

Abstract
Glioblastoma (GBM) is the most common type of primary central nervous system tumor in adults, which has high mortality and morbidity rates, and short survival time, namely <15 months after the diagnosis and application of standard therapy, which includes surgery, radiation therapy and chemotherapy; thus, novel therapeutic strategies are imperative. The activation of the PI3K/AKT signaling pathway plays an important role in GBM. In the present study, U87 and U251 GBM cells were treated with the PI3K/mTORC1/2 inhibitor PQR309, and its effect on glioma cells was investigated. Cell Counting Kit-8 assay, 5-ethynyl-2′-deoxyuridine and colony formation assays revealed dose- and time-dependent cytotoxicity in glioma cells that were treated with PQR309. Flow cytometry and western blotting revealed that PQR309 can significantly induce tumor cell apoptosis and arrest the cell cycle in the G1 phase. Furthermore, the expression levels of AKT, phosphorylated (p)-AKT, Bcl-2, Bcl-xL, Bad, Bax, cyclin D1, cleaved caspase-3, MMP-9 and MMP-2 were altered. In addition, the migration and invasion of glioma cells, as detected by wound healing, migration and Transwell invasion assays, exhibited a marked suppression after treating the cells with PQR309. These results indicated that PQR309 exerts an antitumor effect by inhibiting proliferation, inducing apoptosis, inducing G1 cell cycle arrest, and inhibiting invasion and migration in human glioma cells. The present study provides evidence supportive of further development of PQR309 for adjuvant therapy of GBM.
Keywords: PQR309, glioblastoma, apoptosis, proliferation, invasion
Introduction
Glioblastoma (GBM) remains one of the most common types of primary brain tumors in the adult central nervous system (CNS), with ≤5% of patients living >5 years after the first time of diagnosis (1,2). GBM exhibits an evidently increased incidence rate according to a study on Chinese patients with glioma from 2000 to 2011, and cancer of the brain and the CNS are among the 10 most common cancer types in China (3). The percentage of gliomas diagnosed in the USA among all brain and CNS tumors and malignant brain and CNS tumors is 31 and 81%, respectively (4). Currently, we see an increase in the incidence of GBM, which is the highest grade of glioma classification, ranging from 0.59 to 3.69 per 100,000 individuals and with the highest increase observed in ages ranging from ~75–84 years old. In contrast to GBM, the highest incidence of oligodendroglia and oligoastrocytomas [World Health Organization (WHO) grade III] is in patients 35–44 years of age (2,5,6). The highly invasive feature of GBM cells contributes to the migration of glioma cells into the surrounding brain parenchyma excluding surgical resection treatment, which may explain its dismal prognosis, with a median overall survival of only 15 months, and tumor recurrence (7). Recently, WHO modified the Classification of Tumors of the Central Nervous System based on molecular parameters in addition to histology of tumors (6,8). Grades I–IV are assigned to GBM, generally with an increasing malignancy from grade I to grade IV, and ~55% of all gliomas are classified as grade IV GBM, despite being relatively rare (2–3 cases per 100,000 adults in the USA and Europe annually) (9). As aforementioned, 81% of the CNS malignant types in USA are gliomas, and almost half of them are highly aggressive GBMs according to the list of brain tumors produced by WHO (1,8,10). Surgery, radiotherapy and treatment with temozolomide accompanied by tumor histopathology is the current standard therapy for GBM (11–13). This highly malignant tumor creates a serious social and economic burden, and is associated with high mortality and morbidity (14). Thus, improved therapeutic strategies or drugs are required.
Previous studies have highlighted the great importance of the PI3K/AKT/mTOR signaling pathways in GBM (15,16), which are mainly the consequence of the loss of PTEN in >50% of GBMs (17). Certain inhibitors have exhibited great inhibition effects in glioma cells (17,18). Class I PI3Ks, which consist of the catalytic subunits p110α, p110β, p110δ and PI3Kγ, are lipid kinases that, alongside mTOR, play critical roles in a variety of cellular processes, including differentiation, metabolism, migration and survival in physiological and pathophysiological conditions, and can be activated by cell surface receptors such as receptor tyrosine kinases (RTKs), immunoglobulin receptors and G-protein-coupled receptors (19–22). The PI3K family is overactive in multiple types of tumors, including glioma, and has three different subtypes, which are classified by sequence homology and substrate specificity. The PI3K family can activate AKT [also known as protein kinase B (PKB)] and mTOR (23). PKB/AKT and 3′-phosphoinositide-dependent kinase-1 can be activated by PI3K through their pleckstrin-homology domains.
