

Abstract
Vitamin D deficiency has been associated with increased risk for aggressive prostate cancer (PCa). Prostate epithelium has a unique metabolism compared to other tissues. Normal prostate exhibits low levels of mitochondrial respiration and there is a metabolic switch to increased oxidative phosphorylation in PCa. 25-hydroxyvitamin D (25(OH)D) is the major circulating form of vitamin D and is used clinically to determine vitamin D status. Activation of 25(OH)D to the transcriptionally active form, 1,25(OH)2D occurs via a reduction-oxidation (redox) reaction within the mitochondria that is catalyzed by the P450 enzyme, CYP27B1. We sought to determine if hydroxylation of 25(OH)D by CYP27B1 contributes to non-genomic activity of vitamin D by altering the redox-dependent state of the mitochondria in benign prostate epithelial cells. Exposure to 25(OH)D produced a transient pro-oxidant effect and change in mitochondrial membrane potential that was dependent on CYP27B1. Extended exposure ultimately suppressed mitochondrial respiration, consistent with a protective effect of 25(OH)D in supporting benign prostate metabolism. To model physiologically relevant changes in vitamin D, cells were cultured in constant 25(OH)D then changed to high or deficient concentrations. This model also incurred a biphasic effect with a pro-oxidant shift after short exposure followed by decreased respiration after 16 hours. Several genes involved in redox cycling and mitochondrial health were regulated by 25(OH)D in these cells. These results indicate a secondary non-genomic mechanism for vitamin D to contribute to prostate cell health by supporting normal mitochondrial respiration.
Keywords: Prostate, vitamin D, metabolism
INTRODUCTION
Prostate cancer (PCa) is the most commonly diagnosed cancer and the second leading cause of cancer-related mortality in men [1]. Benign cellular metabolism, as well as disease-induced changes in cellular metabolism, are uniquely different in the prostate [2, 3]. Unlike most other cells of the body, benign prostate epithelial cells predominantly utilize glycolysis for energetic demands with the majority of metabolic substrates utilized to produce citrate [3–7]. The maintenance of glycolytic metabolism results from the zinc-mediated inhibition of m-aconitase, which suppresses the Kreb’s cycle and subsequently decreases respiratory efficiency [8–10]. These events result in low levels of oxidative phosphorylation and high levels of citrate, which are secreted from prostatic epithelial cells into the seminal fluid [3–7].
Epidemiological and experimental data has established a chemopreventive effect for vitamin D in the prostate [11–13]. The VITAL trial, which assessed the impact of vitamin D on prostate cancer mortality, showed a 25% reduction in cancer deaths when the first two years of trial were excluded [14]. Inverse associations have also been shown between aggressive PCa and vitamin D status, with vitamin D supplementation shown to decrease prostate-specific antigen levels [13, 15] and the number of positive biopsies [16–18]. Experimental in vivo and in vitro data further supports these positive effects of vitamin D with mouse PCa models having a delay in initiation and preneoplastic lesion formation [19–21], while cell culture models show an inhibition of cancer-related phenotypes [11, 22–24].
Several human populations are prone to vitamin D deficiency and are more susceptible to a multitude of diseases including diabetes, cardiovascular disease, and cancer (including PCa) [25, 26]. Among them are the elderly, who are often exposed to less sunlight and may have altered activity of CYP27B1, the vitamin D-activating enzyme, and CYP24A1, the vitamin D-inactivating enzyme, as indicated by studies in rats [27]. Consequently, vitamin D deficiency combined with attenuated 25(OH)D activation and elevated 1,25(OH)2D inactivation drives susceptibility to the aforementioned diseases [27–29]. In addition to a lack of sun exposure, the amount of melanin in the skin inhibits vitamin D production, resulting in high rates of deficiency among African American populations [12, 30–32]. Pigmentation protects skin from UV rays; however exposure to UVB light is necessary for the synthesis of prehormone D3 from 7-dehydrocholesterol [33, 34].
Vitamin D is intertwined with mitochondrial metabolism by two distinct mechanisms. Metabolism of the circulating pro-hormone, 25(OH)D, to the active hormone, 1,25-dihydroxyvitamin D (1,25(OH)2D), occurs within the mitochondria via a redox reaction [34–36]. This hydroxylation is catalyzed by CYP27B1 through a series of ferredoxin/ferrireductase reactions to facilitate the transfer of electrons from NADPH to the eventual 1α-hydroxylation (and activation) of 25(OH)D to 1,25(OH)2D. 1,25(OH)2D is the ligand of the vitamin D receptor (VDR) which heterodimerizes with the retinoic acid X receptor to specifically bind DNA and regulate transcription of target genes. This genomic transcriptional regulation is the second mechanism by which vitamin D regulates mitochondrial metabolism, as vitamin D metabolites have been shown to regulate metabolism-related genes [34]. Ryan ZC et al. reported that 25(OH)D and 1,25(OH)2D can have opposing effects on mitochondrial respiration and have implicated several genes that may be involved in reshaping mitochondria in human skeletal muscle cells [37], but this has not been studied in the prostate.
Vitamin D deficiency is associated with a number of metabolism-related diseases including diabetes and cancer, and suggests a role for adequate levels of vitamin D in the maintenance and function of prostatic mitochondria [26, 38–40]. The role of vitamin D in mitochondrial function in the context of the distinct metabolism of normal prostate cells has not been studied. Mitochondria are dynamic organelles that respond to endogenous and exogenous stimuli, adapting their structure, redox states, and functions to the needs of the cell [41–45]. Here we examined both non-genomic and genomic actions of vitamin D on mitochondrial health and metabolism of prostate cells.
MATERIALS & METHODS
Cell Culture
957E/hTERT cells were generously supplied by Dr. Donald Vander Griend. These cells were cultured as described in Yasunaga et al. [46]. Cells were maintained in Keratinocyte-Serum Free Media (KSFM) supplemented with bovine pituitary extract and epidermal growth factor (17-005-042, Fisher Scientific, Hampton, NH). Cells were used between passage 5 and 21, using TrypLE to passage the cells.
Primary prostate epithelial cells (PrE) were isolated from de-identified benign areas of prostatectomy tissue as previously described by our group [47] via University of Illinois at Chicago Institutional Review Board approved protocol #2007–0694. PrE cells were maintained in PrEGM media (Lonza, Basel, Switzerland) and used for one passage on collagen-coated plates.
Seahorse Mito Stress Test
Cells (104 cells/well) were cultured on Seahorse XFe24 plates for 24 hours prior to assay. Cells were seeded in technical quadruplicates and each experiment was repeated three or more times. The assay cartridge was hydrated with Agilent’s equilibration buffer and incubated in a 0% CO2 incubator at 37°C. One hour prior to assay, culture medium on the cells was replaced with Seahorse Assay medium (Seahorse base medium supplemented with 1mM sodium pyruvate, 2mM glutamine, and 10mM glucose, pH of 7.4) and the cells were placed in the CO2-free incubator at 37°C. Assay was run according to manufacturer’s protocol using a Seahorse XFe24 (Agilent, California). Basal respiration was established as the oxygen consumption rate (OCR) prior to oligomycin treatment. ATP levels were determined as the difference in OCR before and after oligomycin treatment.
ATP Assay
Intracellular ATP was quantified by a luminescence-based assay (ab113849, Abcam, Cambridge, MA). Cells were grown to 80% confluency in a white well, clear bottom plate prior to lysis for the assay. Measurements were obtained using the Biotek Synergy LX (Winooski, VT) multi-mode reader.
RT-qPCR
RNA was collected from cells by Trizol (10296028, Thermo Fisher Scientific) per the manufacturer’s instructions. RNA was quantified using the absorbance at 260nm using the NanoDrop 1000 (Thermo Fisher Scientific, Hampton, NH). Synthesis of cDNA was performed on 500ng of RNA using the Highseq cDNA synthesis kit (4368814, Thermo Fisher Scientific). Quantification was achieved using SYBR Green ROX master mix (4385612, Applied Biosciences) and performed on Quantstudio6 (Thermo Fisher Scientific, Hampton, NH). Results were analyzed by ΔΔCt and compared to two control genes, RLP13A and HPRT.
