PCR and confirmed that Nuclear Factor Kappa B Subunit 1 (Nfkb1) was downregulated by 2.9-fold (P < 0.0005); DNA fragmentation factor subunit B (Dffb) was downregulated by 2.3-fold (P < 0.0005); Baculoviral IAP Repeat Containing 3 (Birc3) was downregulated by 1.7-fold (P < 0.005) and Calpain 9 (Capn9) was downregulated by 16-fold (P < 0.0005) (Fig. 5A). The significant downregulation of NFκB1 (1.5-fold, P < 0.05) and cIAP1 (encoded by Birc3, 1.9-fold, P < 0.05) proteins was also confirmed using Western blot and densitometry analyses (Fig. 5B and C).
Table 1.
Ingenuity pathway analysis of apoptosis-associated genes and somatostatin regulation of proliferation-associated genes shown to be dysregulated in JQ1-treated AtT20 cells, when compared to JQ1-treated AtT20 cells by RNA-sequence analyses.
| Gene | Fold change |
|---|---|
| Apoptosis-associated | |
| Capn6 | 7.57 |
| Endog | 3.03 |
| Rras2 | 2.95 |
| Map3k14 | 2.57 |
| Tnfrsf1b | 2.23 |
| Nfkbie | 2.03 |
| Bcl2l11 | 2.03 |
| Sptan1 | 2.00 |
| Nfkb1a | −2.01a |
| Dffba | −2.10a |
| Birc3a | −2.68a |
| Capn9a | −12.13a |
| Somatostatin 2-associated proliferation | |
| Gng3 | 100.43 |
| Rras2 | 2.95 |
| Srca | −2.95a |
| Gng4a | −3.32a |
| Sstr2a | −3.34a |
| Nos1a | −3.53a |
| Npr1a | −3.56a |
| Gnb4a | −4.26a |
| Gnb3a | −5.78a |
| Gucy2ca | −10.13a |
aDownregulated genes.
Figure 5.

Validation of RNA-Seq data from AtT20 cells treated with JQ1- or JQ1. Transcription of the apoptosis associated genes Capn9, Dffb, Birc3 and Nfkb1 was examined using qRT-PCR, after 48 h JQ1 treatment (A). Expression of cIAP2 (encoded by Birc3) and Nfkb1 after JQ1 treatment was confirmed by Western blot analysis (B) and was significantly decreased compared to control treated cells, as indicated by densitometry analysis (C). Transcription of the SSTR2 proliferation pathway associated genes Sstr2, Npr1, Gnb4, Gng4, Gnb3 and Nos1 was examined using qRT-PCR, after JQ1 treatment (D). The expression of SSTR2 after JQ1 treatment was confirmed using Western blot analysis (E) and was significantly downregulated, compared to control treated cells, as indicated by densitometry analysis (F). For all experiments untreated (UT), vehicle only (DMSO) and JQ1- were used as negative controls, and Gapdh (a housekeeper gene) was used as a loading control. All qRT-PCR experiments were performed in n = 4 technical and n = 4 biological replicates, and Western blots performed in n = 5 biological replicates. Statistical significance is relative to DMSO vehicle only treatment with * P < 0.05, ** P < 0.005 and *** P < 0.0005.