Changes in cell surface receptors, such as HER2, oncogenes, protein tyrosine phosphatase non-receptor type 12, presence of activating hotspot mutations in PIK3CA, and inactivation of the lipid phosphatase PTEN can also lead to the activation of the PI3K, PKB/AKT and mTOR signaling pathways (23,24). Previous studies have revealed that certain PI3K/AKT/mTOR pathway inhibitor monotherapies or in combination with other drugs suppressed GBM proliferation in vitro and in xenografts (17,18,24). Overactivation of the PI3K/mTOR signaling pathway not only promotes tumor cell growth and angiogenesis, but also is associated with resistance to chemotherapy related to RTK inhibitors (23,25).
PQR309 is a PI3K/mTORC1/2 targeted inhibitor. Its inhibitory concentration towards PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ is 33, 661, 708 and 451 nM, respectively. It has been revealed that PQR309 may be a major inhibitor of PI3Kα, which is the most effective therapeutic target in human tumors (17,23). PQR309 exhibited a great tumor inhibition effect and minor side effects in phase II clinical trials for different solid tumors such as Ewing sarcoma, colorectal, breast, ovarian, Bartholin's gland and lung carcinomas, mesothelioma, endometrial carcinoma, squamous cell cancer of the tongue, thyme carcinoma, sinonasal carcinoma and cervical carcinoma. Patients who tried this drug had positive results, and reported common adverse events such as fatigue, hyperglycaemia, nausea, diarrhoea, constipation, rash, anorexia and vomiting (22). Considering the small number of patients in the study, the conclusion is not convincing. Its antitumor activity in refractory lymphoma in combination with other drugs has made PQR309 a novel and promising compound that is worth developing in the clinical setting of lymphoma (26). In addition, PQR309 has a great ability to cross the blood brain barrier (BBB), it is orally bioavailable, and has exhibited great pharmacokinetic parameters and an antitumor proliferative effect in mice, rats and dogs, both in vitro and in vivo (23). The great efficiency of this molecule to suppress the activation of malignant tumors accompanied by its safety profile and pharmacokinetic parameters, reveals the great potential of PQR309 to be applied to brain tumors (23). However, the effect of PQR309 in human GBM cells has not been studied to date. Thus, the present study treated U87 cells, which have no mutant PTEN, and U251 cells, which is the most common type of GBM cell line exhibiting mutant PTEN, with PQR309 to evaluate whether PQR309 has an effect on GBM cells.
Materials and methods
Cell culture
Human GBM cell lines (U87 and U251) were purchased from the Cell Bank Type Culture Collection of the Chinese Academy of Sciences. All the cell lines in our laboratory were identified by short tandem repeat profiling by Procell Life Science & Technology Co., Ltd. In addition, the U87 cell line used in the present study is of the ATCC type, and has been reported to be a glioblastoma of unknown origin. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GINOM Co., Ltd.) containing 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich; Merck KGaA) at 37°C in a humidified atmosphere containing 5% carbon dioxide. PQR309 (Fig. 1) was purchased from Selleckchem, and was dissolved in dimethyl sulfoxide (DMSO; Merck KGaA).
Figure 1.

(A) Molecular structure of PQR309. (B) Cell viability for U87 and U251 cells after being treated with PQR309 with various concentrations. (C) The colony formation rates of U87 and U251 cells in various concentration groups. Scale bar, 50 µm. (D) Each cell line was treated at different time-points with the IC50 values of PQR309 for the CCK-8 assay. Each cell line was analyzed in triplicate.
Antibodies
The antibodies used were as follows: Anti-Akt (product no. 4691) and phospho-Akt (product no. 4060; both from Cell Signaling Technology, Inc.), Bcl-2 (cat. no. GTX100064; GeneTex, Inc.), Bcl-xL (product no. 2764), Bad (product no. 9239), Bax (product no. 5023) and cyclin D1 (product no. 2978; all from Cell Signaling Technology, Inc.), cleaved caspase-3 (product code ab32042; Abcam), MMP-9 (product no. 13667), MMP-2 (product no. 40994) and GAPDH (product no. 5174; all from Cell Signaling Technology, Inc.).