Qiagen’s RT2 Profiler™ PCR Array Human Mitochondria, PAHS-087ZC-2, was used to determine the levels of 84 transcripts relative to mRNA produced from five housekeeping genes. Changes in transcription are expressed as fold change of 25(OH)D treated to vehicle control. Analysis was completed using Qiagen’s Data Analysis Center.
Mitochondrial Health Assay
The mitochondrial health assay (H10295, Thermo Fisher Scientific) utilizes a combination of three dyes to assess mitochondrial functionality: a fluorescent dye that accumulates in mitochondria in a membrane potential-dependent manner (used 1:700), image-IT green to identify dead cells (1:2000), and Hoescht blue nuclear stain (1:2000). Live cells were treated with the dye for 30 minutes while being maintained in standard culture conditions. Images and quantification were obtained using the Cellinsight Cx7 (Thermofisher, CA, USA). Cells grown in standard culture conditions were also treated with 5μM CellROX (C10422, Thermo Fisher Scientific) for one hour, then washed, fixed, and counterstained with Hoescht for imaging and quantification on the Cellinsight Cx7.
siRNA knockdown of CYP27B1 and VDR
957E/hTERT cells were plated in 60 mm dishes and transiently transfected with siRNA pools (3–5 distinct 25bp siRNA sequences per target) to negative control, CYP27B1 or VDR (sc-60479 and sc-45920, Santa Cruz Biotech). Cells were grown for 48 hours, then collected for protein isolation or transferred to Seahorse plate for assay.
RESULTS
Vitamin D induced opposing immediate and late effects on the mitochondria of benign prostate epithelial cell culture models
Energy use and metabolism is a highly dynamic process centered around the mitochondria that is tightly coordinated with cellular functions. Since the pro-hormone 25(OH)D is metabolized to the active hormone within the mitochondria (Figure 1A), we assessed mitochondrial function after treatment with 25(OH)D. 957E/hTERT cells (a benign, immortalized prostate cell line) and primary prostatic epithelial cells (PrE) were used for these experiments. Both cell types express CYP27B1 to hydroxylate and activate 25(OH)D, a function lost in PCa cell lines (Figure 1B–C) [24, 48, 49]. 957E/hTERT and PrE cells express the ferredoxin enzymes necessary for CYP27B1 activity and VDR (Supplementary Figure 1A). 957E/hTERT cells provided the benefits of an immortalized cell while primary prostate-derived epithelial cells provided a more clinically relevant model.
Figure 1. 25(OH)D exhibits temporal and differential effects on the mitochondria in benign prostate epithelial cell culture models.
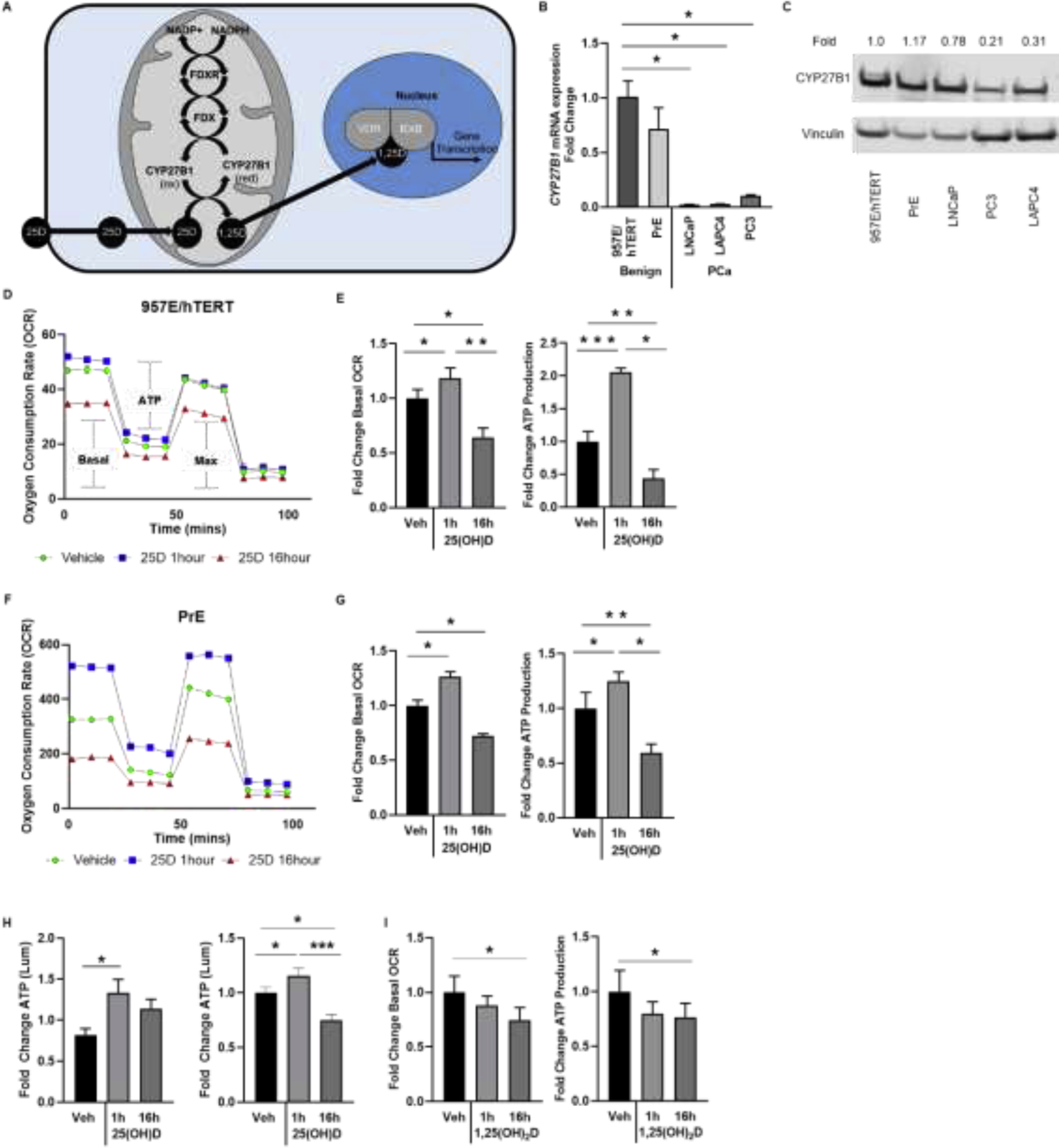
A, Diagram of previously characterized metabolism of 25(OH)D within mitochondria utilizing ferredoxin (FDX) and ferredoxin reductase (FDXR) to transfer electrons from NADPH to CYP27B1 for vitamin D hydroxylation. B-C, Gene expression by RT-qPCR and protein levels by western blot of CYP27B1 in benign (957E/hTERT and PrE) and prostate cancer (LNCaP, LAPC4, and PC3) cell lines shown as relative quantification (RQ) and fold change normalized to housekeeper (HPRT and Vinculin respectively). Seahorse output of oxygen consumption rate (OCR) over time to detect changes in mitochondrial respiration in D-E, 957E/hTERT and F-G, primary prostatic epithelial (PrE) cells treated with 50nM 25(OH)D. OCR tracings for the Mitostress test: 1 hour (blue) and 16 hours (red) relative to vehicle control (green). Bar graphs show mean (± SEM) basal respiration and ATP production (D and F) for three replicates. H, ATP colorimetric assay in 957E/hTERT and PrE cells shown relative to vehicle control. I, Seahorse output of OCR over time in PrE cells treated with 50nM 1,25(OH)2D. Graphs show mean of N=3 biological replicates (4 technical replicates within each experiment) with standard error. Student’s t-test: P<.05= * P<.00%= ** P<.0005= ***.
