

Abstract
Introduction:
Advanced melanoma has recently been transformed by the advent of immune checkpoint inhibitors. These agents have altered the prognosis of this disease from a median survival of <1 year to recent data showing a 5-year survival surpassing 50%. Combination regimens combining PD-1 and CTLA-4 blockade are associated with superior response and progression-free survival at the cost of increased toxicities.
Areas covered:
In this review, we discuss the clinical and investigational utility of predictive biomarkers of immune checkpoint inhibitor treatment in melanoma. Topics include tumor-intrinsic biomarkers, tumor microenvironment biomarkers, and host characteristic biomarkers. We also discuss biomarkers of immune-related adverse events and how biomarkers may be used to personalize selection of immune checkpoint inhibition in patients.
Expert opinion:
The decisions confronting oncologists when choosing treatment is increasing in complexity. Biomarkers may aid in these treatment decisions and are growing in importance.
Keywords: Melanoma, Immunotherapy, PD-1, Biomarkers, CTLA-4
1. INTRODUCTION
Melanoma, the most deadly form of skin cancer, is a major health concern in the United States, with approximately 90,000 new cases diagnosed annually. It represents the fifth- and sixth-most common cancer diagnosis in men and women, respectively. While early stage (I-II) disease has a 90+% five-year survival rate with surgical excision alone, disease which has spread to lymph nodes or distant organs requires systemic treatment and portends a much poorer five-year survival rate. Patients with distant sites of disease at the time of diagnosis historically expected a <20% five-year survival [1, 2]. Until 2017, the incidence of melanoma deaths had been increasing [3].
In the past decade, the treatment of metastatic melanoma has changed dramatically [4]. Prior to 2011, the mainstay of treatment for metastatic disease was high-dose Interleukin-2, a therapy associated with significant and often course-limiting toxicity due to non-specific immune stimulation (including multiorgan dysfunction and systemic inflammatory response syndrome). Patients treated with IL-2 could expect a 20% response rate, but a small subset of these patients (5–8%) went on to have complete responses sustained for decades, representing one of the few curative treatment modalities in metastatic solid tumors [5]. Within the past decade, there has been an explosion of treatment options due to the development of targeted therapies and immune checkpoint inhibitors [6].
Immune checkpoint inhibitors are a novel class of therapeutics that inhibit cytotoxic T lymphocyte antigen-4 (CTLA-4) or programmed death-1 receptor/ligand (PD-1/PD-L1) to potentiate anti-tumor immune response. Members of these targeted drug classes have been used alone and in combination with impressive responses. Currently, there are three immune checkpoint inhibitors (ICIs) FDA-approved for use in patients with metastatic melanoma: ipilimumab, an inhibitor of CTLA-4, and pembrolizumab and nivolumab, both inhibitors of PD-1. These therapies have demonstrated prolonged progression-free survival and overall survival, with response rates as high as 45% for patients treated with anti-PD-1 monotherapy, and 60% treated with combination CTLA-4/PD-1 blockade [7]. Ipilimumab monotherapy was first demonstrated to improve overall survival from 6.4 months in gp100-treated patients to 10.1 months in ipilimumab-treated patients; overall response rates for ipilimumab monotherapy range from 10–20% [8, 9]. This led to the FDA approval of ipilimumab for the treatment of melanoma in 2011. Nivolumab is a humanized IgG4 monoclonal PD-1 antibody; multiple phase II-III studies have demonstrated an objective response rate of 28–40% with nivolumab monotherapy [10–12]. Similarly, pembrolizumab, also a humanized IgG4 monoclonal PD-1 antibody was FDA approved in 2014 after demonstrating a 38% objective response rate in advanced melanoma patients, with improved toxicity profile when compared to ipilimumab [13–15].
2. CLINICAL AND INVESTIGATIONAL BIOMARKER ULTILITY
In an age of precision cancer medicine, the need for validated clinical biomarkers cannot be overstated. This review explores the current state of biomarkers for melanoma in the setting of both clinical and investigational immune checkpoint inhibitor use. Biomarkers can be classified as either prognostic or predictive; a prognostic biomarker provides treatment-agnostic information about a patient’s overall cancer outcome, while a predictive biomarker provides information about the effect of a particular therapeutic intervention [16]. First and foremost, biomarkers enable clinicians to align the most optimal treatment with a specific patient. We currently rely on front-line testing in multiple tumor types to ensure optimal therapy choice, such as targeted sequencing of NSCLC to identify EGFR-mutant tumors, portending significant benefit from EGFR-directed tyrosine kinase inhibitors, or BRAF mutations to inform use of BRAF and MEK inhibitors in melanoma. In addition to stratifying individual patients for specific interventional benefits, biomarkers also augment research efforts. Biomarkers have allowed novel approaches to clinical trials, such as “basket” trials, which test a single intervention in a broad range of tumor histologies all bearing a single positive biomarker, and “umbrella” trials, which test multiple interventions matched with multiple biomarkers within a single tumor histology [17]. In addition, understanding biomarkers can provide insight into the underlying biology of a tumor and facilitate hypothesis generation to both optimize response and overcome innate and/or acquired resistance to a specific biomarker-associated therapy.
In current clinical practice, first-line treatment options for patients presenting with metastatic melanoma include anti-PD-1 monotherapy, combined checkpoint blockade with anti-PD-1/anti-CTLA4 antibodies, and combination targeted therapy in patients whose tumors harbor BRAF V600E mutations. PD-L1 testing by immunohistochemistry is described as controversial at this time, emphasizing the need for more research into this and other biomarkers of melanoma [18]. Biomarkers for ICI response in melanoma can be divided into three subgroups: tumor intrinsic, tumor microenvironment, host factors. This review will evaluate these potential biomarkers for ICI in advanced melanoma.