Cell viability
Cell viability was assessed using Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc.), to determine the inhibitory effect of PQR309 on U87 and U251 cells according to the manufacturer's instructions. Approximately 5×103 cells were seeded in a volume of 100 µl DMEM with 10% FBS, and PQR309 was added into each well of a 96-well plate. Various concentrations of PQR309 (0, 1, 5, 10, 20, 50 and 100 µM), as well as a certain concentration applied for different time-points (24, 48, 72 and 96 h) were evaluated. Next, 10 µl CCK-8 was added, and the cells were incubated for 1 h at 37°C. The absorbance value of every well was measured with a spectrophotometric plate reader at 450 nm. Each group was assessed in triplicate.
Colony formation assay
U87 and U251 cells (~1,000) were plated on a 6-well plate and cultured in 2 ml DMEM with 10% FBS. Then, the cells were treated with different concentrations of PQR309 (0, 1, 5 and 10 µM) for ~2 weeks at 37°C with 5% CO2. The cells were fixed with 2 ml 5% paraformaldehyde at room temperature for 15 min and then stained with 0.5% crystal violet at room temperature for 30 min. Then, each well was photographed with a camera.
5-Ethynyl-2′-deoxyuridine (Edu) incorporation assay
A Cell-Light Edu DNA Cell Proliferation kit was purchased from Guangzhou RiboBio Co., Ltd. Approximately 5×103 cells were seeded in a volume of 100 µl DMEM containing 10% FBS into each well of a 96-well plate and treated with various concentrations of PQR309 (0, 1, 5, 10 and 20 µM) for 72 h. Then, the cells were cultured with 50 µM Edu for 1 h at 37°C in the presence of 5% CO2, and fixed with 4% paraformaldehyde for 30 min at room temperature, according to the manufacturer's protocol. The cells were then treated with 0.5% Triton X-100 for 20 min and washed with PBS (3 times/5 min each). Then, the cells were incubated with 100 µl 1X Apollo® reaction cocktail for 30 min, and the cell nuclei were stained for 30 min with 5 µg/ml Hoechst 33342. Fluorescence images were visualized under a fluorescence microscope at an ×200 magnification (Olympus BX51; Olympus Corporation).
Cell cycle distribution analysis
Flow cytometry was used to determine the cell cycle distribution using a cell cycle kit with PI staining (BD Biosciences). U87 and U251 cells were plated in 6-well plates and treated with various concentrations (0, 1, 5, 10 and 20 µM) of PQR309 for 72 h. Then, the cells were collected by centrifugation at 167.7 × g for 5 min at room temperature. Subsequently, the cells were washed and fixed with PBS and cold 70% ethanol for 24 h at 4°C. Then, the cells were treated with 50 µl 100 µg/ml RNase at 37°C, washed twice with PBS, centrifuged at 167.7 × g for 5 min and stained with 5 µl PI (50 mg/ml stock solution). The results were analyzed by BD FACSAria (BD Biosciences). The data were quantified using ModFit LT 4.0 (Verity Software House, Inc.).
Wound-healing assay
U87 and U251 cells were seeded into 6-well plates at an appropriate density for ~70–80% confluence as a monolayer. Then, the cells were scratched with a pipette tip to create straight wound lines in each well. Subsequently, the floating cells were removed by PBS. Various concentrations of PQR309 (0, 5 or 10 µM) were added to 2 ml DMEM supplemented with 1% FBS for an additional 24 or 48 h. The images were visualized under a fluorescence microscope (magnification, ×100; Olympus BX51; Olympus Corp.). The wound distance was evaluated by ImageJ software (version 1.8.0; National Institutes of Health).
Cell migration and invasion assays
Migration and invasion assays were performed using a Transwell chamber with an 8.0-µm pore polycarbonate membrane. U87 and U251 cells were seeded into the top chambers, where FBS had not been added to DMEM, and were either coated with Matrigel or not coated with Matrigel for invasion and migration, respectively. Then, the chambers were placed into a 24-well plate, and medium containing 10% FBS was added. After incubation for 24 h, the cells were fixed and stained with 0.5% crystal violet at room temperature for 30 min, which penetrated the underside surfaces of the membranes, while the cells that had not crossed the membranes were gently removed with cotton swabs. Subsequently, the cells were quantified under a fluorescence microscope (magnification, ×100; Olympus BX51; Olympus Corporation).