Mitochondrial respiration was measured by extracellular flux analysis with the Seahorse XFe24 using the Mitostress test. Both cell types treated for 1 hour with 25(OH)D had a marked increase in basal respiration as determined by the oxygen consumption rate (OCR) (Figures 1D–G). As calculated from the OCR plot, which compares the difference in respiration before and after ATP synthase is inhibited by oligomycin, ATP production increased as well in response to 25(OH)D. These changes occurred in the context of non-significant changes in extracellular acidification rates (ECAR) (Supplementary Figure 1B). Maximum respirations emulated basal respiration in prostate cells for all experiments (data not shown). 16 hours was used as a later time point at which primary metabolism of 25(OH)D is complete and the phenotype includes transcription-mediated effects [50, 51]. Treatment with 50nM 25(OH)D for 16 hours suppressed basal mitochondrial respiration (Figure 1D–G), which is consistent with prior reports showing low respiratory capacity and energy production efficiency in the prostate [52–54]. 25(OH)D treatment did not change the structure or number of mitochondria when assessed by immunofluorescence using Mitotracker (data not shown). When the Mitostress test was performed on a PCa cell line (LNCaP) lacking CYP27B1, the cells did not exhibit an increase in the basal respiration rate, but rather a decrease (Supplementary Figure 1C). The change in ATP levels observed in 957E/hTERT and PrE cells was also confirmed by a second, luminescence-based method, which also showed an increase in ATP levels at 1 hour 25(OH)D, followed by a sharp decline at the 16 hour time point (Figure 1H) in both cell types. To determine whether this phenotype was specific to the activation of 25(OH)D and CYP27B1, cells were treated with the active hormone 1,25(OH)2D, which does not require CYP27B1 activation. The PrE cells did not have a significant response to 1,25(OH)2D at 1 hour (Figure 1I). Therefore, 25(OH)D exposure results in opposing effects on prostate epithelial cells, initially promoting oxygen consumption and ATP synthesis and subsequently suppressing mitochondrial respiration below baseline.
1 hour 25(OH)D treatment increased mitochondrial membrane potential and oxidation
In order to determine if changes in mitochondrial potential accompanied the 25(OH)D-induced changes in respiration, the mitochondrial health assay was performed (Figure 2A–D). In both 957E/hTERT and PrE cells, there was an increase in mitochondrial potential at 1 hour of 25(OH)D treatment that diminished after 16 hours, dropping beneath those values observed in the vehicle-treated cells. Since the activation of 25(OH)D by CYP27B1 requires a series of redox reactions, we tested if a potent anti-oxidant could alter the observed 25(OH)D-induced phenotypes. An increase in oxidation was observed after 1 hour of 25(OH)D by the CellROX fluorescence-based assay (Figure 2E,F). Pretreatment with the anti-oxidant compound, N-acetylcysteine (NAC), effectively blocked the 25(OH)D-increase in CellROX, without inducing a change in signal on its own. Treatments with 25(OH)D and vehicle did not change cell viability (data not shown). These data indicate that 25(OH)D exposure results in a transient pro-oxidant burst prior to suppression of mitochondrial respiration.
Figure 2. Short-term effects of 25(OH)D on mitochondrial membrane potential and oxidation in benign prostate epithelial cells.
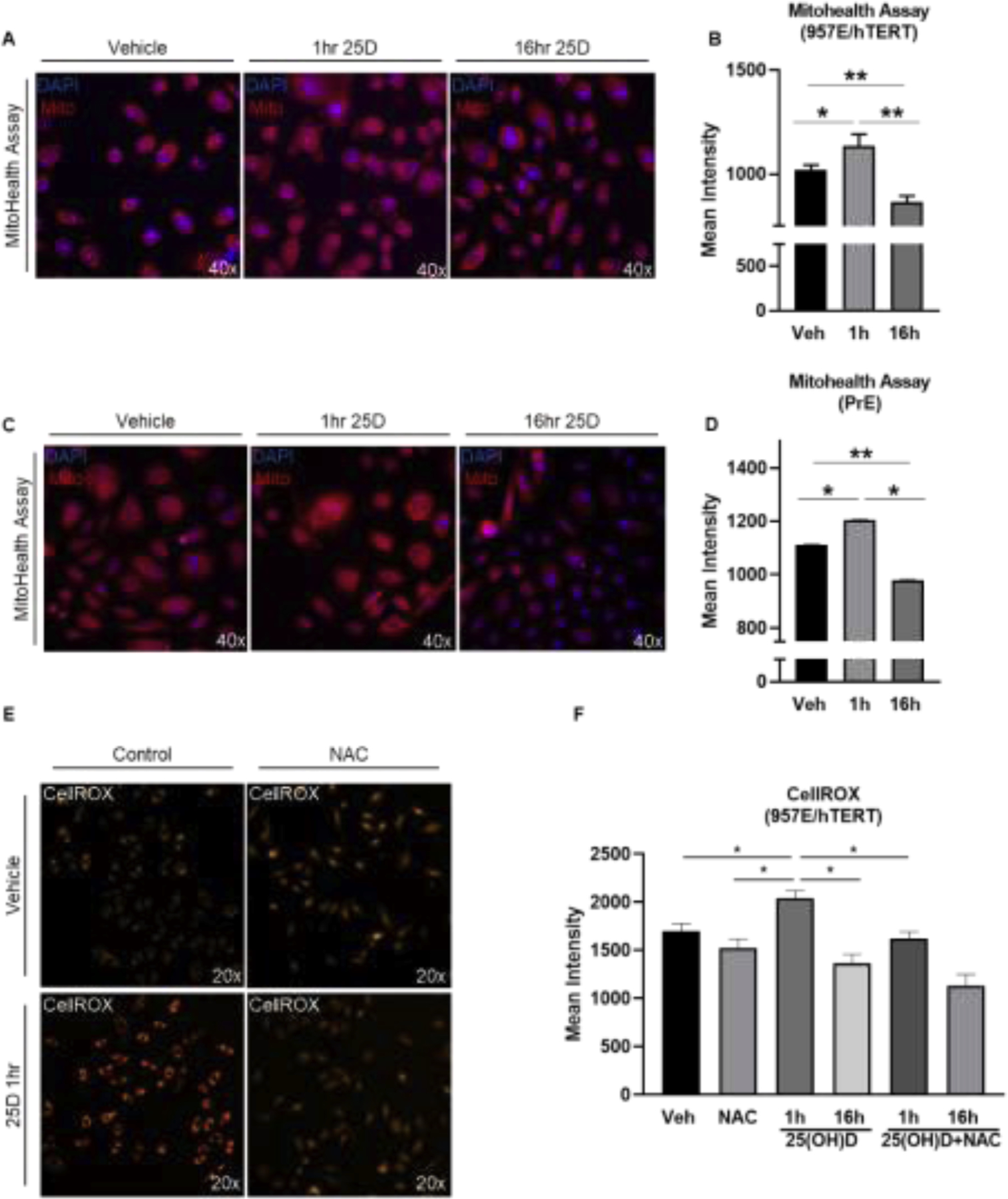
Representative images and quantification of fluorescence from mitohealth assay on 957E/hTERT cells A-B and PrE cells C-D. Cells treated with 50nM 25(OH)D or vehicle control for 1 hour or 16 hours and mitochondrial membrane potential (red) was quantified using the 40x objective on the Cellinsight Cx7 A-D. Graphs show the average intensity in mitochondrial fluorescence (B and D). CellROX staining (orange) and quantification of 957E/hTERT cells treated with 50nM 25(OH)D, vehicle, or 5mM NAC and exposed for 1 hour or 16 hours. Graphs show mean of N=3 biological replicates (3 technical replicates within each experiment) with standard error. Student’s t-test: P<.05=* P<.005= ** P<.0005= ***.
25(OH)D-induced changes at the 1 hour time point were mediated through CYP27B1
The observation that NAC attenuated the oxidative response to 25(OH)D indicated that CYP27B1 activity was involved in the pro-oxidant response. CYP27B1 levels were reduced using siRNA in the 957E/hTERT cells, resulting in a more than 50% reduction in protein levels (Figure 3A). The 1 hour change in basal OCR and ATP was significantly attenuated in the siCYP27B1 cells relative to the scramble cells (Figure 3B,C). Similarly, the siCYP27B1 25(OH)D-treated cells did not respond to 25(OH)D in the mitochondrial health assay at 1 hour as compared to control or scramble-transfected cells, thus supporting the role of CYP27B1 in contributing to short-term responses to 25(OH)D (Figure 3D,E).
Figure 3. CYP27B1-dependent changes in mitochondrial respiration observed after 1 hour treatment with 25(OH)D.