GSEA and IPA analyses also revealed that the only proliferation-associated pathway significantly altered after JQ1 treatment was the ‘somatostatin receptor 2 (SSTR2) anti-proliferative pathway’. This is of interest as somatostatin analogues (e.g. octreotide and lanreotide) inhibit proliferation and hormone secretion by pituitary tumours, such as somatotrophinomas, but not corticotrophinomas (Yates et al. 2015, Cuevas-Ramos et al. 2016, Walls et al. 2016). IPA revealed that JQ1 treatment significantly dysregulated ten genes from this SSTR2 pathway (two upregulated and eight downregulated) when compared to JQ1- treated cells (Table 1 and Supplementary Fig. 4). Analysis by qRT-PCR confirmed downregulation of six of the eight genes that comprised Sstr2 (by -1.5-fold (P < 0.005)); guanine nucleotide binding protein (G protein), gamma 4 (Gng4) (by -4.1-fold (P < 0.0005)); Nitric oxide synthase 1 (Nos1) (by -11.3-fold (P < 0.0005)); Natriuretic peptide receptor 1 (Npr1) (by -1.8-fold (P < 0.0005)); Guanine nucleotide binding protein (G protein) beta 4 (Gng4) (by -3.4-fold (P < 0.0005)) and Guanine nucleotide-binding protein (G protein) beta 3 (Gnb4) (by -6.6-fold (P < 0.005)) (Fig. 5D). The remaining two genes proto-oncogene tyrosine kinase Src (Src) and guanylate cyclase 2C (Gucy2c) were found to have undetectable expression. The significant downregulation of SSTR2 (2.3-fold, P < 0.05) was also confirmed using Western blot (Fig. 5E) and densitometry analyses (Fig. 5F). Octreotide did not significantly alter the proliferation of AtT20 cells, either alone or in combination with JQ1 treatment (Supplementary Fig. 5).
JQ1 treatment reduces pro-opiomelanocortin (POMC) expression and alters ACTH secretion of AtT20 cells
RNA-Seq analysis indicated that expression of Pomc, which encodes the precursor of ACTH, was significantly decreased in JQ1 treated AtT20 cells, compared to JQ1- treated cells, by -11-fold (P = 7.31E-320, Fig. 4A and B). This was confirmed using qRT-PCR, which showed that Pomc expression was significantly reduced in JQ1 treated AtT20 cells by ~16.7-fold (P < 0.005), when compared to vehicle only, and JQ1- treated cells (Fig. 6A). Western blot analysis confirmed that POMC expression was decreased after JQ1 treatment, with significant reduction observed after 24 h (4.4-fold, P < 0.0005), 48 h (6.3-fold, P < 0.0005) and 72 h (5.1-fold, P < 0.005), compared to control JQ1- treated cells (Fig. 6B and C). The reduced POMC expression was associated with significant decreases in the levels of ACTH in cell media at 48 h (1.4-fold, P < 0.0005) and 72 h (1.2-fold, P < 0.05) after JQ1 treatment, when compared to JQ1- treatment (Fig. 6D).
Figure 6.

POMC expression and ACTH secretion after JQ1 treatment. QRT-PCR analysis confirmed the significant down-regulation of Pomc transcription after 48 h JQ1 treatment (A). Down-regulation of POMC protein expression was also confirmed by Western blot analyses at 24 h, 48 h and 72 h after JQ1 treatment, compared to JQ1- treated cells; POMC is observed as two bands corresponding to pre-pro-POMC and pro-POMC (calnexin was used as a loading control) (B). Densitometry analysis confirmed that POMC expression was significantly down-regulated at 24 h, 48 h and 72 h (C). Concentrations of ACTH in the media from the same cells, which reflected ACTH secretion by these cells, significantly decreased at 48 h and 72 h after treatment with JQ1, when compared to JQ1- treated cells (D). DMSO or JQ1-treated cells were used as negative controls, with qRT-PCR experiments performed in n = 4 technical and n = 4 biological replicates, and Western blot and ELISAs performed in n = 4 biological replicates. Statistical significance is relative to JQ1- treatment with *P < 0.05, **P < 0.005 and ***P < 0.0005.
Discussion
Our studies have revealed that JQ1 treatment significantly decreased proliferation and increased apoptosis of the ACTH-secreting pituitary tumour cells AtT20. Furthermore, our results demonstrate that the BET protein family regulates the expression of genes in the ‘somatostatin receptor 2 anti-proliferative signalling pathway’ and anti-apoptotic genes, as well as Pomc, which encodes the ACTH precursor in these AtT20 cells. Thus, our data indicate that inhibitors of the BET protein family may provide a novel therapeutic strategy for the treatment of ACTH-secreting pituitary NETs.