Flow cytometric analysis of apoptosis with PE/7-amino-actinomycin (7-ADD) staining and TUNEL assay
An Apoptosis Annexin V-PE/7-AAD kit (BD Biosciences) was used to analyze the apoptosis of cells treated with various concentrations (0, 1, 5, 10 and 20 µM) of PQR309 for 72 h. The cells, including those floating in DMEM, were washed twice in PBS after being collected by centrifugation at 167.7 × g for 5 min at room temperature, according to the manufacturer's protocol. Then, the cells were suspended in 100 µl 1X binding buffer (0.1 mM HEPES/NaOH, 1.4 M NaCl and 25 mM CaCl2, pH 7.4) and stained with 5 µl PE-Annexin V and 5 µl 7-ADD for 15 min in the dark at room temperature. Then, 400 µl 1X binding buffer was added to each tube. Analysis of the results was carried out with BD FACSAria (BD Biosciences). Data were quantified with FlowJo software (version 10.4.0; FlowJo LLC), while the sum of the upper right and lower right quadrants was used for calculating total apoptosis rates and subjected to statistical analysis. A TUNEL assay was performed to detect DNA fragmentation in apoptotic cells according to the manufacturer's protocol (Roche Molecular Diagnostics). Images were obtained with an Olympus BX51 fluorescence microscope (Olympus Corp.).
Mitochondrial membrane potential (ΔΨm) assay
Considering that the loss of the ΔΨm of the mitochondrial membrane is a hallmark event of early-stage apoptosis, JC-1 staining [Yeasen Biotechnology (Shanghai) Co., Ltd.] was used to assess the ΔΨm. Cells were plated in 6-well plates which consisted of a glass slide placed at the bottom of each well, and treated with or without PQR309 for 72 h. Next, an Olympus BX51 fluorescence microscope (Olympus Corp.) was used to photograph the cells, according to the manufacturer's instructions. A decrease in the ratio of red/green fluorescence intensity detected by flow cytometry and microscopy indicated loss of ΔΨm.
Western blot analysis
Cells were treated with 0, 5, 10 or 20 µM PQR309 for 72 h and then lysed in RIPA buffer (Shanghai Yeasen Biotechnology, Co., Ltd.) for ~20 min on ice. BCA was used to test the concentrations of each sample then, the protein samples were loaded onto 10 or 12% SDS-PAGE for 40 µg per lane and electro-transferred to PVDF membranes (Merck KGaA) for 60 or 90 min. After the transfer, the membrane was blocked with 5% skim milk and then incubated with the primary antibody (all used at 1:1,000) overnight at 4°C. Subsequently, the membranes were incubated with Alexa Fluor 680/790-labelled goat anti-rabbit or goat anti-mouse IgG secondary antibodies (cat. nos. 926-68021 and 926-68020; Li-COR Biosciences) (all used at 1:1,000; LI-COR Biosciences) for 1 h. The bands were visualized using the LI-COR Odyssey Infrared Imaging System (Li-COR Biosciences) and the results were normalized to GAPDH.
Statistical analysis
Statistical analyses were conducted with SPSS 19.0 software (IBM Corp.) and GraphPad Prism 6.0 software (GraphPad Software, Inc.). Data from the experiments are presented as the mean ± SD. The comparisons among the different groups (>2 groups) were analyzed by one-way ANOVA and Tukey's post hoc test, while Student's t-test was used for comparisons between 2 groups. The results presented are representative of 3 independent experiments. P<0.05 was considered to indicate a statistically significant difference.
Results
PQR309 suppresses the proliferation of U87 and U251 cells
The results of a CCK-8 assay revealed a significant suppressive effect of PQR309 on U87 and U251 cells. The results indicated that the viability of the cells was significantly (P<0.05) suppressed in a dose- and time-dependent manner after the cells were treated with PQR309 (0, 1, 5, 10, 50 and 100 µM) after 72 h (Fig. 1B). The colony formation rates of treated U87 and U251 cells decreased in various concentration groups compared to the control (Fig. 1C-D). According to these results, the IC50 values of PQR309 were 7.104 (95% CI, 5.6–8.5) and 11.986 (95% CI, 10.6–13.4) in U87 and U251 cells, respectively.