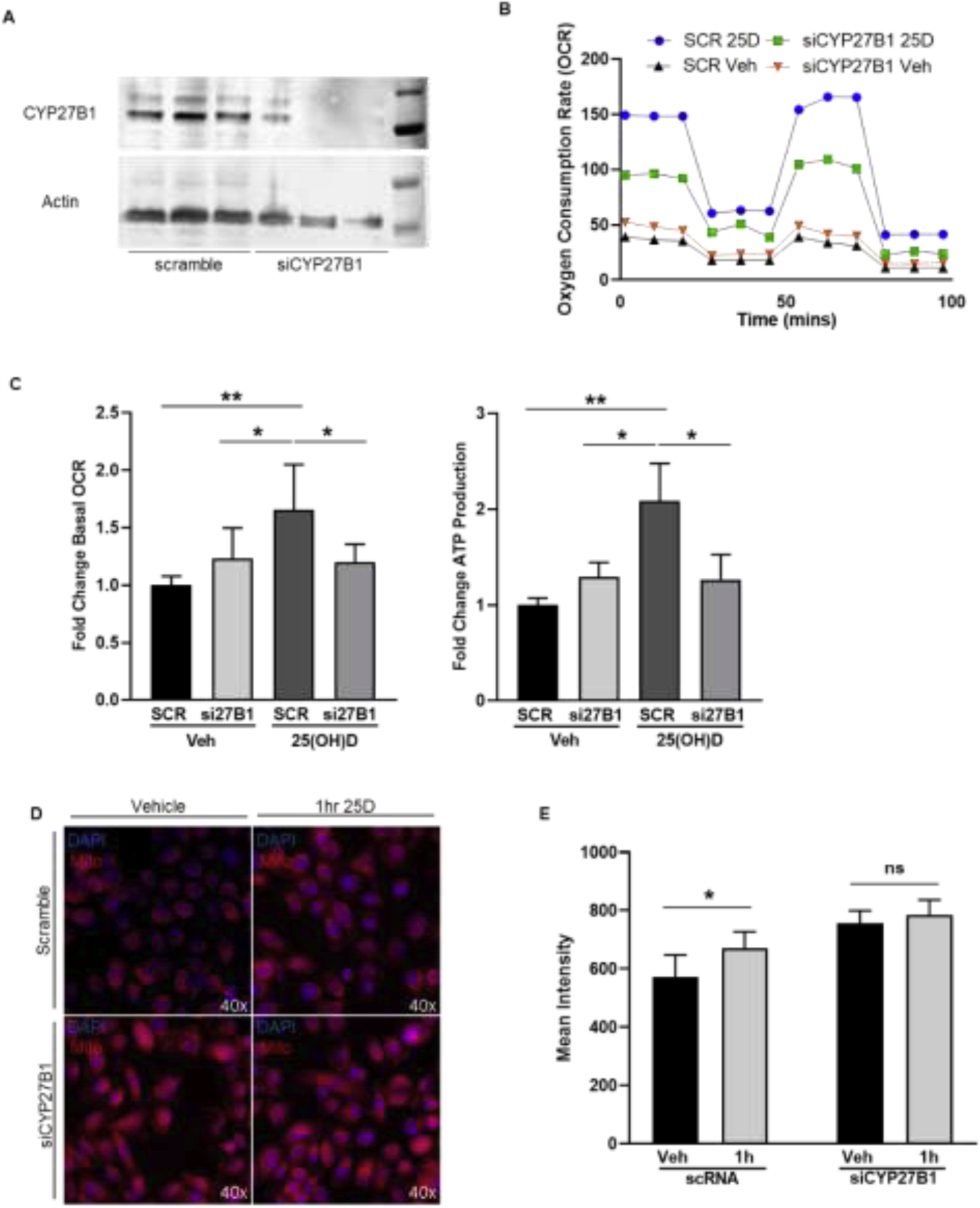
A, Transfection of 957E/hTERT cells with an siRNA for CYP27B1 induced a 50% knockdown in CYP27B1 protein levels. B-C, Seahorse XFe24 output of OCR over time from 957E/hTERT cells with a knockdown of CYP27B1. OCR tracings: si27B1 (green) and scramble (blue) treated for 1 hour with 25(OH)D relative to si27B1 (orange) and scramble (black) treated with vehicle control for 1 hour. Bar graphs show mean (±SEM) change in basal respiration and ATP production, C. D-E, Representative images and quantification of fluorescence from mitohealth assay on siCYP27B1 or scramble cells treated with 50nM 25(OH)D for 1 hour and mitochondrial membrane potential (red) was quantified. Graphs show mean of N=3 biological replicates (3 technical replicates within each experiment) with standard error. Student’s t-test: P<.05=* P<.005= ** P<.0005= ***.
Changes in 25(OH)D concentrations induced mitochondrial alterations in an in vitro model of vitamin D deficiency
Physiologically, the levels of vitamin D in humans oscillate with seasons and sun exposure, but we are never completely devoid of vitamin D. To mimic vitamin D deficiency, 957E/hTERT cells were maintained in 5nM 25(OH)D then adjusted to a level representing deficient (100pM), sufficient (5nM), and high (50nM) 25(OH)D conditions (Figure 4A). A change in 25(OH)D in both deficient and high conditions resulted in a transient spike in mitochondrial respiration at the 1 hour time point (Figure 4B,C). Vitamin D deficiency appeared to cause increased dysregulation in these cells with ECAR also increasing relative to sufficient or high levels of vitamin D (Supplementary Figure 3). Conversely, only the cells exposed to high levels of 25(OH)D exhibited suppression of oxygen consumption at the 16 hour time point. This pattern of responses was emulated with the mitochondrial health assay in which the levels of 25(OH)D were associated with an increase in mitochondrial potential after 1 hour, and only the high dose was protective (supporting normal prostate respiration) at 16 hours (Figure 4D,E). Therefore, in addition to a setting lacking 25(OH)D for the majority of culture, maintaining cells in sufficient levels and changing to an increased level still induced an initial pro-oxidant shift in prostatic mitochondria before the subsequent suppression of mitochondrial respiration.
Figure 4. Modeling vitamin D deficiency in vitro shows alteration of 25(OH)D increased mitochondrial stress at 1 hour.