In our study, we demonstrate that AtT20 cells express the BET family members Brd2, Brd3 and Brd4, with Brd2 being the most abundant, which is similar to data reported for MEN1-associated mouse pancreatic NETs and human pancreatic and bronchial NET cell lines (Lines et al. 2017). Thus, the BET family, and specifically Brd2, likely play an important role in NETs, and inhibiting the activity of these proteins may provide a novel therapeutic target. Indeed, our study reveals that the small molecule inhibitors of the BET family, JQ1 and PFI-1, significantly reduced proliferation of AtT20 cells and that JQ1 also significantly increased apoptosis of AtT20 cells. Furthermore, flow cytometry analysis and cellular senescence data showed that JQ1-treated AtT20 cells did not arrest during cell cycle progression, but instead were detected in the sub G1 phase of the cell cycle. Moreover, AtT20 cells did not resume proliferating after removal of JQ1 from the media, indicating that JQ1 also has a major pro-apoptotic effect on AtT20 cells. This is consistent with our data from human NET cell lines, which indicate that JQ1 is more efficacious than PFI-1, and this difference may be due to variations in potency, cell permeability and bioavailability of the two inhibitors, when used at the same concentration (Muller et al. 2011, Fish et al. 2012, Picaud et al. 2013, Lines et al. 2017). JQ1 treatment almost completely abolished AtT20 cell proliferation and was therefore more efficacious in the AtT20 cells than in pancreatic NET (BON-1) and broncho-pulmonary NET (H727 and H720) cell lines, as reported in our previous studies (Lines et al. 2017). Thieno-diazepines similar to JQ1 are currently undergoing clinical trials for a number of different cancers (Matzuk et al. 2012, Benito et al. 2017) and therefore may have potential as a novel therapeutic compound for corticotrophinomas, although the specific pathways altered by this pan-BET inhibitor, especially in the pituitary, still remain to be fully elucidated (Andrieu et al. 2016).
Our study also demonstrates that JQ1 treatment resulted in a significant dysregulation of apoptosis-associated genes. Of note, one of the major transcriptional targets of BRD4 is Myc, and downregulation of Myc is considered to be one of the key anti-tumour actions of JQ1 (Delmore et al. 2011, Puissant et al. 2013, Bandopadhayay et al. 2014, Wong et al. 2014, Coude et al. 2015, Li et al. 2016). However, in pancreatic and bronchial NET cells, Myc did not appear to be a key target (Lines et al. 2017), and in our current study, JQ1 treatment of AtT20 cells did not decrease Myc expression but instead was associated with increased Myc expression. These findings support the hypothesis that different mechanisms may be responsible for the efficacy of JQ1 in NETs, and these may involve the apoptotic pathway, as indicated by our RNA-Seq analysis that revealed four apoptosis-associated genes (Capn9, Dffb, Birc3 and Nfkb1) to be downregulated by JQ1 treatment. Birc3 encodes cIAP2, which is an E3 ubiquitin protein ligase that is a member of the inhibitors of apoptosis proteins (IAP) family, and increases cIAP2 expression that can lead to evasion of caspase-mediated apoptosis in different cancers including gastric and gallbladder cancers and glioblastoma (Wang et al. 2016, Jiang et al. 2017, Gowda Saralamma et al. 2018). In addition, cIAP2 has been reported to activate NFκB (encoded by Nfkb1) signalling, which also has roles in apoptosis regulation (Tchoghandjian et al. 2013, Jiang et al. 2017). Moreover, previous studies have linked BET proteins to the regulation of NFκB signalling, via direct NFκB transcriptional regulation as well as by the binding of Brd4 to acetylated NFκB co-activating proteins (reviewed by Hajmirza et al. 2018). These observations therefore suggest that modulation of NFκB signalling may be an important mechanism of action of JQ1 in NETs.