3. TUMOR-INTRINSIC BIOMARKERS
3.1. Tumor Mutational Burden
Tumor intrinsic biomarkers, including tumor mutational burden (TMB) and tumor neoantigen profiling, have been fairly well-studied across a wide array of solid tumor histologies. Tumor mutational burden refers to the ratio of somatic mutation rate in tumor DNA as compared to matched normal germline DNA. While research studies typically use whole-exome sequencing data to determine total mutation load, in clinical practice, we typically rely on commercially-available targeted genomic sequencing assays (such as FoundationOne, Tempus, MSK-IMPACT). These assays identify somatic exonic mutations in a predefined subset of cancer-related genes (typically 300–600 genes) by comparison of tumor-derived and matched germline DNA. Targeted sequencing assays, which represent about 2–3% of the total coding exome, have been validated to highly correlate with total mutational burden as measured by whole-exome sequencing approaches [19].
At this time, it remains speculative why high TMB correlates with response to immune checkpoint inhibitors. The dominant hypothesis is that increasing numbers of mutations correlate with increased neoantigen generation, which increases the likelihood of generating immunogenic peptides that can be targeted by T cells (e.g. more mutations = more likelihood of generating a “foreign appearing” protein). The evidence of this hypothesis is as follows. In general, tumors with higher TMB are also, in general, the ones which respond to checkpoint inhibitors [20]. Within tumor types, more evidence exists. Using a broad cohort of advanced cancer patients treated with immune checkpoint blockade whose tumors were profiled using a targeted sequencing assay (MSK-IMPACT), Samstein et al defined TMB percentile subgroups ranging from the top 10–50% which conferred improved survival within each specific tumor type. This study included bladder, breast, colorectal, esophagogastric, glioma, head and neck, melanoma, non-small cell lung cancer, and renal cell carcinoma and represents the largest such study to date with 1,662 ICI-treated patients included. Importantly, this study also evaluated a similar group of 5,371 advanced cancer patients with targeted sequencing data who were not treated with immune checkpoint blockade; this analysis found no survival benefit in tumors with subgroup-defined high TMB, indicating that the survival benefit depends of high-TMB tumors response to immune checkpoint blockade therapy [21]. Because primary tumor usually requires invasive biopsy to obtain, there have also been efforts to mine cell-free DNA (cfDNA) accessible from a simple peripheral blood draw to obtain this information. Georgiadis et al recently developed a targeted plasma panel of five mutations corresponding to a mutation rate of 51 mutations/Mgbp sequenced that can serve as a marker of exceptionally high TMB related to mismatch repair deficiencies. While this was validated in a small panel of mostly advanced gastrointestinal malignancies (and no melanomas), such targeted sequencing of cfDNA may certainly be translatable to melanoma in the future, though the specific algorithm may need refinement given the stronger association of MMR deficiency with GI malignancy than melanoma [22].
Using a similar tumor-agnostic approach, Miao at el sought to further refine the predictive power of TMB by combining TMB with other genetic data, including clonal mutations and neoantigens, mutations and copy number alterations, and affecting particular genes and signaling pathways and overall tumor aneuploidy. A subgroup analysis of melanoma tumors was stratified by mutational signatures of UV light (S7), prior exposure to alkylating agents (S11), and other signatures not clearly associated with environmental exposures (S1 and S5). After stratifying, no significant difference in mutational burden was observed between responders and non-responders to immune checkpoint blockade. Given the very high tumor burden associated with melanoma generally, the authors hypothesize that TMB in melanoma may simply be a surrogate marker of underlying biology related to pathway alterations that promote tumor immunogenicity and somatic mutations [23]. NCCN guidelines for treatment of cutaneous melanoma describe TMB testing as investigational at this time. A number of other studies have suggested a correlation between TMB and response in various tumor types [24–29].
Additionally, a number of pre-clinical and clinical studies have suggested that particular mutations may influence immune responses through immune cell exclusion, altered metabolism, or other immune cell-tumor cell interactions. These include mutations in NRAS [30], CTNNB1 pathway [31], MYC [32], PTEN [33], and LKB1 [34]. However, these studies have, in general, not been verified in larger cohorts and are not used to guide therapy at this time.
3.2. Tumor Neoantigen Profiles
Tumor neoantigen profiles refer to groups of tumor-specific mutant epitopes/peptides associated with T-cell reactivity. This was first examined in melanoma in in 2013, when van Rooij et al published a case report of tumor exome analysis of a melanoma patient with exceptional response to ipilimumab. In this patient, two neoantigens (mutant epitopes of the ataxia telangiectasia and RAD3 related gene product) were found to be enriched after treatment [35]. In 2014, Snyder et al compared two cohorts of malignant melanoma patients, one of which experienced long-term clinical benefit from treatment with CTLA-4 blockade (ipilimumab or tremelimumab) and one of which minimal or no clinical benefit. Whole exome sequence conducted on the two groups identified that mutation load was associated with response but not sufficient to predict benefit. Further analysis of whole exome sequencing data identified a somatic neoantigen-derived tetrapeptide signature that predicted a good response to CTLA-4 blockade that was validated to have strong correlation with survival. Interestingly, analysis of the predictive neoepitope group identified a close resemblance of epitopes predicting clinical benefit from CTLA-4 inhibition to epitopes seen on viral pathogens such as human cytomegalovirus [25]. However, larger studies failed to recapitulate this finding or identify any recurrent neoantigen patterns with predictive power [36]. In addition to specific neoantigen profiles, microsatellite instability has been suggested as a surrogate marker for general high neoantigen load based on successful predictive power in colorectal cancer. Le et al validated the predictive power of a commercially available assay for mismatch repair deficiency in an expanded panel of 12 solid tumor types; however, this analysis did not include melanoma [29].