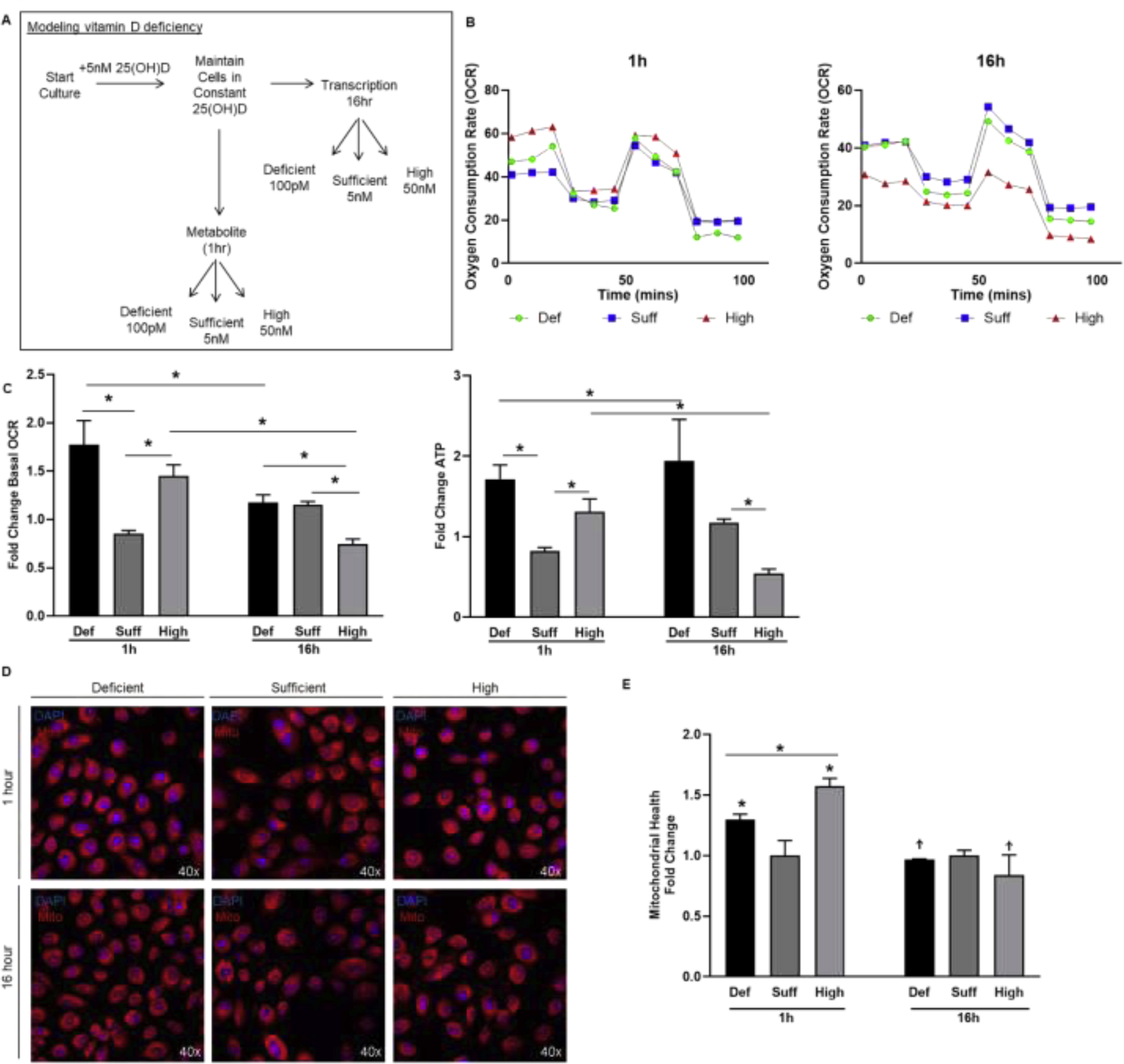
A, Diagram of physiologically-relevant cell culture model to examine mitochondrial responses to alteration in 25(OH)D status. B-C, Seahorse XFe24 output of OCR over time in 957E/hTERT cells maintained in 25(OH)D and switched to a deficient dose of 100pM (green), maintained at sufficient dose of 5nM (blue), or changed to a high dose of 50nM (red) for 1 hour (left panel) and 16 hours (right panel). Bar graphs show mean (±SEM) change in basal respiration and ATP production relative to sufficient 25(OH)D. D-E, Representative images and quantification of fluorescence from the mitohealth assay on 957E/hTERT cells treated with deficient, sufficient, or high dose of 25(OH)D for 1 hour and 16 hours and mitochondrial membrane potential (red) was quantified. Graphs show mean of N=3 biological replicates (3 technical replicates within each experiment) with standard error. Student’s t-test: P<.05=* P<.005= ** P<.0005= ***.
25(OH)D-induced transcriptional changes of NADPH-producing genes, mitochondrial structural genes, and mitochondrial transport genes
Distinct from the short-term oxidative burst by 1 hour 25(OH)D treatment is the opposing and consistent reduction in mitochondrial respiration and membrane potential observed at 16 hours. This phenotype supports an overall role for 25(OH)D in maintaining the low mitochondrial respiration of benign prostate epithelium. These 16 hour effects are the culmination of non-genomic and transcriptional regulation by VDR as evidenced by the Mito Stress test on VDR knockdown cells (Figure 5A–B). Cells deficient in VDR did not have 25(OH)D-induced suppression of mitochondrial respiration. Previous reports in breast cancer cells have shown that 1,25(OH)2D upregulates IDH2, an enzyme that replenishes the cellular NADPH pools [55–58]. NADPH is a crucial electron donor for ferredoxin cycling and CYP27B1-mediated 25(OH)D hydroxylation [34]. 25(OH)D increased IDH2 mRNA levels in 957E/hTERT and PrE cells (Figure 5A,B) as well as IDH2 protein levels in a dose-dependent manner (Figure 5C–D).
Figure 5. Transcriptional regulation of metabolism and mitochondrial function by 25(OH)D.
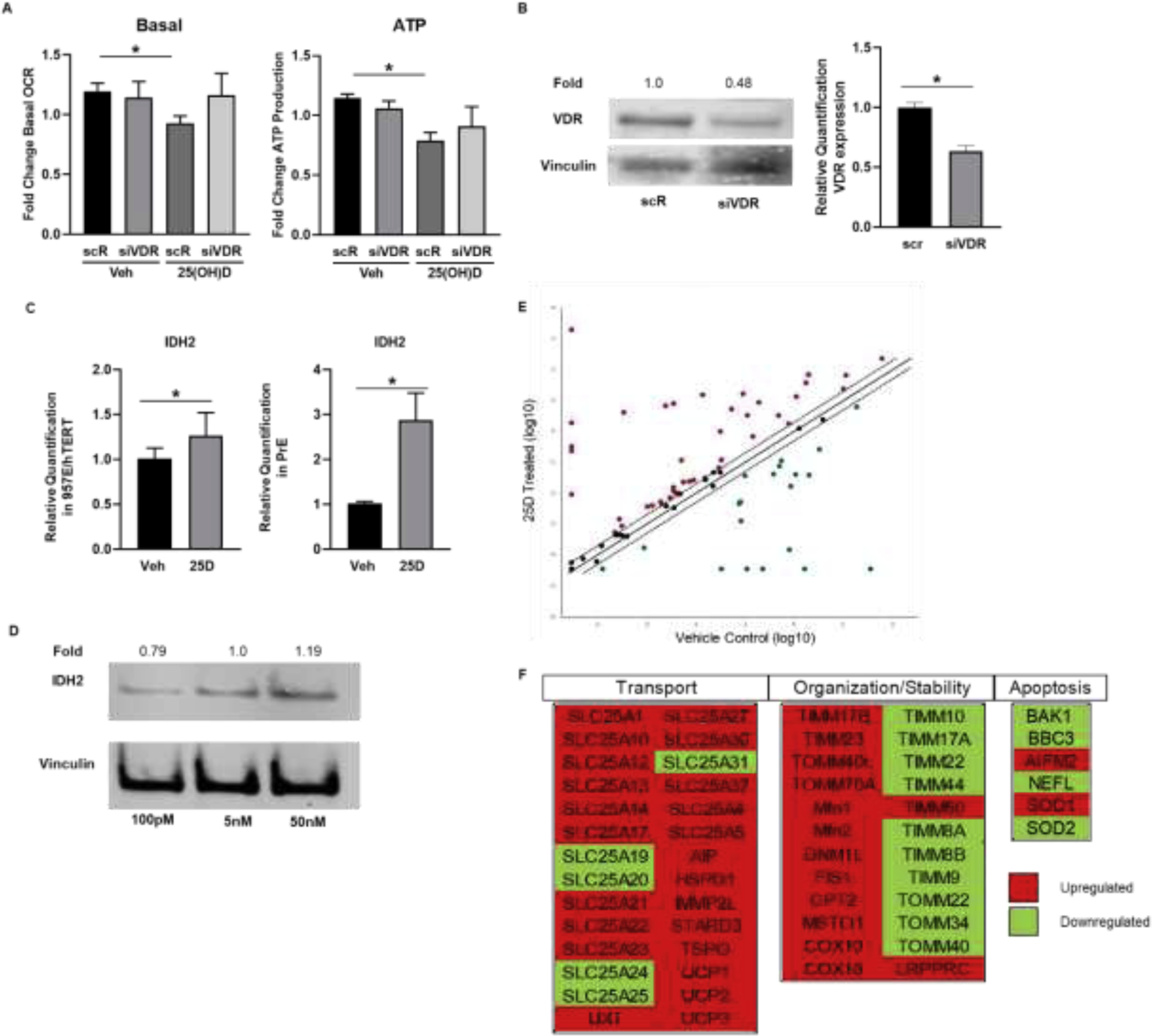
A, Seahorse XFE24 output of OCR over time from 957E/hTERT cells with a knockdown of VDR. Bar graphs show mean (± SEM) change in basal respiration and ATP production. B, Transfection of 957E/hTERT cells with siRNA for VDR induced a 50% knockdown in VDR protein and RNA levels. Gene expression by RT-qPCR of metabolic regulatory gene, IDH2, in 957E/hTERT cells and PrE cells (C) treated with 50nM 25(OH)D for 16 hours. Graphs show mean relative quantification (RQ) shown normalized to HPRT of N=3 biological replicates (3 technical replicates within each experiment) with standard error. Student’s t-test: P<.05= * P<.00%= ** P<.0005= ***. D, Protein expression of IDH2 after 5nM 25(OH)D constant culture and treatment changed to 100pM, 5nM, or 50nM for 16 hours. Fold change relative to 5nM. E and F, Results from mitochondrial gene RT-qPCR panel from PrE cells treated with 50nM 25(OH)D for 16 hours. Data shown by scatter plot and gene table of mitochondrial dynamic, transport, and apoptotic genes. Gene table denotes upregulation (red) and downregulation (green) of 25(OH)D treated cells relative to vehicle control cells. Scatterplot denotes significant change at +/−2-fold change.