Our study also revealed that the only proliferation-associated pathway significantly altered by JQ1 treatment of AtT20 cells was the anti-proliferative SSTR2 pathway. This indicates that the predominant action of JQ1 is likely through apoptosis; however, somatostatin analogues that target somatostatin receptors are effective in the treatment of somatotrophinomas, a pituitary NET, although their efficacy in treating corticotrophinomas is limited (Feelders et al. 2018). There are five somatostatin receptors, SSTR1–5, and SSTR2 and 5 are predominantly involved in hormone secretion, whilst SSTR1, 2, 3, 4 and 5 have roles in apoptosis and proliferation (Grozinsky-Glasberg et al. 2008). Pasireotide that has high affinity for SSTR1, 2, 3 and 5 has been reported to decrease cortisol levels in patients with Cushing’s disease, although this was associated with hyperglycaemia-related adverse events in approximately 75% of patients (Colao et al. 2012). In contrast, octreotide, which predominantly targets SSTR2 that is one of the most frequently expressed SSTRs in pituitary adenomas, has been reported to have no effect on ACTH secretion in patients with Cushing’s disease or proliferation of AtT20 cells. This is likely because SSTR5 is the most abundant SSTR in corticotrophinomas; however, both AtT20 cells and human corticotrophinomas have been show to express SSTR2, 3 and 5 (Murasawa et al. 2014, Behling et al. 2018). Thus, our results suggest that using a somatostatin analogue that targets multiple SSTR’s, for example, paseriotide, may be preferable to octreotide, that predominantly targets SSTR2. In addition, our results, which demonstrate that JQ1 treatment can significantly decrease AtT20 cell proliferation and downregulate expression of SSTR2, appear to be paradoxical because SSTR2 is reported to exert anti-proliferative actions (Panetta & Patel 1995, Grozinsky-Glasberg et al. 2008). A possible explanation for this apparent paradox may be provided by the reported formation of heterodimers by SSTRs (Duran-Prado et al. 2008). For example, SSTR3 is reported to form heterodimers with SSTR2, and as SSTR3 is pro-apoptotic, the observed downregulation of SSTR2 by JQ1 could reduce SSTR2-SSTR3 heterodimer formation but enhance SSTR3-SSTR3 homodimer formation that will increase SSTR3-induced apoptosis. (Theodoropoulou & Stalla 2013). Hence, JQ1 treatment could provide a novel medical approach for modulation of apoptosis-associated somatostatin signalling in patients with corticotrophinomas.
In addition to increasing apoptosis, our results showing that JQ1 can reduce the hypersecretion of ACTH may also provide a possible treatment for Cushing’s syndrome, which is one of the key morbidities of corticotrophinomas (Ntali et al. 2013). Thus, JQ1 treatment of AtT20 cells that have been reported to actively secrete ACTH (Buonassisi et al. 1962, Gong et al. 2014, Asari et al. 2017) significantly decreased levels of ACTH in cell media by 48 h and this was preceded by a significant reduction in the expression of Pomc, which encodes pre-pro-opiomelanocortin (pre-pro-POMC) that is cleaved to give rise to multiple peptide hormones, including ACTH (Rousseau et al. 2007). Therefore, JQ1 may downregulate ACTH secretion by suppressing expression of the protein it is cleaved from in a BET-mediated mechanism. In addition, our results showed a significant decrease in POMC expression after 24 h JQ1 treatment, but secretion of ACTH was not significantly reduced until 48 h after treatment. This is likely because of the time required for the cells to process the POMC protein into ACTH and the fact that the data collected was cumulative, with total ACTH in cell media measured at each time point. Therefore these observations suggest that JQ1 may have a potential to decrease the growth of corticotrophinomas, as well as reduce their ability to secrete ACTH. However, it is important to note that all these studies were performed in an in vitro cell line model, and hence these findings require further confirmation by in vivo or ex vivo investigations before considering a direct clinical utility for treating JQ1 in corticotrophinomas.
Overall, our studies indicate that JQ1 can significantly decrease proliferation and increase apoptosis of the ACTH-secreting (corticotrophinoma) cell line, AtT20, as well as reduce its ACTH secretion. Thus, BET inhibition may provide a novel therapeutic approach for corticotrophinomas possibly through transcriptional regulation of genes including NFκB, SSTR2 and POMC.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was funded by the United Kingdom Medical Research Council (MRC) program Grant G1000467 (K E L, M S and R V T) and R V T is a Wellcome Trust Senior Investigator. P F, S K, S M and C B are supported by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda and the Wellcome Trust. H L, B W and D B are supported by a Wellcome Trust Core award to the Wellcome Trust Centre for Human Genetics (090532/Z/09/Z).