An emerging approach to understanding response to checkpoint blockade relies on insights into the immune metabolism. Harel et al recently performed proteomic profiling of advanced melanoma clinical samples, performing high-resolution mass spectrometry on ~4,500 proteins per sample. Statistical analysis led to the finding that ICI-sensitive tumors had higher oxidative phosphorylation and lipid metabolism as compared to ICI-resistant tumors. Molecular studies demonstrated that increased lipid metabolism increased antigen presentation, thus providing a mechanistic link to ICI sensitivity [37].
3.3. Tumor size
In addition to tumor genetic profiling, a more clinically measurable surrogate of baseline tumor size has been validated as a predictive biomarker in melanoma patients Joseph et al evaluated a cohort of 583 melanoma patients with measurable disease. Patients with baseline tumor size below the median of 10.2cm was associated with higher overall response rate and overall survival upon treatment with anti-PD-1 antibody (pembrolizumab). A multivariate analysis of a 459-patients subset also demonstrated that patients with lower baseline tumor size and high PD-L1 expression (as defined by immunohistochemical staining >1% of tumor/tumor-associated lymphocytes) was independently associated with higher overall response rate and longer OS [38]. While the authors acknowledge that their analysis cannot differentiate between the predictive versus prognostic effect of this finding, it is certainly reasonable to hypothesize that lower baseline tumor size corresponds to increased immune response in these smaller tumors [39]. However, they did not find a difference in PD-L1 positivity between low and high baseline tumor size cohorts. This may speak more to sampling variability and the dynamic nature of PD-L1 as a marker.
4. TUMOR MICROENVIRONMENT BIOMARKERS
There has been much interest in determining how the tumor microenvornment, particularly the immunomicroenvironment, might affect checkpoint blockade treatment response.
4.1. PD-L1
Given the mechanism of anti-PD-1 antibodies, PD-L1 seems to be the most “obvious” choice as a predictive biomarkers for these therapies. Early clinical trials of PD-1 inhibitors have included assay of baseline PD-L1 status across all tumor types, including melanoma, non-small cell lung cancer, renal cell carcinoma, prostate cancer, and colorectal cancer. In current clinical practice, PD-L1 status is readily obtainable with immunohistochemistry, but the threshold for PD-L1 positivity is poorly defined, ranging from 1–50% in various studies. This is further complicated by variance among epitope specificity of available antibodies for IHC, including numerous proprietary companion diagnostic assays commonly used in the clinical setting [40]. The biology of PD-L1 itself also makes this a challenging biomarker: it is regulated by numerous pathways, including many of which are perturbed in malignant transformation (MAPK, PI3K, Akt, HIF1, STAT3, NFkB), it is transient and subject to significant sampling error (even within a single patient), and it can be expressed by immune cells in the microenvironment [41]. Most practically, we know that patients whose tumors do not express PD-L1 on IHC staining can still respond to immune checkpoint blockade. The CHECKMATE-067 trial demonstrated response lower but substantial response rates among patients treated with mono- or combination checkpoint blockade with negative PD-L1 expression [42]. At this time, PD-L1 is used to help select patients for ICI in certain cancer types, including NSCLC, triple negative breast cancer, head and neck squamous cell carcinoma, and others, but has limited value in other cancers such as melanoma and renal cell carcinoma.
4.2. Tumor Infiltrating Lymphocytes
Multiple studies have demonstrated that response to PD-1 inhibition in melanoma can be predicted by higher number and spatial distribution of T cells within the invasive border of a tumor. Defined as “hot” or immunogenic, such T cells infiltrates tend to show higher levels of PD-1+ and CD8+ as well as increased T-cell receptor clonality [43–45]. Such “hot” tumors demonstrate increased response to immune checkpoint blockade, and the topic of how “cold” tumor can be influenced to become “hot” is a major area of research in the field.
Many groups have worked to further characterize the molecular underpinnings of “hot” tumors. For example, T cell gene expression profiles have been analyzed by Ayres et al; this study identified an interferon-Ƴ signature containing genes related to antigen presentation, chemokine expression, cytotoxic activity and adaptive immune resistance which were necessary but not always sufficient for clinical benefit [46]; a clinical-grade assay is currently in development. Sade-Feldmen et al have used single-cell RNA of immune cells of 48 different melanoma tumor samples to stratified CD8+ T cells into two distinct groups associated with tumor regression or progression. Their analysis identified a key transcription factor, TCF7, as a marker of tumor response to PD-1 inhibition independent of total CD8+ T cell infiltration. This transcription factor is part of the Wnt/β-catenin signaling pathway is crucial for differentiation, self-renewal, and persistence of CD8+ T cells. Avogadri et al previously described the upregulation of non-cytotoxic T-cell (CD4+Foxp3-PD-1+, termed 4PD1hi) upon treatment with CTLA4 inhibition and postulated that this phenotype, which is very similar to that of follicular helper T cells, may be associated with tumor progression [47]. More recently, their group elucidated that combination checkpoint blockade with CTLA-4 and PD-1 inhibition actually has an opposing modulatory effect on this phenotype [48]. From this, it is reasonable to hypothesize that assaying the 4PD1hi status of a tumor either before ICI treatment or upon progression during mono- or combined checkpoint blockade may be a useful approach to guide choice of ICI treatment. Prognostic studies are warranted to validate the 4PD1hi signature in clinical specimens.