PQR309 induces EdU incorporation decrease in glioma cells
To further assess the inhibitory effect of PQR309 in U87 and U251 GBM cells, the DNA replication activity was assessed by EdU incorporation assay. The results revealed a significant suppression of cell proliferation in PQR309-treated U87 and U251 cells in a dose-dependent manner. The higher the concentration of PQR309, the fewer cell nuclei with thymidine analog incorporation were observed (Fig. 2A and C). The total percentage of stained nuclei in cells treated with PQR309 was lower than that in cells treated with DMSO (Fig. 2B and D). This indicated that DNA replication was inhibited by PQR309. These results, along with the viability data, confirmed the anti-proliferative effect of PQR309 on glioma cells.
Figure 2.

The nucleus was stained with Hoechst 33342 (blue) and the proliferative cells were stained red with EdU. Scale bar, 20 µm. The two cell lines were treated with various concentrations of PQR309 for 72 h. (A and C) The percentage of Edu-positive cells after being treated with PQR309. (B and D) The total percentage of stained nuclei in cells treated with PQR309 (*P<0.05).
PQR309 causes cell cycle arrest in glioma cells
Flow cytometry was performed to analyze the cell cycle distribution (Fig. 3). PQR 309 induced a significant arrest at the G1 phase of the cell cycle in U87 and U251 cells (Fig. 3A and B). The cell cycle is associated with the viability of cells. When cells were treated with various concentrations of PQR309 for 72 h, the number of EdU-positive cells decreased, while the percentage of cells in G1 phase was increased compared with that in the control groups treated with DMSO, while the percentage of cells in the S and G2 phases was decreased (Fig. 3C and D). Notably, the percentage of cells in the S and G2 phases, and the level of expression of cyclin D1 and p-AKT decreased in a dose-dependent manner in GBM cells (Fig. 4). Another PI3K-AKT inhibitor used in our previous study revealed a similar phenomenon; therefore, PQR309 may induce G1 arrest in U87 and U251 cells via the PI3K/AKT signaling pathway (27).
Figure 3.

Cell cycle arrest assay. (A and B) Cell cycle distribution analysis by flow cytometry of U87 and U251 cells after being treated with various concentrations of PQR309 (0–20 µM) for 72 h. (C and D) The percentage of the cells in G1 and S+G2 phases of U87 and U251 cells.
Figure 4.
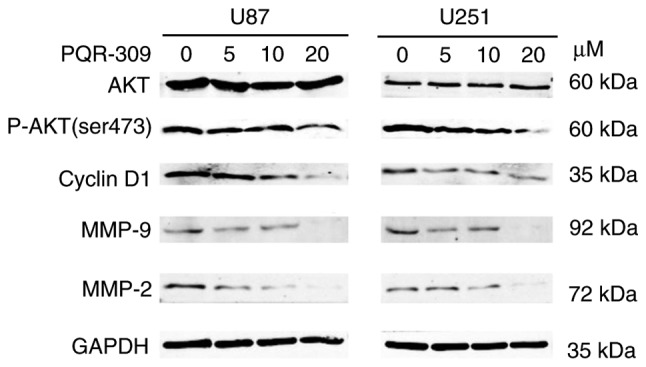
AKT, p-AKT (ser 473), cyclin D1, MMP-2 and MMP-9 expression in U87 and U251 cells after treatment with PQR309. A representative experiment is presented. Each protein was analyzed in triplicate.
PQR309 inhibits the migration and invasion of U87 and U251 cells
Wound-healing, migration and invasion assays were performed to investigate the effect of PQR309 on glioma metastasis. U87 and U251 cells were treated with various concentrations (0, 5, 10 and 20 µM) of PQR309, and the group treated only with DMSO was used as a control for comparisons. As revealed in Fig. 5, the PQR309-treated group of U87 and U251 cells exhibited less wound closure compared with that of the untreated group after 48 h. Furthermore, the migration and invasion abilities of GBM cells were inhibited after treatment with PQR309 (Fig. 6). These results were further confirmed using western blot analyses. The expression levels of MMP-9 and MMP-2 in glioma cells were gradually decreased with increasing concentrations of PQR309 (Fig. 4).
Figure 5.

Wound closure assay. (A and B) Cell lines were treated with PQR309 for 48 h and the untreated controls with 0 µM of PQR309. Scale bar, 50 µm. (C and D) The rate of wound closure of U87 and U251 cells after being treated with PQR309. *P<0.05.