Acknowledgements
The authors thank the High-Throughput Genomics Group, including Sarah Lamble, Hubert Slawinski, Sophia Ward and Chris Allan at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z) for the generation of sequencing data. They thank John Broxholme and Eshita Sharma of the Bioinformatics and Statistical Genetics Core group at Wellcome Trust Centre for Human Genetics for developing and running the RNA-Seq data processing pipeline.
References
- Anders S, Pyl PT, Huber W. 2015. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics 166–169. ( 10.1093/bioinformatics/btu638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu G, Belkina AC, Denis GV. 2016. Clinical trials for BET inhibitors run ahead of the science. Drug Discovery Today: Technologies 45–50. ( 10.1016/j.ddtec.2016.06.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asari Y, Kageyama K, Nakada Y, Tasso M, Takayasu S, Niioka K, Ishigame N, Daimon M. 2017. Inhibitory effects of a selective Jak2 inhibitor on adrenocorticotropic hormone production and proliferation of corticotroph tumor att20 cells. OncoTargets and Therapy 4329–4338. ( 10.2147/OTT.S141345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al 2014. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clinical Cancer Research 912–925. ( 10.1158/1078-0432.CCR-13-2281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley AH, Stirnweiss A, Ferrari E, Endersby R, Howlett M, Failes TW, Arndt GM, Charles AK, Cole CH, Kees UR. 2014. Comparative drug screening in NUT midline carcinoma. British Journal of Cancer 1189–1198. ( 10.1038/bjc.2014.54) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behling F, Honegger J, Skardelly M, Gepfner-Tuma I, Tabatabai G, Tatagiba M, Schittenhelm J. 2018. High expression of somatostatin receptors 2A, 3, and 5 in corticotroph pituitary adenoma. International Journal of Endocrinology 1763735 ( 10.1155/2018/1763735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Ramachandran B, Schroeder H, Schmidt G, Urbanke H, Burkhardt S, Capece V, Dean C, Fischer A. 2017. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Translational Psychiatry e1239 ( 10.1038/tp.2017.202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B 289–300. ( 10.1111/j.2517-6161.1995.tb02031.x) [DOI] [Google Scholar]
- Buonassisi V, Sato G, Cohen AI. 1962. Hormone-producing cultures of adrenal and pituitary tumor origin. PNAS 1184–1190. ( 10.1073/pnas.48.7.1184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Schoenherr U, Mills D, Salgado LR, Biller BM, et al 2012. A 12-month phase 3 study of pasireotide in Cushing’s disease. New England Journal of Medicine 914–924. ( 10.1056/NEJMoa1105743) [DOI] [PubMed] [Google Scholar]
- Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME, et al 2015. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 17698–17712. ( 10.18632/oncotarget.4131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas-Ramos D, Lim DST, Fleseriu M. 2016. Update on medical treatment for Cushing’s disease. Clinical Diabetes and Endocrinology 16 ( 10.1186/s40842-016-0033-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AF, Tichomirowa MA, Beckers A. 2009. The epidemiology and genetics of pituitary adenomas. Best Practice and Research: Clinical Endocrinology and Metabolism 543–554. ( 10.1016/j.beem.2009.05.008) [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 904–917. ( 10.1016/j.cell.2011.08.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Prado M, Malagon MM, Gracia-Navarro F, Castano JP. 2008. Dimerization of G protein-coupled receptors: new avenues for somatostatin receptor signalling, control and functioning. Molecular and Cellular Endocrinology 63–68. ( 10.1016/j.mce.2007.12.006) [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols 1184–1191. ( 10.1038/nprot.2009.97) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eachkoti R, Reddy MV, Lieu YK, Cosenza SC, Reddy EP. 2014. Identification and characterisation of a novel heat shock protein 90 inhibitor ONO4140. European Journal of Cancer 1982–1992. ( 10.1016/j.ejca.2014.04.