4.3. MHC-I/II
Another important biomarker of response to immune checkpoint blockade in melanoma are the major histocompatibility complex I and II (MHC-I/II). These protein complexes are responsible for tumor antigen presentation for recognition by T cells (MHC-I:CD8+ and MHC-II:CD4+) and thus represent an essential step of tumor targeting by the immune system. MHC-II expression has been positively correlated with improved response to PD-1 blockade, as well as prolonged overall survival and response rate, which was confirmed in a larger cohort in the context of a multiplexed panel integrating MHC-II with PD-L1 immunofluorescence [49]. Unsurprisingly, higher MHC-II expression in this study was also correlated with increased CD4+ and CD8+ T cell infiltrate in the immune microenvironment [50]. Further exploration of the role of MHC-II has elucidated a mechanism for this: tumors with high levels of MHC-II positivity are more dependent of both PD-1 and Lag-3, a known MHC-II inhibitory receptor. Upon development of acquired resistance to PD-1 therapy, Lag-3 was found to be upregulated in MHC-II positive tumors [51]. More recently, a study by Rodig et al evaluated MHC-I and MHC-II expression in pretreatment biopsy samples among patients enrolled in CHECKMATE-064. This analysis demonstrated that patients with MHC-II expression >1% correlated with increased likelihood of stable disease, partial or complete response as compared with progressive disease upon treatment with PD-1 inhibition (nivolumab). MHC-II expression did not predict response to CTLA-4 inhibition with ipilumumab. However, lower levels of MHC-I expression (defined as <30%) did correlate with increased likelihood of progressive disease upon treatment with ipilimumab with an impressive negative predictive value of 100% [52]. Using a panel of over 1500 patient samples comprised of ~1/3 melanoma tumors, Chowell et al determined that maximal heterozygosity at MHC-I loci improved overall survival upon treatment with checkpoint blockade. In a melanoma-specific cohort, they identified two HLA supertypes associated with CTLA-4 inhibition treatment outcome: the HLA-B44 supertype was associated with prolonged survival, while the HLA-B62 supertype was associated with decreased survival. Based on molecular dynamic simulations, this negative association was explored and is thought to be related to the HLA-B*15:01 allele, though mutations in this gene were not found to change overall survival [53].
4.4. Immune Metabolism
An emerging approach to understanding response to checkpoint blockade relies on insights into the immune metabolism. Harel et al recently performed proteomic profiling of advanced melanoma clinical samples, performing high-resolution mass spectrometry on ~4,500 proteins per sample. Statistical analysis led to the finding that ICI-sensitive tumors had higher oxidative phosphorylation and lipid metabolism as compared to ICI-resistant tumors. Molecular studies demonstrated that increased lipid metabolism increased antigen presentation, thus providing a mechanistic link to ICI sensitivity [37].
5. HOST BIOMARKERS
5.1. Gut Microbiome
Higher gastrointestinal microbiodiversity is associated with positive response to ICI [54]. This has been demonstrated in a panel of metastatic melanoma patients undergoing treatment with PD-1 inhibition, where both higher alpha diversity (defined as within-sample diversity) and relative abundance of bacteria of the Ruminococcaceae family were seen in responders as compared to non-responders to PD-1 inhibition. The authors hypothesize that this effect is due to enhanced systemic and antitumor immune responses mediated by increased antigen presentation and improved effector T cell function. As proof-of-concept, they conducted fecal transplant from stool samples of either ICI responder or non-responder patients in mice and showed that animals receiving fecal transplant from responder patients had improved response to anti-PD-1 treatment after xenografting with BRAFV600E melanoma cell line [55]. This finding was recapitulated by Matson et al; evaluation of stool samples from metastatic melanoma patients prior to ICI treatment identified bacterial species Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium to be enriched in responders as compared to non-responders [56].
Given the association with microbiome diversity, it is unsurprising that patients who consume greater dietary fiber and thus have increase diversity of gut flora have better responses to ICI. Spencer et al showed that patients who consumed a high-fiber diet (as assessed by self-report) were five times more likely to respond to anti-PD-1 treatment as compared to patients who consume low-fiber diets (AACR abstract 2019 2838/24). This is also supported by the observation that patients undergoing treatment with PD-1/PD-L1 inhibition had worse outcomes by nearly double if they had prior antibiotic treatment (defined as antibiotic use >30 days prior to initiation of ICI); this finding was independent of antibiotic class. No association was found with concurrent ICI and antibiotic treatment was found. The authors hypothesize that this observation suggests a “priming” effect of the gut microbiome in response to ICI that is altered with prior but not concurrent antibiotic use [57].
5.2. Stress
Murine models seeking to elucidate the effect of stress on treatment response identified that psychologic distress caused elevated plasma corticosterone and upregulated the expression of glucocorticoid-inducible factor Tsc22d3, which ultimately blocked interferon-gamma related T cell activation. Mice were xenografted with a NSCLC murine solid tumor cell line, then treated with anti-PD-1 monoclonal antibody and subjected to stress via social defeat (controlled encounter with an aggressor mouse). As compared to mice that did not undergo such stress, the socially defeated mice showed less response to PD-1 inhibition [58].
5.3. Peripheral Blood Markers
Numerous markers obtained on routine peripheral blood checks have also been evaluated. Elevated LDH, neutrophil count, and neutrophil/lymphocyte ratio correlate with low rates of response to either single-agent PD-1 inhibition or combination checkpoint blockade [59, 60]. However, this may be more representative of the general prognostic nature of these biomarkers rather than any predicative power specific to ICI. Both low absolute lymphocyte count and lack of increased absolute lymphocyte count upon treatment correlate with poor response to ipilimumab monotherapy [61].
6. BIOMARKERS OF IMMUNE MEDIATED ADVERSE EVENTS
While application of ICI has dramatically improved patient outcomes in metastatic melanoma, it has also brought a new set of treatment-related toxicities, termed immune-related adverse events (irAEs). These toxicities most commonly manifest in as rash (dermatitis), hepatitis and colitis, but nearly every organ system in the human body is known to at risk for irAE. Some rare toxicities, including myocarditis and neuritis, can be fulminant and lethal [62, 63]. It is estimated that irAE of grade 3 or higher leads to the discontinuation of ICI in 4–45% of patients, with lower rates of toxicity-related discontinuation observed in single-agent anti-PD-1 treatment and higher rates seen in dual CTLA4/PD-1 blockade [64].