Figure 6.

Migration and invasion abilities of U87 and U251 cells after treatment with PQR309. (A and C) The migration and the invasion abilities of glioblastoma cells. after being treated with PQR309 at various concentrations. (B and D) Quantification of the results of A and C. *P<0.05 and **P<0.01.
PQR309 induces apoptosis in U87 and U251 cells
PQR309 was revealed to play a marked role in the inhibition of glioma cells. The present study demonstrated its ability to kill cells. Glioma cells were treated with various concentrations (0, 1, 5, 10 and 20 µM) of PQR309 to observe its influence on cell apoptosis. The results revealed that the apoptotic cell population was increased with increasing concentrations of PQR309 (Fig. 7). The decrease in ΔΨm of the mitochondrial membrane accompanies the early stage of apoptosis. Changes in ΔΨm were assessed by JC-1 staining according to the manufacturer's instructions after treating the cells for 72 h with various concentrations of PQR309. The flow cytometric results and images are presented in Fig. 8. The results revealed that the change in the ratio of red/green fluorescence intensity indicated the loss of ΔΨm (Fig. 8). Moreover, the western blot results revealed that the expression levels of Bcl-2 (Fig. 9) and p-AKT (Fig. 4) were downregulated, in contrast to those of Bax, Bad, cleaved caspase-3 which were increased (Fig. 9) with increasing drug concentration. These results revealed that PQR309 had the ability to cause apoptosis of glioma cells through the PI3K/AKT signaling pathway in U87 and U251 cells, as revealed in Fig. 9. In addition, the results of TUNEL assay support the apoptosis effect of PQR309 on U87 and U251 cells (Fig. 10).
Figure 7.

PQR309 induces apoptosis in U87 and U251 cells stained with 7-ADD/PE. (A and B) Apoptosis of U87 and U251 cells treated with increasing concentrations of PQR309 for 72 h. (C and D) Apoptosis rates of U87 and U251 cells treated with different concentrations of PQR309. *P<0.05 and **P<0.01.
Figure 8.

ΔΨm of (A) U87 cells (treated with 5 µ) and (B) U251 cells (treated with 10 µ) after being treated with PQR309 according to the manufacturer's protocol.
Figure 9.

Expression of Bcl-2, Bcl-xL, Bax, Bad and cleaved caspase-3 and GAPDH in U87 and U251 cells after being treated with PQR309 with various concentrations (0, 5, 10, 20 µM).
Figure 10.

TUNNEL trail of U87 and U251 cells (B) after being treated with PQR309 with IC50 concentrations and (A) without any treatment, respectively.
Discussion
GBM has a poor prognosis, with a median survival time of 15 months, and only 27% of patients survive >2 years (28). Although tremendous efforts have been made, the exact pathogenesis of glioma remains unknown (14). Loss of the tumor-suppressor PTEN and activation of the RTKs, such as EGF receptor, c-Met, PDGF receptor and VEGF receptor, contribute to molecular dysfunctions associated with glioma malignancy (29). The PI3K signaling pathway is one of the most critical signaling pathways involved in the development of human GBM pathogenesis (16,30). PI3K activation initiates a signaling transduction pathway that stimulates differentiation, metabolism, migration, cell proliferation and survival (22). One of the principal downstream effectors of PI3K is mTOR, which is an important regulator of cell growth and proliferation (31,32). In summary, different cell signaling pathways, including activation of RTKs, constitutive recruitment and activation by Ras, activation of the p110a subunit (PI3KCA), loss or inactivating mutations of PTEN, G-protein-coupled receptors and chemotherapy resistance, are the result of dysregulation of the PI3K/mTOR signaling pathway, which may be a potential therapy for glioma via targeted PI3K/mTOR inhibitors, such as PQR309.
A previous study reported that PQR309 is orally available, crosses the BBB and is a PI3K/mTORC1/2 inhibitor (23). Although a phase 1 trial evaluated its clinical activity and revealed that patients benefit from PQR309 trial medication in other solid tumors, such as breast cancer and lung carcinoma (22). However, the sample size was too small to show its clinical benefit for patients. The present study revealed for the first time that this small molecule has a great clinical significance on GBM, and is a potential new anti-glioma drug.