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feelders RA, Newell-Price J, Pivonello R, Nieman LK, Hofland LJ, Lacroix A. 2018. Advances in the medical treatment of Cushing’s syndrome. Lancet: Diabetes and Endocrinology 7 300–312. ( 10.1016/S2213-8587(18)30155-4) [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al 2010. Selective inhibition of BET bromodomains. Nature 1067–1073. ( 10.1038/nature09504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish PV, Filippakopoulos P, Bish G, Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones H, Knapp S, et al 2012. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. Journal of Medicinal Chemistry 9831–9837. ( 10.1021/jm3010515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YY, Liu YY, Yu S, Zhu XN, Cao XP, Xiao HP. 2014. Ursolic acid suppresses growth and adrenocorticotrophic hormone secretion in AtT20 cells as a potential agent targeting adrenocorticotrophic hormone-producing pituitary adenoma. Molecular Medicine Reports 2533–2539. ( 10.3892/mmr.2014.2078) [DOI] [PubMed] [Google Scholar]
- Gorvin CM, Wilmer MJ, Piret SE, Harding B, van den Heuvel LP, Wrong O, Jat PS, Lippiat JD, Levtchenko EN, Thakker RV. 2013. Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients. PNAS 7014–7019. ( 10.1073/pnas.1302063110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda Saralamma VV, Lee HJ, Raha S, Lee WS, Kim EH, Lee SJ, Heo JD, Won C, Kang CK, Kim GS. 2018. Inhibition of IAP’s and activation of p53 leads to caspase-dependent apoptosis in gastric cancer cells treated with scutellarein. Oncotarget 5993–6006. ( 10.18632/oncotarget.23202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinsky-Glasberg S, Shimon I, Korbonits M, Grossman AB. 2008. Somatostatin analogues in the control of neuroendocrine tumours: efficacy and mechanisms. Endocrine-Related Cancer 701–720. ( 10.1677/ERC-07-0288) [DOI] [PubMed] [Google Scholar]
- Hajmirza A, Emadali A, Gauthier A, Casasnovas O, Gressin R, Callanan MB. 2018. BET family protein BRD4: an emerging actor in NFkappaB signaling in inflammation and cancer. Biomedicines E16 ( 10.3390/biomedicines6010016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida CT, Bianchetti E, Shu C, Halatsch ME, Westhoff MA, Karpel-Massler G, Siegelin MD. 2017a. BH3-mimetics and BET-inhibitors elicit enhanced lethality in malignant glioma. Oncotarget 29558–29573. ( 10.18632/oncotarget.16365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida CT, Shu C, Halatsch ME, Westhoff MA, Altieri DC, Karpel-Massler G, Siegelin MD. 2017b. Mitochondrial matrix chaperone and c-myc inhibition causes enhanced lethality in glioblastoma. Oncotarget 37140–37153. ( 10.18632/oncotarget.16202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Li C, Lin B, Hong H, Jiang L, Zhu S, Wang X, Tang N, Li X, She F, et al 2017. cIAP2 promotes gallbladder cancer invasion and lymphangiogenesis by activating the NF-kappaB pathway. Cancer Science 1144–1156. ( 10.1111/cas.13236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology R36 ( 10.1186/gb-2013-14-4-r36) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. 2017. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Letters 76–87. ( 10.1016/j.canlet.2017.02.021) [DOI] [PubMed] [Google Scholar]
- Lee DH, Qi J, Bradner JE, Said JW, Doan NB, Forscher C, Yang H, Koeffler HP. 2015. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. International Journal of Cancer 2055–2064. ( 10.1002/ijc.29269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, et al 2016. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2462–2474. ( 10.18632/oncotarget.6275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines KE, Stevenson M, Filippakopoulos P, Muller S, Lockstone HE, Wright B, Grozinsky-Glasberg S, Grossman AB, Knapp S, Buck D, et al 2017. Epigenetic pathway inhibitors represent potential drugs for treating pancreatic and bronchial neuroendocrine tumors. Oncogenesis e332 ( 10.1038/oncsis.2017.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, et al 2012. Small-molecule inhibition of BRDT for male contraception. Cell 673–684. ( 10.1016/j.cell.2012.06.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Filippakopoulos P, Knapp S. 2011. Bromodomains as therapeutic targets. Expert Reviews in Molecular Medicine e29 ( 10.1017/S1462399411001992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murasawa S, Kageyama K, Sugiyama A, Ishigame N, Niioka K, Suda T, Daimon M. 2014. Inhibitory effects of SOM230 on adrenocorticotropic hormone production and corticotroph tumor cell proliferation in vitro and in vivo. Molecular and Cellular Endocrinology 37–46. ( 10.1016/j.mce.2014.07.001) [DOI] [PubMed] [Google Scholar]