6.1. B Lymphocytes
A 2014 genetic analysis of four families with heterozygous CTLA4 mutations demonstrated T cell and B cell dysregulation associated with increased risk of autoimmunity (Kuehn et al). While the effects of T-cell mediated autoimmunity has well-studied in the context of irAE, B-cell changes are also implicated. Das et al evaluated a cohort of advanced melanoma patients receiving single-agent or combination checkpoint blockade and observed changes in absolute B cell counts before and after combination therapy. Within their cohort, the severity of B cell decline after treatment was correlated to both time of toxicity onset and grade of maximal toxicity (i.e. greater decrease in B cells correlated with more toxicities). They have developed a profile of B cell change after combination checkpoint blockade which includes percentage change of B cell number after therapy (≤70% baseline) with a two-fold or greater increase in either CD21lo B cells or plasmablasts. The median time by which B cell changes preceded clinical irAE was 3 weeks [65]. Unlike T cell changes, B cell changes correlated with irAE only and not tumor response to treatment.
6.2. Cytokines
Because of their importance in immune cell activation and recruitment, numerous cytokines have been evaluated for their ability to predict irAE. Fujimura et al evaluated levels of tumor-associated macrophage associated chemokine CXCR5 and soluble CD163 in a cohort of 46 Japanese patients with metastatic melanoma treated with anti-PD-1 inhibitor nivolumab; about half (45%) of these patient developed irAE, consistent with larger trials. These proteins were chosen for study given their validated association with other autoimmune disease such as rheumatoid arthritis, atherosclerosis, pemphigus vulgaris, and bullous pemphigoid. Serum analysis in melanoma patients of CXCL5 and sCD163 prior to nivolumab treatment and at day 42 showed a significant increase in sCD163 levels in patients with irAE as compared to those without adverse events (sensitivity 72%, specificity 75%). Similar studies have also correlated baseline serum IL-17 levels with development of grade 3 colitis in melanoma patients treated with ipilimumab [66] and baseline IL-6 and IL-8 levels with generalized irAE [67]. More recently, Lim et al developed and validated a cytokine toxicity score (CYTOX) integrating expression data of 11 cytokines as baseline and during early treatment (within first 14 days) to predict irAE in patients treated with combination PD-1/CTLA-4 checkpoint blockade [68].
6.3. Autoantibodies
Specific autoantibodies have positively associated with organ-specific manifestations of irAE. For example, higher baseline levels of antithyroglobulin antibody was associated with increased likelihood of nivolumab-induced thyroid dysfunction [69]; baseline presence of diabetes autoantibodies was associated with earlier presentation of irAE after treatment initiation in patients treated with PD-1 inhibitors [70]. An interesting area of future research will be understanding paraneoplastic autoantibody profiles in patients treated with ICI, particularly given the recent approval of atezolizumab in small cell lung cancer, where as many as 10% of patients may develop paraneoplastic syndrome.
7. PERSONALIZATION OF IMMUNE CHECKPOINT INHIBITOR SELECTION
As a primary goal in the implementation of biomarkers is to select the best possible treatment for an individual patient, we consider three clinical scenarios of advanced melanoma in which such biomarkers stand to improve patient care.
7.1. Monotherapy versus Combination Checkpoint Blockade
Although seemingly the most accessible decision point, developing reliable markers to guide this decision has been challenging. Initial reports from the phase III study of nivolumab vs. nivolumab + ipilimumab suggested that patients with high PD-L1 experienced equivalent outcomes with both monotherapy and combination therapy. However, with longer follow up, and with incorporating multiple relevant endpoints (PFS, OS, response rate), using PD-L1 as a stratifying factor has at least largely fallen out of favor. Other molecular markers (TMB, MHC-II expression) may be useful but have not been thoroughly studied in combination treated patients compared with single agent; thus the relative benefit remains unclear. At this time, many clinicians use a composite of clinical biomarkers, including high LDH, increased tumor bulk, non-cutaneous (vs. cutaneous) primaries, and presence of brain or liver metastases (as well as good performance status) as factors pushing toward combination therapy.
7.2. Targeted Therapy versus Immune Checkpoint Inhibition
At least two biomarkers can help make this decision: BRAF and KIT mutations, which confer sensitivity to BRAF/MEK and KIT inhibitors, respectively. However, the choice between first-line targeted vs. ICI remains an area of great controversy. Currently, a phase III study (DREAMSEQ; ) is comparing combination ipilimumab and nivolumab with dabrafenib and trametinib (with crossover to the other arm at progression) in patients with untreated BRAF V600 mutant melanoma. Many clinicians favor ICI front-line in most patients without rapidly progressive or symptomatic disease, given the durable responses observed. However, more recent data suggests that patients who have excellent clinical outcomes to BRAF/MEK inhibitor therapy are typically those with low-volume disease [71]. Moreover, we have shown that patients who have poor outcomes to front-line ICI also tend to do poorly with second line BRAF/MEK inhibitor therapy [72]. Thus, populations who do well with both immune and targeted therapy likely at least partially overlap.
7.3. Adjuvant Treatment
Despite the dearth of reliable data in the metastatic setting, the adjuvant setting remains even more poorly understood from a biomarker standpoint. Very little molecular work has been done to understand correlates of outcomes in patients receiving adjuvant anti-PD-1 therapy, although one might expect that these would be overlapping with markers of benefit in the metastatic setting. Clinically, anti-PD-1 therapy was associated with benefit in essentially all subgroup analyses (including by PD-L1 levels) in the phase III studies leading to their approval. Similarly, it remains unclear how to determine whether adjuvant anti-PD-1 or BRAF/MEK inhibitor should be used in resected BRAF mutant stage III disease.