To query for other changes in expression of 25(OH)D-regulated mitochondrial metabolism genes, an 84 gene RT-qPCR panel was examined. The levels of approximately half of the genes (22 upregulated, 19 downregulated) were altered by 25(OH)D exposure at 16 hours (Figure 5 E,F). The expression of SLC25A12, a gene responsible for glutamate transport [59], was upregulated in response to 25(OH)D treatment, supporting its likely role in normal prostate epithelial cells by replenishing intermediates necessary for citrate production. The levels of expression of genes involved in maintaining mitochondrial structure were upregulated, including FIS1, MSTO1, and DNM1L. Conversely, many of the genes whose products promote apoptosis were downregulated after vitamin D treatment, including BAK1, BBC3, and NEFL. These observations lead us to suggest a role for vitamin D in promoting mitochondrial reorganization in prostate epithelial cells to promote citrate-producing metabolic activity without inducing apoptosis in noncancerous cells. Additional targets observed to be upregulated also facilitate the transport of substrates such as iron, and amino acids, SLC25A37 and SLC25A13 respectively. Transcriptional changes for genes in this array corresponding with structural and transporter proteins supported further metabolic reorganization after 25(OH)D treatment with significant increases in many mitochondrial transporters (Figure 5 E,F). Therefore, changes in transcription suggest changes in mitochondrial structure and composition that contribute to the citrate-producing function of prostate epithelial cells.
DISCUSSION
We report a biphasic mechanism by which 25(OH)D contributes to the cellular metabolism of prostate epithelial cells. This cell model translates to regulation of mitochondrial respiration by vitamin D that involves both non-genomic and genomic actions. Previous studies have focused on the active hormone, 1,25(OH)2D, and VDR’s ability to regulate cellular metabolism in several cell types in addition to prostate, including human skeletal muscle cells, keratinocytes, and peripheral blood mononuclear cells [37, 41, 42, 60–62]. Our data indicate that final mitochondrial function is a culmination of the process of redox-mediated metabolism of 25(OH)D within the mitochondria shortly after exposure, as well as transcriptional regulation of mitochondrial structural genes.
Consistent with the previously known protective effects of 25(OH)D on the prostate [52, 53], we observed that 16 hour treatment of prostate cells with 25(OH)D potentiated the low respiratory phenotype of normal prostate. This was observed in low basal respiration as well as a corresponding decrease in ATP production, thus confirming a decreased penchant for energy production in benign prostate epithelium. Others have observed that 16 hour treatment with 25(OH)D also suppressed mitochondrial respiration in LNCaP cells which have significantly less CYP27B1, supporting vitamin D’s role in promoting benign prostate epithelial cell metabolism [24, 48]. In skeletal muscle 1,25(OH)2D increased mitochondrial respiration, though skeletal muscle cells have high energetic demands and are therefore more adept at producing ATP than glycolytic prostate cells [37, 62]. Thus, in supporting the basal respiratory phenotype of prostatic cells, those cells treated with 25(OH)D were not anticipated to increase mitochondrial respiration [54]. Instead, as evidenced by our data, 25(OH)D further reduced oxidative phosphorylation, consistent with the highly glycolytic phenotype of benign prostate. Vitamin D likely works in coordination with other hormones, such as testosterone, which has been shown to facilitate energy-efficient citrate production instead of oxidation when co-treated with 1,25(OH)2D in PCa cell lines, encouraging the predominant method of metabolism in healthy prostate tissue [7].
The same pattern of respiratory changes was not observed at short exposures to 25(OH)D, which instead stimulated mitochondrial oxidation and respiration. We used the short 1 hour time point as an experimental tool to observe this biphasic burst in respiration that cannot be distinguished in vivo, as the overall phenotype is a culmination of all actions of vitamin D metabolites. Opposing effects of 25(OH)D have been seen in other cell types, such as skeletal muscle cells, though the differences observed there were attributed to differences in the vitamin D isoform, not exposure time [37]. In their study, Ryan et al. observed the inverse relationship to that seen in prostate epithelial cells, with 25(OH)D suppressing mitochondrial respiration while 1,25(OH)2D (isoform circumventing CYP27B1) increasing mitochondrial respiration. This could also be a cell type-specific response to vitamin D and could highlight the importance of vitamin D regulation in the prostate, which has unique metabolic demands and is specifically predisposed to disease aggression in the setting of vitamin D deficiency.
Cytochrome P450 activity is generally tightly regulated due to the damaging effects of redox reaction byproducts like reactive oxygen species [63–65]. The rapid response of mitochondria observed in our benign prostate models indicates that these changes may be due to alterations in the redox balance generated as a consequence of CYP27B1 activity. Much of the research attributed to redox biology involving CYP-enzymes has focused on other members of the P450 family. However, the effects of these enzymes could explain some of the non-genomic changes stipulated for CYP27B1, particularly as it pertains to vitamin D activity [43–45]. A study by Favus et al. in 1986 observed similar non-genomic-effect findings in kidney proximal tubules where 25(OH)D-induced changes in metabolism and mitochondrial function, leading the authors to hypothesize these were CYP27B1-mediated effects [43]. These data in conjunction with our own findings suggest an initial priming effect induced by 25(OH)D on the mitochondrial phenotype.
The priming of the mitochondria in this context is a redox shift that allows further activity that may involve other effects as well, such as promoting further 25(OH)D activation, remodeling and reinforcing mitochondrial and other organelle landscapes [66], or providing an adaptive mechanism for oscillating levels of nutrients. Of note, treatment of these prostatic cells with the active, 1,25(OH)2D did not induce a change in mitochondrial activity at 1 hour. This suggests that the effects are specific to the activation of vitamin D and supports its continued contribution to maintaining normal prostate epithelial cell function.
We modeled vitamin D deficiency by culturing cells in deficient, sufficient, or high vitamin D, to determine the translational relevance of our findings to patients with extended periods of vitamin D deficiency, such as the elderly and African Americans [67]. Physiologically, cells are never completely deprived of vitamin D as 1,25(OH)2D levels are tightly regulated in the tissue and are crucial for a number of cellular processes [34]. The conservative threshold for vitamin D deficiency is set at 25(OH)D levels in the serum under 30nmol/L as deficient and levels under 50nmol/L as inadequate for overall health [68]. All doses used in these studies are within the physiological range [68]. The mitochondrial response at 1 hour to increased 25(OH)D was similar, albeit a marginally attenuated response, indicating that any change in 25(OH)D levels alter the mitochondria. Vitamin D deficiency also appeared to cause general metabolic dysregulation with ECAR also elevated in these cells. We did see that cells grown in prolonged deficiency retained an increased oxidative state that may have detrimental effects on the mitochondria and further supports the importance of sufficient 25(OH)D entering the tissue.
Studies on the role of vitamin D have revealed a number of phenotypes that may relate back to mitochondrial metabolism and are accentuated in mouse models lacking the VDR receptor [69]. A knockdown of VDR in 957E/hTERT cells showed that treatment with 25(OH)D no longer suppressed mitochondrial respiration when VDR levels were depleted. In our gene panel, the group of genes found to be the most upregulated in response to 25(OH)D treatment were those responsible for import of substrates and proteins into the mitochondria. Most notably is upregulation of the amino acid transporter, SLC25A12, indicating vitamin D’s possible contribution to preserving benign activity of prostatic mitochondria by transporting additional reducing equivalents from the cytoplasm to the mitochondria, maintaining redox balance [59]. Further supporting the coupling of vitamin D activity to cellular redox state, one of the genes known to replenish NADPH, IDH2, was upregulated in response to 25(OH)D [55–58, 61]. The upregulation of additional transporters that replenish iron and cholesterol, important for CYP activity and vitamin D intake, support vitamin D’s role in preparing prostatic mitochondria for further activity. Additionally, SOD1 was upregulated after 25(OH)D treatment, which is consistent with prior reports suggesting a role for SOD1 in processing redox changes in response to vitamin D treatment [70]. In opposition, apoptosis-related genes were down-regulated suggesting these cells are not experiencing stress or damage from these treatments. When taken together, vitamin D creates a microenvironment promoting physiological prostatic mitochondrial function in addition to the pleiotropic effects controlled through transcriptional regulation of known VDR target genes.