In the present study, U87 and U251 cells were treated with PQR309 to examine the effect of PQR309 on GBM cells. PQR309 inhibited the proliferation of U87 and U251 cells in a dose- and time-dependent manner, with IC50 values of 7.104 and 11.986 µm, respectively. The concentration of PQR309 in blood 24 h later was >2 µM after oral administration in female rats, while no signs of liver toxicity were observed, accompanied with antiproliferative action in vitro and antitumor activity in vivo (23). PQR309 also promoted marked G1 arrest and cell apoptosis in a dose-dependent manner, and cell migration and invasion abilities were suppressed too. The wound-healing, cell migration and cell invasion assays confirmed that the migration and invasion of human glioma cell lines were reduced by PQR309. These results were supported by the expression of p-AKT, AKT, Bcl-2, Bcl-xL, Bax, Bad, cleaved caspase-3, MMP-2, MMP-9 and cyclin D1, as determined by western blotting. There are various signaling pathways involved in the anti-proliferation effect of PI3K/mTOR inhibitors, including the NF-κB, ERK/MAPK and PI3K/AKT signaling pathways, which can affect cell survival, proliferation and apoptosis (33–35). There are numerous molecules with antiproliferative activity against GBM which exhibit similar effects to PQR309; however, the majority of them may not have the ability to cross the BBB or may have a significant toxicity in vitro and in vivo at the working concentration. Thus, PQR309 should be investigated in clinical trials.
Although it was evident that PQR309 suppressed the proliferation and induced the apoptosis of GBM U87 and U251 cells in the present study, several limitations exist, including the lack of animal experiments to assess whether PQR309 is orally available and crosses the BBB in a GBM model. Despite exhibiting favorable pharmacokinetic parameters in mice, rats and dogs, a trial on patients with glioma has not been performed yet; thus, a larger number of clinical trials on CNS metastasis and and patients with GBM are required. To the best of our knowledge, the present study is the first to report that PQR309 suppresses cell proliferation and invasion, and induces apoptosis and G1 cell cycle arrest in glioma cells. The present results strongly support further clinical investigation of PQR309 in GBM (NCT02850744).
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81572489).
Availability of data and materials
The datasets used during the present study are available from the corresponding author upon reasonable request.
Authors' contributions
KY and QXC conceived and designed the study. KY conducted the experiments. KY, XJT, FFX, LG, QS, YQT, XD, BHL and JHL performed the statistical analysis. KY wrote the manuscript. XJT, YQT, XD, BHL and QXC reviewed and edited the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Zanders ED, Svensson F, Bailey DS. Therapy for glioblastoma: Is it working? Drug Discov Today. 2019;24:1193–1201. doi: 10.1016/j.drudis.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318:2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 4.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;15(Suppl 2):ii1–ii56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS. Epidemiology of gliomas. Cancer Treat Res. 2015;163:1–14. doi: 10.1007/978-3-319-12048-5_1. [DOI] [PubMed] [Google Scholar]
- 6.Santosh V, Sravya P, Gupta T, Muzumdar D, Chacko G, Suri V, Epari S, Balasubramaniam A, Radotra BD, Chatterjee S, et al. ISNO consensus guidelines for practical adaptation of the WHO 2016 classification of adult diffuse gliomas. Neurol India. 2019;67:173–182. doi: 10.4103/0028-3886.253572. [DOI] [PubMed] [Google Scholar]
- 7.Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019;15:405–417. doi: 10.1038/s41582-019-0220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 9.Verdecchia A, De Angelis G, Capocaccia R. Estimation and projections of cancer prevalence from cancer registry data. Stat Med. 2002;21:3511–3526. doi: 10.1002/sim.1304. [DOI] [PubMed] [Google Scholar]
- 10.Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392:432–446. doi: 10.1016/S0140-6736(18)30990-5. [DOI] [PubMed] [Google Scholar]
- 11.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 12.Linz U. Commentary on effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466) Cancer. 2010;116:1844–1846. doi: 10.1002/cncr.24950. [DOI] [PubMed] [Google Scholar]
- 13.Anjum K, Shagufta BI, Abbas SQ, Patel S, Khan I, Shah SAA, Akhter N, Hassan SSU. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed Pharmacother. 2017;92:681–689. doi: 10.1016/j.biopha.2017.05.125. [DOI] [PubMed] [Google Scholar]