8. EXPERT OPINION
Advanced melanoma has been transformed by the advent of effective immunotherapy, but some patients fail to respond. The lack of response in some patients, coupled with the toxicities of combination immunotherapy, suggests that biomarkers will play a large role in tailoring therapy moving forward. Biomarkers could stratify patients into those that will respond to single agent anti-PD-1, those that require combination ipilimumab and nivolumab, and those that need alternative strategies (either immune or non-immune therapies).
To date, in our opinion, the most promising biomarkers for melanoma have been tumor-based, and have primarily related to those focusing on the tumor and microenvironment. First, PD-L1, which has been very useful in many tumor types to select anti-PD-1 or anti-PD-L1 therapy, is only weakly (but consistently) correlated with response in advanced melanoma [6]. While not likely to have great utility on its own, PD-L1 expression could potentially be integrated with other biomarkers to help predict response more broadly. Second, tumor mutational burden has been correlated with outcomes to anti-PD-1 monotherapy, although the relation with combination therapy remains to be seen, and no prospective validation has been performed in advanced melanoma [16]. The drawbacks to mutation burden are somewhat obvious, in that it requires an expensive test with longer turnaround time. However, as testing becomes cheaper and more efficient, and with the need to genomically profile tumors for actionable targets, this may be a feasible approach as well. Third, the expression of MHC-II on tumor cells has now been validated in several studies, both with our group (with anti-PD-1 monotherapy [42], in one case integrated with PD-L1 expression [65]) and as a correlative analysis of a prospective study of nivolumab and ipilimumab given sequentially [44]. MHC-II has also correlated with response in Hodgkin lymphoma [66], suggesting that it may have value in other cancer types. Many other approaches, including those based on flow cytometry, gene expression profiling, and multiplexed immunofluorescence have also demonstrated predictive value, although the clear “winner” remains to be seen [67].
Microbiome and blood-based biomarkers also hold promise, although the predictive capacity of these modalities remains somewhat less clear. Microbiome based analysis is particularly intriguing, given the potential of modulating microbiome composition with therapeutic interventions. At this time, the distinct flora associated with response in different studies, and the lack of prospective validation precludes specific microbiome interventions (e.g., probiotics) outside the context of a clinical trial.
One other hurdle for biomarker development is the need to supersede clinical factors, many of which have some predictive value on their own. Tumor bulk, LDH, metastatic stage, performance status, and prior therapies have all correlated with outcomes [31, 68–70]. Thus, often expensive and time consuming tests must add value to those generated by clinical factors. The value of clinical factors, however, is often in patients with extremes of presentations (e.g., extremely bulky liver and/or brain metastases compared with patients with very low volume lung metastases), and the great majority of patients with more “intermediate” presentations are unable to be effectively stratified simply with clinical factors. Thus, the role of biomarkers will likely continue to increase in the era of immunotherapy, both in melanoma and in other cancers.
Figure 1.
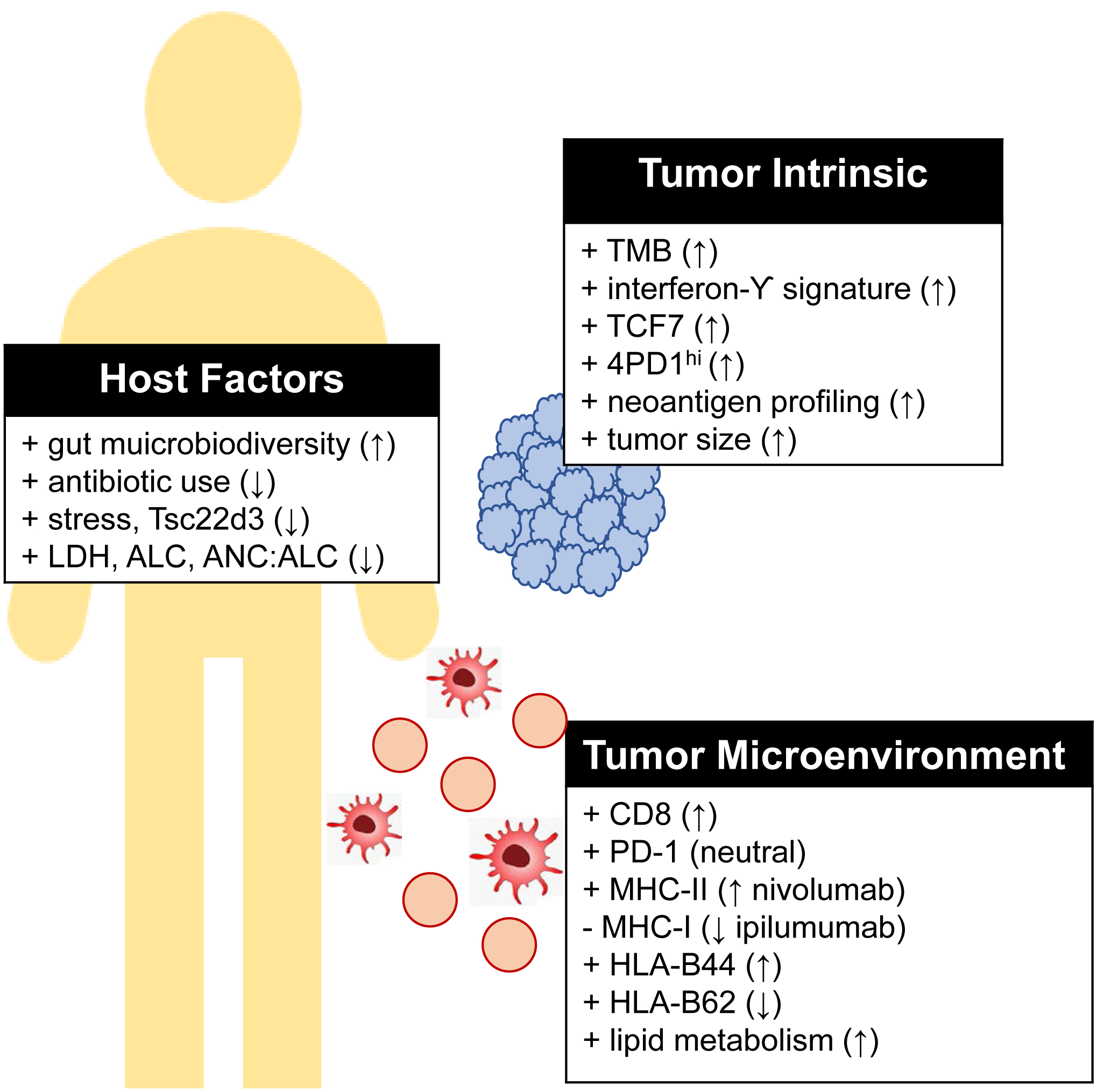
Predictive biomarkers of melanoma response to checkpoint inhibition.