CONCLUSION
Assessing our findings as a whole, deficiency in vitamin D may induce an altered metabolic state (favoring increased oxidation), increasing the risk of metabolic changes that permit PCa growth by contributing to energetic dysregulation [25, 26]. The bioactivation of vitamin D within the mitochondria cannot be uncoupled from its effects as the process contributes to the state of the mitochondria. Therefore, patients with sufficient 25(OH)D levels reinforce the benign metabolic phenotype of prostatic epithelial cells, making them less susceptible to carcinogenesis, further supporting the coupling of vitamin D activity to the redox balance. This action compliments the well-established transcriptionally-mediated effects of vitamin D. Sufficient vitamin D levels may now be considered crucial not only for proper regulation of transcription-mediated cell processes but also for maintaining proper mitochondrial functionality. These findings also suggest that patients with dysfunctional mitochondria or those experiencing metabolic disorders may be less able to convert 25(OH)D to 1,25(OH)2D thus perpetuating vitamin D deficiency.
Supplementary Material
Highlights.
Conversion of 25(OH)D to 1,25(OH)2D changes local redox balance in prostatic cells
CYP27B1 is important in the maintenance of prostatic mitochondrial function
Vitamin D impacts mitochondrial composition through transcriptional regulation
ACKNOWLEDGEMENTS
We thank Dr. Alan Diamond for his insight and proofreading of the manuscript. This work was funded by the UIC Center for Clinical and Translation Science Pre-doctoral Education for Clinical and Translational Scientists (PECTS) Program (Blajszczak). We thank the UIC Biorepository members, Dr. Klara Valyi-Nagy and Alex Susma, and the urologists, Drs. Michael Abern, Daniel Moreira, and Simone Crivallero, for facilitation of the tissue acquisition for the primary cell cultures. We thank the UIC Urology patients for donating their tissue to research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Der T, et al. , Vitamin D and prostate cancer survival in veterans. Mil Med, 2014. 179(1): p. 81–4. [DOI] [PubMed] [Google Scholar]
- 2.Muntzing J, et al. , Comparison and significance of respiration and glycolysis of prostatic tissue from various species. J Med Primatol, 1975. 4(4): p. 245–51. [DOI] [PubMed] [Google Scholar]
- 3.Costello LC and Franklin RB, The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Mol Cancer, 2006. 5: p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franklin RB, et al. , The effect of testosterone on citrate synthesis and citrate oxidation and a proposed mechanism for regulation of net citrate production in prostate. Horm Metab Res, 1986. 18(3): p. 177–81. [DOI] [PubMed] [Google Scholar]
- 5.Kavanagh JP, Isocitric and citric acid in human prostatic and seminal fluid: implications for prostatic metabolism and secretion. Prostate, 1994. 24(3): p. 139–42. [DOI] [PubMed] [Google Scholar]
- 6.Dittrich R, et al. , Assessing prostate cancer growth with citrate measured by intact tissue proton magnetic resonance spectroscopy. Prostate Cancer Prostatic Dis, 2012. 15(3): p. 278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, et al. , Vitamin D and testosterone co-ordinately modulate intracellular zinc levels and energy metabolism in prostate cancer cells. J Steroid Biochem Mol Biol, 2019. [DOI] [PubMed] [Google Scholar]
- 8.Costello LC, et al. , Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem, 1997. 272(46): p. 28875–81. [DOI] [PubMed] [Google Scholar]
- 9.Costello LC, et al. , Role of zinc in the pathogenesis and treatment of prostate cancer: critical issues to resolve. Prostate Cancer Prostatic Dis, 2004. 7(2): p. 111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costello LC, et al. , Terminal oxidation and the effects of zinc in prostate versus liver mitochondria. Mitochondrion, 2004. 4(4): p. 331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnan AV, et al. , The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin North Am, 2010. 39(2): p. 401–18, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards Z, et al. , Prostatic compensation of the vitamin D axis in African American men. JCI Insight, 2017. 2(2): p. e91054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie DD, et al. , Low vitamin D status is associated with inflammation in patients with prostate cancer. Oncotarget, 2017. 8(13): p. 22076–22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manson JE, et al. , Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N Engl J Med, 2019. 380(1): p. 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo TC, et al. , Pilot study: potential role of vitamin D (Cholecalciferol) in patients with PSA relapse after definitive therapy. Nutr Cancer, 2005. 51(1): p. 32–6. [DOI] [PubMed] [Google Scholar]
- 16.Murphy AB, et al. , Vitamin D deficiency predicts prostate biopsy outcomes. Clin Cancer Res, 2014. 20(9): p. 2289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall DT, et al. , Vitamin D3 supplementation at 4000 international units per day for one year results in a decrease of positive cores at repeat biopsy in subjects with low-risk prostate cancer under active surveillance. J Clin Endocrinol Metab, 2012. 97(7): p. 2315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollis BW, et al. , Vitamin D3 supplementation, low-risk prostate cancer, and health disparities. J Steroid Biochem Mol Biol, 2013. 136: p. 233–7. [DOI] [PubMed] [Google Scholar]
- 19.Banach-Petrosky W, et al. , Vitamin D inhibits the formation of prostatic intraepithelial neoplasia in Nkx3.1;Pten mutant mice. Clin Cancer Res, 2006. 12(19): p. 5895–901. [DOI] [PubMed] [Google Scholar]
- 20.Fleet JC, et al. , Vitamin D Signaling Suppresses Early Prostate Carcinogenesis in TgAPT121 Mice. Cancer Prev Res (Phila), 2019. 12(6): p. 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swami S, et al. , Dietary vitamin D(3) and 1,25-dihydroxyvitamin D(3) (calcitriol) exhibit equivalent anticancer activity in mouse xenograft models of breast and prostate cancer. Endocrinology, 2012. 153(6): p. 2576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz GG, et al. , 1 alpha,25-Dihydroxyvitamin D (calcitriol) inhibits the invasiveness of human prostate cancer cells. Cancer Epidemiol Biomarkers Prev, 1997. 6(9): p. 727–32. [PubMed] [Google Scholar]
- 23.van den Bemd GJ, Pols HA, and van Leeuwen JP, Anti-tumor effects of 1,25-dihydroxyvitamin D3 and vitamin D analogs. Curr Pharm Des, 2000. 6(7): p. 717–32. [DOI] [PubMed] [Google Scholar]
- 24.Banks M and Holick MF, Molecular Mechanism(s) Involved in 25-Hydroxyvitamin D’s Antiproliferative Effects in CYP27B1-transfected LNCaP Cells. Anticancer Res, 2015. 35(7): p. 3773–9. [PubMed] [Google Scholar]
- 25.Ahonen MH, et al. , Prostate cancer risk and prediagnostic serum 25-hydroxyvitamin D levels (Finland). Cancer Causes Control, 2000. 11(9): p. 847–52. [DOI] [PubMed] [Google Scholar]
- 26.Bratchikov OI, Artishchev SO, and Tyuzikov IA, [Vitamin D deficiency, metabolic syndrome, and prostate adenoma: current epidemiological trends and pathophysiological mechanisms of interaction]. Urologiia, 2018(4): p. 179–185. [PubMed] [Google Scholar]
- 27.de Jongh RT, van Schoor NM, and Lips P, Changes in vitamin D endocrinology during aging in adults. Mol Cell Endocrinol, 2017. 453: p. 144–150. [DOI] [PubMed] [Google Scholar]
- 28.Boucher BJ, The problems of vitamin d insufficiency in older people. Aging Dis, 2012. 3(4): p. 313–29. [PMC free article] [PubMed] [Google Scholar]
- 29.Terabe Y, et al. , Vitamin D deficiency in elderly women in nursing homes: investigation with consideration of decreased activation function from the kidneys. J Am Geriatr Soc, 2012. 60(2): p. 251–5. [DOI] [PubMed] [Google Scholar]