- Ntali G, Asimakopoulou A, Siamatras T, Komninos J, Vassiliadi D, Tzanela M, Tsagarakis S, Grossman AB, Wass JA, Karavitaki N. 2013. Mortality in Cushing’s syndrome: systematic analysis of a large series with prolonged follow-up. European Journal of Endocrinology 715–723. ( 10.1530/EJE-13-0569) [DOI] [PubMed] [Google Scholar]
- Panetta R, Patel YC. 1995. Expression of mRNA for all five human somatostatin receptors (hSSTR1-5) in pituitary tumors. Life Sciences 333–342. ( 10.1016/0024-3205(94)00956-2) [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research e45 ( 10.1093/nar/29.9.e45) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, Owen DR, et al 2013. PFI-1, a highly selective protein interaction inhibitor, targeting BET bromodomains. Cancer Research 3336–3346. ( 10.1158/0008-5472.CAN-12-3292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al 2013. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery 308–323. ( 10.1158/2159-8290.CD-12-0418) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 139–140. ( 10.1093/bioinformatics/btp616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau K, Kauser S, Pritchard LE, Warhurst A, Oliver RL, Slominski A, Wei ET, Thody AJ, Tobin DJ, White A. 2007. Proopiomelanocortin (POMC), the ACTH/melanocortin precursor, is secreted by human epidermal keratinocytes and melanocytes and stimulates melanogenesis. FASEB Journal 1844–1856. ( 10.1096/fj.06-7398com) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev AA. 2016. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. ( 10.1101/060012) [DOI] [Google Scholar]
- Tchoghandjian A, Jennewein C, Eckhardt I, Rajalingam K, Fulda S. 2013. Identification of non-canonical NF-kappaB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death and Disease e564 ( 10.1038/cddis.2013.70) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoropoulou M, Stalla GK. 2013. Somatostatin receptors: from signaling to clinical practice. Frontiers in Neuroendocrinology 228–252. ( 10.1016/j.yfrne.2013.07.005) [DOI] [PubMed] [Google Scholar]
- Walls GV, Reed AA, Jeyabalan J, Javid M, Hill NR, Harding B, Thakker RV. 2012. Proliferation rates of multiple endocrine neoplasia type 1 (MEN1)-associated tumors. Endocrinology 5167–5179. ( 10.1210/en.2012-1675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls GV, Stevenson M, Soukup BS, Lines KE, Grossman AB, Schmid HA, Thakker RV. 2016. Pasireotide therapy of multiple endocrine neoplasia Type 1-associated neuroendocrine tumors in female mice deleted for an Men1 allele improves survival and reduces tumor progression. Endocrinology 1789–1798. ( 10.1210/en.2015-1965) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Berglund A, Kenchappa RS, Forsyth PA, Mule JJ, Etame AB. 2016. BIRC3 is a novel driver of therapeutic resistance in Glioblastoma. Scientific Reports 21710 ( 10.1038/srep21710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C, Laddha SV, Tang L, Vosburgh E, Levine AJ, Normant E, Sandy P, Harris CR, Chan CS, Xu EY. 2014. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death and Disease e1450 ( 10.1038/cddis.2014.396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu D, Gao X, Xie F, Tao D, Xiao X, Wang L, Jiang G, Zeng F. 2017. Inhibition of BRD4 suppresses cell proliferation and induces apoptosis in renal cell carcinoma. Cellular Physiology and Biochemistry 1947–1956. ( 10.1159/000472407) [DOI] [PubMed] [Google Scholar]
- Yates CJ, Lines KE, Thakker RV. 2015. Molecular genetic advances in pituitary tumor development. Expert Review of Endocrinology and Metabolism 35–53. ( 10.1586/17446651.2015.955795) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.