↑ indicates evidence supporting increased response to checkpoint blockade, ↓ indicates decreased response to checkpoint blockade.
Article highlights:
Immune checkpoint inhibition has dramatically improved outcomes for patients with advanced melanoma, but biomarkers predictive of clinical response are still lacking
Given the complex mechanism of ICIs, biomarkers of advanced melanoma response to treatment go beyond the tumor-intrinsic properties
Other biomarkers that have been explored include assay of the tumor-immune microenvironment as well as broader host-specific factors
While it is strongly associated with response to ICI in other tumor types, PD-L1 expression in melanoma is weakly correlated at best
More research is needed to develop clinically meaningful biomarkers predictive of response to ICI in melanoma
Further work is also needed to develop widely-applicable biomarkers of irAE
Funding
This paper was supported in part by NIH/NCI K23 CA204726 (DBJ), James C. Bradford Jr. melanoma fund (DBJ), NIH/NCI T32 CA217834-01A1 (CAN), Vanderbilt-Ingram Cancer Center Brock Family Fellowship (CAN).
Declaration of interest
Douglas B Johnson has served on the Advisory boards for Array Biopharma, BMS, Genoptix, Incyte, Novartis, Merck, and has received research support from BMS. Douglas B Johnson also has a patent pending on employing MHC-II as a biomarker of response to immune therapy. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
REFERENCES
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2018. CA Cancer J Clin, 2018. 68(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, N. A, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds), SEER Cancer Statistics Review, 1975–2016, in National Cancer Institute. [Google Scholar]
- 3.Kohler BA, et al. , Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst, 2011. 103(9): p. 714–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CS, Thomas CM, and Ng KE, An Overview of the Changing Landscape of Treatment for Advanced Melanoma. Pharmacotherapy, 2017. 37(3): p. 319–333. [DOI] [PubMed] [Google Scholar]
- 5.Atkins MB, et al. , High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol, 1999. 17(7): p. 2105–16. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DB and Sosman JA, Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol, 2015. 1(3): p. 380–6. [DOI] [PubMed] [Google Scholar]
- 7.Domingues B, et al. , Melanoma treatment in review. Immunotargets Ther, 2018. 7: p. 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodis E, et al. , A landscape of driver mutations in melanoma. Cell, 2012. 150(2): p. 251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, et al. , Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med, 2010. 363(8): p. 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robert C, et al. , Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med, 2015. 372(4): p. 320–30. [DOI] [PubMed] [Google Scholar]
- 11.Topalian SL, et al. , Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol, 2014. 32(10): p. 1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber J, et al. , Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N Engl J Med, 2017. 377(19): p. 1824–1835. [DOI] [PubMed] [Google Scholar]
- 13.Hamid O, et al. , Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol, 2019. 30(4): p. 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamid O, et al. , Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med, 2013. 369(2): p. 134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert C, et al. , Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med, 2015. 372(26): p. 2521–32. [DOI] [PubMed] [Google Scholar]
- 16.Oldenhuis CN, et al. , Prognostic versus predictive value of biomarkers in oncology. Eur J Cancer, 2008. 44(7): p. 946–53. [DOI] [PubMed] [Google Scholar]
- 17.Garralda E, et al. , New clinical trial designs in the era of precision medicine. Mol Oncol, 2019. 13(3): p. 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coit DG, et al. , Cutaneous Melanoma, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw, 2019. 17(4): p. 367–402. [DOI] [PubMed] [Google Scholar]
- 19.Chalmers ZR, et al. , Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med, 2017. 9(1): p. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yarchoan M, Hopkins A, and Jaffee EM, Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med, 2017. 377(25): p. 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samstein RM, et al. , Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet, 2019. 51(2): p. 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgiadis A, et al. , Noninvasive Detection of Microsatellite Instability and High Tumor Mutation Burden in Cancer Patients Treated with PD-1 Blockade. Clin Cancer Res, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miao D, et al. , Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet, 2018. 50(9): p. 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson DB, et al. , Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol Res, 2016. 4(11): p. 959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.*.Snyder A, et al. , Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med, 2014. 371(23): p. 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]; o This article elegantly elucidates the tumor-intrinsic property of TMB as necessary but not specific to predict cinical benefit from CTLA-4 inhibition in advanced melanoma, and further identifies a predictive neopitode group with homology to known viral epitope, but this neoantigen pattern did not hold predictive power in an expanded cohort.