- 30.Han Y, et al. , Prostate Cancer Susceptibility in Men of African Ancestry at 8q24. J Natl Cancer Inst, 2016. 108(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batai K, et al. , Race and BMI modify associations of calcium and vitamin D intake with prostate cancer. BMC Cancer, 2017. 17(1): p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossberg W, et al. , Human Pigmentation, Cutaneous Vitamin D Synthesis and Evolution: Variants of Genes (SNPs) Involved in Skin Pigmentation Are Associated with 25(OH)D Serum Concentration. Anticancer Res, 2016. 36(3): p. 1429–37. [PubMed] [Google Scholar]
- 33.Holick MF, Tian XQ, and Allen M, Evolutionary importance for the membrane enhancement of the production of vitamin D3 in the skin of poikilothermic animals. Proc Natl Acad Sci U S A, 1995. 92(8): p. 3124–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bikle DD, Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol, 2014. 21(3): p. 319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zehnder D, et al. , Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab, 2001. 86(2): p. 888–94. [DOI] [PubMed] [Google Scholar]
- 36.Giangreco AA, et al. , Differential expression and regulation of vitamin D hydroxylases and inflammatory genes in prostate stroma and epithelium by 1,25-dihydroxyvitamin D in men with prostate cancer and an in vitro model. J Steroid Biochem Mol Biol, 2015. 148: p. 156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan ZC, et al. , 1alpha,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J Biol Chem, 2016. 291(3): p. 1514–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scragg R, et al. , Serum 25-hydroxyvitamin D, diabetes, and ethnicity in the Third National Health and Nutrition Examination Survey. Diabetes Care, 2004. 27(12): p. 2813–8. [DOI] [PubMed] [Google Scholar]
- 39.Hypponen E, et al. , 25-hydroxyvitamin D, IGF-1, and metabolic syndrome at 45 years of age: a cross-sectional study in the 1958 British Birth Cohort. Diabetes, 2008. 57(2): p. 298–305. [DOI] [PubMed] [Google Scholar]
- 40.Aljabri KS, Bokhari SA, and Khan MJ, Glycemic changes after vitamin D supplementation in patients with type 1 diabetes mellitus and vitamin D deficiency. Ann Saudi Med, 2010. 30(6): p. 454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calton EK, et al. , Prevailing vitamin D status influences mitochondrial and glycolytic bioenergetics in peripheral blood mononuclear cells obtained from adults. Redox Biol, 2016. 10: p. 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricca C, et al. , Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int J Mol Sci, 2018. 19(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Favus MJ and Langman CB, Evidence for calcium-dependent control of 1,25-dihydroxyvitamin D3 production by rat kidney proximal tubules. J Biol Chem, 1986. 261(24): p. 11224–9. [PubMed] [Google Scholar]
- 44.Berridge MJ, Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos Trans R Soc Lond B Biol Sci, 2016. 371(1700). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dowd DR and MacDonald PN, The 1,25-dihydroxyvitamin D3-independent actions of the vitamin D receptor in skin. J Steroid Biochem Mol Biol, 2010. 121(1–2): p. 317–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasunaga Y, et al. , A novel human cell culture model for the study of familial prostate cancer. Cancer Res, 2001. 61(16): p. 5969–73. [PubMed] [Google Scholar]
- 47.Nonn L, et al. , Inhibition of p38 by vitamin D reduces interleukin-6 production in normal prostate cells via mitogen-activated protein kinase phosphatase 5: implications for prostate cancer prevention by vitamin D. Cancer Res, 2006. 66(8): p. 4516–24. [DOI] [PubMed] [Google Scholar]
- 48.Susa T, et al. , Without 1alpha-hydroxylation, the gene expression profile of 25(OH)D3 treatment overlaps deeply with that of 1,25(OH)2D3 in prostate cancer cells. Sci Rep, 2018. 8(1): p. 9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma JF, et al. , Mechanisms of decreased Vitamin D 1alpha-hydroxylase activity in prostate cancer cells. Mol Cell Endocrinol, 2004. 221(1–2): p. 67–74. [DOI] [PubMed] [Google Scholar]
- 50.Cassan N, Lagoutte B, and Setif P, Ferredoxin-NADP+ reductase. Kinetics of electron transfer, transient intermediates, and catalytic activities studied by flash-absorption spectroscopy with isolated photosystem I and ferredoxin. J Biol Chem, 2005. 280(28): p. 25960–72. [DOI] [PubMed] [Google Scholar]
- 51.Theobald J, et al. , In vitro metabolic activation of vitamin D3 by using a multi-compartment microfluidic liver-kidney organ on chip platform. Sci Rep, 2019. 9(1): p. 4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Costello LC and Franklin RB, Concepts of citrate production and secretion by prostate. 1. Metabolic relationships. Prostate, 1991. 18(1): p. 25–46. [DOI] [PubMed] [Google Scholar]
- 53.Costello LC and Franklin RB, Concepts of citrate production and secretion by prostate: 2. Hormonal relationships in normal and neoplastic prostate. Prostate, 1991. 19(3): p. 181–205. [DOI] [PubMed] [Google Scholar]
- 54.Cutruzzola F, et al. , Glucose Metabolism in the Progression of Prostate Cancer. Front Physiol, 2017. 8: p. 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Handy DE and Loscalzo J, Redox regulation of mitochondrial function. Antioxid Redox Signal, 2012. 16(11): p. 1323–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noun A, Garabedian M, and Monet JD, Stimulatory effect of 1,25-dihydroxyvitamin D3 on the glucose-6-phosphate dehydrogenase activity in the MCF-7 human breast cancer cell line. Cell Biochem Funct, 1989. 7(1): p. 1–6. [DOI] [PubMed] [Google Scholar]
- 57.Simmons KM, et al. , Gene Signatures of 1,25-Dihydroxyvitamin D3 Exposure in Normal and Transformed Mammary Cells. J Cell Biochem, 2015. 116(8): p. 1693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beaudin S and Welsh J, 1,25-Dihydroxyvitamin D induces the glutamate transporter SLC1A1 and alters glutamate handling in non-transformed mammary cells. Mol Cell Endocrinol, 2016. 424: p. 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jalil MA, et al. , Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem, 2005. 280(35): p. 31333–9. [DOI] [PubMed] [Google Scholar]
- 60.Consiglio M, et al. , The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation. PLoS One, 2014. 9(12): p. e115816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bao BY, et al. , Protective role of 1 alpha, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. Int J Cancer, 2008. 122(12): p. 2699–706. [DOI] [PubMed] [Google Scholar]
- 62.Sinha A, et al. , Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J Clin Endocrinol Metab, 2013. 98(3): p. E509–13. [DOI] [PubMed] [Google Scholar]
- 63.Sevrioukova IF, et al. , Structure of a cytochrome P450-redox partner electron-transfer complex. Proc Natl Acad Sci U S A, 1999. 96(5): p. 1863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Daff SN, et al. , Redox control of the catalytic cycle of flavocytochrome P-450 BM3. Biochemistry, 1997. 36(45): p. 13816–23. [DOI] [PubMed] [Google Scholar]
- 65.Sevrioukova I, et al. , Equilibrium and transient state spectrophotometric studies of the mechanism of reduction of the flavoprotein domain of P450BM-3. Biochemistry, 1996. 35(22): p. 7058–68. [DOI] [PubMed] [Google Scholar]
- 66.Hoitzing H, Johnston IG, and Jones NS, What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. Bioessays, 2015. 37(6): p. 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Y, et al. , Suppression of RelB-mediated manganese superoxide dismutase expression reveals a primary mechanism for radiosensitization effect of 1alpha,25-dihydroxyvitamin D(3) in prostate cancer cells. Mol Cancer Ther, 2007. 6(7): p. 2048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bouillon R and Carmeliet G, Vitamin D insufficiency: Definition, diagnosis and management. Best Pract Res Clin Endocrinol Metab, 2018. 32(5): p. 669–684. [DOI] [PubMed] [Google Scholar]
- 69.Chung I, et al. , Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res, 2009. 69(3): p. 967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chaiprasongsuk A, et al. , Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB-induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol, 2019. 24: p. 101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.