- 14.Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129:829–848. doi: 10.1007/s00401-015-1432-1. [DOI] [PubMed] [Google Scholar]
- 15.Park AK, Kim P, Ballester LY, Esquenazi Y, Zhao Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019;21:59–70. doi: 10.1093/neuonc/noy120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wojtas B, Gielniewski B, Wojnicki K, Maleszewska M, Mondal SS, Nauman P, Grajkowska W, Glass R, Schüller U, Herold-Mende C, Kaminska B. Gliosarcoma Is driven by alterations in PI3K/AKT, RAS/MAPK pathways and characterized by collagen gene expression signature. Cancers (Basel) 2019;11(pii):E284. doi: 10.3390/cancers11030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Yang JA, Liu BH, Liao JM, Yuan FE, Tan YQ, Chen QX. TGX-221 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol Rep. 2017;38:2836–2842. doi: 10.3892/or.2017.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu PF, Yang JA, Liu JH, Yang X, Liao JM, Yuan FE, Liu BH, Chen QX. PI3Kβ inhibitor AZD6482 exerts antiproliferative activity and induces apoptosis in human glioblastoma cells. Oncol Rep. 2019;41:125–132. doi: 10.3892/or.2018.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 20.Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–128. doi: 10.1016/j.gene.2019.02.076. [DOI] [PubMed] [Google Scholar]
- 21.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wicki A, Brown N, Xyrafas A, Bize V, Hawle H, Berardi S, Cmiljanović N, Cmiljanović V, Stumm M, Dimitrijević S, et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13) Eur J Cancer. 2018;96:6–16. doi: 10.1016/j.ejca.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Beaufils F, Cmiljanovic N, Cmiljanovic V, Bohnacker T, Melone A, Marone R, Jackson E, Zhang X, Sele A, Borsari C, et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a potent, brain-penetrant, orally bioavailable, pan-class I PI3K/mTOR inhibitor as clinical candidate in oncology. J Med Chem. 2017;60:7524–7538. doi: 10.1021/acs.jmedchem.7b00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Mei L, Zhou L, Shen X, Guo C, Zheng Y, Zhu H, Zhu Y, Huang L. PTEN restoration and PIK3CB knockdown synergistically suppress glioblastoma growth in vitro and in xenografts. J Neurooncol. 2011;104:155–167. doi: 10.1007/s11060-010-0492-2. [DOI] [PubMed] [Google Scholar]
- 25.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarantelli C, Gaudio E, Arribas AJ, Kwee I, Hillmann P, Rinaldi A, Cascione L, Spriano F, Bernasconi E, Guidetti F, et al. PQR309 is a novel dual PI3K/mTOR inhibitor with preclinical antitumor activity in lymphomas as a single agent and in combination therapy. Clin Cancer Res. 2018;24:120–129. doi: 10.1158/1078-0432.CCR-17-1041. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Li XM, Bai Z, Chi BX, Wei Y, Chen X. Curcumol induces cell cycle arrest in colon cancer cells via reactive oxygen species and Akt/ GSK3β/cyclin D1 pathway. J Ethnopharmacol. 2018;210:1–9. doi: 10.1016/j.jep.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 28.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: A clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 29.Teodorczyk M, Martin-Villalba A. Sensing invasion: Cell surface receptors driving spreading of glioblastoma. J Cell Physiol. 2010;222:1–10. doi: 10.1002/jcp.21901. [DOI] [PubMed] [Google Scholar]
- 30.Vastrad B, Vastrad C, Godavarthi A, Chandrashekar R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med Oncol. 2017;34:182. doi: 10.1007/s12032-017-1043-x. [DOI] [PubMed] [Google Scholar]
- 31.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of AKT/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 32.Huang J, Manning BD. A complex interplay between AKT, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Xu Y, Zhou Q, Chen M, Zhang Y, Liang H, Zhao J, Zhong W, Wang M. PI3K in cancer: Its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018;14:665–674. doi: 10.2217/fon-2017-0588. [DOI] [PubMed] [Google Scholar]
- 34.Hanker AB, Kaklamani V, Arteaga CL. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019;9:482–491. doi: 10.1158/2159-8290.CD-18-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yap TA, Bjerke L, Clarke PA, Workman P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr Opin Pharmacol. 2015;23:98–107. doi: 10.1016/j.coph.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used during the present study are available from the corresponding author upon reasonable request.