- 26.Rizvi NA, et al. , Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 2015. 348(6230): p. 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellmann MD, et al. , Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell, 2019. 35(2): p. 329. [DOI] [PubMed] [Google Scholar]
- 28.Hellmann MD, et al. , Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med, 2018. 378(22): p. 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le DT, et al. , Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 2017. 357(6349): p. 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson DB, et al. , Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res, 2015. 3(3): p. 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spranger S, Bao R, and Gajewski TF, Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature, 2015. 523(7559): p. 231–5. [DOI] [PubMed] [Google Scholar]
- 32.Casey SC, et al. , MYC regulates the antitumor immune response through CD47 and PD-L1. Science, 2016. 352(6282): p. 227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng W, et al. , Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov, 2016. 6(2): p. 202–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koyama S, et al. , STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res, 2016. 76(5): p. 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Rooij N, et al. , Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol, 2013. 31(32): p. e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Allen EM, et al. , Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science, 2015. 350(6257): p. 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harel M, et al. , Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell, 2019. 179(1): p. 236–250 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph RW, et al. , Baseline Tumor Size Is an Independent Prognostic Factor for Overall Survival in Patients with Melanoma Treated with Pembrolizumab. Clin Cancer Res, 2018. 24(20): p. 4960–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis EJ, et al. , Clinical Correlates of Response to Anti-PD-1-based Therapy in Patients With Metastatic Melanoma. J Immunother, 2019. 42(6): p. 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel SP and Kurzrock R, PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther, 2015. 14(4): p. 847–56. [DOI] [PubMed] [Google Scholar]
- 41.Sanlorenzo M, et al. , BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti-PD-1 Antibody. Clin Cancer Res, 2018. 24(14): p. 3377–3385. [DOI] [PubMed] [Google Scholar]
- 42.*.Larkin J Hodi FS, and Wolchok JD, Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med, 2015. 373(13): p. 1270–1. [DOI] [PubMed] [Google Scholar]; o This landmark trial demonstrated that PD-L1 expression is a weak biomarker in melanoma, with lower but substantial response rates among patients treated with mono- or combination checkpoint blockade with negative PD-L1 expression
- 43.Chen PL, et al. , Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov, 2016. 6(8): p. 827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daud AI, et al. , Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest, 2016. 126(9): p. 3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tumeh PC, et al. , PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 2014. 515(7528): p. 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayers M, et al. , IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest, 2017. 127(8): p. 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avogadri F, et al. , Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol Res, 2014. 2(5): p. 448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zappasodi R, et al. , Non-conventional Inhibitory CD4(+)Foxp3(−)PD-1(hi) T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell, 2018. 33(6): p. 1017–1032 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson DB, et al. , Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of Anti-PD-1 Therapies in Metastatic Melanoma. Clin Cancer Res, 2018. 24(21): p. 5250–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson DB, et al. , Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun, 2016. 7: p. 10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson DB, et al. , Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight, 2018. 3(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodig SJ, et al. , MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med, 2018. 10(450). [DOI] [PubMed] [Google Scholar]
- 53.Chowell D, et al. , Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science, 2018. 359(6375): p. 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zitvogel L, et al. , The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science, 2018. 359(6382): p. 1366–1370. [DOI] [PubMed] [Google Scholar]
- 55.*.Gopalakrishnan V, et al. , Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science, 2018. 359(6371): p. 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]; o This article highlights the host-specific gut microbiome as a predictor of response to PD-1 inhibition, highlighting the importance of symbiotic relationships between host, tumor and bacterial microbiome.
- 56.Matson V, et al. , The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science, 2018. 359(6371): p. 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinato DJ, et al. , Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang H, et al. , Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med, 2019. 25(9): p. 1428–1441. [DOI] [PubMed] [Google Scholar]
- 59.Ferrucci PF, et al. , Baseline neutrophils and derived neutrophil-to-lymphocyte ratio: prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann Oncol, 2016. 27(4): p. 732–8. [DOI] [PubMed] [Google Scholar]
- 60.Rosner S, et al. , Peripheral blood clinical laboratory variables associated with outcomes following combination nivolumab and ipilimumab immunotherapy in melanoma. Cancer Med, 2018. 7(3): p. 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martens A, et al. , Increases in Absolute Lymphocytes and Circulating CD4+ and CD8+ T Cells Are Associated with Positive Clinical Outcome of Melanoma Patients Treated with Ipilimumab. Clin Cancer Res, 2016. 22(19): p. 4848–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson DB, et al. , A case report of clonal EBV-like memory CD4(+) T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat Med, 2019. 25(8): p. 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang DY, et al. , Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol, 2018. 4(12): p. 1721–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boutros C, et al. , Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol, 2016. 13(8): p. 473–86. [DOI] [PubMed] [Google Scholar]
- 65.Das R, et al. , Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest, 2018. 128(2): p. 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tarhini AA, et al. , Baseline circulating IL-17 predicts toxicity while TGF-beta1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer, 2015. 3: p. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanaka R, et al. , Serum level of interleukin-6 is increased in nivolumab-associated psoriasiform dermatitis and tumor necrosis factor-alpha is a biomarker of nivolumab recativity. J Dermatol Sci, 2017. 86(1): p. 71–73. [DOI] [PubMed] [Google Scholar]
- 68.Lim SY, et al. , Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-Based Immunotherapy. Clin Cancer Res, 2019. 25(5): p. 1557–1563. [DOI] [PubMed] [Google Scholar]
- 69.Kimbara S, et al. , Association of antithyroglobulin antibodies with the development of thyroid dysfunction induced by nivolumab. Cancer Sci, 2018. 109(11): p. 3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stamatouli AM, et al. , Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes, 2018. 67(8): p. 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robert C, et al. , Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med, 2019. 381(7): p. 626–636. [DOI] [PubMed] [Google Scholar]
- 72.Johnson DB, et al. , Sequencing Treatment in BRAFV600 Mutant Melanoma: Anti-PD-1 Before and After BRAF Inhibition. J Immunother, 2017. 40(1): p. 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]